
Transgenerational Interaction of Alzheimer’s Disease with Schizophrenia through Amyloid Evolvability
Abstract
Alzheimer’s disease (AD), the most common neurodegenerative dementia, leads to memory dysfunction due to widespread neuronal loss associated with aggregation of amyloidogenic proteins (APs), while schizophrenia (SCZ) represents a major psychiatric disorder characterized by delusions, hallucinations, and other cognitive abnormalities, the underlying mechanisms of which remain obscure. Although AD and SCZ partially overlap in terms of psychiatric symptoms and some aspects of cognitive impairment, the causal relationship between AD and SCZ is unclear. Based on the similarity of APs with yeast prion in terms of stress-induced protein aggregation, we recently proposed that evolvability of APs might be an epigenetic phenomenon to transmit stress information of parental brain to cope with the stressors in offspring. Although amyloid evolvability may be beneficial in evolution, AD might be manifested during parental aging as the mechanism of antagonistic pleiotropy phenomenon. Provided that accumulating evidence implicates stress as an important factor in SCZ, the main objective of this paper is to better understand the possible connection of AD and SCZ through amyloid evolvability. Hypothetically, the delivery of information of stress by APs may be less efficient under the decreased evolvability conditions such as disease-modifying treatment, leading to SCZ in offspring. Conversely, the increased evolvability conditions including gene mutations of APs are supposed to be beneficial for offspring, but might lead to AD in parents. Collectively, AD and SCZ might transgenerationally interfere with each other through amyloid evolvability, and this could explain why both AD and SCZ have not been selected out through evolution.
INTRODUCTION
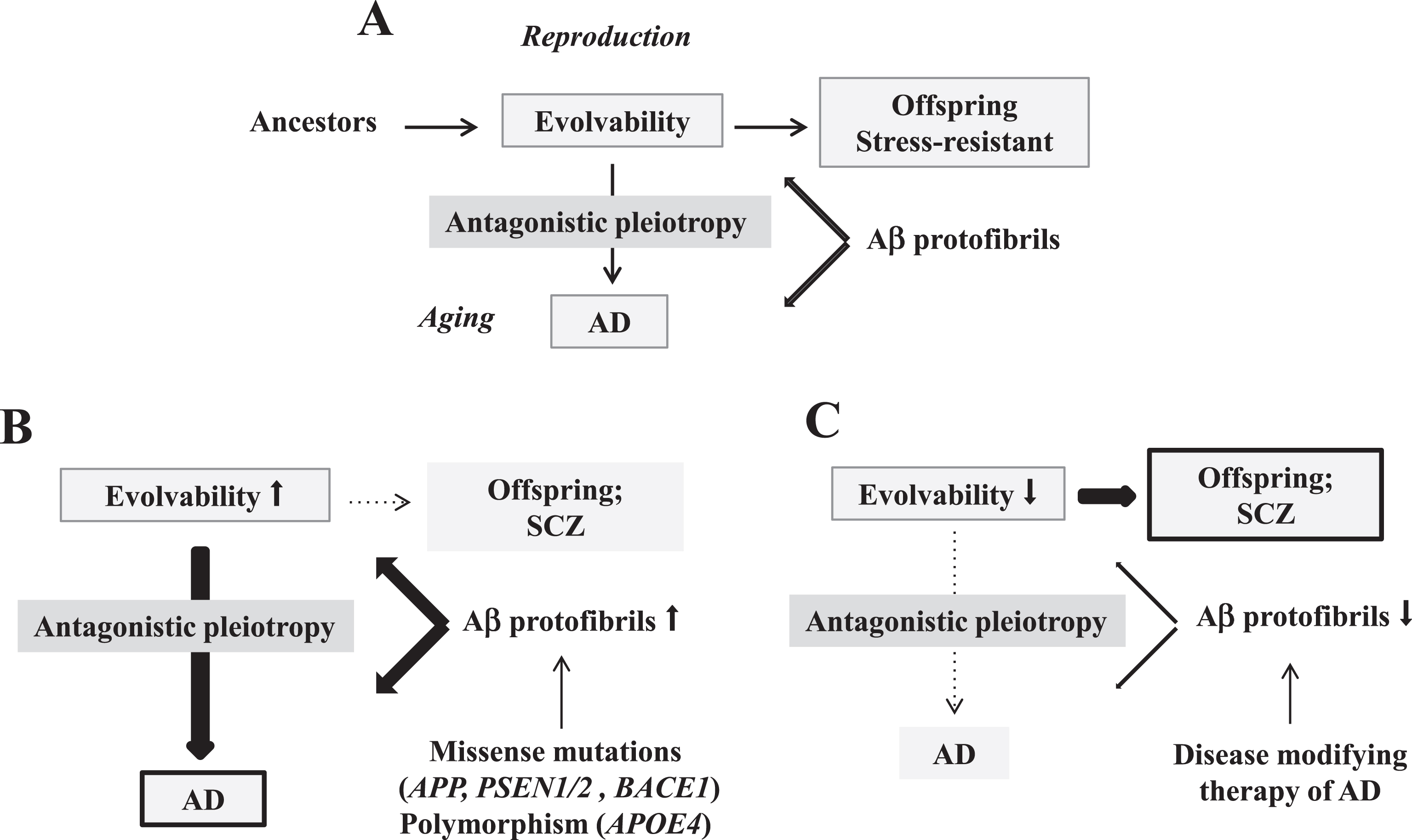
At present, Alzheimer’s disease (AD) remains the most common form of neurodegenerative dementia, demonstrating progressive memory loss and cognitive and functional decline [1]. It most often occurs in those over the age of 65 years, although ∼5% of all cases are due to early-onset familial mutations [2]. Although an understanding of neurodegenerative disease mechanisms has progressed remarkably, the physiological functions of amyloidogenic proteins (APs), including amyloid-β (Aβ) and α-synuclein (αS), are still elusive [3]. In this regard, based on the structural diversity of AP aggregates, we recently proposed that evolvability of APs could be defined as an epigenetic phenomenon transgenerationally transmitted to offspring to cope with the forthcoming diverse stressors in the offspring’s brain (Fig. 1A) [4]. Although evolvability may be beneficial for offspring during reproductive life, it manifests during aging as neurodegeneration, possibly through an antagonistic pleiotropy mechanism (Fig. 1A) [5]. It remains obscure, however, why disorders such as AD have escaped natural selection during postmenopausal aging.
Fig.1
Schematic of disease manifestation caused by alterations in evolvability. A) Evolvability of APs might be an epigenetic phenomenon transmitted transgenerationally to confer resistance against the stressors such as Aβ protofibrils in offspring during reproduction, which may be beneficial in evolution. However, evolvability might lead to AD during parental aging through the antagonistic pleiotropy mechanism. B) Increased evolvability of Aβ caused by missense mutations (APP, PSEN1/2, BACE1) and polymorphism (APOE4) may result in an efficient delivery of information of stresses for offspring, leading to less frequency of SCZ in offspring (thin dot line) and increased frequency of AD might in parents (thick bold line). C) In contrast, inefficient delivery of the information of stresses due to decrease of Aβ evolvability caused by disease-modifying therapy may result in the increased frequency of SCZ in offspring (thick bold line) and the decreased frequency of AD in parents (thin dot line).
On the other hand, the neuropathological mechanisms underlying schizophrenia (SCZ), a mental disorder characterized by delusions, hallucinations, and other cognitive difficulties, remains obscure [6]. Given that SCZ typically occurs between late adolescence and the early twenties, which overlaps with the period of human reproduction and rearing of children, and because it can often be a lifelong disabling condition, SCZ may be considered evolutionarily disadvantageous [7]. Nonetheless, SCZ has not been selected out by evolution, and moreover, epidemiologically, the global prevalence of SCZ is approximately 0.2∼0.5% of the population [8]. Therefore, the biological reasons as to why SCZ has not been selected out by evolution, have, to the best of our knowledge, never been adequately explained.
Since AD and SCZ often overlap in terms of various psychiatric symptoms and, to a lesser extent, in cognitive dysfunction, a similar pathogenic mechanism might be a common basis for these conditions. Given that recent study suggests that stress may play an important role in the pathogenesis of SCZ [9, 10], the main objective of this paper is to discuss the possible link of AD and SCZ through evolvability. Specifically, we hypothesize that the effects of evolvability may be differential between AD and SCZ. According to the prevailing view, Aβ burden may be not associated with cognitive impairment in SCZ [11, 12]. However, it was indeed shown that mice with a deletion of Disrupted-in-Schizophrenia-1 (DISC1), a familial risk factor of SCZ [13], exhibited decreased expression of Aβ [14]. Furthermore, we suggest that AD and SCZ may interfere with each other through evolvability during evolution. Finally, we predict that our hypothesis might provide a unique insight into therapeutic strategies for these conditions. Collectively, this framework could explain why both AD and SCZ, over evolutionary time, have persisted despite the pressures of natural selection.
CURRENT VIEW OF THE RELATIONSHIP BETWEEN AD AND SCZ
Although AD and SCZ often overlap in terms of altered behaviors, including psychiatric symptoms and cognitive impairment, the causal relationship between AD and SCZ has been yet to be clarified [15, 16].
Psychiatric symptom
Indeed, the psychiatric symptoms, such as delusions and hallucinations, represent mere symptoms that are not specific to AD and SCZ, but can be found in most neurodegenerative diseases, including Parkinson’s and Huntington’s diseases [17, 18]. Furthermore, these symptoms are relatively rare in AD, whereas are pathognomotic in SCZ.
Cognitive decline
AD is the main cause of dementia among the elderly, and severe cognitive impairment is also observed in elderly patients with SCZ [19]. Whereas cognitive decline represents the first and the pathognomotic symptomatology required for diagnosis of AD, cognitive decline does not represent the first nor the pathognomotic symptomatology required for diagnosis of SCZ [20]. Thus, it is important to recognize that the pattern of cognitive decline differs significantly.
In this context, aggregation and accumulation of Aβ are early events that precede the onset of cognitive impairment in AD [21], whereas the prevailing view is that Aβ burden may be not associated with cognitive impairment in SCZ [11, 12]. In support of the latter view, there was no evidence suggesting that Aβ levels differ in patients with SCZ from elderly control subjects or patients with other psychiatric illnesses [11], while the levels of cortical Aβ in patients with SCZ were lower than those in patients with AD (Fig. 2A). Furthermore, it was recently shown that there were no associations between Aβ and cognitive impairment in SCZ [12]. Given that the previous studies were performed using small sample sizes, and Aβ was assessed in cerebrospinal fluid (CSF) rather than cortical region in brain, additional analyses of different brain regions may be required [12].
Fig.2
Alteration of Aβ expression in SCZ. A) Mean postmortem levels of total Aβ, Aβx–40, and Aβx–42 in the dorsolateral prefrontal cortex of normal elderly comparison subjects, patients with AD, patients with SCZ only, and patients with SCZ plus mild AD pathology. aSignificantly different from all other groups (Newman-Keuls tests, p < 0.01). bSignificantly different from all other groups (Newman-Keuls tests, p < 0.001). Reprinted from Religa et al. [11] with permissions. B) AβPP processing is dysregulated in DISC1 locus mutants of 3XTg mice. Loss of DISC1 in homozygous DISC1 locus impairment (– /–) mice leads to alteration of AβPP processing, compared to wild-type (+/+) controls. a–c) Analysis of cortical lysates by ELISA shows increased sAβPPα and decreased Aβ42 and Aβ40 levels in – /– mice. Bars represent the mean±SEM, n = 5–6 mice per group. **p < 0.01, ***p < 0.001. d) Neurons from – /– mice display higher levels of surface AβPP than those from+/+mice. Scale bar, 10μm. Reprinted from Shahani et al. [14] with permission.
![Alteration of Aβ expression in SCZ. A) Mean postmortem levels of total Aβ, Aβx–40, and Aβx–42 in the dorsolateral prefrontal cortex of normal elderly comparison subjects, patients with AD, patients with SCZ only, and patients with SCZ plus mild AD pathology. aSignificantly different from all other groups (Newman-Keuls tests, p < 0.01). bSignificantly different from all other groups (Newman-Keuls tests, p < 0.001). Reprinted from Religa et al. [11] with permissions. B) AβPP processing is dysregulated in DISC1 locus mutants of 3XTg mice. Loss of DISC1 in homozygous DISC1 locus impairment (– /–) mice leads to alteration of AβPP processing, compared to wild-type (+/+) controls. a–c) Analysis of cortical lysates by ELISA shows increased sAβPPα and decreased Aβ42 and Aβ40 levels in – /– mice. Bars represent the mean±SEM, n = 5–6 mice per group. **p < 0.01, ***p < 0.001. d) Neurons from – /– mice display higher levels of surface AβPP than those from+/+mice. Scale bar, 10μm. Reprinted from Shahani et al. [14] with permission.](https://content.iospress.com:443/media/jad/2019/68-2/jad-68-2-jad180986/jad-68-jad180986-g002.jpg)
Notably, a limited number of studies showed that levels of CSF Aβ1 - 42 in elderly SCZ patients were significantly lower compared to those in healthy elders but higher than those in AD patients [22, 23]. Consistent with these results, it was shown that Aβ1 - 42 and, to a lesser extent, Aβ1 - 40, were reduced in the AD model mice deleted with DISC1 (Fig. 2B), suggesting that the processing of the amyloid-β protein precursor might be altered in SCZ [13, 14]. Further investigations are warranted to explore this intriguing possibility.
EMERGENCE OF CLINICAL DISEASE DUE TO ALTERED EVOLVABILITY
Accumulating evidence suggests that APs, such as Aβ and αS, exhibit a diversity of strains due to structural variation [24–26]. Given that APs are intrinsically disordered protein [27], it is predicted that the structural diversity of APs might correspond to diverse stresses. Furthermore, we speculate that unstable monomer of APs becomes more stable through oligomerization which are feasible for the transgenerational transmission to offspring [4]. Because the evolvability of APs may be attributed to their protofibrillar forms [4], it is assumed that an increased level of the Aβ protofibrils may result, during reproduction, in increased transmission of information regarding brain stressors from parent to offspring, and in turn, promote resistance against neuronal stressors and reduce the occurrence of SCZ in offspring (Fig. 1B). However, it is also probable that the accumulation of Aβ protofibrils in parental brain may also lead to AD and neurodegeneration through the mechanism of antagonistic pleiotropy during aging (Fig. 1B) [5]. This scenario would be predicted based on familial early-onset AD with missense mutations, including amyloid precursor protein (APP), presenilin (PSEN) 1/2, and β-site APP cleaving enzyme (BACE) 1 [28–30], and for polymorphism of APOE4 [31].
What then might be the consequence of reduced evolvability of Aβ during reproduction? Because reduced information on forthcoming stressors from parental brain is available to be delivered to offspring, an increased vulnerability to stressors may lead to neuronal toxicity, dysfunction and perhaps a pathologic phenotype in offspring. On the other hand, such reduced action of evolvability might inversely reduce the risk of AD and neurodegeneration (Fig. 1C). Such inverse relationships could also apply to a number of developmental brain disorders, including autism spectrum disorder, epilepsy, and psychiatric diseases, such as SCZ, whose precise etiologies are yet to be determined [32]. Notably, it was previously shown that mice with knockdown/deletion of DISC1, a genetic risk factor for a wide range of mental disorders, including SCZ, major depression, and bipolar disorders [13], exhibited altered processing of AβPP and reduced Aβ production in mouse brain (Fig. 2) [14], consistent with our concept that evolvability through Aβ protofibrils might be decreased in the setting of SCZ.
Interaction of parental AD and SCZ in offspring through evolvability
Our hypothesis suggests that the presence of AD in aging parents might transgenerationally interact with the occurrence of SCZ in their offspring (Fig. 3). Given that APs are intrinsically disordered protein [27], it is predicted that the structural diversity of APs might correspond to diverse stresses. Furthermore, it is speculated that unstable monomer of APs becomes more stable through oligomerization which are feasible for the transgenerational transmission to offspring [4].
Fig.3
Schematic representation of the reciprocal transgenerational relationship between AD and SCZ through evolvability. Given that transmission of APs may confer the information on stressors from parental brain, resulting in suppression of SCZ, evolvability of the APs protofibrils would be beneficial for offspring. Therefore, AD would be inevitable in which the minimum of evolvability is required to reduce the risk of SCZ in offspring (Emin). The activity of evolvability may gradually increase, but AD is not manifested earlier before the postmenopausal period because of the law of natural selection. Accordingly, it is predicted that evolvability has a maximal point (Emax) in which the incidence of SCZ is compromised. Currently, the epidemiological data suggest the risk of SCZ is estimated to be 0.2∼0.5% [33], while the risk of AD is approximately ∼20% (Ecurr) [34]. It is suggested that the activity of the evolvability derived from familial AD should be higher than that derived from sporadic AD, being closer to Emax (Efad), and foretells that the incidence of SCZ among familial AD should be below the average. In contrast, the activity of evolvability might be decreased in AD patients treated with disease-modifying therapy (Edmt), in which the risk of SCZ in offspring might be increased. Thus, it is theoretically predicted that parental AD and SCZ in offspring might exist in a reciprocal relationship through evolvability. •: Current average of sporadic AD, ∘: familial AD, □: AD patients treated with disease-modifying therapy.
![Schematic representation of the reciprocal transgenerational relationship between AD and SCZ through evolvability. Given that transmission of APs may confer the information on stressors from parental brain, resulting in suppression of SCZ, evolvability of the APs protofibrils would be beneficial for offspring. Therefore, AD would be inevitable in which the minimum of evolvability is required to reduce the risk of SCZ in offspring (Emin). The activity of evolvability may gradually increase, but AD is not manifested earlier before the postmenopausal period because of the law of natural selection. Accordingly, it is predicted that evolvability has a maximal point (Emax) in which the incidence of SCZ is compromised. Currently, the epidemiological data suggest the risk of SCZ is estimated to be 0.2∼0.5% [33], while the risk of AD is approximately ∼20% (Ecurr) [34]. It is suggested that the activity of the evolvability derived from familial AD should be higher than that derived from sporadic AD, being closer to Emax (Efad), and foretells that the incidence of SCZ among familial AD should be below the average. In contrast, the activity of evolvability might be decreased in AD patients treated with disease-modifying therapy (Edmt), in which the risk of SCZ in offspring might be increased. Thus, it is theoretically predicted that parental AD and SCZ in offspring might exist in a reciprocal relationship through evolvability. •: Current average of sporadic AD, ∘: familial AD, □: AD patients treated with disease-modifying therapy.](https://content.iospress.com:443/media/jad/2019/68-2/jad-68-2-jad180986/jad-68-jad180986-g003.jpg)
Because the transmission of APs may confer information regarding stressors from parental brain, evolvability of AP protofibrils should be beneficial for offspring. Therefore, AD should be inevitable at the point at which minimum evolvability is required to reduce the risk of SCZ in offspring (Emin). Since evolvability may be beneficial for offspring, it gradually increases until incident AD occurs increasingly early, prior to the postmenopausal period in humans, which then would interfere with reproduction and contravene the law of natural selection. Accordingly, evolvability should also demonstrate a maximal point (Emax) in which SCZ incidence is markedly reduced to a low level, but does not reach zero. Based on the epidemiological data in which the risk of SCZ currently is estimated to be ∼0.5% [33], while that of AD is approximately ∼20% [34], the activity of evolvability corresponding to sporadic AD may be situated between Emin and Emax (Espor). Extrapolation of this concept would predict that the level of evolvability activity derived from familial AD (Efad) due to missense gene mutations such as APP, PSEN1/2, and BACE1, should be higher than Espor and closer to Emax. At the point of Efad, the incidence of SCZ in the familial AD cohort is predicted to be below the observed average. In contrast, as described later, the activity of evolvability might be reduced if AD were treated with an effective disease-modifying therapy at the prodromal stage (Ethe), where the risk of SCZ in offspring might be increased. In other words, theoretically, parental AD and SCZ in their offspring might exist in a relative inverse relationship through evolvability. Thus, regulated or constrained activity of evolvability might account for the observed incidences of AD and SCZ, as well as their resistance against the constant pressures of evolution. It follows that factors influencing the activity of evolvability, which have been discussed previously but remain unclear, may be important in common disease pathogenesis of AD and SCZ [4].
Although SCZ is clinically heterogeneous [35], the precise mechanism remains poorly understood. In this regard, different SCZ subtypes might be in part associated with other APs and their evolvability states. Certainly, it is well known that other forms of aging-associated neurodegenerative diseases, aside from AD, are accompanied by psychiatric behaviors. Naturally, it is predicted that SCZ may be similarly affected by the evolvability of these APs. For instance, α-synucleinopthies, such as Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy, are characterized by motor-and non-motor symptoms which may be derived from evolvability of αS. Because neurodegenerative disorders such as AD and α-synucleinopthies are manifested during postmenopausal senescence, which is a human-specific phenomenon by virtue of the absence of natural predators and abundant nutritional security [5], it is plausible that transgenerational relationships between parental neurodegenerative disease and SCZ in offspring has emerged relatively recently in evolutionary history.
However, a unique situation with regards to AP evolvability may exist for Huntington’s disease and other polyglutamine (polyQ) diseases. Since disease onset appears prior to menopause, it is possible that evolvability of polyQ might be less potent than those of other APs, such as Aβ and αS. Indeed, the psychiatric symptoms associated with polyQ diseases, often paranoia, have less resemblance to SCZ and are perhaps less prominent compared to those in other neurodegenerative conditions, including AD and α-synucleinopthies [36]. Based on the expression of polyQ in microorganisms and plants [37, 38], it is predicted that polyQ may be more primitive relative to other APs across evolution, and might play an important role for the evolution of amyloidgenic evolvability [39]. Thus, it is tempting to speculate that the old prototype of SCZ could be observed in the polyQ evolvability.
Indeed, biological evidence exists which might support this view. For instance, various senescent features are rapidly induced in the spawning salmon, including brain amyloid deposition, which has been known as the “salmon model of AD” [40]. In this situation, the demise of parents due to AD may in turn be beneficial for offspring by ensuring availability of nutrition. Furthermore, in “the grandmother effect,” the rearing of grandchildren by a grandmother experiencing post-menopausal aging may benefit their daughter(s) who can birth further children [41], suggesting that aging can evolve as beneficial for offspring. Although aberrant behaviors in AD are clearly not beneficial for offspring, it is probable that the pathology of AD might be beneficial for offspring indirectly through evolvability. Indeed, such a novel concept may be worthy of further investigation.
THERAPEUTIC IMPLICATIONS
If this theoretical framework proves correct, then a critical issue will be the reduced evolvability of APs connected to the treatment of neurodegenerative disorders with disease-modifying therapies during prodromal disease stages. Regarding the progress of disease-modifying therapies for AD, contrary to the positive preclinical findings, the results of active amyloid immunotherapy for this condition in early-stage clinical trials have been disappointing and accompanied by serious complications, such as meningoencephalitis [42]. Subsequent focus on passive AD immunotherapy using anti-Aβ monoclonal antibodies has also not fully demonstrated to be therapeutically effective [3]. The phase III clinical trial (NCT02008357; patients of 55–90 years old), for instance, failed to demonstrate reproducible efficacy [43], and, on a cautious note, it was recently reported that an ongoing early phase II trial using the anti-protofibrillar Aβ antibody, BAN2401 by Eisai, Inc./Biogen, Inc. in mild cognitive impairment and early AD patients, demonstrated non-futility, but little cognitive improvement at 18 months [44]. Still, the need exists for even earlier intervention in the disease course [3]. As such, secondary prevention trials such as Dominantly inherited Alzheimer Network-Trial Unit (DIAN-TU) (NCT01760005; patients of 18 years and older) have already started to assess the potential of new protocols [45, 46] to prevent the disease progression in autosomal-dominant AD individuals without cognitive dysfunction and in cognitively healthy subjects at risk of developing sporadic AD [45].
Again, a key point is that if intervention using disease-modifying therapies during prodromal AD becomes a reality, such treatment may alter the pace of evolvability during the reproductive time of life. It follows that passive immunotherapy using anti-Aβ monoclonal antibodies could suppress evolvability, leading to the compromised resistance against stressors in the brains of offspring. Thus, it is imperative that such potential risks of therapeutic intervention must be considered to establish working disease-modifying therapy in AD.
The current hypothesis also provides an important insight into potential therapeutic strategies for SCZ. Despite significant advances in SCZ pharmacotherapy [47], treatment remains suboptimal, with many patients having persisting deficits, especially in cognitive and social functioning [48, 49]. Given the differential effects of evolvability on AD and SCZ, one might propose that stimulating the evolvability of relevant APs might significantly reverse such evolvability deficits, improving clinical efficacy for patients. To achieve this, we propose the up-regulation of APs, either directly or indirectly, as a therapeutic intervention. As for direct upregulation, Aβ or its derivative could be administered, although side effects such as meningoencephalitis may be a limiting factor based on the previous complications of active Aβ immunotherapy in AD [42]. Alternatively, augmenting the synthesis and/or secretion of Aβ might be more effective, where a DISC1 knockout mouse model may be an ideal tool to achieve this. A major drawback, however, to using the DISC1 knockout mouse model to demonstrate the above, would be the markedly truncated postmenopausal senescent period in rodents relative to humans [5]. Furthermore, caution must be paid to the possibility that AD risk might be increased in offspring in the future in either case. Taking consideration, however, that protein degradation systems, such as autophagy and ubiquitin proteasome system, are more robust in younger individuals compared to the elderly [50], further exploration of these interesting possibilities is warranted.
CONCLUSION
Currently, no definite conclusions have yet been reached as to the causal relationship between AD and SCZ. As described above, our concept of evolvability may provide one plausible explanation for this. To summarize it parental AD and SCZ in offspring may transgenerationally interact with each other, which may explain why AD and SCZ have not been selected out through evolutionary natural selection. Our current hypothesis, although fascinating, requires a demonstration of the protofibrils of APs being involved in evolvability.
Recently, a number of gene-based studies in SCZ, such as genome-wide association studies, and the mapping of several regions have extensively been performed and have led to the identification of a number of putative susceptibility genes, including DISC1, neuregulin-1, dysbindin, and catechol-o-methyltransferase [51–54]. In particular, neuregulin-1 has been identified as a β-secretase substrate, suggesting a link between SCZ and AD [55]. At present, however, no single gene has been demonstrated to be sufficient to cause the SCZ phenotype by itself emphasizing the multifactorial nature of this condition. Although still controversial, the current dominant view of SCZ falls under the ‘common disease-common variant’ hypothesis, suggesting that the genetic component to most common disorders is due to a relatively large number of disease-causing alleles that occur relatively often in the population [56–58]. It is particularly interesting to note that AD with psychosis is associated with polygenic risk for a set of novel loci related to endosomal trafficking, autophagy, and calcium channel signaling, which is inversely associated with polygenic risk for SCZ [59]. Together with the current hypothesis, we suggest that both genetic and epigenetic mechanisms may cooperate in the pathogenesis of SCZ. Thus, a better understanding of the interaction of AD in parents and SCZ in offspring may potentially shed light on novel therapies for both AD and SCZ.
ACKNOWLEDGMENTS
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (University of Chicago).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0986r1).
REFERENCES
[1] | Masliah E ((1995) ) Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol 10: , 509–519. |
[2] | Blennow K , de Leon MJ , Zetterberg H ((2006) ) Alzheimer’s disease. Lancet 368: , 387–403. |
[3] | Takamatsu Y, Ho G, Koike W , Sugama S , Takenouchi T , Waragai M , Wei J , Sekiyama K , Hashimoto M ((2017) ) Combined immunotherapy with "anti-insulin resistance" therapy as a novel therapeutic strategy against neurodegenerative diseases. NPJ Parkinsons Dis 3: , 4. |
[4] | Hashimoto M , Ho G , Sugama S , Takamatsu Y , Shimizu Y , Takenouchi T , Waragai M , Masliah E ((2018) ) Evolvability of amyloidogenic proteins in human brain. J Alzheimers Dis 62: , 73–83. |
[5] | Hashimoto M , Ho G , Takamatsu Y , Shimizu Y , Sugama S , Takenouchi T , Waragai M , Masliah E ((2018) ) Evolvability and neurodegenerative disease: Antagonistic pleiotropy phenomena derived from amyloid aggregates. J Parkinsons Dis 8: , 405–408. |
[6] | Schultz SK , Andreasen NC ((1999) ) Schizophrenia. Lancet 353: , 1425–1430. |
[7] | Polimeni J , Reiss JP ((2003) ) Evolutionary perspectives on schizophrenia. Can J Psychiatry 48: , 34–39. |
[8] | Jablensky A ((2000) ) Epidemiology of schizophrenia: The global burden of disease and disability. Eur Arch Psychiatry Clin Neurosci 250: , 274–285. |
[9] | Itokawa M , Miyashita M , Arai M , Miyata T ((2014) ) Carbonyl stress in schizophrenia. Biochem Soc Trans 42: , 468–472. |
[10] | Emiliani FE , Sedlak TW , Sawa A ((2014) ) Oxidative stress and schizophrenia: Recent breakthroughs from an old story. Curr Opin Psychiatry 27: , 185–190. |
[11] | Religa D , Laudon H , Styczynska M , Winblad B , Naslund J , Haroutunian V ((2003) ) Amyloid beta pathology in Alzheimer’s disease and schizophrenia. Am J Psychiatry 160: , 867–872. |
[12] | Chung JK , Nakajima S , Plitman E , Iwata Y , Uy D , Gerretsen P , Caravaggio F , Chakravarty MM , Graff-Guerrero A ((2016) ) Beta-amyloid burden is not associated with cognitive impairment in schizophrenia: A systematic review. Am J Geriatr Psychiatry 24: , 923–939. |
[13] | Millar JK , Wilson-Annan JC , Anderson S , Christie S , Taylor MS , Semple CA , Devon RS , St Clair DM , Muir WJ , Blackwood DH , Porteous DJ ((2000) ) Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet 9: , 1415–1423. |
[14] | Shahani N , Seshadri S , Jaaro-Peled H , Ishizuka K , Hirota-Tsuyada Y , Wang Q , Koga M , Sedlak TW , Korth C , Brandon NJ , Kamiya A , Subramaniam S , Tomoda T , Sawa A ((2015) ) DISC1 regulates trafficking and processing of APP and Abeta generation. Mol Psychiatry 20: , 874–879. |
[15] | White KE , Cummings JL ((1996) ) Schizophrenia and Alzheimer’s disease: Clinical and pathophysiologic analogies. Compr Psychiatry 37: , 188–195. |
[16] | Murray PS , Kumar S , Demichele-Sweet MA , Sweet RA ((2014) ) Psychosis in Alzheimer’s disease. Biol Psychiatry 75: , 542–552. |
[17] | Fredericks D , Norton JC , Atchison C , Schoenhaus R , Pill MW ((2017) ) Parkinson’s disease and Parkinson’s disease psychosis: A perspective on the challenges, treatments, and economic burden. AmJ Manag Care 23: , S83–S92. |
[18] | Tsuang D , DiGiacomo L , Lipe H , Bird TD ((1998) ) Familial aggregation of schizophrenia-like symptoms in Hunting-ton’s disease. Am J Med Genet 81: , 323–327. |
[19] | Arnold SE , Trojanowski JQ ((1996) ) Cognitive impairment in elderly schizophrenia: A dementia (still) lacking distinctive histopathology. Schizophr Bull 22: , 5–9. |
[20] | Cai L , Huang J ((2018) ) Schizophrenia and risk of dementia: A meta-analysis study. Neuropsychiatr Dis Treat 14: , 2047–2055. |
[21] | Hampel H ((2013) ) Amyloid-beta and cognition in aging and Alzheimer’s disease: Molecular and neurophysiological mechanisms. J Alzheimers Dis 33: (Suppl 1), S79–86. |
[22] | Albertini V , Benussi L , Paterlini A , Glionna M , Prestia A , Bocchio-Chiavetto L , Amicucci G , Galluzzi S , Adorni A , Geroldi C , Binetti G , Frisoni GB , Ghidoni R ((2012) ) Distinct cerebrospinal fluid amyloid-beta peptide signatures in cognitive decline associated with Alzheimer’s disease and schizophrenia. Electrophoresis 33: , 3738–3744. |
[23] | Frisoni GB , Prestia A , Geroldi C , Adorni A , Ghidoni R , Amicucci G , Bonetti M , Soricelli A , Rasser PE , Thompson PM , Giannakopoulos P ((2011) ) Alzheimer’s CSF markers in older schizophrenia patients. Int J Geriatr Psychiatry 26: , 640–648. |
[24] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[25] | Theillet FX , Binolfi A , Bekei B , Martorana A , Rose HM , Stuiver M , Verzini S , Lorenz D , van Rossum M , Goldfarb D , Selenko P ((2016) ) Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530: , 45–50. |
[26] | Qiang W , Yau WM , Lu JX , Collinge J , Tycko R ((2017) ) Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature 541: , 217–221. |
[27] | Bartels T , Ahlstrom LS , Leftin A , Kamp F , Haass C , Brown MF , Beyer K ((2010) ) The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys J 99: , 2116–2124. |
[28] | Selkoe DJ ((2011) ) Alzheimer’s disease. Cold Spring Harb Perspect Biol 3: , a004457. |
[29] | De Strooper B , Iwatsubo T , Wolfe MS ((2012) ) Presenilins and gamma-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006304. |
[30] | Yan R , Vassar R ((2014) ) Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 13: , 319–329. |
[31] | Potter H , Wefes IM , Nilsson LN ((2001) ) The inflammation-induced pathological chaperones ACT and apo-E are necessary catalysts of Alzheimer amyloid formation. Neu-robiol Aging 22: , 923–930. |
[32] | van Karnebeek CD , Bowden K , Berry-Kravis E ((2016) ) Treatment of neurogenetic developmental conditions: From 2016 into the future. Pediatr Neurol 65: , 1–13. |
[33] | McGrath J , Saha S , Chant D , Welham J ((2008) ) Schizophrenia: A concise overview of incidence, prevalence, and mortality. Epidemiol Rev 30: , 67–76. |
[34] | Carrillo MC , Bain LJ , Frisoni GB , Weiner MW ((2012) ) Worldwide Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 8: , 337–342. |
[35] | Tsuang MT , Lyons MJ , Faraone SV ((1990) ) Heterogeneity of schizophrenia. Conceptual models and analytic strategies. Br J Psychiatry 156: , 17–26. |
[36] | Jellinger KA ((2012) ) Cerebral correlates of psychotic syndromes in neurodegenerative diseases. J Cell Mol Med 16: , 995–1012. |
[37] | Michael TP , Park S , Kim TS , Booth J , Byer A , Sun Q , Chory J , Lee K ((2007) ) Simple sequence repeats provide a substrate for phenotypic variation in the Neurospora crassa circadian clock. PLoS One 2: , e795. |
[38] | Lindqvist C , Laakkonen L , Albert VA ((2007) ) Polyglutamine variation in a flowering time protein correlates with island age in a Hawaiian plant radiation. BMC Evol Biol 7: , 105. |
[39] | Hashimoto M , Ho G , Takamatsu Y , Wada R , Sugama S , Takenouchi T , Masliah E , Waragai M ((2018) ) Possible role of the polyglutamine elongation in evolution of amyloidrelated evolvability. J Huntingtons Dis 7: , 297–307. |
[40] | Maldonado TA , Jones RE , Norris DO ((2000) ) Distribution of beta-amyloid and amyloid precursor protein in the brain of spawning (senescent) salmon: A natural, brain-aging model. Brain Res 858: , 237–251. |
[41] | Lahdenpera M , Lummaa V , Helle S , Tremblay M , Russell AF ((2004) ) Fitness benefits of prolonged post-reproductive lifespan in women. Nature 428: , 178–181. |
[42] | Schenk D ((2002) ) Amyloid-beta immunotherapy for Alzheimer’s disease: The end of the beginning. Nat Rev Neurosci 3: , 824–828. |
[43] | Honig LS , Vellas B , Woodward M , Boada M , Bullock R , Borrie M , Hager K , Andreasen N , Scarpini E , Liu-Seifert H , Case M , Dean RA , Hake A , Sundell K , Poole Hoffmann V , Carlson C , Khanna R , Mintun M , DeMattos R , Selzler KJ , Siemers E ((2018) ) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378: , 321–330. |
[44] | Eisai Co., Ltd. and Biogen Inc. ((2018) ) Eisai and Biogen announce positive topline results of the final analysis for BAN2401 at 18 months. URL: http://investors.biogen.com/news-releases/news-release-details/eisai-and-biogen-announce-positive-topline-results-final. |
[45] | Panza F , Seripa D , Lozupone M , Solfrizzi V , Imbimbo BP , Barulli MR , Tortelli R , Capozzo R , Bisceglia P , Dimitri A , Stallone R , Dibello V , Quaranta N , Daniele A , Bellomo A , Greco A , Logroscino G ((2018) ) The potential of solanezumab and gantenerumab to prevent Alzheimer’s disease in people with inherited mutations that cause its early onset. Expert Opin Biol Ther 18: , 25–35. |
[46] | Siemers ER , Sundell KL , Carlson C , Case M , Sethuraman G , Liu-Seifert H , Dowsett SA , Pontecorvo MJ , Dean RA , Demattos R ((2016) ) Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 12: , 110–120. |
[47] | Krause M , Zhu Y , Huhn M , Schneider-Thoma J , Bighelli I , Chaimani A , Leucht S ((2018) ) Efficacy, acceptability, and tolerability of antipsychotics in children and adolescents with schizophrenia: A network meta-analysis. Eur Neuropsychopharmacol 28: , 659–674. |
[48] | Stefanopoulou E , Manoharan A , Landau S , Geddes JR , Goodwin G , Frangou S ((2009) ) Cognitive functioning in patients with affective disorders and schizophrenia: A meta-analysis. Int Rev Psychiatry 21: , 336–356. |
[49] | Meesters PD , Stek ML , Comijs HC , de Haan L , Patterson TL , Eikelenboom P , Beekman AT ((2010) ) Social functioning among older community-dwelling patients with schizophrenia: A review. Am J Geriatr Psychiatry 18: , 862–878. |
[50] | Martinez-Vicente M , Sovak G , Cuervo AM ((2005) ) Protein degradation and aging. Exp Gerontol 40: , 622–633. |
[51] | Jia P , Wang L , Fanous AH , Pato CN , Edwards TL , International Schizophrenia C , Zhao Z ((2012) ) Network-assisted investigation of combined causal signals from genome-wide association studies in schizophrenia. PLoS Comput Biol 8: , e1002587. |
[52] | Agim ZS , Esendal M , Briollais L , Uyan O , Meschian M , Martinez LA , Ding Y , Basak AN , Ozcelik H ((2013) ) Discovery, validation and characterization of Erbb4 and Nrg1 haplotypes using data from three genome-wide association studies of schizophrenia. PLoS One 8: , e53042. |
[53] | Maier W , Zobel A , Rietschel M ((2003) ) Genetics of schizophrenia and affective disorders. Pharmacopsychiatry 36: Suppl 3, S195–202. |
[54] | Zhao J , Jin L , Xiong M ((2006) ) Nonlinear tests for genomewide association studies. Genetics 174: , 1529–1538. |
[55] | Vassar R ((2004) ) BACE1: The beta-secretase enzyme in Alzheimer’s disease. J Mol Neurosci 23: , 105–114. |
[56] | Glessner JT , Hakonarson H ((2009) ) Common variants in polygenic schizophrenia. Genome Biol 10: , 236. |
[57] | Francks C , Tozzi F , Farmer A , Vincent JB , Rujescu D , St Clair D , Muglia P ((2010) ) Population-based linkage analysis of schizophrenia and bipolar case-control cohorts identifies a potential susceptibility locus on 19q13. Mol Psychiatry 15: , 319–325. |
[58] | Doherty JL , O'Donovan MC , Owen MJ ((2012) ) Recent genomic advances in schizophrenia. Clin Genet 81: , 103109. |
[59] | DeMichele-Sweet MAA , Weamer EA , Klei L , Vrana DT , Hollingshead DJ , Seltman HJ , Sims R , Foroud T , Hernandez I , Moreno-Grau S , Tarraga L , Boada M , Ruiz A , Williams J , Mayeux R , Lopez OL , Sibille EL , Kamboh MI , Devlin B , Sweet RA ((2018) ) Genetic risk for schizophrenia and psychosis in Alzheimer disease. Mol Psychiatry 23: , 963–972. |


