
Ceramide Accumulation Is Associated with Declining Verbal Memory in Coronary Artery Disease Patients: An Observational Study
Abstract
Background:
Biomarkers in cognitively vulnerable populations, like those with coronary artery disease (CAD), may inform earlier intervention in vascular neurodegeneration. Circulating ceramide C18:0 (CerC18:0) is associated with changes in verbal memory in early neurodegeneration and CAD progression.
Objective:
To investigate whether plasma CerC18:0 accumulation is associated with longitudinal declines in verbal memory performance in CAD.
Methods:
In addition to total CerC18:0, we assessed its relative abundance to its precursors as ratios: CerC18:0 to monohexosylceramide C18:0 (MHxCer18:0), CerC18:0 to sphingomyelin C18:0 (SM18:0), and CerC18:0 to sphingosine-1-phosphate (S1P). Verbal memory was assessed using the California Verbal Learning Test 2nd Ed. Using mixed models in 60 CAD participants, we evaluated associations between baseline CerC18:0 ratios and changes in verbal memory performance, adjusting for age, body mass index, and education. Given that cognitive decline is more rapid following onset of deficits, these associations were compared between those with possible mild vascular neurocognitive disorder (MVND).
Results:
Increased baseline CerC18:0 concentrations correlated with worse verbal memory performance over time (b[SE] = – 0.91[0.30], p = 0.003). Increased baseline CerC18:0/SM18:0 (b[SE] = – 1.11[`], p = 0.03) were associated with worse verbal memory performance over time. These associations were not mediated by whether or not patients had possible MVND at baseline.
Conclusion:
These findings support aberrant CerC18:0 metabolism as an early neurobiological change in vascular neurodegeneration. Future studies should measure enzymes responsible for conversion of sphingolipid precursors into CerC18:0 to assess enzymatic activity.
INTRODUCTION
It is estimated that half of the patients with vascular cognitive impairment progress to dementia within 5 years [1], which results in high institutionalization rates and poor quality of life. Because there are no recommended drugs to prevent this progressive vascular neurodegeneration, early identification and active control of cardiovascular risk factors is currently considered the only way to modify the course of this disease [2, 3]. As such, there is significant interest in the development of biological markers to detect disease signatures in cognitively vulnerable populations. One such population is those with coronary artery disease (CAD); CAD patients experience increased brain atrophy [4, 5], increased white matter lesions [6], and an increased risk of memory impairment [7]. Consequently, this clinical population is approximately two times more likely to develop incipient neurodegenerative disorders such as mild vascular neurocognitive disorder (MVND) [8], which may progress to dementia [9]. Therefore, those with CAD are an ideal prodromal population to identify markers of early cognitive changes and identify potential therapeutic targets.
Compelling evidence suggests that changes in verbal memory may be one of the earliest events in cognitive decline caused by vascular pathology. A previous study elucidated the neuropsychological profiles of patient populations along the spectrum of cognitive decline due to vascular risks: healthy elders, those at-risk of cerebrovascular disease, those with MVND, and those with vascular dementia. Those with MVND had significantly worse verbal memory compared to the healthy participants, while performing within normative ranges in other cognitive domains [10]. Cognitive deterioration from the time of CAD diagnosis to clinically significant cognitive impairment generally occurs over several years, making proper assessment a challenge. As such, longitudinal data with a sensitive biological marker, in association with the cognitive performance of CAD patients would improve our etiologic and prognostic understanding of neurodegeneration.
Perturbed sphingolipid metabolism has been associated with both cardiovascular and neurodegenerative diseases. This is consistently and specifically reported in the ceramide species with an acyl chain length of 18 carbons (CerC18:0). Plasma levels of CerC18:0 are elevated in coronary plaques, a hallmark of CAD pathophysiology [11]. Evidence from numerous studies also implicates CerC18:0 in the biochemical profile of various neurodegenerative diseases. In adults at-risk of Alzheimer’s disease (AD), cerebrospinal fluid (CSF) CerC18:0 was associated with higher CSF amyloid-β and higher CSF tau levels [12]. Notably, these associations were stronger among individuals aged 54 years and older, an age range during which the prevalence of CAD increases. Among subjects infected with the human immunodeficiency virus (HIV), CSF CerC18:0 was a strong predictor of poorer performance in multiple cognitive domains [13]. In a cognitively asymptomatic population, higher levels of serum CerC18:0 were associated with an increased risk of incident memory impairment over 9 years of follow-up [14]. Moreover, we previously found that greater plasma CerC18:0 levels were associated with less improvement in verbal memory performance among CAD patients over 6 months [15]. Collectively, this evidence coupled with the abundance of CerC18:0 in brain tissue [16], demonstrates the potential and feasibility of CerC18:0 as a biomarker of the pre-symptomatic phases of cognitive impairment.
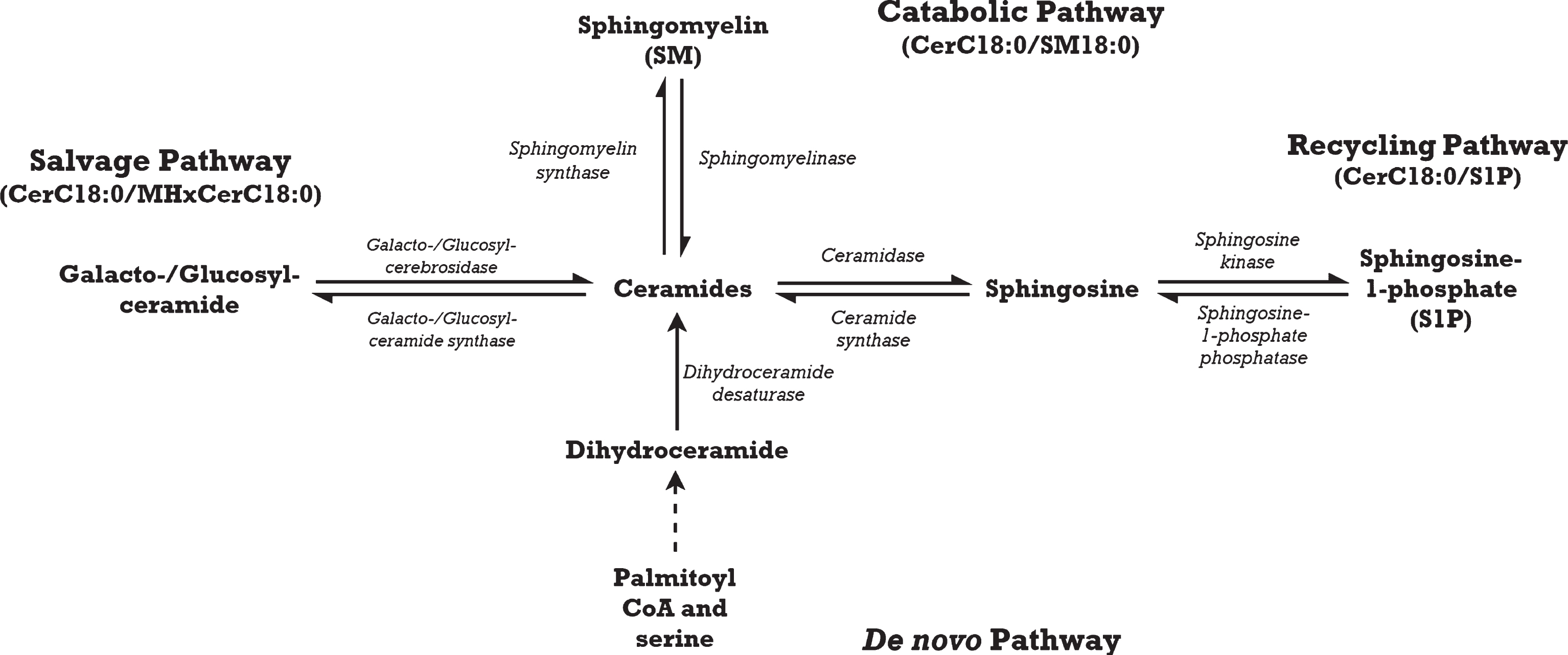
The present study builds upon our previous work [15] by evaluating the relationship between circulating ceramides and verbal memory performance among CAD subjects over a longer period of time, in which cognitive deterioration may be more apparent. Although cognitive trajectories have been investigated in those with mild cognitive impairment [17], it has not been studied in those with vascular risk factors. Sphingolipid abnormalities in neurodegenerative processes are likely the result of enzymatic imbalance as the majority of sphingolipids are synthesized within the body [18, 19]. We postulate that the balance of sphingolipid metabolism is skewed toward the accumulation of CerC18:0 in various biosynthesis pathways, resulting in apoptosis [20] and eventual neurodegeneration. We explored the relative abundance of CerC18:0 to its precursors as ratios; changes in these ratios may indicate perturbations in CerC18:0 biosynthesis pathways. In addition, sphingolipid ratios have been reported to be stronger predictors of cognitive decline than individual sphingolipids [21], but this has not been investigated in vascular cognitive impairment. Briefly, ceramides are generated through four biosynthesis pathways [22] (Fig. 1). In the catabolic pathway, sphingomyelinase breaks sphingomyelin (SM) down into ceramides and a by-product, phosphocholine. While the salvage and recycling pathways are distinct, they coordinate in ceramide biosynthesis. The salvage pathway uses glucocerebrosidase or galactocerebrosidase to catabolize glucosylceramides such as monohexosylceramides (MHxCer) into sphingosine for ceramide biosynthesis. The recycling pathway then phosphorylates sphingosine to make sphingosine-1-phosphate (S1P), which is then converted into ceramides. Lastly, the de novo pathway synthesizes ceramides from palmitoyl coenzyme A and serine via action of serine palmitoyltransferase. Given that the salvage pathway accounts for 50–90% of sphingolipid generation [22], our primary hypothesis is that higher baseline ratios of CerC18:0 to MHxCerC18:0 would be associated with greater decline in verbal memory performance as dysregulation in this pathway would presumably have the greatest clinical impact. Our secondary hypotheses were that higher baseline ratios of CerC18:0/SM18:0 and CerC18:0/S1P would also predict greater verbal memory decline. We did not conduct analysis in the de novo pathway as we did not measure a precursor metabolite specific to that pathway. In an exploratory analysis, we hypothesized that the associations between baseline CerC18:0 ratios and change in verbal memory performance will be significantly different between those with possible MVND and those without possible MVND. Our exploratory hypothesis focused on study participants with possible MVND rather than MVND, because the neuropathology may be too advanced by the time the stricter neurocognitive criteria of MVND is met [23]. Therapeutic intervention or cardiac rehabilitation may be more beneficial in early prodromal states as a more preventative approach [24]. Moreover, evidence shows that cognitive decline is more rapid following onset of deficits [25].
Fig.1
Ceramide with an acyl chain length of 18 carbons (CerC18:0) is synthesized through four main pathways: the catabolic, salvage, recycling, and de novo pathways.
MATERIALS AND METHODS
Study participants
This study was approved by the research ethics boards at both Sunnybrook Health Sciences Centre and the University Health Network. All study participants were recruited from a cardiac rehabilitation program at the Rumsey Centre of University Health Network Toronto Rehabilitation Institute and provided written, informed consent to participate before study enrolment and assessment for inclusion/exclusion criteria.
All study participants had evidence of CAD based on previous hospitalization for myocardial infarction, prior revascularization procedure, and/or angiographic evidence of at least 50% stenosis in a major coronary artery. All participants had dyslipidemia, were taking statins, were between the ages of 50–75, and spoke and understood English. Participants were excluded if they had any of the following comorbidities that could affect the interpretation of outcomes: a history of a neurodegenerative disease, active cancer, schizophrenia, bipolar disorder, surgery planned within 12 months, or dementia. The Standardized Mini Mental Status Examination (sMMSE) was used to screen for dementia and consequently, those with a sMMSE score <24 were excluded [26]. Participants with possible MVND were defined as those with composite verbal memory or executive function Z-scores of ≤1, as previously described [27].
Demographics and clinical characteristics
A comprehensive medical history, including demographic information, co-morbidities, and vascular risk factors (hypertension, smoking, hypercholesterolemia, obesity, and diabetes) were collected from participant interviews. Anthropometric data such as height, weight, body mass index (BMI) were obtained from patient charts at the Rumsey Centre or obtained by trained researchers. Lipid profile including total cholesterol and triglycerides were measured using standard clinical assays.
The Structured Clinical Interview for Depression (SCID) was used to diagnose depression at baseline. It was administered by a trained researcher under the supervision of the study psychiatrist, to determine if participants met the Diagnostic and Statistical Manual, Version 4 (DSM-IV) criteria for minor or major depressive disorder (MDD).
Cognitive and behavioral assessments
Study participants were assessed at the beginning of cardiac rehabilitation (baseline), 3-months, 6-months, and after 12 months. The interval between baseline and the post 12-month visit was variable. A 30-min standardized battery of tests was used to assess cognitive performance due to vascular causes as recommended by the National Institute of Neurological Disorders and Stroke-Canadian Stroke Network (NINDS-CSN) at baseline, 3, 6, and post-12 months [28]. A trained researcher administered the battery at a standardized time (0930 h±30 min) and participants refrained from eating or drinking any caffeine-containing beverages for at least 4 h before testing. As the primary outcome, verbal memory was assessed using the California Verbal Learning Test 2nd Ed. (CVLT-II). The CVLT-II yields multiple measures of verbal memory function, including verbal learning (recall of a word list over 5 learning trials), short-delay free recall (recall of a word list after an interfering list), and long-delay free recall (recall of a word list after 20 min) [28]. It has been shown to be able to distinguish elderly adults with mild cognitive impairment from cognitively normal elderly individuals, because it assesses learning acrossmultiple trials [29].
MVND patients also have deficits in other cognitive domains along a continuum of severity. Measures of executive function included the Trail-Making Test Part B [30] and Stroop Color-Word Interference Test. Performance in visuospatial memory was assessed using the Brief Visuospatial Test-Revised (BVMT-R), which presents a measure of visual learning and delayed recall [31]. The domain of processing speed was assessed using the Trail-Making Test Part A [30] and the Digit Symbol-Coding task, a measure of complex attention and psychomotor speed from the Wechsler Adult Intelligence Scale 3rd Edition [32].
Z-scores were determined from age, gender and education-matched norms [33]. Z-scores from tests that involve the same cognitive domain were summed into a composite Z-score to provide a more stable representation of performance in that particular domain and to avoid multiple comparisons [34, 35]. For verbal memory, the three Z-scores from the CVLT-II: learning, short-delay free recall, and long-delay free recall were summed into a composite score. For executive function, the Z-scores from the Trails-Making Test Part B and Stroop Color-Word Interference Test were summed into a composite score.
Measurement of plasma sphingolipids
At baseline, blood was drawn from participants who were fasting at 0900 h±30 min on the same day as the cognitive testing and then centrifuged at 1000 g for 10 min at 4°C. The resulting plasma was immediately isolated and stored at – 80°C until analysis. High performance liquid chromatography coupled electrospray ionization tandem mass spectrometry (LC/MS/MS) using multiple reaction monitoring was used to quantify individual sphingolipid species and processed by the Analyst 1.4.2 software package, as previously described [36].
A crude lipid extraction from plasma was performed using a modified Bligh and Dyer procedure (Avanti Polar Lipids, Alabaster, AL, USA). Plasma extracts were dried and then re-suspended in methanol before analysis. High performance liquid chromatography (PerkinElmer, MA, USA) with a reverse phase C18 column (Phenomenex, Torrance, CA, USA) was used for temporal resolution of compounds. The eluted samples were then injected into an electrospray ion source coupled to a triple-quadrupole mass spectrometer (API3000, AB Sciex Inc, Thornhill, Ontario, Canada) and analyses were conducted by multiple reaction monitoring. Eight-point calibration curves (0.1–1000 ng/mL) were constructed by plotting area under the curve for different sphingolipids for each calibration standard normalized to the internal standard. The final sphingolipid concentrations (ng/mL) were determined by fitting the identified species to standard curves based on acyl chain length. Instrument control and quantification of spectral data were performed using Analyst 1.4.2 and MultiQuant software (AB Sciex Inc, Thornhill, Ontario, Canada).
Statistical analyses
Ratios of CerC18:0 relative to its precursors were used to explore accumulation of CerC18:0 (Fig. 1). Ratios of CerC18:0/SM18:0, CerC18:0/MHxCerC18:0, and CerC18:0/S1P were calculated using raw sphingolipid concentrations. The resulting CerC18:0 ratios and CerC18:0 were log-transformed to obtain a normal distribution prior to analyses. Only sphingolipid ratios with identical acyl chain lengths were included, because bi-directional conversions between ceramides and its precursors primarily modify the head group and not the acyl chain length [13].
Linear regressions were used to assess cross-sectional relationships between clinical characteristics of the study population and verbal memory performance. To characterize our participant population, regression models were used to evaluate bivariate relationships between sociodemographic features at baseline and change in verbal memory. These analyses were conducted in two populations: the overall study population and those with possible MVND.
For our primary outcome, mixed models were used to evaluate longitudinal relationships between baseline C18:0 concentrations, baseline ratios of C18:0 to its precursors or metabolites, and changes in verbal memory over time. For our exploratory outcome, we used mixed models from the main analyses and included a possible MVND and CerC18:0 biomarker interaction term as a covariate. This evaluated whether associations between baseline sphingolipid concentrations and change in verbal memory were significantly different between those with possible MVND and those without.
Mixed model analyses were conducted using the MIXED procedure in SAS University Edition statistical software (SAS Institute Inc., North Carolina, USA) with a two-tailed significance level of p≤0.05. The magnitude of association is presented in terms of change in standard deviation per unit change in the independent variable. A spatial power covariance structure was used to adjust for the varying time intervals between study visits. Age, BMI, and years of education were included as potential confounders a priori based on previous reports in the literature [14, 36], while the number of days since baseline was added as a random effect.
RESULTS
Bivariate associations between baseline characteristics and cognitive performance
In the present study, 60 study participants met inclusion criteria and were recruited at the beginning of their cardiac rehabilitation program. The majority of these study participants were male (n = 49, 83.1%) and Caucasian (n = 47, 79.7%), with an average age of 65±6 years and 14 (23.3%) of these individuals had possible MVND at baseline. There were no significant cross-sectional associations between plasma CerC18:0 and any of the four cognitive domains at baseline.
Study participants were followed for a median of 3.5 years from their baseline visit (inter-quartile range: 1.1 years). In bivariate longitudinal associations (Table 1), greater years of education (b[SE] = 0.05[0.02], p = 0.04) and greater VO2peak (b[SE] = 0.03[0.01], p = 0.01) at baseline were associated with a greater improvement in verbal memory. Conversely, having depression (b[SE] = – 0.54[0.24], p = 0.03) was associated with worse verbal memory performance over time (Table 1). In those with possible MVND, diabetes at baseline (b[SE] = 0.68[0.24], p = 0.01) was associated with less improvement in verbal memory. Across all participants, verbal memory performance did not change over time (b = – 0.0001, p = 0.32).
Table 1
Baseline clinical and demographic characteristics in association with verbal memory z-score over a median of 3.5 years
| Overall study participants (n = 60) | Participants with possible MVND at baseline (n = 14) | |||||
|---|---|---|---|---|---|---|
| Characteristic | Mean (±SD) or n (%) | b | p | Mean (±SD) or n (%) | b [SE] | p |
| Sociodemographics | ||||||
| Age (y) | 64.6 (6.0) | 0.004 [0.01] | 0.74 | 62.5 (5.6) | 0.02 [0.02] | 0.44 |
| Female | 10 (16.7) | – 0.24 [0.21] | 0.26 | 3 (21.4) | 0.23 [0.32] | 0.49 |
| BMI (kg/m2) | 29.4 (5.6) | 0.01 [0.01] | 0.49 | 29.0 (3.5) | 0.0008 [0.0009] | 0.38 |
| Education (y) | 16.6 (3.2) | 0.05 [0.02] | 0.04* | 16.1 (3.6) | – 0.02 [0.04] | 0.59 |
| Married | 49 (81.7) | – 0.04 [0.20] | 0.83 | 13 (92.9) | – 0.58 [0.48] | 0.25 |
| Has a smoking history | 32 (53.3) | 0.05 [0.15] | 0.76 | 7 (50.0) | – 0.12 [0.26] | 0.64 |
| Has an APOE4 allele | 11 (18.3) | – 0.05 [0.20] | 0.81 | 2 (14.2) | – 0.39 [0.35] | 0.29 |
| CAD Severity | ||||||
| Cumulative stenosis (%) | 152.3 (71.4) | 0.002 [0.001] | 0.05 | 117.8 (65.4) | – 0.003 [0.002] | 0.29 |
| Lipid profile and Hb1AC | ||||||
| LDL (mmol/L) | 1.6 (0.6) | 0.07 [0.12] | 0.59 | 1.7 (0.9) | – 0.07 [0.21] | 0.74 |
| HDL (mmol/L) | 1.3 (0.3) | – 0.16 [0.23] | 0.48 | 1.3 (0.3) | – 0.05 [0.30] | 0.86 |
| Cholesterol (mmol/L) | 3.5 (0.8) | – 0.13 [0.09] | 0.17 | 3.6 (1.1) | 0.08 [0.14] | 0.60 |
| Triglycerides (mmol/L) | 1.2 (0.6) | – 0.23 [0.12] | 0.07 | 1.4 (0.7) | 0.06 [0.17] | 0.71 |
| Hemoglobin A1C | 0.06 (0.01) | – 17.13 [11.77] | 0.15 | 0.06 (0.01) | – 20 [27.26] | 0.46 |
| Fitness parameters | ||||||
| VO2peak (mL/kg/min) | 20.7 (5.8) | – 0.03 [0.01] | 0.01* | 18.2 (5.1) | – 0.03 [0.02] | 0.32 |
| Comorbidities | ||||||
| Depressed | 8 (13.3) | – 0.54 [0.24] | 0.03* | 3 (21.4) | – 0.45 [0.32] | 0.18 |
| Diabetes mellitus | 10 (16.7) | – 0.05 [0.21] | 0.81 | 5 (35.7) | 0.68 [0.24] | 0.01* |
BMI, body mass index; APOE4, apolipoprotein E4; LDL, low density lipoprotein; HDL, high density lipoprotein; S.D, standard deviation; SE, standard error; CAD, coronary artery disease. *indicates statistically significant associations (p < 0.05).
Raw and log-transformed concentrations of sphingolipids are presented in Table 2. CerC18:0, SM18:0, MHxCerC18:0, and S1P plasma concentrations were log-transformed to obtain a normal distribution for mixed model analyses, so that their concentrations were standardized to a notionally common scale. CerC18:0 ratios were calculated using the log-transformed sphingolipid ratios.
Table 2
Distribution of plasma sphingolipid concentrations and ratios at baseline and their log-transformed values
| Species (ng/mL) | Mean (SD) | Range |
|---|---|---|
| CerC18:0 | 7.60 (5.14) | 1.45 to 30.89 |
| SM18:0 | 123505 (27762) | 65300 to 205000 |
| MHxCerC18:0 | 8.33 (4.66) | 1.84 to 28.86 |
| S1P | 197.70 (178.16) | 16.32 to 878.95 |
| Log CerC18:0 | 0.80 (0.27) | 0.16 to 1.49 |
| Log SM18:0 | 5.08 (0.10) | 4.81 to 5.31 |
| Log MHxCerC18:0 | 0.87 (0.22) | 0.27 to 1.46 |
| Log S1P | 2.13 (0.39) | 1.21 to 2.94 |
| Log CerC18:0/MHxCerC18:0 | – 0.07 (0.19) | – 0.62 to – 0.43 |
| Log CerC18:0/SM18:0 | – 4.28 (0.22) | – 4.84 to – 3.82 |
| Log CerC18:0/S1P | – 1.34 (0.42) | – 2.39 to – 0.11 |
C18, acyl chain of 18 carbons; Cer, ceramide; MHxCer, monohexosylceramide; S1P, sphingosine-1-phosphate; SD, standard deviation; SM, sphingomyelin; MVND, mild vascular neurocognitive disorder.
Association between baseline CerC18:0 and change in verbal memory
Longitudinally, baseline plasma CerC18:0 was significantly associated with decline in verbal memory performance, after adjustment for age, BMI, years of education, and days since baseline (Table 3). Each log-unit increase in baseline plasma CerC18:0 concentrations correlated with a 0.91 standard deviation (SD) decline in verbal memory performance (b [SE] = – 0.91 [0.30], p = 0.003).
Table 3
Association between plasma CerC18:0 at baseline and change in verbal memory performance over the course of the study in participants with CAD (n = 60), adjusted for age, BMI, years of education, and days since baseline
| Variable | Estimate (b) [95% CI] | SE | df | t-value | Significance (p) |
|---|---|---|---|---|---|
| Intercept | – 0.95 [– 3.09, 1.19] | 1.07 | 55 | – 0.89 | 0.38 |
| Days since baseline | – 0.0002 [– 0.0004, 0.00008] | 0.0001 | 177 | – 1.29 | 0.20 |
| Age | 0.02 [– 0.01, 0.05] | 0.01 | 55 | 1.45 | 0.15 |
| BMI | – 0.01 [– 0.03, 0.02] | 0.01 | 55 | – 0.36 | 0.72 |
| Years of education | 0.04 [– 0.00555, 0.09] | 0.02 | 55 | 1.77 | 0.08 |
| CerC18:0 | – 0.91 [– 1.51, 0.32] | 0.30 | 55 | – 3.08 | 0.003* |
BMI, body-mass index; C18:0, acyl chain length of 18 carbons; CAD, coronary artery disease; Cer, ceramide; CI, confidence interval; df, degrees of freedom; SE, standard error.
Associations between baseline CerC18:0 ratios and change in verbal memory
Baseline plasma MHxCerC18:0 concentrations were not associated with change in verbal memory over time (b [SE] = – 0.46 [0.38], p = 0.22), after adjusting for age, BMI, years of education and days since baseline visit. In contrast, baseline CerC18:0/MHxCerC18:0 ratios were associated with change in verbal memory performance, after adjusting for age, BMI, years of education, and days since baseline (Table 4). Each log unit increase in this ratio correlated with a 0.90 SD decrease in verbal memory performance (b [SE] = – 0.90 [0.40], p = 0.03).
Table 4
Association between plasma CerC18:0/MHxCerC18:0 at baseline and change in verbal memory performance over the course of the study in participants with CAD (n = 60), adjusted for age, BMI, years of education, and days since baseline
| Variable | Estimate (b) [95% CI] | SE | df | t-value | Significance (p) |
|---|---|---|---|---|---|
| Intercept | – 1.29 [– 3.53, 0.94] | 1.12 | 55 | – 1.16 | 0.25 |
| Days since baseline | – 0.0002 [– 0.0004, 0.0001] | 0.0001 | 177 | – 1.27 | 0.20 |
| Age | 0.01 [– 0.02, 0.04] | 0.01 | 55 | 0.78 | 0.44 |
| BMI | – 0.004 [– 0.03, 0.02] | 0.01 | 55 | – 0.30 | 0.76 |
| Years of education | 0.05 [0.002, 0.10] | 0.02 | 55 | 2.10 | 0.04* |
| CerC18:0/MHxCerC18:0 | – 0.90 [– 1.70, – 0.09] | 0.40 | 55 | – 2.23 | 0.03* |
BMI, body-mass index; C18:0, acyl chain length of 18 carbons; CAD, coronary artery disease; Cer, ceramide; CI, confidence interval; df, degrees of freedom; MHxCer, monohexosylceramide; SE, standard error.
Plasma concentrations of SM18:0 were not associated with change in verbal memory over time after adjusting for age, BMI, years of education, and days since baseline (b [SE] = – 0.90 [0.84], p = 0.29). However, baseline ratios of CerC18:0/SM18:0 were significantly associated with decline in verbal memory performance, after adjusting for age, BMI, years of education, and days since baseline (Table 5). For instance, each log-unit increase in CerC18:0/SM18:0 ratio correlated with a 1.11 SD decline in verbal memory performance (b [SE] = – 1.11 [0.36], p = 0.004).
Table 5
Association between plasma CerC18:0/SM18:0 at baseline and change in verbal memory performance over the course of the study in participants with CAD, adjusted for age, BMI, years of education, and days since baseline
| Variable | Estimate (b) [95% CI] | SE | df | t-value | Significance (p) |
|---|---|---|---|---|---|
| Intercept | – 6.30 [– 10.54, – 2.06] | 2.11 | 55 | – 2.98 | 0.004 |
| Days since baseline | – 0.00016 [– 0.0004, 0.0009] | 0.0001 | 177 | – 1.27 | 0.20 |
| Age | 0.02 [– 0.01, 0.05] | 0.01 | 55 | 1.41 | 0.16 |
| BMI | – 0.01 [– 0.04, 0.02] | 0.01 | 55 | – 0.62 | 0.54 |
| Years of education | 0.04 [– 0.01, 0.09] | 0.02 | 55 | 1.75 | 0.09 |
| CerC18:0/SM18:0 | – 1.11 [– 1.84, – 0.38] | 0.36 | 55 | – 3.05 | 0.004* |
BMI, body-mass index; C18:0, acyl chain length of 18 carbons; CAD, coronary artery disease; Cer, ceramide; CI, confidence interval; df, degrees of freedom; SE, standard error; SM, sphingomyelin.
Concentrations of S1P (b [SE] = – 0.41 [0.20], p = 0.05) were not associated with change in verbal memory over time in multivariable models. Likewise, CerC18:0/S1P was not associated with change in verbal memory over time, after adjustment for age, BMI, years of education, and days since baseline (b [SE] = 0.01 [0.21], p = 0.97; Table 6).
Table 6
Association between plasma CerC18:0/S1P at baseline and change in verbal memory performance over the course of the study in participants with CAD, adjusted for age, BMI, years of education, and days since baseline
| Variable | Estimate (b) [95% CI] | SE | df | t-value | Significance (p) |
|---|---|---|---|---|---|
| Intercept | – 1.04 [– 3.41, 1.32] | 1.18 | 52 | – 0.88 | 0.38 |
| Days since baseline | – 0.0002 [– 0.0004, 0.0001] | 0.0001 | 168 | – 1.13 | 0.26 |
| Age | 0.01 [– 0.02, 0.03] | 0.01 | 52 | 0.52 | 0.60 |
| BMI | – 0.001 [– 0.03, 0.03] | 0.01 | 52 | – 0.10 | 0.92 |
| Years of education | 0.05 [– 0.001, 0.10] | 0.02 | 52 | 1.98 | 0.05 |
| CerC18:0/S1P | 0.01 [– 0.41, 0.42] | 0.21 | 52 | 0.04 | 0.97 |
BMI, body-mass index; C18:0, acyl chain length of 18 carbons; CAD, coronary artery disease; Cer, ceramide; CI, confidence interval; df, degrees of freedom; S1P, sphingosine-1-phosphate; SE, standard error.
Differences in associations between those with possible MVND and those with no possible MVND
Although the presence of possible MVND was significantly associated with decline in verbal memory as a main effect, it did not significantly affect the association between baseline CerC18:0 and change in verbal memory performance (b [SE] = 0.10 [0.56], p = 0.86), after adjusting for age, years of education, days since baseline, possible MVND, and baseline CerC18:0. Likewise, the presence of possible MVND did not significantly affect the association between baseline CerC18:0/MHxCerC18:0 and change in verbal memory performance (b [SE] = 1.08 [0.74], p = 0.15), after adjusting for age, years of education, days since baseline, possible MVND, and baseline CerC18:0/MHxCerC18:0. Similarly, the association between baseline CerC18:0/SM18:0 and change in verbal memory performance was not significantly different between those with possible MVND and those with no possible MVND (b [SE] = – 0.28 [0.67], p = 0.68), after adjusting for age, years of education, days since baseline, possible MVND, and baseline CerC18:0/SM18:0. Lastly, the presence of possible MVND did not significantly affect the association between baseline CerC18:0/S1P and change in verbal memory performance (b [SE] = 0.65 [0.55], p = 0.25), after adjusting for age, years of education, days since baseline, possible MVND, and baseline CerC18:0/S1P. Overall, none of the above associations were mediated by the presence of possible MVND. A summary of these results is presented in Table 7.
Table 7
Associations between baseline CerC18:0 biomarkers and change in verbal memory in the overall CAD population (n = 60), adjusted for age, BMI, and years of education
| b [SE], p | Without interaction term | With interaction term* |
|---|---|---|
| (b [SE], p) | (b [SE], p) | |
| CerC18:0 | – 0.91 [0.30], 0.003* | 0.10 [0.56], 0.86 |
| CerC18:0/MHxCerC18:0 | – 0.90 [0.40], 0.03* | 1.08 [0.74], 0.15 |
| CerC18:0/SM18:0 | – 1.11 [0.36], 0.004* | – 0.28 [0.67], 0.68 |
| CerC18:0/S1P | 0.01 [0.21], 0.97 | 0.68 [0.44], 0.13 |
*An interaction term of baseline CerC18:0 biomarker and possible MVND. C18:0, acyl chain length of 18 carbons; CAD, coronary artery disease; Cer, ceramide; MHxCer, monohexosylceramide; S1P, sphingosine-1-phosphate; SM, sphingomyelin; SE, standard error; MVND, mild vascular neurocognitive disorder.
DISCUSSION
This study found a relationship between markers of aberrant sphingolipid metabolism and cognitive decline, corroborating our previous finding that baseline plasma CerC18:0 is a robust marker of verbal memory performance in a cohort of CAD patients [15]. We investigated the longitudinal relationships between the equilibrium of baseline CerC18:0 biosynthesis and verbal memory performance over time. In both the catabolic and salvage pathways, an increased baseline ratio of CerC18:0 to its respective precursor was significantly correlated with worse verbal memory performance over time. In post-hoc analyses of the catabolic pathway, this relationship was significant among participants with possible MVND at baseline, but not in those without.Collectively, our results show that ratios of CerC18:0 to its precursors are a sensitive marker of cognitive change in the early stages of vascular cognitive impairment.
Consistent with previous reports in the literature, we did not find a significant cross-sectional association between CerC18:0 and any of the four cognitive domains [21, 36]. A possible explanation for the lack of cross-sectional findings is that our CAD participants are generally cognitively intact. Conversely, baseline CerC18:0 was associated with change in verbal memory in longitudinal analyses. As such, we postulate that differences in ceramide concentrations are associated with the rate of cognitive change over time, rather than inter-individual differences in cognition at a certain timepoint. Moreover, given that sphingolipids ratios may be more predictive of cognitive decline than individual sphingolipids [21], we used similar CerC18:0 ratios as an exploratory marker of shifts in these metabolic pathways.
The emphasis on rate of cognitive change indicates aberrations in enzymatic activity. An increased ratio of CerC18:0/SM18:0 suggests increased activity of sphingomyelinase in the catabolic pathway, which catabolizes SM18:0 into CerC18:0. In support of this, plasma levels of secretory acid sphingomyelinase and ceramides have been found to be elevated in CAD patients [37]. This increase in ceramides causes aggregation of low-density lipoproteins, which leads to formation of atherosclerotic plaque [38]. However, the role of sphingomyelinase in cognition remains less clear. A study investigating gene expression in sphingolipid metabolism found that sphingomyelinase gene expression did not differ between cohorts along the cognitive impairment spectrum [39]. In contrast, a recent paper reported that an induced deficiency of neutral sphingomyelinase-2 improved cognition in an animal model of AD [40]. As such, the influence of sphingomyelinase activity on cognitive performance in CAD patients remains unclear and warrants further investigation.
We also report a novel association between higher baseline CerC18:0/MHxCer18:0 ratio and decline in verbal memory performance over time. In the salvage pathway, galactocerebrosidase and glucosylcerebrosidase catalyze the conversion from monohexosylceramides into ceramides. Given that the salvage pathway accounts for 50–90% of sphingolipid generation [22], its dysregulation may have significant clinical impact. This is no more evident than in individuals with Krabbe disease; a disease characterized by infantile-onset neurologic deterioration due to deficient galactocerebrosidase activity, ranging from 5–10% of normal levels [41]. Interestingly, in adults diagnosed with late-onset Krabbe disease, a case series demonstrated a clinical profile similar to that of cerebrovascular disease, reporting progressive cognitive impairment and white matter hyperintensities not unlike those associated with vascular cognitive impairment [42]. Furthermore, galactocerebrosidase gene expression is increased with increasing severity of cognitive impairment in temporal regions, which are functional brain regions that are involved in verbal memory [39]. As evident in Krabbe disease, severe dysregulation of the salvage pathway has significant clinical consequences, but the extent of perturbation in this pathology likely exceeds that observed in our study population. Despite this, our findings suggest that chronic, mild dysregulation may have a cumulative, clinical impact.
In our CAD participants, elevated plasma S1P was associated with worse verbal memory performance over time. This was surprising, because numerous studies report S1P to be neuroprotective, particularly in AD pathology [43, 44]. However, S1P signaling induces adhesion molecule expression and subsequent atherogenesis [45], implicating its involvement in CAD pathogenesis. Furthermore, peripheral levels of S1P are elevated in CAD patients and predict occurrence and severity of coronary stenosis [46, 47]. Given this evidence, peripheral S1P levels in our participants may be more representative of CAD pathology due to its proximity to the anatomical location of CAD. As such, we propose that greater CAD severity due to elevated peripheral S1P levels is associated with more severe cognitive impairment. In light of the contrasting relationships of S1P with neurodegeneration and CAD, this highlights the importance of anatomical location and S1P balance as recently suggested by Karunakaran [48].
Although the de novo pathway may only be responsible for a small portion of sphingolipids due to its slow rate of synthesis, dysregulation of enzymes within this pathway may potentially disrupt sphingolipid balance. For instance, inhibition of dihydroceramide synthase using fumonisins results in sphinganine accumulation [49], which may have implications for lipotoxic disease [50]. However, study of the de novo pathway in terms of its clinical impact has not been thoroughly elucidated and warrant further investigation.
Since previous evidence suggests that cognition in patients with MVND may decline at a rate greater than those without MVND [25], we investigated whether these associations between ceramide ratios and cognition would be stronger in participants with MVND at baseline. This was not as apparent likely due to the small number of patients with possible MVND in our exploratory analysis, which possibly limited the power of our analysis. This warrants further investigation in a larger sample of patients with possible MVND.
Premature death of neurons and oligodendrocytes is a hallmark of progressive and irreversible cognitive deterioration as these terminally differentiated cells are not easily replaced. Several hypotheses have been proposed to explain how ceramides contribute to neuronal death [51]. Given a plethora of evidence and the role of ceramides in multiple biochemical pathways, there are likely multiple mechanisms by which this occurs. Ceramides are consistently associated with excessive apoptosis; increased level of ceramides induces release of tumor necrosis factor alpha, which triggers cell death [52], while its depletion hinders apoptotic progression [53]. Alternatively, it may amplify signals in the programmed cell death pathway through spatial reorganization of the plasma membrane [51].
Our present study has numerous strengths. We adjusted for the varying time period between study visits by using a spatial covariance structure in our mixed models. In addition, we also investigated the possible perturbations of three CerC18:0 biosynthesis pathways to ensure a comprehensive analysis of CerC18:0 metabolism. Although metabolites specific for the de novo pathway were not assessed, ceramide synthesis through this pathway accounts for only a minority of brain sphingolipids. Furthermore, we adjusted for pertinent covariates, particularly age. Our findings remained significant after adjustment for age, contributing to evidence that altered sphingolipid metabolism is independent of the aging process [41].
There are some limitations in our analysis. First, our limited sample size cannot accommodate the addition of other covariates such as baseline cardiopulmonary fitness or APOE4 genotype that have been shown to be associated with plasma ceramide levels or verbal memory performance [54, 55]. Future studies should strive to adjust for these covariates if there is an adequate sample size. Secondly, we were not able to elucidate the independent influence of CAD on plasma ceramides levels as we did not have a healthy control group. However, this population of CAD patients with their perturbed lipid metabolism and no clinical cognitive impairment is the ideal population to investigate our hypotheses as they are representative of the pre-symptomatic stage. That said, our analyses should be investigated in a larger, independent population in validation studies. Furthermore, given that perturbed CerC18:0 ratios suggest change in enzymatic activity, future investigations should directly measure levels of enzymatic activity using biochemical assays and compare levels of activity. Relevant transporters should also be taken into consideration as well. Lastly, the mass spectroscopy method used was not able to isolate glucosylceramides from galactosylceramides, because of the technical difficulties in separation of these lipids [56]. As such, more extensive techniques should be used to separate them in future studies to see if there are clinically impactful differences between these two related isomers.
In summary, higher baseline ratios of plasma CerC18:0/SM18:0 and CerC18:0/MHxCer18:0 were correlated with declining verbal memory performance in CAD participants. These ratios may indicate perturbations within the catabolic pathway and salvage pathway of ceramides biosynthesis, respectively. In the catabolic pathway, post-hoc analyses replicated this association in participants with possible MVND, but not in those with no possible MVND. Our present findings suggest that altered sphingolipid metabolism may be among the earliest neurobiological changes associated with vascular neurodegeneration and invite further research to determine whether its metabolic pathways may be a clinically relevant target for drug discovery. Future studies should measure the enzymes responsible for conversion of sphingolipid precursors into CerC18:0 to directly assess enzymatic activity and correlate change in ceramide levels with cognitive change.
ACKNOWLEDGMENTS
This study was supported by a research grant from the Canadian Institutes of Health Research (LanctotMOP-114913). The Canadian Institutes of Health Research did not have any role in the study design, analysis, and interpretation of data. NJH is supported by the National Institutes of Health (MH105280, MH075673, DA040390, MH096630, and MH110246). MMM receives research support from the National Institutes of Health (R01 AG49704, P50 AG44170, U01 AG06786 RF1 AG55151), Department of Defense (W81XWH-15-1), and unrestricted research grants from Biogen, Roche, and Lundbeck.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0030r1).
REFERENCES
[1] | Wentzel C , Rockwood K , MacKnight C , Hachinski V , Hogan DB , Feldman H , Østbye T , Wolfson C , Gauthier S , Verreault R , McDowell I ((2001) ) Progression of impairment in patients with vascular cognitive impairment without dementia. Neurology 57: , 714–716. |
[2] | Rockwood K ((2002) ) Vascular cognitive impairment and vascular dementia. J Neurol Sci 203: , 23–27. |
[3] | Ikeda M ((2003) ) Prevention and early intervention for vascular dementia in community dwelling elderly: Findings from the Nakayama study. Psychogeriatrics 3: , 17–20. |
[4] | Koschack J , Irle E ((2005) ) Small hippocampal size in cognitively normal subjects with coronary artery disease. Neurobiol Aging 26: , 865–871. |
[5] | Barekatain M , Askarpour H , Zahedian F , Walterfang M , Velakoulis D , Maracy MR , Jazi MH ((2014) ) The relationship between regional brain volumes and the extent of coronary artery disease in mild cognitive impairment. J Res Med Sci 19: , 739–745. |
[6] | Debette S , Bombois S , Bruandet A , Delbeuck X , Lepoittevin S , Delmaire C , Leys D , Pasquier F ((2007) ) Subcortical hyperintensities are associated with cognitive decline in patients with mild cognitive impairment. Stroke 38: , 2924–2930. |
[7] | Vinkers DJ , Stek ML , van der Mast RC , de Craen AJ , Le Cessie S , Jolles J , Westendorp RG , Gussekloo J ((2005) ) Generalized atherosclerosis, cognitive decline, and depressive symptoms in old age. Neurology 65: , 107–112. |
[8] | Roberts RO , Knopman DS , Geda YE , Cha RH , Roger VL , Petersen RC ((2010) ) Coronary heart disease is associated with non-amnestic mild cognitive impairment. Neurobiol Aging 31: , 1894–1902. |
[9] | van Oijen M , de Jong FJ , Witteman JC , Hofman A , Koudstaal PJ , Breteler MM ((2007) ) Atherosclerosis and risk for dementia. Ann Neurol 61: , 403–410. |
[10] | Garrett KD , Browndyke JN , Whelihan W , Paul RH , DiCarlo M , Moser DJ , Cohen RA , Ott BR ((2004) ) The neuropsychological profile of vascular cognitive impairment–no dementia: Comparisons to patients at risk for cerebrovascular disease and vascular dementia. Arch Clin Neuropsychol 19: , 745–757. |
[11] | Uchida Y , Uchida Y , Kobayashi T , Shirai S , Hiruta N , Shimoyama E , Tabata T ((2017) ) Detection of ceramide, a risk factor for coronary artery disease, in human coronary plaques by fluorescent angioscopy. Circ J 81: , 1886–1893. |
[12] | Mielke MM , Haughey NJ , Bandaru VVR , Zetterberg H , Blennow K , Andreasson U , Johnson SC , Gleason CE , Blazel HM , Puglielli L , Sager MA , Asthana S , Carlsson CM ((2014) ) Cerebrospinal fluid sphingolipids, β-amyloid, and tau in adults at risk for Alzheimer’s disease. Neurobiol Aging 35: , 2486–2494. |
[13] | Mielke MM , Bandaru VV , McArthur JC , Chu M , Haughey NJ ((2010) ) Disturbance in cerebral spinal fluid sphingolipid content is associated with memory impairment in subjects infected with the human immunodeficiency virus. J Neurovirol 16: , 445–456. |
[14] | Mielke MM , Bandaru VV , Haughey NJ , Rabins PV , Lyketsos CG , Carlson MC ((2010) ) Serum sphingomyelins and ceramides are early predictors of memory impairment. Neurobiol Aging 31: , 17–24. |
[15] | Saleem M , Herrmann N , Dinoff A , Mielke MM , Oh PI , Shammi P , Cao X , Venkata SLV , Haughey NJ , Lanctôt KL ((2017) ) A lipidomics approach to assess the association between plasma sphingolipids and verbal memory performance in coronary artery disease patients undertaking cardiac rehabilitation: A C18:0 signature for cognitive response to exercise. J Alzheimers Dis 60: , 829–841. |
[16] | Filippov V , Song MA , Zhang K , Vinters HV , Tung S , Kirsch WM , Yang J , Duerksen-Hughes PJ ((2012) ) Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J Alzheimers Dis 29: , 537–547. |
[17] | Mielke MM , Haughey NJ , Bandaru VV , Schech S , Carrick R , Carlson MC , Mori S , Miller MI , Ceritoglu C , Brown T , Albert M , Lyketsos CG ((2010) ) Plasma ceramides are altered in mild cognitive impairment and predict cognitive decline and hippocampal volume loss. Alzheimers Dement 6: , 378–385. |
[18] | Lee JT , Xu J , Lee JM , Ku G , Han X , Yang DI , Chen S , Hsu CY ((2004) ) Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol 164: , 123–131. |
[19] | Jana A , Pahan K ((2007) ) Oxidative stress kills human primary oligodendrocytes via neutral sphingomyelinase: Implications for multiple sclerosis. J Neuroimmune Pharmacol 2: , 184–193. |
[20] | Jana A , Hogan EL ((2009) ) Pahan K. Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci 278: , 5–15. |
[21] | Mielke MM , Haughey NJ , Bandaru VV , Weinberg DD , Darby E , Zaidi N , Pavlik V , Doody RS , Lyketsos CG ((2011) ) Plasma sphingomyelins are associated with cognitive progression in Alzheimer’s disease. J Alzheimers Dis 27: , 259–269. |
[22] | Kitatani K , Idkowiak-Baldys J , Hannun YA ((2008) ) The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal 20: , 1010–1018. |
[23] | Gorelick PB , Scuteri A , Black SE , Decarli C , Greenberg SM , Iadecola C , Launer LJ , Laurent S , Lopez OL , Nyenhuis D , Petersen RC , Schneider JA , Tzourio C , Arnett DK , Bennett DA , Chui HC , Higashida RT , Lindquist R , Nilsson PM , Roman GC , Sellke FW , Seshadri S , American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia ((2011) ) Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42: , 2672–713. |
[24] | Intzandt B , Black SE , Lanctôt KL , Herrmann N , Oh P , Middleton LE ((2015) ) Is cardiac rehabilitation exercise feasible for people with mild cognitive impairment? Can Geriatr J 18: , 65–72. |
[25] | Stern RG , Mohs RC , Davidson M , Schmeidler J , Silverman J , Kramer-Ginsberg E , Searcey T , Bierer L , Davis KL ((1994) ) A longitudinal study of Alzheimer’s disease: Measurement, rate, and predictors of cognitive deterioration. Am J Psychiatry 151: , 390–396. |
[26] | Molloy DW , Standish TI ((1997) ) A guide to the standardized Mini-Mental State Examination. Int Psychogeriatr 9: (Suppl 1), 87–94; discussion 143-150. |
[27] | Suridjan I , Herrmann N1 , Adibfar A , Saleem M , Andreazza A , Oh PI , Lanctôt KL ((2017) ) Lipid peroxidation markers in coronary artery disease patients with possible vascular mild cognitive impairment. J Alzheimers Dis 58: , 885–896. |
[28] | Hachinski V , Iadecola C , Petersen RC , Breteler MM , Nyenhuis DL , Black SE , Powers WJ , DeCarli C , Merino JG , Kalaria RN , Vinters HV , Holtzman DM , Rosenberg GA , Wallin A , Dichgans M , Marler JR , Leblanc GG ((2006) ) National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37: , 2220–2241. |
[29] | Rabin LA , Paré N , Saykin AJ , Brown MJ , Wishart HA , Flashman LA , Santulli RB ((2009) ) Differential memory test sensitivity for diagnosing amnestic mild cognitive impairment and predicting conversion to Alzheimer’s disease. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn 16: , 357–376. |
[30] | Gaudino EA , Geisler MW , Squires NK ((1995) ) Construct validity in the Trail Making Test: What makes Part B harder? J Clin Exp Neuropsychol 17: , 529–535. |
[31] | Benedict RHB , Groninger L , Schretlen D , Dobraski M , Shpritz B ((1996) ) Revision of the brief visuospatial memory test: Studies of normal performance, reliability, and, validity. Psychol Assess 8: , 145–153. |
[32] | Joy S , Kaplan E , Fein D ((2004) ) Speed and memory in the WAIS-III Digit Symbol–Coding subtest across the adult lifespan. Arch Clin Neuropsychol 19: , 759–767. |
[33] | Delis DC , Kramer JH , Kaplan E , Ober BA ((2000) ) Manual for the California Verbal Learning Test, (CVLT-II). The Psychological Corporation. San Antonio, TX. |
[34] | Weuve J , Kang JH , Manson JE , Breteler MM , Ware JH , Grodstein F ((2004) ) Physical activity, including walking, and cognitive function in older women. JAMA 292: , 1454–1461. |
[35] | Harrison J , Minassian SL , Jenkins L , Black RS , Koller M , Grundman M ((2007) ) A neuropsychological test battery for use in Alzheimer disease clinical trials. Arch Neurol 64: , 1323–1329. |
[36] | Mielke MM , Bandaru VV , Han D , An Y , Resnick SM , Ferrucci L , Haughey NJ ((2015) ) Factors affecting longitudinal trajectories of plasma sphingomyelins: The Baltimore Longitudinal Study of Aging. Aging Cell 14: , 112–121. |
[37] | Pan W , Yu J , Shi R , Yan L , Yang T , Li Y , Zhang Z , Yu G , Bai Y , Schuchman EH , He X , Zhang G ((2014) ) Elevation of ceramide and activation of secretory acid sphingomyelinase in patients with acute coronary syndromes. Coron Artery Dis 25: , 230–235. |
[38] | Tabas I , Williams KJ , Borén J ((2007) ) Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 116: , 1832–1844. |
[39] | Katsel P , Li C , Haroutunian V ((2007) ) Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: A shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem Res 32: , 845–856. |
[40] | Dinkins MB , Enasko J , Hernandez C , Wang G , Kong J , Helwa I , Liu Y , Terry AVJr, Bieberich E ((2016) ) Neutral sphingomyelinase-2 deficiency ameliorates Alzheimer’s disease pathology and improves cognition in the 5XFAD mouse. J Neurosci 36: , 8653–8667. |
[41] | Suzuki K ((2003) ) Globoid cell leukodystrophy (Krabbe’s disease): Update. J Child Neurol 18: , 595–603. |
[42] | Malandrini A , D’Eramo C , Palmeri S , Gaudiano C , Gambelli S , Sicurelli F , Berti G , Formichi P , Kuqo A , Dotti MT , Federico A ((2013) ) Peripheral neuropathy in late-onset Krabbe disease: Report of three cases. Neurol Sci 34: , 79–83. |
[43] | Couttas TA , Kain N , Daniels B , Lim XY , Shepherd C , Kril J , Pickford R , Li H , Garner B , Don AS ((2014) ) Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol Commun 2: , 9. |
[44] | Ceccom J , Loukh N , Lauwers-Cances V , Touriol C , Nicaise Y , Gentil C , Uro-Coste E , Pitson S , Maurage CA , Duyckaerts C , Cuvillier O , Delisle MB ((2014) ) Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta Neuropathol Commun 2: , 12. |
[45] | Xia P , Gamble JR , Rye KA , Wang L , Hii CS , Cockerill P , Khew-Goodall Y , Bert AG , Barter PJ , Vadas MA ((1998) ) Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci U S A 95: , 14196–14201. |
[46] | Deutschman DH , Carstens JS , Klepper RL , Smith WS , Page MT , Young TR , Gleason LA , Nakajima N , Sabbadini RA ((2003) ) Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am Heart J 146: , 62–68. |
[47] | Sattler KJ , Elbasan S , Keul P , Elter-Schulz M , Bode C , Gräler MH , Bröcker-Preuss M , Budde T , Erbel R , Heusch G , Levkau B ((2010) ) Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol 105: , 821–832. |
[48] | Karunakaran I , van Echten-Deckert G ((2017) ) Sphingosine 1-phosphate - A double edged sword in the brain, Biochim Biophys Acta 1859: (9 Pt B), 1573–1582. |
[49] | MerrillJr AH, Sullards MC , Wang E , Voss KA , Riley RT ((2001) ) Sphingolipid metabolism: Roles in signal transduction and disruption by fumonisins. Environ Health Perspect 109: (Suppl 2), 283–289. |
[50] | Unger RH ((2002) ) Lipotoxic diseases. Annu Rev Med 53: , 319–336. |
[51] | Mencarelli C , Martinez-Martinez P ((2013) ) Ceramide function in the brain: When a slight tilt is enough. Cell Mol Life Sci 70: , 181–203. |
[52] | Obeid LM , Linardic CM , Karolak LA , Hannun YA ((1993) ) Programmed cell death induced by ceramide. Science 259: , 1769–1771. |
[53] | Bose R , Verheij M , Haimovitz-Friedman A , Scotto K , Fuks Z , Kolesnick R ((1995) ) Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 82: , 405–414. |
[54] | Fabbri E , Yang A , Simonsick EM , Chia CW , Zoli M , Haughey NJ , Mielke MM , Ferrucci L , Coen PM ((2016) ) Circulating ceramides are inversely associated with cardiorespiratory fitness in participants aged 54-96 years from the Baltimore Longitudinal Study of Aging. Aging Cell 15: , 825–831. |
[55] | Bandaru VV , Troncoso J , Wheeler D , Pletnikova O , Wang J , Conant K , Haughey NJ ((2009) ) ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol Aging 30: , 591–599. |
[56] | Savica R , Murray ME , Persson XM , Kantarci K , Parisi JE , Dickson DW , Petersen RC , Ferman TJ , Boeve BF , Mielke MM ((2016) ) Plasma sphingolipid changes with autopsy-confirmed Lewy body or Alzheimer’s pathology. Alzheimers Dement (Amst) 3: , 43–50. |


