
Multimodal Neuroimaging in Alzheimer’s Disease: Early Diagnosis, Physiopathological Mechanisms, and Impact of Lifestyle
Abstract
Over the last ten years, we have conducted research in Alzheimer’s disease (AD) using multimodal neuroimaging techniques to improve diagnosis, further our understanding of the pathological mechanisms underlying the disease, and support the development of innovative non-pharmacological preventive strategies. Our works emphasized the interest of hippocampal subfield volumetry in early diagnosis and the need for further development in this field including optimization, standardization, and automatization of the techniques. Also, we conducted several studies in cognitively intact at-risk elderly (e.g., subjective cognitive decline patients and APOE4 carriers) to better identify biomarkers associated with increased risk of developing AD. Regarding the physiopathological mechanisms, specific multimodal neuroimaging techniques allowed us to highlight the relevance of diaschisis, the mismatch between neurodegeneration and local Aβ deposition and the regional variation in the mechanisms underlying structural or functional alterations. Further works integrating other biomarkers known to play a role in the physiopathology of AD (tau, TDP-43, inflammation, etc.) in a longitudinal design would be useful to get a comprehensive understanding of their relative role, sequence, and causal relationships. Our works also highlighted the relevance of functional connectivity in further understanding the specificity of cognitive deficits in AD and how connectivity differentially influences the propagation of the different AD biomarkers. Finally, we conducted several studies on the links between lifestyle factors and neuroimaging biomarkers to unravel mechanisms of reserve. Further efforts are needed to better understand which lifestyle factor, or combination of factors, impact on AD pathology, and when, to help translating our knowledge to training programs that might prevent or delay brain and cognitive changes leading to AD dementia.
INTRODUCTION
Over the last twenty years, neuroimaging has increasingly contributed to major advances in Alzheimer’s disease (AD), especially for the clinical diagnosis and to improve our understanding of the pathophysiological mechanisms of the disease.
The contribution of neuroimaging to advances in AD diagnosis is well illustrated by the fact that the three most established neuroimaging markers for AD (hippocampal atrophy, temporo-parietal hypometabolism, and cortical amyloid-β (Aβ) deposition) have been recently included in the revised criteria for AD [1–5]; their presence increases the likelihood of AD etiology, including in predementia stages, e.g., in mild cognitive impairment (MCI). Nevertheless, progress is still needed to understand the use of these biomarkers and how they should be combined in the different stages of the disease. Moreover, more refined biomarkers are needed to increase the specificity and the sensitivity of the diagnosis, especially in early stages.
The pathophysiological mechanisms of AD are not fully understood. The two main neuropathological landmarks of AD are Aβ and tau-neurofibrillary tangles pathologies. Their relative role and sequence is still debated. The main hypothesis is that Aβ accumulation is the (only) causative agent of AD pathology and that tau-neurofibrillary tangles, neuronal dysfunction, cell loss, vascular damage, cognitive deterioration, and dementia follow as a direct result of this initial Aβ deposition [6, 7]. This position is yet challenged by recent neuroimaging evidence with Aβ PET imaging (e.g., using PIB or Florbetapir) highlighting Aβ-independent tau-related neuronal injury [8–10]. Another key element in the study of AD mechanisms—and especially when considering the topography and propagation of the lesions—is to consider the structural and functional architecture of the normal brain. Indeed, neuroimaging studies have shown that neurodegenerative diseases initially target, and then spread within pre-existing brain networks (i.e., interconnected brain regions), which leads to a novel concept called the network degeneration hypothesis [11–13].
Over and above clinical diagnosis and mechanisms understanding, the development of new therapeutic strategies is urgently needed. As mentioned above, AD is a multifactorial disease that likely results from the complex interplay of multiple pathological processes, under the influence of internal and external determinants. The repeated failure of clinical trials strengthens the need to develop global strategies that may prevent, delay, and/or downregulate several of these AD pathological processes. In this context, there is a growing interest in the impact of modifiable environmental or lifestyle factors not only on AD but also more generally on cognition, mental health, and wellbeing in the aging population. Neuroimaging participates in these rising developments by providing tools to test the relationships between these factors and biomarkers of aging and AD and to monitor the effects of interventions based on lifestyle changes.
I have been asked to contribute an article focused on the implications of my work on AD, and where I see, or would like to see, the field moving in the future. The three following sections will thus give an overview of the contribution of the research I conducted with my team to the three main areas of investigation in AD research mentioned above, namely, early diagnosis of AD, elucidation of its pathophysiological mechanisms, and assessment of lifestyle factors for development of intervention strategies. These works and more generally recent advances in the field has led to new perspectives and questions that pave the way of future research. The last section will be dedicated to my perspective on the future of AD research and gives examples of ongoing and future research projects that we are running or would like to run in my laboratory.
EARLY DIAGNOSIS OF AD
Hippocampal subfield volumetry
Hippocampal atrophy is well-known as an early biomarker of AD, but it lacks specificity as it is also observed in many different situations, such as normal aging and several neurologic and psychiatric disorders including other neurodegenerative diseases (e.g., frontotemporal dementia, including semantic dementia (SD) [14]). Neuropathological studies have shown that hippocampal subfields (subiculum, CA1-4, and dentate gyrus) are differentially vulnerable to AD; hippocampal subfield volumetry may thus prove to be more accurate than global hippocampal volumetry to detect AD. This has been confirmed in an early work where we used a voxelwise analyses coupled with 3D hippocampal surface mapping to illustrate the discrepancies between the effects of AD versus normal aging on hippocampal subfield volumes, with a preferential involvement of the CA1 subfield versus the subiculum, respectively [15] (Fig. 1A. B). The same approach allowed us to illustrate the specificity of the relationships between hippocampal subfield volumes and episodic memory deficits in MCI patients [16]. These findings encouraged us to optimize a proton density sequence for very high resolution acquisition of the hippocampus and to develop guidelines for hippocampal subfield delineation [17] (Fig. 1C-E). Using this improved technology, we showed the differential involvement of the hippocampal subfields in normal aging, MCI, AD, and SD [17–19]. Thus, a linear effect of normal aging was observed on the subiculum from 20 to 90 years old, while the effect on CA1 volume was non-linear with a decrease starting from 50 years old only [19] (Fig. 1F). CA1 was the most sensitive subfield in early AD with higher accuracy to discriminate between MCI and cognitively normal elderly than the whole hippocampus [18]. By comparison, SD was characterized by an hemispheric and antero–posterior asymmetry, significantly more marked than in AD, with greater involvement of the left and anterior hippocampal subfields [18]. Coupled with resting-state functional MRI, this approach also allowed us to highlight the specificities in hippocampal subfield intrinsic connectivity with the cerebral cortex in healthy elderly as well as their changes in patients with amnestic MCI [19].
Fig.1
Differential alteration of hippocampal subfields in AD versus normal aging. The hippocampal subfields can be distinguished on 3D hippocampal surface views (A), and this technique showed predominant atrophy of the CA1 subfield in AD (B). Compared to standard resolution T1 MRI (C), a high-resolution proton density MRI sequence allows to visualize the hippocampus fine anatomy (D) and thus to delineate the different hippocampal subfields (E). This approach is promising for early AD diagnosis as it allows to distinguish the effects of AD from that of other conditions such as normal aging (F).
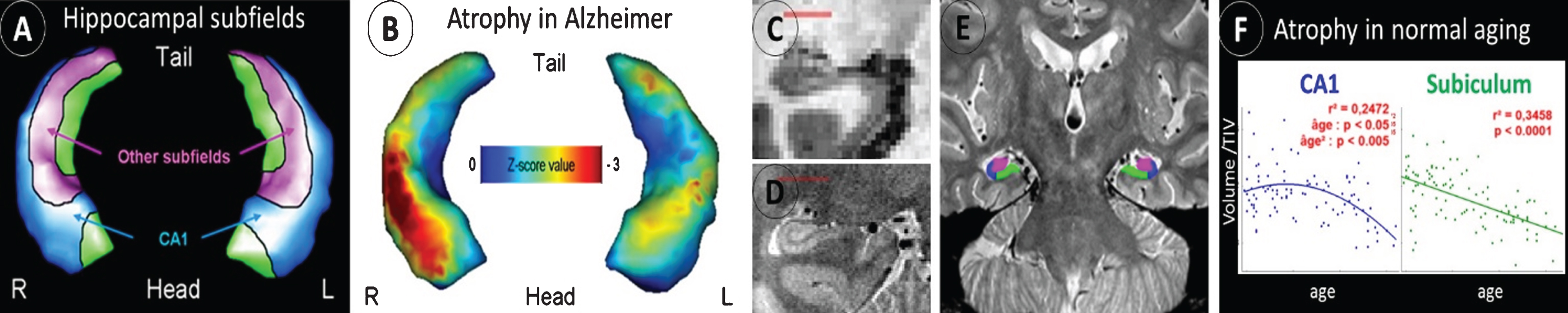
Altogether, these studies and other works conducted worldwide on hippocampal subfields in AD highlight the relevance of high-resolution hippocampal acquisition in the early and differential diagnosis of AD. An international collaborative project has developed, the Hippocampal Subfields Group, aiming at standardizing hippocampal subfield delineation and promoting research on this field [20].
AD diagnosis and multimodal neuroimaging
Over and above hippocampal atrophy, other neuroimaging measures are known to be altered in AD. The most recognized ones, which have been integrated in the revised AD criteria [1–5], are hypometabolism in posterior cingulate and temporoparietal areas as measured with FDG-PET, and cortical Aβ deposition measured with PET and different Aβ-binding tracers such as florbetapir. According to the amyloid cascade hypothesis, Aβ deposition is supposed to appear first, then followed by atrophy and/or hypometabolism (both considered as markers of neurodegeneration) [5, 8]. We have, however, proposed an alternative perspective of the neuropathological processes of the disease which has implications for the use of the neuroimaging biomarkers of AD [21–23] (Fig. 2). In our perspective, all neuroimaging biomarkers should be considered to the same degree (rather than sequentially), their presence being associated with an incremental increase in the risk of AD pathophysiology and of progression to AD dementia. To test this hypothesis, we used neuropsychological, structural MRI, FDG-PET, and florbetapir-PET data in cognitively intact elderly individuals [24]. We showed that atrophy and hypometabolism biomarkers provide independent and complementary rather than redundant information, and that cognitively normal elderly tend to have either neurodegeneration or Aβ deposition but not both, suggesting additive rather than sequential/causative links between AD neuroimaging biomarkers. These works argue for the use of neuroimaging biomarkers as partly independent evidences increasing the likelihood of AD etiology.
Fig.2
Hypothetical model illustrating the links between the main AD biomarkers and the underlying neuropathological processes. In this multidetermined perspective of the disease, Aβ and tau pathologies appear as at least partly independent processes, under the influence of genetic and environmental factors, and interact to lead to AD disease. Other neuropathological processes, some of which are still unknown, are likely also involved in the physiopathology of the disease. Adapted from [21, 22].
![Hypothetical model illustrating the links between the main AD biomarkers and the underlying neuropathological processes. In this multidetermined perspective of the disease, Aβ and tau pathologies appear as at least partly independent processes, under the influence of genetic and environmental factors, and interact to lead to AD disease. Other neuropathological processes, some of which are still unknown, are likely also involved in the physiopathology of the disease. Adapted from [21, 22].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g002.jpg)
Subjective cognitive decline (SCD)
The challenge in AD research is to diagnose the disease as early as possible to be able to assess the earliest pathophysiological processes and to intervene when the neurodegenerative process is still limited. The field has thus progressively moved towards the earliest stages such as subjective cognitive decline (SCD). Specific processes could be highlighted in this early stage; for instance, using data from the AIBL study in Melbourne, we found a specific relationship between atrophy and Aβ deposition in patients with SCD, but not in controls, MCI, or AD patients [25]. In an independent cohort of patients from the IMAP+ study in Caen, we showed that SCD was associated with hippocampal atrophy only when recruited from a memory clinic [26]. We also found that the same patients showed a profile of hippocampal subfield atrophy similar to that observed in AD and different from cognitively intact elderly (see above; [27]). Finally, we showed that detailed evaluation of SCD could provide accessible indication of the presence of cerebral Aβ or cognitive deficits [28]. Thus, we showed that specific SCD items (notably related with temporal disorientation) were associated with the presence of memory deficits in patients consulting at a memory clinic, and that stronger SCD (including for memory and attention) was associated with the presence of cortical Aβ deposition only in the asymptomatic elderly.
In sum, our research using neuroimaging biomarkers strengthens the view that SCD may represent a predementia stage of AD. We showed that the profile of brain atrophy associated with SCD resembles that observed in AD. However, all SCD patients will not develop AD and the challenge of future research will be to detect the pre-AD SCD. Developments in this field will be facilitated by the international initiative on SCD recently developed by expert researchers in the field [29].
Asymptomatic elderly carrying the APOE4 allele
The ɛ4 allele of the APOE (APOE4) is the major known genetic risk factor for late-onset AD [30]. Assessing brain changes in APOE4 carriers versus non-carriers in presymptomatic stages might help identifying early AD biomarkers. Based on a review of previous literature in the field, we proposed that APOE4 has a graded effect on the different AD biomarkers, with a predominant effect on Aβ deposition over brain structure and glucose metabolism [31] (Fig. 3). This view was supported by a study where we measured the effects of APOE, age, and the interaction between age and APOE on structural-MRI, FDG-PET, and Florbetapir-PET to provide a comprehensive and comparative assessment of APOE4 effects across the lifespan [32]. Thus, although decreases in brain volume and glucose metabolism with age tended to be stronger in noncarriers than in carriers, the difference between groups was not significant, while Aβ deposition was significantly higher, and increased faster with age, in carriers than non-carriers. These results reinforce the view that APOE4 mainly influences Aβ deposition, while the effects on neurodegeneration are at best subtle.
Fig.3
Schematic representation of the graded effect of APOE4 on structural MRI (atrophy), FDG-PET (metabolism), and molecular (Aβ deposition) cortical changes. APOE4 effects clearly predominate on Aβ deposition (thick arrows), while the effects are more modest on cortical metabolism and volume (thin arrows). This figure also illustrates that APOE4 operates through both Aβ-dependent and Aβ-independent processes. From [31].
![Schematic representation of the graded effect of APOE4 on structural MRI (atrophy), FDG-PET (metabolism), and molecular (Aβ deposition) cortical changes. APOE4 effects clearly predominate on Aβ deposition (thick arrows), while the effects are more modest on cortical metabolism and volume (thin arrows). This figure also illustrates that APOE4 operates through both Aβ-dependent and Aβ-independent processes. From [31].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g003.jpg)
UNDERSTANDING AD PATHOPHYSIOLOGICAL MECHANISMS
Multimodal imaging provides a unique opportunity to investigate the temporal and topographical relationship between distinct pathological variables, and thus improve our understanding of pathophysiological interactions in vivo. Working with multimodal neuroimaging techniques for 15 years, we developed original analysis techniques to take full advantage of the complementarity of the different neuroimaging modalities.
Role of cortical Aβ deposition
Working on the AIBL data together with Victor Villemagne and Christopher Rowe in Melbourne, we conducted a series of studies aiming to further our understanding of the role of Aβ deposition in AD pathophysiology, throughout different stages of the disease and in relation with atrophy and cognitive deficits. We showed that Aβ deposition was only poorly related with local atrophy (i.e., only in SCD patients in the posterior cingulate cortex) [25]. We found a reverse relationship in cognitively intact elderly such that those with Aβ deposition tended to have greater temporal volume (which might reflect brain reserve, see also below) [33]. Moreover, we showed that hippocampal atrophy and neocortical Aβ deposition both independently predicted episodic memory performances in non-demented individuals [34]. The presence of Aβ deposition in the neocortex of cognitively normal elderly was also associated with increased rate of brain cortical atrophy within the next two years [35] (Fig. 4). Finally, we demonstrated that the rate of Aβ accumulation varied according to the initial amount of Aβ deposition (i.e., higher rate was found in Aβ-positive compared to negative individuals) but not according to the cognitive state [36]. This series of works demonstrate that Aβ deposition only poorly explains local atrophy, but is associated with increased rate of atrophy over time. In other words, the presence of Aβ deposition is associated with a worse prognosis but the relationship between Aβ deposition and neurodegeneration is complex and indirect.
Fig.4
Comparison of the rate of atrophy over two years between cognitively intact older adults with (PIB+) and without (PIB-) Aβ deposition in their brain (as measured with PIB-PET) (Left). This study shows a greater rate of atrophy in PIB+ individuals, especially in the temporal neocortex and posterior and middle cingulate cortex (Right). From [35].
![Comparison of the rate of atrophy over two years between cognitively intact older adults with (PIB+) and without (PIB-) Aβ deposition in their brain (as measured with PIB-PET) (Left). This study shows a greater rate of atrophy in PIB+ individuals, especially in the temporal neocortex and posterior and middle cingulate cortex (Right). From [35].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g004.jpg)
Interesting, using data acquired in Caen in asymptomatic young to middle age adults, we were able to show that a physiological accumulation of Aβ starting from young adulthood and predominating in temporal lobes superimposed to the well-known medial frontal and parietal Aβ accumulation in late adulthood and AD [37].
Finally, in a collaborative work including data from Caen, Melbourne, Amsterdam, and San Francisco, we were able to study a series of 40 patients with a pre-scan clinical diagnosis of AD dementia but who had a negative Aβ PET scan [38]. We assessed their clinical and demographic features, patterns of brain atrophy and hypometabolism, and longitudinal clinical trajectories compared to a group of Aβ-positive AD and Aβ-negative controls. The main conclusions were that 1) the diagnosis was changed after the Aβ PET scan in almost all non-amnestic Aβ-negative AD cases and the individual profiles of atrophy and glucose metabolism helped to find an alternative diagnosis, which was most often confirmed by the clinical follow-up; 2) in the amnestic Aβ-negative AD cases, however, an alternative diagnosis could not be found in almost half of the cases as, although they had no Aβ, they mimic AD dementia in their clinical presentation and trajectory. These cases could thus not be classified as AD based on the neuropathologic definition of this disease. This study emphasizes the need to define a clinical framework and terminology for the classification of these patients, who likely represent a mixed population of limbic-predominant AD-mimics.
Regional variations in the relative degree of the different AD biomarkers
In a series of works, we compared the relative degree of the different alterations using a method that we specifically developed for the purpose of multimodal neuroimaging analyses [39, 40]. This allowed us to highlight differences in the degree of atrophy, hypometabolism, and Aβ deposition across brain regions. We thus found that the hippocampus showed disproportionate atrophy (intermediate level of hypometabolism and almost no Aβ deposition), posterior associative temporal and parietal cortical areas showed disproportionate hypometabolism compared with atrophy (and important degree of Aβ deposition), while the frontal cortex was characterized by very high Aβ deposition and relatively weak atrophy and hypometabolism (Fig. 5). Interestingly, we showed in a more recent work that the expression of these patterns varied across different groups of patients at-risk for AD [41]. Thus, in SCD patients only the atrophy-predominant pattern was detected, while APOE4 carriers only demonstrated the frontal amyloid-predominant pattern. These findings altogether suggest that there might be different underlying mechanisms, and maybe different sequences, in the different groups of brain regions and across different at-risk populations.
Fig.5
Regional variation in the degree of biomarkers. Some regions show predominant atrophy (left panel), others have higher hypometabolism than atrophy (middle panel), and Aβ deposition predominates in other areas (right panel). This suggests differences in the underlying pathophysiological mechanisms. From [39].
![Regional variation in the degree of biomarkers. Some regions show predominant atrophy (left panel), others have higher hypometabolism than atrophy (middle panel), and Aβ deposition predominates in other areas (right panel). This suggests differences in the underlying pathophysiological mechanisms. From [39].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g005.jpg)
Local and distant relationships between the different neuroimaging biomarkers
In a series of studies, we investigated the local and distant relationships between the different biomarkers. This allowed us to demonstrate that hypometabolism correlates with local atrophy (by contrast to Aβ deposition) suggesting that both alterations share at least partly common underlying mechanisms [40]. A significant proportion of hypometabolism and atrophy remains unrelated though [42]. We showed that disproportionate hypometabolism at least partly reflects diaschisis mechanisms, i.e., long distant effect of hippocampal atrophy on disconnected brain areas [43]. In a follow-up study, we used longitudinal neuroimaging data to provide support for the sequence of events and their causality [44] (Fig. 6).
Fig.6
Distant relationships between atrophy and hypometabolism in AD. Using original methods especially developed for this purpose, we showed that hippocampal atrophy (red) was at least partly responsible for the disruption of white matter fibers (the perforant path in blue and the uncinate fasciculus in yellow) (1) itself responsible for hypometabolism in the posterior cingulate (green) and medial orbitofrontal cortex (purple and light blue) (2). From [42].
![Distant relationships between atrophy and hypometabolism in AD. Using original methods especially developed for this purpose, we showed that hippocampal atrophy (red) was at least partly responsible for the disruption of white matter fibers (the perforant path in blue and the uncinate fasciculus in yellow) (1) itself responsible for hypometabolism in the posterior cingulate (green) and medial orbitofrontal cortex (purple and light blue) (2). From [42].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g006.jpg)
The role of intrinsic connectivity in the pathophysiology of AD and SD
In addition to tau and Aβ pathologies, another key element in the pathophysiology of AD is brain structural and functional connectivity. Over and above diaschisis mechanisms mentioned above, other works conducted in our laboratory offered evidence for this purpose. First, we showed in cognitively intact individuals that functional connectivity measured with resting-state fMRI (within the default mode network that is especially relevant in AD) was related to cognitive performance, specifically in autobiographical memory, and not with inner experience [45]. We also showed the relevance of functional connectivity to explain certain cognitive manifestation of AD such as anosognosia, i.e., the lack of consciousness of cognitive (memory) deficits [46]. Thus, we showed that anosognosia in AD results from a disruption of the communication between memory-related and the self-related brain networks. Using multiple imaging techniques including functional connectivity with resting-state fMRI, we also investigated the paradox of SD, i.e., the intriguing relative preservation of episodic memory in SD despite similar degree of hippocampal atrophy compared with AD [12]. We found that both diseases affect brain regions that are connected to the hippocampus, but only the connectivity with brain regions affected in AD are important for episodic memory in healthy individuals. We also showed that both diseases target different hippocampal networks, probably because they differentially affect the anterior versus posterior parts of the hippocampus, which are known to be connected to different brain regions [12]. This hypothesis found support in a recent evidence showing that the atrophy common to both AD and SD is associated with alterations in different white matter tracts, i.e., mainly the cingulum and corpus callosum in AD versus the uncinate and inferior longitudinal fasciculi in SD [47] (Fig. 7).
Fig.7
Relationships between medial temporal lobe atrophy common to AD and SD (center panel) and whole-brain white matter density maps in patients with AD (top panel) and semantic dementia (bottom panel). From [45].
![Relationships between medial temporal lobe atrophy common to AD and SD (center panel) and whole-brain white matter density maps in patients with AD (top panel) and semantic dementia (bottom panel). From [45].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g007.jpg)
There is growing recognition for the relevance of connectivity in the propagation of the disease; thus, neuroimaging studies have shown that neurodegenerative diseases target brain networks (i.e., interconnected brain regions), which leads to a novel concept called the network degeneration hypothesis [11–13]. We also explored how much connectivity influences the topography and propagation of lesions in AD. We contributed to this area in showing that the influence of brain connectivity in AD lesion propagation depends on the neuroimaging modality. Thus, first using a cross sectional design, we showed that atrophy and intrinsic connectivity disruption were only present in the ventral posterior cingulate cortex (PCC) in MCI patients and spread to the dorsal PCC network in AD patients, while hypometabolism was present in both networks since the aMCI stage, possibly reflecting not only local disruption but also distant synaptic dysfunction [48]. Then we used longitudinal multimodal neuroimaging data in AD patients and showed that atrophy spread in regions with high specific connectivity, consistently with the transneuronal propagation hypothesis, while hypometabolism propagated in areas showing high global connectivity (that were also more vulnerable to Aβ deposition), in line with the hypothesis of higher vulnerability of hubs to hypometabolism and Aβ deposition [49].
LIFESTYLE VERSUS AD NEUROIMAGING BIOMARKERS
The alternative model we proposed on the pathophysiological mechanisms of AD [21, 22] (Fig. 2) recognizes the influence of environmental factors on AD pathophysiological mechanisms and processes. As a matter of fact, there is growing evidence in the literature that we could modify the course of the disease, and brain and mental health in general, by modifying our lifestyle [50–53]. We showed for example that higher education was able to counteract the effects of APOE ɛ4 on metabolism independently of Aβ deposition, as increased metabolism with education was found in APOE ɛ4 carriers in critical regions that sustain episodic memory performance [54]. In another study, we assessed the links between lifestyle factors and different neuroimaging measures including markers of AD. Thus, assessing the relationships between years of education and brain volume, metabolism, and connectivity, we showed that, in healthy elderly with no evidence for Aβ deposition, there was a positive relationship between education and brain volume and metabolism, especially in the anterior cingulate cortex [55]. Moreover, the connectivity of this region increased with increasing years of education especially with the hippocampus and posterior cingulate cortex, two regions particularly important in AD. By contrast, in a collaborative project, we found negative relationships between education and brain metabolism and connectivity in asymptomatic older adults including individuals with Aβ deposition [56]. We think that these apparently discrepant findings with positive versus negative relationships, also found in the literature, reflect the progression from neuroprotective to compensation processes over the course of the disease, which we summarized in an integrative model [57] (Fig. 8). Thus, in individuals without AD lesions, education is related with increased brain performances while when AD-related pathology appears, education is related with increased resistance to brain lesions so that at the same level of cognitive impairment, more lesions will be found in those with higher education. This model was supported by a recent study where we showed that higher education was associated with lower Aβ deposition in normal older adults but with higher Aβ deposition in MCI [58]. Moreover, in the same study we found increased FDG-PET uptake with education in MCI patients within the regions of higher Florbetapir-PET uptake, suggesting a compensatory increase in glucose metabolism. The findings suggest that early intellectual enrichment before the onset of dementia may be associated with protection in healthy asymptomatic elderly, and then with compensation from Aβ at the symptomatic stage.
Fig.8
Schematic theoretical representation of the differential expression of reserve mechanisms (neuroprotection versus compensation) across the spectrum from cognitively normal healthy adults to AD dementia. We propose that neuroprotection and brain maintenance predominates in healthy elderly while compensation processes predominate as AD progresses to dementia (probably up to a certain stage where compensation is not possible anymore). [54, 55].
![Schematic theoretical representation of the differential expression of reserve mechanisms (neuroprotection versus compensation) across the spectrum from cognitively normal healthy adults to AD dementia. We propose that neuroprotection and brain maintenance predominates in healthy elderly while compensation processes predominate as AD progresses to dementia (probably up to a certain stage where compensation is not possible anymore). [54, 55].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179920/jad-64-jad179920-g008.jpg)
Another relevant aspect to be further investigated in this area is the relative impact of different lifestyle factors. We started to assess this question by investigating the specific relationships between cognitive versus physical activity engagement during late-adulthood and gray matter volume in normal older adults. We showed independent relationships of the two lifestyle factors in both common and distinct brain areas, and found that the effects of late life cognitive and physical activity were independent from early cognitive engagement as reflected by years of education [59]. Further works are needed to understand the specific and synergic effects of different lifestyle factors, in different lifetime periods, as this information is crucial to design optimal non-pharmacological (preventive and therapeutic) intervention programs.
FUTURE DIRECTIONS
There is more and more acknowledgment that AD is a multifactorial disease resulting from the contribution of, and interaction between, several pathological factors. Hence, we need to consider developing further specific markers of these pathologies; tau-PET imaging is particularly challenging but significant progress has been made [60]. Specific markers of other pathological processes including TDP-43, inflammation, α-synuclein, etc., are also awaited. This is important both to improve the diagnosis of neurodegenerative diseases, but also to understand the physiopathological processes leading to the disease. More specifically, applying the multimodal analysis methods described above to multiple neuroimaging techniques targeting specific pathological processes, especially within longitudinal design, would allow us to obtain a comprehensive picture of their relative role, sequence, and causal relationships. Further works on the role of brain connectivity in the propagation of the different pathological processes are needed to understand the relative contribution of several predictors. This offers relevant lines for future research as it might help us to develop strategies to slow down or even stop the propagation process.
In parallel, efforts should continue to develop biomarkers that are more widely available and less expensive than PET and that do not necessitate radioactivity exposure, especially for clinical application. For instance, previous works point toward the potential for MRI-based biomarkers including hippocampal subfield volumetry and MRI-based proxies of brain metabolism and Aβ deposition (e.g., perfusion MRI and susceptibility-weighted imaging) as promising biomarkers for AD. Research efforts are needed for the optimization, validation, and standardization of these approaches for clinical use. This is on-going for instance with the international Hippocampal Subfields Group (http://www.hippocampalsubfields.com).
For improvement of early diagnosis, the field has progressively moved from the MCI to the SCD stage. More studies are needed in this direction to better understand and differentiate the several possible causes for SCD based on neuroimaging but also on refined cognitive, self-assessment, and psycho-affective measures. These developments are crucial not only for early AD diagnosis, but also more generally to provide better care for those elderly who are more and more worried about getting AD. The international SCD Initiative would help promoting these developments [29].
The development of treatments is still a priority for future research to prevent, delay, slow down, or halt the degenerative process. Innovative strategies should be developed, new paths should be considered, and pharmacological therapeutics should take into account the multifactorial dimension of the disease instead of treating one of the element. Besides, as we recognize the impact of environmental, lifestyle, and psychoaffective factors on AD risk, we should seize the opportunity to translate our knowledge to training programs that might prevent or delay brain and cognitive changes leading to AD dementia. The growing interest for these approaches is reflected in the recent advent of international initiatives, groups of experts, and meetings to promote research in this field (e.g., ISTAART professional interest area on Reserve, Resilience and Protective Factors and on non-pharmacological interventions: https://act.alz.org/site/SPageServer?pagename=ISTAART_PIA; 1st International Conference on Cognitive Reserve in the Dementias (ResDem): http://resdem2017.com/). Cognitive training programs, but also interventions based on physical or artistic activities, are being developed. Psychoaffective factors are less often considered, although stress, anxiety, and depression, all related to cognitive and sleep difficulties, are associated with increased risk for AD [61–63]. There is increased acknowledgment in the role for sleep in the physiopathology of the disease and we contributed to this knowledge showing specific relationships with neuroimaging markers [64], though more research is needed in this direction. Mental training for stress reduction and emotion regulation through meditation practice for instance might thus be particularly beneficial to elderly populations in reducing AD risk. In a pilot study, we showed that elderly expert meditators had higher gray matter volume and/or FDG metabolism compared to age-matched non-meditators in several frontal and parietal areas particularly sensitive to aging or AD effects [65]. These findings are encouraging as they suggest that meditation practice could reduce age-associated structural and functional brain changes. We are running a large European project including clinical trials assessing the effects of short and long-term meditation practice versus English learning and health education programs in elderly populations at-risk for AD (https://silversantestudy.fr/). Further works are needed to understand the specific and synergic effects of different lifestyle factors, in different lifetime periods, as this information is crucial to design optimal non-pharmacological (preventive and therapeutic) intervention programs.
ACKNOWLEDGMENTS
I would like to thank all the persons who contributed to these works: Florence Mézenge, Brigitte Landeau, Renaud La Joie, Julie Gonneaud, Audrey Perrotin, Eider Arenaza-Urquijo, Alexandre Bejanin, Robin de Flores, Clémence Tomadesso, Justine Mutlu, Nicolas Villain, Marine Fouquet, Katell Mevel, Francis Eustache, Béatrice Desgranges, Stéphanie Egret, Vincent de La Sayette, Jean-Claude Baron, Fausto Viader, Alice Pélerin, Malo Gaubert, Géraldine Poisnel, Géraldine Rauchs, Anne Quillard, Anne Chocat, Ahmed Abbas, Louisa Barré, Alain Manrique, Denis Guilloteau, Florence Pasquier, Serge Belliard, Christopher Rowe, Victor Villemagne, Antoine Lutz, the Cyceron staff members and the volunteers who were included in these studies.
Our works were supported by Institut National de la Santé et de la Recherche Médicale (Inserm), Fondation Plan Alzheimer (Alzheimer Plan 2008-2012); Programme Hospitalier de Recherche Clinique (PHRCN 2011-A01493-38 and PHRCN 2012 12-006-0347); Agence Nationale de la Recherche (LONGVIE 2007); Région Basse-Normandie; Association France Alzheimer et maladies apparentées, Fondation Vaincre Alzheimer.
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/17-9920).
REFERENCES
[1] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[2] | Dubois B , Feldman HH , Jacova C , Cummings JL , Dekosky ST , Barberger-Gateau P , Delacourte A , Frisoni G , Fox NC , Galasko D , Gauthier S , Hampel H , Jicha GA , Meguro K , O’Brien J , Pasquier F , Robert P , Rossor M , Salloway S , Sarazin M , de Souza LC , Stern Y , Visser PJ , Scheltens P ((2010) ) Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol 9: , 1118–1127. |
[3] | Dubois B , Hampel H , Feldman HH , Scheltens P , Aisen P , Andrieu S , Bakardjian H , Benali H , Bertram L , Blennow K , Broich K , Cavedo E , Crutch S , Dartigues J-F , Duyckaerts C , Epelbaum S , Frisoni GB , Gauthier S , Genthon R , Gouw AA , Habert M-O , Holtzman DM , Kivipelto M , Lista S , Molinuevo J-L , O’Bryant SE , Rabinovici GD , Rowe C , Salloway S , Schneider LS , Sperling R , Teichmann M , Carrillo MC , Cummings J , Jack CR , Proceedings of the Meeting of the International Working Group (IWG) and the American Alzheimer’s Association on “The Preclinical State of AD”, July 23, 2015, Washington DC, USA ((2016) ) Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement 12: , 292–323. |
[4] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CRJr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[5] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CRJr , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[6] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[7] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[8] | Jack CR , Wiste HJ , Weigand SD , Knopman DS , Lowe V , Vemuri P , Mielke MM , Jones DT , Senjem ML , Gunter JL , Gregg BE , Pankratz VS , Petersen RC ((2013) ) Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 81: , 1732–1740. |
[9] | Jagust WJ , Landau SM ((2012) ) Apolipoprotein E, not fibrillar β-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci 32: , 18227–18233. |
[10] | Knopman DS , Jack CR , Wiste HJ , Weigand SD , Vemuri P , Lowe VJ , Kantarci K , Gunter JL , Senjem ML , Mielke MM , Roberts RO , Boeve BF , Petersen RC ((2013) ) Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann Neurol 73: , 472–480. |
[11] | Seeley WW , Crawford RK , Zhou J , Miller BL , Greicius MD ((2009) ) Neurodegenerative diseases target large-scale human brain networks. Neuron 62: , 42–52. |
[12] | La Joie R , Landeau B , Perrotin A , Bejanin A , Egret S , Pélerin A , Mézenge F , Belliard S , de La Sayette V , Eustache F , Desgranges B , Chételat G ((2014) ) Intrinsic connectivity identifies the hippocampus as a main crossroad between Alzheimer’s and semantic dementia-targeted networks. Neuron 81: , 1417–1428. |
[13] | Raj A , Kuceyeski A , Weiner M ((2012) ) A network diffusion model of disease progression in dementia. Neuron 73: , 1204–1215. |
[14] | Geuze E , Vermetten E , Bremner JD ((2005) ) MR-based in vivo hippocampal volumetrics: 2. Findings in neuropsychiatric disorders. Mol Psychiatry 10: , 160–184. |
[15] | Chételat G , Fouquet M , Kalpouzos G , Denghien I , De la Sayette V , Viader F , Mézenge F , Landeau B , Baron JC , Eustache F , Desgranges B ((2008) ) Three-dimensional surface mapping of hippocampal atrophy progression from MCI to AD and over normal aging as assessed using voxel-based morphometry. Neuropsychologia 46: , 1721–1731. |
[16] | Fouquet M , Desgranges B , La Joie R , Rivière D , Mangin J-F , Landeau B , Mézenge F , Pélerin A , de La Sayette V , Viader F , Baron JC , Eustache F , Chételat G ((2012) ) Role of hippocampal CA1 atrophy in memory encoding deficits in amnestic mild cognitive impairment. Neuroimage 59: , 3309–3315. |
[17] | La Joie R , Fouquet M , Mézenge F , Landeau B , Villain N , Mevel K , Pélerin A , Eustache F , Desgranges B , Chételat G ((2010) ) Differential effect of age on hippocampal subfields assessed using a new high-resolution 3T MR sequence. Neuroimage 53: , 506–514. |
[18] | La Joie R , Perrotin A , de La Sayette V , Egret S , Doeuvre L , Belliard S , Eustache F , Desgranges B , Chételat G ((2013) ) Hippocampal subfield volumetry in mild cognitive impairment, Alzheimer’s disease and semantic dementia. Neuroimage Clin 3: , 155–162. |
[19] | de Flores R , La Joie R , Landeau B , Perrotin A , Mézenge F , de La Sayette V , Eustache F , Desgranges B , Chételat G ((2015) ) Effects of age and Alzheimer’s disease on hippocampal subfields: Comparison between manual and freesurfer volumetry. Hum Brain Mapp 36: , 463–474. |
[20] | Yushkevich PA , Amaral RSC , Augustinack JC , Bender AR , Bernstein JD , Boccardi M , Bocchetta M , Burggren AC , Carr VA , Chakravarty MM , Chételat G , Daugherty AM , Davachi L , Ding S-L , Ekstrom A , Geerlings MI , Hassan A , Huang Y , Iglesias JE , La Joie R , Kerchner GA , LaRocque KF , Libby LA , Malykhin N , Mueller SG , Olsen RK , Palombo DJ , Parekh MB , Pluta JB , Preston AR , Pruessner JC , Ranganath C , Raz N , Schlichting ML , Schoemaker D , Singh S , Stark CEL , Suthana N , Tompary A , Turowski MM , Van Leemput K , Wagner AD , Wang L , Winterburn JL , Wisse LEM , Yassa MA , Zeineh MM , for the Hippocampal Subfields Group (HSG) ((2015) ) Quantitative comparison of 21 protocols for labeling hippocampal subfields and parahippocampal subregions in in vivo MRI: Towards a harmonized segmentation protocol. Neuroimage 111: , 526–541. |
[21] | Chételat G ((2013) ) Alzheimer disease: Aβ-independent processes—rethinking preclinical AD. Nat Rev Neurol 9: , 123–124. |
[22] | Chételat G ((2013) ) Reply: The amyloid cascade is not the only pathway to AD. Nat Rev Neurol 9: , 356. |
[23] | Jack CR , Knopman DS , Chételat G , Dickson D , Fagan AM , Frisoni GB , Jagust W , Mormino EC , Petersen RC , Sperling RA , van der Flier WM , Villemagne VL , Visser PJ , Vos SJB ((2016) ) Suspected non-Alzheimer disease pathophysiology - concept and controversy. Nat Rev Neurol 12: , 117–124. |
[24] | Besson FL , La Joie R , Doeuvre L , Gaubert M , Mézenge F , Egret S , Landeau B , Barré L , Abbas A , Ibazizene M , de La Sayette V , Desgranges B , Eustache F , Chételat G ((2015) ) Cognitive and brain profiles associated with current neuroimaging biomarkers of preclinical Alzheimer’s Disease. J Neurosci 35: , 10402–10411. |
[25] | Chételat G , Villemagne VL , Bourgeat P , Pike KE , Jones G , Ames D , Ellis KA , Szoeke C , Martins RN , O’Keefe GJ , Salvado O , Masters CL , Rowe CC ((2010) ) Relationship between atrophy and beta-amyloid deposition in Alzheimer disease. Ann Neurol 67: , 317–324. |
[26] | Perrotin A , La Joie R , de La Sayette V , Barré L , Mézenge F , Mutlu J , Guilloteau D , Egret S , Eustache F , Chételat G ((2017) ) Subjective cognitive decline in cognitively normal elders from the community or from a memory clinic: Differential affective and imaging correlates. Alzheimers Dement 13: , 550–560. |
[27] | Perrotin A , de Flores R , Lamberton F , Poisnel G , La Joie R , de la Sayette V , Mézenge F , Tomadesso C , Landeau B , Desgranges B , Chételat G ((2015) ) Hippocampal subfield volumetry and 3D surface mapping in subjective cognitive decline. J Alzheimers Dis 48: (Suppl 1), S141–150. |
[28] | La Joie R , Perrotin A , Egret S , Pasquier F , Tomadesso C , Mézenge F , Desgranges B , de La Sayette V , Chételat G ((2016) ) Qualitative and quantitative assessment of self-reported cognitive difficulties in nondemented elders: Association with medical help seeking, cognitive deficits, and β-amyloid imaging. Alzheimers Dement (Amst) 5: , 23–34. |
[29] | Jessen F , Amariglio RE , van Boxtel M , Breteler M , Ceccaldi M , Chételat G , Dubois B , Dufouil C , Ellis KA , van der Flier WM , Glodzik L , van Harten AC , de Leon MJ , McHugh P , Mielke MM , Molinuevo JL , Mosconi L , Osorio RS , Perrotin A , Petersen RC , Rabin LA , Rami L , Reisberg B , Rentz DM , Sachdev PS , de la Sayette V , Saykin AJ , Scheltens P , Shulman MB , Slavin MJ , Sperling RA , Stewart R , Uspenskaya O , Vellas B , Visser PJ , Wagner M , Subjective Cognitive Decline Initiative (SCD-I) Working Group ((2014) ) A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 10: , 844–852. |
[30] | Genin E , Hannequin D , Wallon D , Sleegers K , Hiltunen M , Combarros O , Bullido MJ , Engelborghs S , De Deyn P , Berr C , Pasquier F , Dubois B , Tognoni G , Fiévet N , Brouwers N , Bettens K , Arosio B , Coto E , Del Zompo M , Mateo I , Epelbaum J , Frank-Garcia A , Helisalmi S , Porcellini E , Pilotto A , Forti P , Ferri R , Scarpini E , Siciliano G , Solfrizzi V , Sorbi S , Spalletta G , Valdivieso F , Vepsäläinen S , Alvarez V , Bosco P , Mancuso M , Panza F , Nacmias B , Bossù P , Hanon O , Piccardi P , Annoni G , Seripa D , Galimberti D , Licastro F , Soininen H , Dartigues JF , Kamboh MI , Van Broeckhoven C , Lambert JC , Amouyel P , Campion D ((2011) ) APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol Psychiatry 16: , 903–907. |
[31] | Fouquet M , Besson FL , Gonneaud J , La Joie R , Chételat G ((2014) ) Imaging brain effects of APOE4 in cognitively normal individuals across the lifespan. Neuropsychol Rev 24: , 290–299. |
[32] | Gonneaud J , Arenaza-Urquijo EM , Fouquet M , Perrotin A , Fradin S , de La Sayette V , Eustache F , Chételat G ((2016) ) Relative effect of APOE ɛ4 on neuroimaging biomarker changes across the lifespan. Neurology 87: , 1696–1703. |
[33] | Chételat G , Villemagne VL , Pike KE , Baron J-C , Bourgeat P , Jones G , Faux NG , Ellis KA , Salvado O , Szoeke C , Martins RN , Ames D , Masters CL , Rowe CC ((2010) ) Larger temporal volume in elderly with high versus low beta-amyloid deposition. Brain 133: , 3349–3358. |
[34] | Chételat G , Villemagne VL , Pike KE , Ellis KA , Bourgeat P , Jones G , O’Keefe GJ , Salvado O , Szoeke C , Martins RN , Ames D , Masters CL , Rowe CC ((2011) ) Independent contribution of temporal beta-amyloid deposition to memory decline in the pre-dementia phase of Alzheimer’s disease. Brain 134: , 798–807. |
[35] | Chételat G , Villemagne VL , Villain N , Jones G , Ellis KA , Ames D , Martins RN , Masters CL , Rowe CC ((2012) ) Accelerated cortical atrophy in cognitively normal elderly with high β-amyloid deposition. Neurology 78: , 477–484. |
[36] | Villain N , Chételat G , Grassiot B , Bourgeat P , Jones G , Ellis KA , Ames D , Martins RN , Eustache F , Salvado O , Masters CL , Rowe CC , Villemagne VL ((2012) ) Regional dynamics of amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: A voxelwise PiB-PET longitudinal study. Brain 135: , 2126–2139. |
[37] | Gonneaud J , Arenaza-Urquijo EM , Mézenge F , Landeau B , Gaubert M , Bejanin A , de Flores R , Wirth M , Tomadesso C , Poisnel G , Abbas A , Desgranges B , Chételat G ((2017) ) Increased florbetapir binding in the temporal neocortex from age 20 to 60 years. Neurology 89: , 2438–2446. |
[38] | Chételat G , Ossenkoppele R , Villemagne VL , Perrotin A , Landeau B , Mézenge F , Jagust WJ , Dore V , Miller BL , Egret S , Seeley WW , van der Flier WM , La Joie R , Ames D , van Berckel BNM , Scheltens P , Barkhof F , Rowe CC , Masters CL , de La Sayette V , Bouwman F , Rabinovici GD ((2016) ) Atrophy, hypometabolism and clinical trajectories in patients with amyloid-negative Alzheimer’s disease. Brain 139: , 2528–2539. |
[39] | Chételat G , Desgranges B , Landeau B , Mézenge F , Poline JB , de la Sayette V , Viader F , Eustache F , Baron J-C ((2008) ) Direct voxel-based comparison between grey matter hypometabolism and atrophy in Alzheimer’s disease. Brain 131: , 60–71. |
[40] | La Joie R , Perrotin A , Barré L , Hommet C , Mézenge F , Ibazizene M , Camus V , Abbas A , Landeau B , Guilloteau D , de La Sayette V , Eustache F , Desgranges B , Chételat G ((2012) ) Region-specific hierarchy between atrophy, hypometabolism, and β-amyloid (Aβ) load in Alzheimer’s disease dementia. J Neurosci 32: , 16265–16273. |
[41] | Wirth M , Bejanin A , La Joie R , Arenaza-Urquijo EM , Gonneaud J , Landeau B , Perrotin A , Mézenge F , de La Sayette V , Desgranges B , Chételat G ((2018) ) Regional patterns of gray matter volume, hypometabolism, and beta-amyloid in groups at risk of Alzheimer’s disease. Neurobiol Aging 63: , 140–151. |
[42] | Chételat G , Villain N , Desgranges B , Eustache F , Baron J-C ((2009) ) Posterior cingulate hypometabolism in early Alzheimer’s disease: What is the contribution of local atrophy versus disconnection? Brain 132: , e133; author reply e134. |
[43] | Villain N , Desgranges B , Viader F , de la Sayette V , Mézenge F , Landeau B , Baron J-C , Eustache F , Chételat G ((2008) ) Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer’s disease. J Neurosci 28: , 6174–6181. |
[44] | Villain N , Fouquet M , Baron J-C , Mézenge F , Landeau B , de la Sayette V , Viader F , Eustache F , Desgranges B , Chételat G ((2010) ) Sequential relationships between gray matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer’s disease. Brain 133: , 3301–3314. |
[45] | Mevel K , Landeau B , Fouquet M , La Joie R , Villain N , Mézenge F , Perrotin A , Eustache F , Desgranges B , Chételat G ((2013) ) Age effect on the default mode network, inner thoughts, and cognitive abilities. Neurobiol Aging 34: , 1292–1301. |
[46] | Perrotin A , Desgranges B , Landeau B , Mézenge F , La Joie R , Egret S , Pélerin A , de la Sayette V , Eustache F , Chételat G ((2015) ) Anosognosia in Alzheimer disease: Disconnection between memory and self-related brain networks. Ann Neurol 78: , 477–486. |
[47] | Bejanin A , Desgranges B , La Joie R , Landeau B , Perrotin A , Mézenge F , Belliard S , de La Sayette V , Eustache F , Chételat G ((2017) ) Distinct white matter injury associated with medial temporal lobe atrophy in Alzheimer’s versus semantic dementia. Hum Brain Mapp 38: , 1791–1800. |
[48] | Mutlu J , de Flores R , Tomadesso C , Landeau B , Mézenge F , de La Sayette V , Eustache F , Chételat G ((2015) ) Differential functional disruption, hypometabolism, and atrophy between ventral and dorsal posterior cingulate cortex networks in mild cognitive impairment and Alzheimer disease. Alzheimers Dement 11: (Suppl), P237. |
[49] | Mutlu J , Landeau B , Gaubert M , de La Sayette V , Desgranges B , Chételat G ((2017) ) Distinct influence of specific versus global connectivity on the different Alzheimer’s disease biomarkers. Brain 140: , 3317–3328. |
[50] | Baumgart M , Snyder HM , Carrillo MC , Fazio S , Kim H , Johns H ((2015) ) Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement 11: , 718–726. |
[51] | Bennett DA , Arnold SE , Valenzuela MJ , Brayne C , Schneider JA ((2014) ) Cognitive and social lifestyle: Links with neuropathology and cognition in late life. Acta Neuropathol (Berl) 127: , 137–150. |
[52] | Brookmeyer R , Johnson E , Ziegler-Graham K , Arrighi HM ((2007) ) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 3: , 186–191. |
[53] | Valenzuela MJ , Matthews FE , Brayne C , Ince P , Halliday G , Kril JJ , Dalton MA , Richardson K , Forster G , Sachdev PS , Medical Research Council Cognitive Function and Ageing Study ((2012) ) Multiple biological pathways link cognitive lifestyle to protection from dementia. Biol Psychiatry 71: , 783–791. |
[54] | Arenaza-Urquijo EM , Gonneaud J , Fouquet M , Perrotin A , Mézenge F , Landeau B , Egret S , De la Sayette V , Desgranges B , Chételat G ((2015) ) Interaction between years of education and APOE ɛ4 status on frontal and temporal metabolism. Neurology 85: , 1392–1399. |
[55] | Arenaza-Urquijo EM , Landeau B , La Joie R , Mevel K , Mézenge F , Perrotin A , Desgranges B , Bartrés-Faz D , Eustache F , Chételat G ((2013) ) Relationships between years of education and gray matter volume, metabolism and functional connectivity in healthy elders. Neuroimage 83: , 450–457. |
[56] | Bastin C , Yakushev I , Bahri MA , Fellgiebel A , Eustache F , Landeau B , Scheurich A , Feyers D , Collette F , Chételat G , Salmon E ((2012) ) Cognitive reserve impacts on inter-individual variability in resting-state cerebral metabolism in normal aging. Neuroimage 63: , 713–722. |
[57] | Arenaza-Urquijo EM , Wirth M , Chételat G ((2015) ) Cognitive reserve and lifestyle: Moving towards preclinical Alzheimer’s disease. Front Aging Neurosci 7: , 134. |
[58] | Arenaza-Urquijo EM , Bejanin A , Gonneaud J , Wirth M , La Joie R , Mutlu J , Gaubert M , Landeau B , Sayette V de la , Eustache F , Chételat G ((2017) ) Association between educational attainment and amyloid deposition across the spectrum from normal cognition to dementia: Neuroimaging evidence for protection and compensation. Neurobiol Aging 59: , 72–79. |
[59] | Arenaza-Urquijo EM , de Flores R , Gonneaud J , Wirth M , Ourry V , Callewaert W , Landeau B , Egret S , Mézenge F , Desgranges B , Chételat G ((2017) ) Distinct effects of late adulthood cognitive and physical activities on gray matter volume. Brain Imaging Behav 11: , 346–356. |
[60] | Robertson JS , Rowe CC , Villemagne VL ((2017) ) Tau imaging with PET: An overview of challenges, current progress, and future applications. Q J Nucl Med Mol Imaging 61: , 405–413. |
[61] | Lucey BP , Bateman RJ ((2014) ) Amyloid-β diurnal pattern: Possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiol Aging 35: (Suppl 2), S29–34. |
[62] | Ricci S , Fuso A , Ippoliti F , Businaro R ((2012) ) Stress-induced cytokines and neuronal dysfunction in Alzheimer’s disease. J Alzheimers Dis 28: , 11–24. |
[63] | Wilson RS , Barnes LL , Mendes de Leon CF , Aggarwal NT , Schneider JS , Bach J , Pilat J , Beckett LA , Arnold SE , Evans DA , Bennett DA ((2002) ) Depressive symptoms, cognitive decline, and risk of AD in older persons. Neurology 59: , 364–370. |
[64] | Branger P , Arenaza-Urquijo EM , Tomadesso C , Mézenge F , André C , de Flores R , Mutlu J , de La Sayette V , Eustache F , Chételat G , Rauchs G ((2016) ) Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol Aging 41: , 107–114. |
[65] | Chételat G , Mézenge F , Tomadesso C , Landeau B , Arenaza-Urquijo E , Rauchs G , André C , de Flores R , Egret S , Gonneaud J , Poisnel G , Chocat A , Quillard A , Desgranges B , Bloch J-G , Ricard M , Lutz A ((2017) ) Reduced age-associated brain changes in expert meditators: A multimodal neuroimaging pilot study. Sci Rep 7: , 10160. |


