
Evolvability of Amyloidogenic Proteins in Human Brain
Abstract
Currently, the physiological roles of amyloidogenic proteins (APs) in human brain, such as amyloid-β and α-synuclein, are elusive. Given that many APs arose by gene duplication and have been resistant against the pressures of natural selection, APs may be associated with some functions that are advantageous for survival of offspring. Nonetheless, evolvability is the sole physiological quality of APs that has been characterized in microorganisms such as yeast. Since yeast and human brain may share similar strategies in coping with diverse range of critical environmental stresses, the objective of this paper was to discuss the potential role of evolvability of APs in aging-associated neurodegenerative disorders, including Alzheimer’s disease and Parkinson’s disease. Given the heterogeneity of APs in terms of structure and cytotoxicity, it is argued that APs might be involved in preconditioning against diverse stresses in human brain. It is further speculated that these stress-related APs, most likely protofibrillar forms, might be transmitted to offspring via the germline, conferring preconditioning against forthcoming stresses. Thus, APs might represent a vehicle for the inheritance of the acquired characteristics against environmental stresses. Curiously, such a characteristic of APs is reminiscent of Charles Darwin’s ‘gemmules’, imagined molecules of heritability described in his pangenesis theory. We propose that evolvability might be a physiological function of APs during the reproductive stage and neurodegenerative diseases could be a by-product effect manifested later in aging. Collectively, our evolvability hypothesis may play a complementary role in the pathophysiology of APs with the conventional amyloid cascade hypothesis.
INTRODUCTION
Recently, a number of clinical studies in Alzheimer’s disease (AD) targeting amyloid-β (Aβ), including use of immunotherapy and γ-secretase inhibitors, have failed to demonstrate efficacy [1–3]. Although the reasons for the unsatisfactory outcome remain elusive, we must reflect on our ignorance of the underlying physiological function of Aβ. Given that Aβ aggregates might serve some critical biological functions, therapies targeting Aβ might be a ‘trade-off,’ counteracting the normal physiological action of Aβ. Similarly, clinical trials of α-synuclein (αS) immunotherapy are ongoing in Parkinson’s disease (PD) without a complete understanding of the physiological function of αS [3]. Indeed, the physiological functions of amyloidogenic proteins (APs) relevant to aging-related neurodegenerative diseases, such as AD and PD, are presently obscure.
Notably, the concept of evolvability has been applied to yeast prions expressed in microorganisms such as Saccharomyces cerevisiae. Phenotypic diversity in yeast due to amyloid-regulated genetic variation is a powerful survival strategy against multiple environmental stresses, which is transmissible to offspring during cell division [4]. The concept of a prion as an infectious self-propagating protein isoform was initially proposed to explain prion and related diseases in mammalian brain [5] and later was applied to yeast prion [4]. Indeed, yeast have heritable elements transmitted via proteins, and the “protein only” model of prion transmission was first proven using a yeast prion [4].
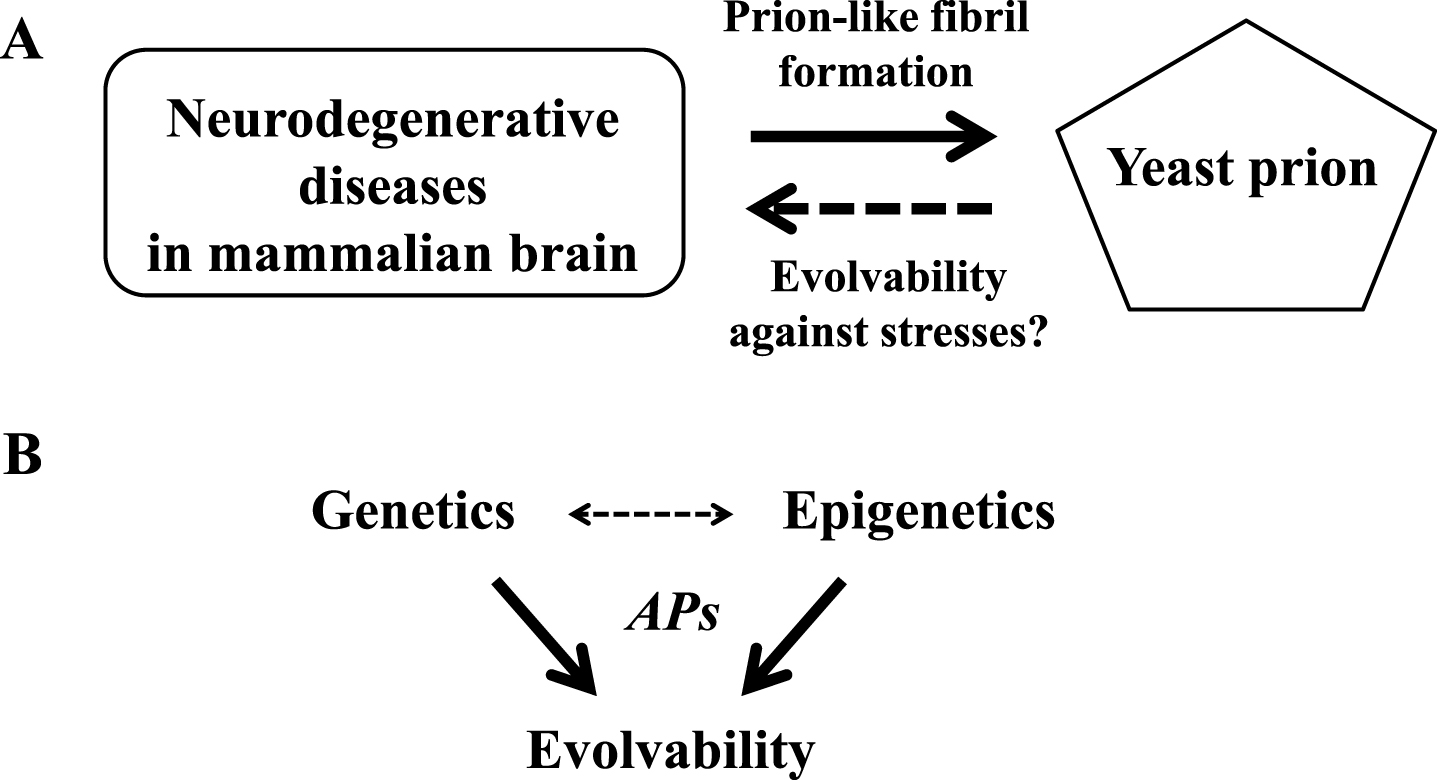
Considering that yeasts are forced to exist in a perpetually fluctuating environment, while the human brain is also frequently confronted by multiple environmental challenges, coping with stress is a critical issue in both circumstances [4]. As such, one might speculate that the findings in yeast prion might to some degree, be applied to mammalian brain. In this context, the objective of this paper is to discuss the possible involvement of evolvability of APs in aging-associated neurodegenerative diseases (Fig. 1). First, we show that APs, including Aβ and αS, originate from gene duplication, suggesting that one function of APs might be to confer an advantage for the survival of offspring. Second, we hypothesize that APs might confer preconditioning against diverse stresses in the brain based on recent evidence demonstrating the heterogeneity of APs in terms of molecular structure and cytotoxicity [6–10]. Furthermore, we speculate that stress-corresponding APs, presumably in their protofibrillar forms, might be transmitted to offspring through the germline. Collectively, APs might be involved in the inheritance of acquired resilience against stresses in human brain during reproduction. According to this hypothetical view, which we tentatively designate as the evolvability hypothesis, a normal physiological function of APs might be to confer evolvability on the human brain at reproduction, whereas neurodegeneration may be regarded as a pleiotropic effect manifested in aging. This view may provide novel insights into several critical issues that are poorly understood in neurodegenerative diseases.
Fig.1
Commonalities between neurodegeneration in mammalian brain and prion activity in yeast. A) The concept of a prion as an infectious self-propagating amyloid fibril was initially proposed to explain neurodegenerative diseases in mammalian brain, and later was applied to yeast prions. In turn, the concept of evolvability by yeast prions against stress could be applicable to mammalian brain. B) Genetics and epigenetics of APs may cooperate to regulate evolvability in human brain and in yeast.
THE ORIGIN OF APs DURING EVOLUTION
From a genetic standpoint, it is apparent that many APs, including Aβ and αS, arose during gene duplication (Fig. 1). According to Susumu Ohno, gene duplication confers mutations and results in the creation of novel functions, leading to the promotion of evolution [11]. Moreover, APs, if only detrimental during the aging process, must have been rapidly eliminated. Therefore, it is reasonable to speculate that APs have survived through evolution against the pressures of natural selection, because APs might function in a manner advantageous for the survival of the species.
Role of gene duplication
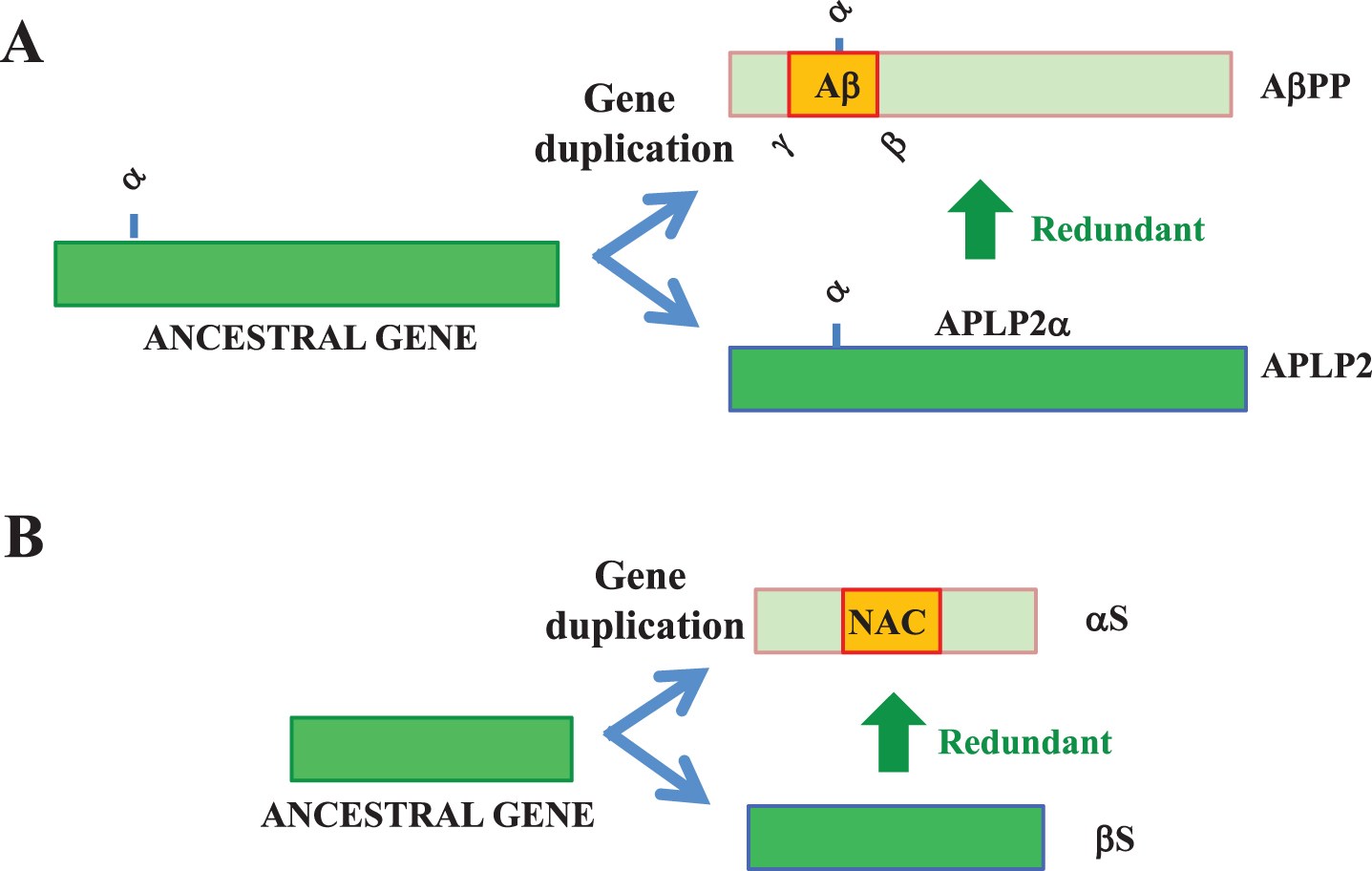
Aβ peptides with 36–43 amino acids derived from amyloid-β protein precursor (AβPP) are the main component of amyloid plaques in the brains of AD patients [12]. AβPP is highly homologous with AβPP-like protein (APLP)-2 and to a lesser extent with APLP-1, both of which lack Aβ-like domains [12]. Given the importance of gene duplication as a source of novelty in evolution [11], Aβ may have arisen as a new functional domain of AβPP during gene duplication, in which APLP-2 played redundant roles for AβPP (Fig. 2A). Since α-secretase sites are common in AβPP family members, but β- and γ-secretase sites that generate Aβ are specific to AβPP, it is estimated that the α-synuclein site was first created in the AβPP-ancestral gene, followed by the appearance of the Aβ domain with β-secretase and γ-secretase sites added to the new functional domain in AβPP during evolution. This is pathologically important because the cleavage of sAβPPα by α-secretase precludes generation of Aβ by both β-secretase and γ-secretase, suggesting that an imbalance of sAβPPα/Aβ may lead to the promotion of neurodegeneration (Fig. 2A).
Fig.2
APs and their homologues. A) Aβ may have been created by gene duplication from an AβPP-ancestral gene into AβPP and APLP-2 genes. APLP2/APLP2α may be redundant for some physiological functions of AβPP/AβPPα that were downregulated during creation of the Aβ domain. While α-secretase sites are common among AβPP family members, β- and γ-secretase sites are specific to AβPP. B) Similarly, βS, a non-amyloidogenic homologue of αS, may be redundant for some physiological functions of αS that were downregulated during gene duplication.
Distinct from a lack of clarity regarding the physiological function of Aβ, AβPP family members have been characterized by a number of physiological roles, including cell adhesion, cell migration, cell development, neurogenesis, neuroprotection, and learning and memory [13–16]. From the standpoint of evolution, one may predict that these functions derived from AβPP/sAβPPα have been downregulated during the process of gene duplication. Consistent with this notion, the neurotrophic and neuroprotective actions of sAβPPβ, the cleavage product of AβPP produced by β-secretase, are weaker than those of sAβPPα [17]. Under such circumstances, APLP-2 would be expected to be redundant. Consistent with this view, AβPP-knockout mice exhibit a minimal phenotype, presumably owing to the redundancy of APLP-2 [18, 19].
A similar situation could be present for αS, an AP predominantly expressed in presynaptic terminals. αS is featured by the presence of a hydrophobic domain, NAC (non-Aβ component of AD amyloid) which is not physiologically cleaved by a protease (Fig. 2B) [20]. In the αS family of peptides, there are two homologous proteins which lack NAC-corresponding domain: β-synuclein (βS) and γ-synuclein (γS) [21]. It has been characterized that βS and γS are inhibitors of αS aggregation under the neurodegenerative condition [22, 23]. From a viewpoint of physiology, functional redundancy of these homologous molecules with αS may be important. Indeed, it has been shown that αS and βS share some biological properties, including chaperone activities and stimulation of phospholipase D [24].
EVOLVABILITY AS A POSSIBLE FUNCTION OF APs IN HUMAN BRAIN
Presently, the normal physiological functions of APs remain unclear. Supposing that APs possess physiological functions during developmental and/or reproductive stages, the question arises as to what these functions are likely to be. Aside from the evolvability of yeast prion as mentioned, physiological functions for APs have been not yet been established. Therefore, it is important to determine whether APs might be related to evolvability in the human brain similar to yeast.
Evolvability of yeast prion: An inheritance of acquired characteristics?
Microorganisms such as yeast are forced to survive and thrive in a fluctuating environment associated with a diverse array of stressors, and phenotypic diversity due to amyloid-regulated genetic variation is a powerful survival tool which is transmissible to offspring by cell division [4]. Supporting this, it is well known that [PSI+] is a prion form of SUP35 protein [4]. [PSI+] is the result of the amyloid fibril-like conformational change of SUP35 that becomes self-propagating and is transmissible to offspring. [PSI+]-aggregates sequester soluble SUP35, resulting in defective translation termination by read-through of translation termination codons, which can be completely reversed by guanidinium chloride [4]. Similar to [PSI+], dozen of other yeast prions relevant to environmental stresses have been reported [25]. Collectively, one interpretation is that the emergence of prion forms would be at least mildly deleterious in most environments, but overall may increase evolvability especially in certain harsh environments.
It is noteworthy that evolvability of yeast prion may exhibit an inheritance of acquired characteristics, suggesting that physiological changes acquired over the life of an organism could be transmitted to offspring, since this evolutionary theory has historically been a critical issue that remains unresolved in the field of biology [26]. Emerging evidence suggests that various mechanisms of epigenetics, including DNA methylation and acetylation, histone modification and non-coding RNAs, might be involved [27, 28]. Furthermore, evidence has accumulated to support a transgenerational epigenetics in mammals via an integration of soma to germline communication with gametic inheritance [27, 28]. In this context, it is tempting to speculate that prion action, one mechanism of epigenetics, might also be relevant in the inheritance of acquired characteristics associated with neurodegenerative disorders in human brain.
Relevance to brain stress resistance?
Although yeast and human brain are very different in most respects, both are somewhat similar in terms of molecular response to stress. Indeed, the human brain is confronted with numerous stressors, including hypoxia, oxidative stress, and inflammation [29, 30]. Therefore, it is reasonable to speculate that APs might play a role in coping with diverse stresses in human brain.
Emerging evidence suggests that APs such as Aβ and αS are composed of structurally heterogeneous populations [8–10]. Given that APs are neurotoxic and that promoting diversity is a fundamental strategy in biology, it is probable that the heterogeneity of APs might exemplify hormesis, and provide resistance against multiple brain stressors (Fig. 3A). Indeed, neurons are equipped with adaptive mechanisms, including the unfolded protein response, the ubiquitin-proteasome system and autophagy, which can be stimulated by preconditioning treatments that confer resistance to a subsequent toxic challenge [30]. As an example, it was shown that Aβ expression in various experimental models of AD, including mouse, nematode, and cultured cells, conferred protection against fungal and bacterial infections, raising the possibility that Aβ may play a protective role in innate immunity [31]. Since infection is a stressful and potentially life-threatening condition for the brain, it is possible that transgenerational transmission of Aβ may be essential to normal brain protective functions.
Fig.3
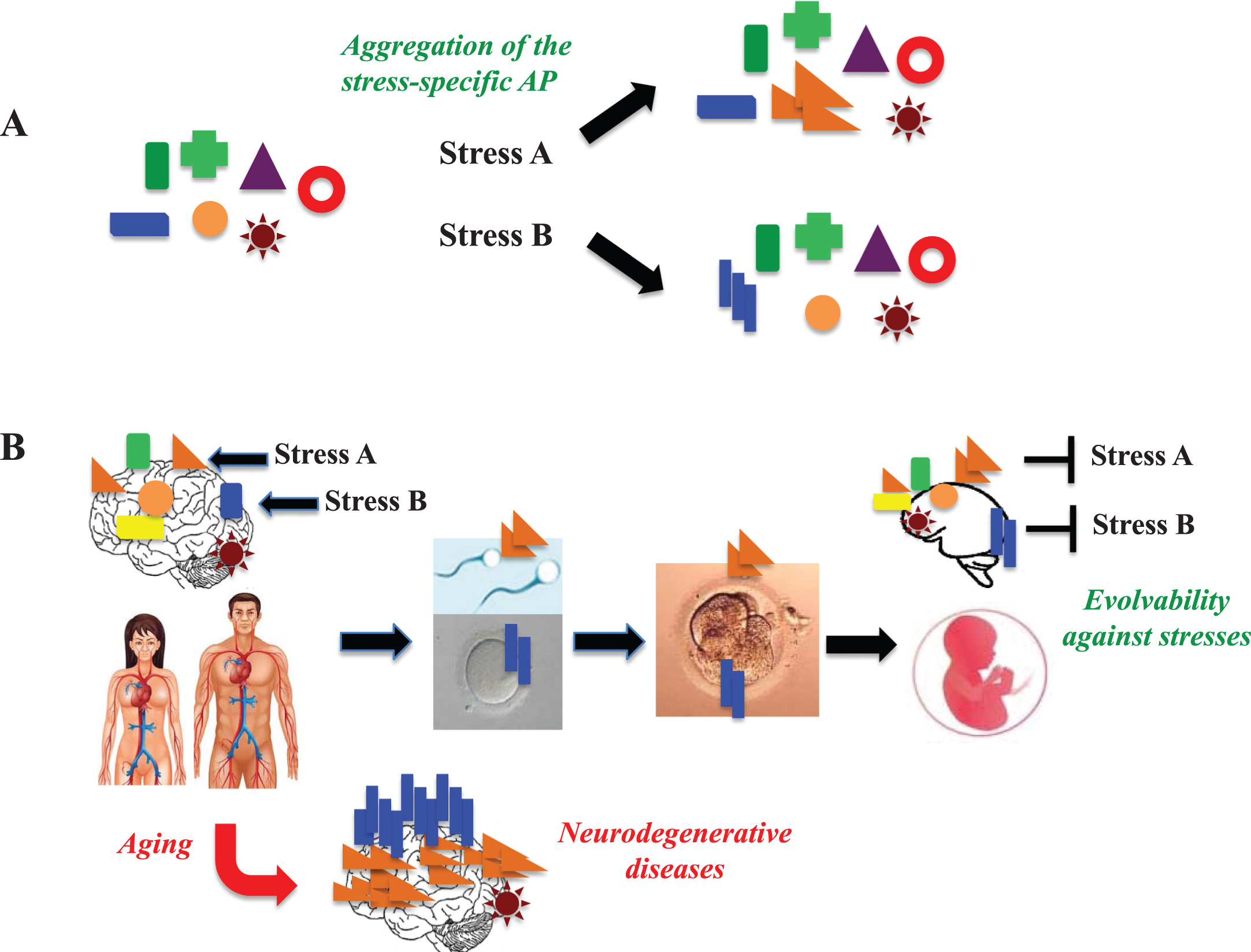
Evolvability hypothesis: a hypothetical mechanism for transmission of APs to offspring. A) APs are composed of heterogeneous populations in terms of structure and neurotoxicity, which might be relevant to the hormesis and resistance against diverse stresses in the brain. In response to specific stresses (Stress A and stress B), misfolding induces aggregation of the specific APs. B) The stress-responsive forms of APs may go through the BBB and enter blood, reaching the reproductive system, thereby passing through the blood and testis (and ovary) barrier. Subsequently, endogenous APs in germ cells (sperm, oocytes) are converted and transmitted to the embryo like a prion-like fashion. Thus, evolvability by APs may occur during the reproductive stage, and neurodegenerative diseases may be manifested as a by-product in aging.
Transgenerational transmission of APs
Evolvability is defined as the capacity of a population of organisms that does not merely generate genetic diversity, but generates adaptive genetic diversity, often overriding natural selection [32]. Similar to evolvability in yeast prion, an intriguing hypothesis is that information on human brain stressors could be transmitted through stress-specific strains of APs from parents to offspring, thus promoting the evolution of the brain. Since yeast is a unicellular microorganism, parental APs can simply be transferred to offspring during cell division [4], but the same exact mechanism does not apply to human brain post-mitotic neurons which largely do not proliferate. Yet, alternatively, recent findings suggest that transmission of APs could indeed be possible from parents to offspring via the germline, even at the level of higher organisms such as humans [33].
According to the current prion hypothesis in neurodegenerative disease, the propagation of APs, might occur trans-neuronally between adjacent cells within brain [34, 35]. This concept could be extended to include the transgenerational transmission of APs. To clarify, propagation of APs can occur in many parts of the body outside the brain, accessing the brain by traversing the blood-brain barrier (Fig. 3B). Outside the brain, Aβ is abundant in serum [36], and also expressed in gonadal tissues, including Sertoli cells and follicles [37]. With this in mind, the stress-specific Aβ strains could propagate into the reproductive system, passing across the blood/gonads barrier, followed by conversion of the endogenous Aβ to a stress-specific misfolded form which is then transmitted from gametes to embryo (Fig. 3B). Alternative and not mutually exclusive possibility is that Aβ could also be transferred passively via a trans-placental route from mother to embryo/fetus during pregnancy. However, transmission through gonadal cells is a more attractive possibility given that dual parental transmission of APs may be more advantageous compared to only maternal transmission. Beyond Aβ, APs could also be transmitted via the germline in relation to other neurodegenerative disorders. Supporting this notion, αS is also expressed in serum and gonadal tissues [38, 39], and it was previously shown that a C-terminal-truncated prion protein isoform is present in mature sperm [40]. Thus, information regarding environmental stress conditions encoded on the altered structure of parental APs, could deliver such survival information to offspring. Such a mechanism would confer offspring preconditioning against forthcoming stresses in the brain and other tissues, which would be beneficial for the survival of neurons. It is therefore reasonable to predict that evolvability mediated by APs in response to diverse brain stressors could be regarded as a natural physiological function. Furthermore, given that APs in neurodegenerative conditions are ubiquitously expressed, it is speculated that the evolvability hypothesis could be applicable not only to brain, but also to a variety of tissues and organs beyond the brain.
Along similar lines, other APs in non-neurodegenerative conditions might also be passed down by transgenerational transmission related to evolvability. For example, it is especially notable that levels of calcitonin, a 32-amino acid polypeptide hormone produced in the parafollicular cells of the thyroid, which also displays amyloidogenic properties, is 30-fold higher in seminal plasma than in peripheral serum [41]. Moreover, serum amyloid P component involved in systemic amyloidosis is also expressed in the male reproductive tract including seminal fluid, on spermatozoa, and in epididymal, seminal vesicle and prostate tissue [42]. Finally, transthyretin, the protein transporter carrying the thyroid hormone, thyroxine, in serum and cerebrospinal fluid, which misfolds to form amyloid fibrils contributing to familial neuropathy and cardiac disease, is immunoreactive in the male reproductive system [43].
Reminiscent of gemmules?
In addition, it is noteworthy that evolvability of yeast prion may exhibit an inheritance of acquired characteristics since this phenomenon has historically been a critical issue that remains unresolved in the field of biology [26]. Emerging evidence suggests that various mechanisms of epigenetics, including DNA methylation, histone modification and non-coding RNAs, might be involved [27, 28]. In this context, it is tempting to speculate that prion actions, one mechanism of epigenetics, might be relevant to the inheritance of acquired characteristics associated with neurodegenerative disorders in human brain.
Notably, the concept of evolvability through APs against brain stressors is reminiscent of Charles Darwin’s ‘gemmules’, a virtual molecule described in his pangenesis theory [44]. Darwin speculated that the ‘gemmule’ is involved in the heritability of acquired characteristics against multiple environmental changes, and further proposed that this molecule should be stable, prone to accumulate and is ubiquitously expressed in various tissues, causing other cells to undergo structural changes. Surprisingly, the concept of ‘gemmules’ partly resembles APs. However, Darwin was unlikely to know Mendel’s law when he proposed the pangenesis theory although Darwin and Mendel lived in the same era [44]. Given modern genetics, in which epigenetics is heavily involved in regulation of many biological phenomena, it is likely that both genetics and epigenetics of APs may cooperate to regulate evolvability (Fig. 1B). Although little attention has been paid to the pangenesis theory underlying the great success of the theory of natural selection, Darwin’s pangenesis concept could be worth revisiting in a modern context of evolvability and neurodegeneration.
MODULATORS OF EVOLVABILITY OF APs IN HUMAN BRAIN
Thus, a prion-like mode of transmission underlying the transgenerational migration of APs would therefore naturally predict that the aggregation of APs is somehow central to the process. Accordingly, various factors that modulate AP aggregation might be critical in regulating evolvability, and findings from neurodegenerative disease research might provide insight into relevant mechanisms.
The role of protein aggregation
It is possible that various mechanisms of AP degradation, such as the ubiquitin-proteasome system and autophagy, might be involved in the negative regulation of transgenerational transmission of APs. Furthermore, proteolytic degradation of APs might also be an important regulator, and in this regard, insulin degrading enzyme and neurosin, proteases which can degrade Aβ and αS, respectively, are abundant in serum [45, 46]. Naturally, it is expected that the post-translational modifications, such as phosphorylation, glycation, and oxidation, may also affect the aggregation properties of APs and regulate evolvability [3, 47, 48].
In addition, heat shock proteins (HSPs) might be particularly important for the regulation of the evolvability by APs given that HSPs are ubiquitously expressed, and have been characterized as molecular chaperones that play crucial roles in regulating protein folding/unfolding. Notably, Susan Lindquist and colleagues demonstrated that HSP90 buffers evolvability caused by environmental stresses in various biological systems, including plants and drosophila [49, 50]. Since other HSPs, such as HSP70, are inducible and abundant in serum, HSPs might cooperate to regulate transgenerational transmission and evolvability of APs.
Homologs of APs
Accumulating evidence suggest that the aggregation of APs are also affected by their homologous proteins. It was previously shown that Aβ production is increased by AβPP homodimerization, whereas AβPP heterodimerization with its homologues APLP-1 and -2 decreased Aβ levels, suggesting that APLP-1 and -2 may negatively regulate evolvability by Aβ in AD [51]. Similarly, microtubule-binding protein, which is homologous with tau [52], may negatively regulate the evolvability of tau through suppression of tau aggregation.
Relevant to PD, the αS homologues, βS and γS, might be important modulators of evolvability by αS. βS, in particular, has been characterized as an inhibitor of αS aggregation and neurodegeneration [24]. However, two missense mutations in βS have been associated with familial dementia with Lewy bodies [53], causing increased αS neuropathology in both cultured cells and in mice [54]. Also, overexpression of wild type βS using adenoviral vector resulted in the formation of inclusion bodies in mouse brain [55]. βS, however, has not been identified in Lewy bodies in human brain, but has been associated with hippocampal axon pathology in PD and in dementia with Lewy bodies [56]. Although these results suggest that βS may either negatively or positively affect evolvability by αS, further investigation is warranted to better understand this phenomenon.
In contrast to abundant expression of α- and βS in the brain, γS is expressed predominantly in the peripheral nervous system and has been implicated in some types of neurodegenerative disorders, including amyotrophic lateral sclerosis and retinal neurodegeneration [21]. Furthermore, it was also shown that γS is involved in neuritic pathology in hippocampus in patients with α-synucleinopathies, similar to βS [56]. Thus, given the analogy of γS with αS [21], γS may contribute to evolvability of the peripheral nervous system and might cooperate with αS to promote evolvability. Along these lines, oxidized γS increases seeding with αS and promotes αS transmission between cells [57]. Furthermore, γS was abundantly expressed in testis and ovary in zebrafish [58], supporting the possibility of γS transmission to offspring.
Notably, γS has been identified as being involved in the progression of a variety of malignancies, including breast, ovary, lung, liver, esophagus, colon, and prostate [21, 59, 60]. As tumors are also subjected to various stressors, including hypoxia, metabolic stress, and chemotherapy-induced toxicity, it is predicted that γS might be involved in evolvability of cancers. Indeed, γS has been shown to be involved in drug resistance in cancer cells [60, 61]. Because malignant tumor cells divide robustly, analogous to yeast cell division, a similar mechanism of transmission might be applicable in these two biological systems.
ADVANTAGES AND DISADVANTAGES OF THE EVOLVABILITY HYPOTHESIS
If AP-mediated evolvability is active during reproductive stages in the parents and is advantageous for the survival of offspring, it might explain why APs are conserved under the pressures of natural selection. Thus, as a consequence of our novel view, neurodegenerative diseases could be no more than a by-product manifested in the post-reproductive stage (Fig. 3B). Furthermore, the evolvability hypothesis may also provide new insights into several key issues in neurodegenerative diseases that remain unresolved. Thus, the evolvability hypothesis may be complementary to the conventional amyloid cascade hypothesis in terms of pathophysiology of APs (Table 1).
Table 1
Comparison of the evolvability hypothesis with the amyloid cascade hypothesis. The evolvability hypothesis was compared with the amyloid cascade hypothesis in terms of ‘physiological function of APs’, ‘mechanism of neurotoxicity’, ‘experimental models’ and other points, such as initiation of protein aggregation, anticipation, missense mutation of APs and therapy
| Evolvability hypothesis | Amyloid cascade hypothesis | |
| Physiological function of APs | Evolvability | Unexplainable |
| Mechanism of neurotoxicity | Unexplainable | Toxic protofibrils of APs |
| Experimental models | Not established | Cells and Animals |
| Initiation of protein aggregation | Explainable | Unexplainable |
| Anticipation | Explainable | Unexplainable |
| Missense mutation of APs | Explainable | Unexplainable |
| Therapy | Still unclear | Targeting of APs |
| (Clinical studies: unsuccessful) |
Practical advantages of evolvability
First, the evolvability hypothesis may have direct implications for the mechanism of protein aggregation in neurodegenerative disease (Table 1). It has been well characterized that protein aggregation is driven by nucleation-dependent polymerization [62]. Although this paradigm applies well to familial neurodegenerative diseases in which misfolding of APs is genetically induced, the trigger for initial conversion of normally soluble proteins into filamentous polymers is elusive in sporadic disease [35]. The evolvability hypothesis thus provides a solution for the latter problem, because it presumes the pre-existence of protofibrillar APs in offspring transmitted from the parental brain. A similar idea can also be expanded to neurodegenerative diseases associated with multiple APs. AD, for instance, is characterized by the aggregation of Aβ and tau, the major components of extracellular senile plaques and intracellular neurofibrillary tangles, respectively. The conventional amyloid cascade hypothesis, which is based on data showing that Aβ stimulates aberrant phosphorylation of tau, remains somewhat controversial [63], whereas the evolvability hypothesis postulates the pre-existence of Aβ and tau protofibrils in brain that are vertically transmitted from the parents. Based on this, it follows that transmission of aggregated tau alone could result in progressive supranuclear palsy, corticobasal degeneration or other tauopathies [64], whereas transmission of aggregated Aβ alone may be asymptomatic given the buffer of cognitive reserve [65]. Similar neurobiological patterns may also occur in other neurodegenerative diseases, such as Parkinsonism-ALS-dementia complex of the Guam and Kii peninsula [66, 67].
Second, the evolvability hypothesis might account for anticipation (Table 1). Anticipation is a phenomenon observed in certain neurogenetic disorders in which the expansion of trinucleotide triplet repeats such as CAG observed in polyQ diseases such as Huntington’s disease and Fragile X Disease, are passed on to the next generation with ever-increasing size, leading to symptoms apparent at an earlier age and with increased severity [68]. Conventionally, it is believed that such phenotypic alterations associated with longer repeat expansions is due to the unstable structure of the gene [68]. Alternatively, however, evolvability would predict that other mechanisms might also be involved such as the transgenerational transmission of APs that are prone to aggregation due to hereditary mutations. Because anticipation has also been suggested in various common disorders, such as AD, type II diabetes mellitus (T2DM) and cancers [69, 70], anticipation in these diseases might, in part, be attributed to transgenerational transmission of various APs. In this context, it is interesting to note that accumulation of misfolded aggregates of the islet amyloid polypeptide are associated with T2DM [71], while γS overexpression is linked to various malignancies, including breast and colon cancer [60].
Third, the evolvability hypothesis might provide an insight into the mechanism of missense mutations in APs and related proteins linked to familial neurodegenerative diseases (Table 1). Although a number of missense mutations of APs result in enhanced aggregation, the fundamental question exists as to why such mutations accumulate causing aggregation in these specific molecules. If AP protofibrils are important players in evolvability, similar to the enhanced infectivity of prion protofibrils compared to mature prion fibrils [72], one possible interpretation is that nature may pursue greater efficacy of evolvability by adjusting the aggregative properties of such proteins. This phenomena could be comparable to the concept of evolving evolvability [73]. As an extension of this, a similar mechanism could account for the distinct evolutionary patterns among species, where the species-specific aggregative properties of APs often depend on only small differences in amino acid sequence. For instance, in mice, wild-type αS is equivalent to the human A53T αS, which is associated with familial PD and is more prone to aggregate than human wild-type αS [74]. On the other hand, human Aβ is prone to aggregate more compared to mouse Aβ due to a difference of only three amino acids [75]. These results are consistent with mice having superior motor function versus humans, but inferior cognitive abilities. It is thus intriguing to speculate that different aggregative properties of APs among species might lead to a profound difference in brain evolution.
Finally, it is expected that the evolvability hypothesis may hold significant therapeutic implications for neurodegenerative and other disorders (Table 1). The disappointing results for Aβ immunotherapy in mild to moderate AD, have given rise to the idea that a neurodegenerative disease-modifying strategy to suppress AP aggregation should start as early as possible, even in the pre-symptomatic stage [76], However, if aggregation of all APs is central to evolvability as defined, then, inhibiting protein aggregates during human reproductive stages may adversely affect the transmission of APs to offspring and interfering with evolvability. Perhaps a more effective strategy would be to take advantage of evolvability, for instance, by targeting γS and suppressing evolvability of tumor cells. One should bear in mind, however, that elimination of γS during the reproductive stage may be not a good strategy considering the potential role of γS in the evolvability of the nervous system.
Disadvantages of evolvability
A major challenge of the evolvability hypothesis, however, would be the difficulty in identifying specific species of APs transmissible from parent to offspring. Based on observations of prion protein infectivity [72], such species are predicted to be protofibrillar in nature. Indeed, the concept of protofibrils was created primarily to explain the neurotoxicity of APs in neurodegenerative diseases, but the studies on fibrillar intermediates, such as oligomers and protofibrils of various APs, have been performed in vitro, and their presence has yet to be firmly established in vivo. Conceivably, further innovations in methodology are required to detect intermediate species of protein aggregates for the demonstration of the evolvability hypothesis.
Alternatively, the evolvability hypothesis could be demonstrated if tagged APs injected intracerebroventricularly in rodent brain in the presence of various stresses, such as neurotoxins, kindling, hyperthermia and physical stress could be transmitted to offspring.
CONCLUSIONS
Although mounting evidence has shown that aggregation of APs is central to neurotoxicity in AD and other neurodegenerative disorders, the failure of clinical trials targeting Aβ in AD raises concern that the current prevailing view may have significant limitations. Exemplifying a weakness in the amyloid cascade hypothesis, the precise physiological roles of APs remain unclear [77]. We suggest that evolvability might be a key physiological function of APs that underlies the inheritance of acquired characteristics for stress response and adaptability in human brain. Provided that evolvability mediated by APs against environmental stressors, is apparently beneficial for the survival of offspring and is active in reproductive stages, APs might have overcome the pressures of natural selection. Furthermore, the evolvability hypothesis provides insights into unsolved issues in the field of neurodegenerative disease that have been difficult to resolve by conventional concepts. If proven, the evolvability hypothesis has the potential to alter the direction of research, leading to more fruitful therapeutic outcomes for neurodegenerative conditions for which no effective therapies currently exist.
ACKNOWLEDGMENTS
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (The University of Chicago).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0894r2).
REFERENCES
[1] | De Strooper B ((2014) ) Lessons from a failed gamma-secretase Alzheimer trial. Cell 159: , 721–726. |
[2] | Goni F , Sigurdsson EM ((2005) ) New directions towards safer and effective vaccines for Alzheimer’s disease. Curr Opin Mol Ther 7: , 17–23. |
[3] | Takamatasu YHG , Koike W , Sugama S , Takenouchi T , Waragai M , Wei J , Sekiyama K , Hashimoto M ((2017) ) Combined immunotherapy with “anti-insulin resistance” therapy as a novel therapeutic strategy against neurodegenerative diseases. NPJ Parkinsons Dis 3: , 4. |
[4] | Wickner RB ((2016) ) Yeast and fungal prions. Cold Spring Harb Perspect Biol 8: , pii: a023531. |
[5] | Watts JC , Prusiner SB ((2017) ) Beta-Amyloid prions and the pathobiology of Alzheimer’s disease. Cold Spring Harb Perspect Med, doi: 10.1101/cshperspect.a023507 |
[6] | Mucke L , Selkoe DJ ((2012) ) Neurotoxicity of amyloid beta-protein: Synatic and network dysfunction. Cold Spring Harb Perspect Med 2: , a006338. |
[7] | Ahmed M , Davis J , Aucoin D , Sato T , Ahuja S , Aimoto S , Elliott JI , Van Nostrand WE , Smith SO ((2010) ) Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat Struct Mol Biol 17: , 561–567. |
[8] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[9] | Qiang W , Yau WM , Lu JX , Collinge J , Tycko R ((2017) ) Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature 541: , 217–221. |
[10] | Theillet FX , Binolfi A , Bekei B , Martorana A , Rose HM , Stuiver M , Verzini S , Lorenz D , van Rossum M , Goldfarb D , Selenko P ((2016) ) Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature 530: , 45–50. |
[11] | Ohno S ((1999) ) Gene duplication and the uniqueness of vertebrate genomes circa 1970-1999. Semin Cell Dev Biol 10: , 517–522. |
[12] | Walsh DM , Minogue AM , Sala Frigerio C , Fadeeva JV , Wasco W , Selkoe DJ ((2007) ) The APP family of proteins: Similarities and differences. Biochem Soc Trans 35: , 416–420. |
[13] | Schubert D , Schroeder R , LaCorbiere M , Saitoh T , Cole G ((1988) ) Amyloid beta protein precursor is possibly a heparan sulfate proteoglycan core protein. Science 241: , 223–226. |
[14] | Thinakaran G , Koo EH ((2008) ) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283: , 29615–29619. |
[15] | Nalivaeva NN , Turner AJ ((2013) ) The amyloid precursor protein: A biochemical enigma in brain develoment, function and disease. FEBS Lett 587: , 2046–2054. |
[16] | Lazarov O , Demars MP ((2012) ) All in the family: How the APPs regulate neurogenesis. Front Neurosci 6: , 81. |
[17] | Nhan HS , Chiang K , Koo EH ((2015) ) The multifaceted nature of amyloid precursor protein and its proteolytic fragments: Friends and foes. Acta Neuropathol 129: , 1–19. |
[18] | Zheng H , Jiang M , Trumbauer ME , Hopkins R , Sirinathsinghji DJ , Stevens KA , Conner MW , Slunt HH , Sisodia SS , Chen HY , Van der Ploeg LH ((1996) ) Mice deficient for the amyloid precursor protein gene. Ann N Y Acad Sci 777: , 421–426. |
[19] | Heber S , Herms J , Gajic V , Hainfellner J , Aguzzi A , Rulicke T , von Kretzschmar H , von Koch C , Sisodia S , Tremml P , Lipp HP , Wolfer DP , Muller U ((2000) ) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20: , 7951–7963. |
[20] | Ueda K , Fukushima H , Masliah E , Xia Y , Iwai A , Yoshimoto M , Otero DA , Kondo J , Ihara Y , Saitoh T ((1993) ) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90: , 11282–11286. |
[21] | George JM ((2002) ) The synucleins. Genome Biol 3: , REVIEWS3002. |
[22] | Hashimoto M , Rockenstein E , Mante M , Mallory M , Masliah E ((2001) ) Beta-Synuclein inhibits alpha-synuclein aggregation: A possible role as an anti-parkinsonian factor. Neuron 32: , 213–223. |
[23] | Uversky VN , Li J , Souillac P , Millett IS , Doniach S , Jakes R , Goedert M , Fink AL ((2002) ) Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem 277: , 11970–11978. |
[24] | Hashimoto M , La Spada AR ((2012) ) Review β-synuclein in the pathogenesis of Parkinson’s disease and related α-synucleinopathies: Emerging roles and new directions. Future Neurol 7: , 155–163. |
[25] | Liebman SW , Chernoff YO ((2012) ) Prions in yeast. Genetics 191: , 1041–1072. |
[26] | Liu Y ((2007) ) Like father like son. A fresh review of the inheritance of acquired characteristics. EMBO Rep 8: , 798–803. |
[27] | Sharma A ((2017) ) Transgenerational epigenetics: Integrating soma to germline communication with gametic inheritance. Mech Ageing Dev 163: , 15–22. |
[28] | Bohacek J , Mansuy IM ((2015) ) Molecular insights into transgenerational non-genetic inheritance of acquired behaviours. Nat Rev Genet 16: , 641–652. |
[29] | Moreira PI , Smith MA , Zhu X , Nunomura A , Castellani RJ , Perry G ((2005) ) Oxidative stress and neurodegeneration. Ann N Y Acad Sci 1043: , 545–552. |
[30] | Mollereau B , Rzechorzek NM , Roussel BD , Sedru M , Van den Brink DM , Bailly-Maitre B , Palladino F , Medinas DB , Domingos PM , Hunot S , Chandran S , Birman S , Baron T , Vivien D , Duarte CB , Ryoo HD , Steller H , Urano F , Chevet E , Kroemer G , Ciechanover A , Calabrese EJ , Kaufman RJ , Hetz C ((2016) ) Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations. Brain Res 1648: , 603–616. |
[31] | Kumar DK , Choi SH , Washicosky KJ , Eimer WA , Tucker S , Ghofrani J , Lefkowitz A , McColl G , Goldstein LE , Tanzi RE , Moir RD ((2016) ) Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8: , 340ra372. |
[32] | Kirschner M , Gerhart J ((1998) ) Evolvability. Proc Natl Acad Sci U S A 95: , 8420–8427. |
[33] | Becker C ((2017) ) Like father, like son. Elife 6: , pii: e25669. |
[34] | Lee SJ , Desplats P , Sigurdson C , Tsigelny I , Masliah E ((2010) ) Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 6: , 702–706. |
[35] | Guo JL , Lee VM ((2014) ) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20: , 130–138. |
[36] | Liu L , Tapiola T , Herukka SK , Heikkila M , Tanila H ((2003) ) Abeta levels in serum, CSF and brain, and cognitive deficits in APP+PS1 transgenic mice. Neuroreport 14: , 163–166. |
[37] | Beer J , Masters CL , Beyreuther K ((1995) ) Cells from peripheral tissues that exhibit high APP expression are characterized by their high membrane fusion activity. Neurodegeneration 4: , 51–59. |
[38] | van Geel WJ , Abdo WF , Melis R , Williams S , Bloem BR , Verbeek MM ((2008) ) A more efficient enzyme-linked immunosorbent assay for measurement of alpha-synuclein in cerebrospinal fluid. J Neurosci Methods 168: , 182–185. |
[39] | Ltic S , Perovic M , Mladenovic A , Raicevic N , Ruzdijic S , Rakic L , Kanazir S ((2004) ) Alpha-synuclein is expressed in different tissues during human fetal development. J Mol Neurosci 22: , 199–204. |
[40] | Shaked Y , Rosenmann H , Talmor G , Gabizon R ((1999) ) A C-terminal-truncated PrP isoform is present in mature sperm. J Biol Chem 274: , 32153–32158. |
[41] | Badr O , Imam A , Monieb H , Khalifa A , al Ahmady O , Abdallah MA ((1989) ) Concentration of calcitonin in seminal plasma of infertile men. Andrologia 21: , 416–422. |
[42] | Malm J , Sonesson A , Hellman J , Bjartell A , Frohm B , Hillarp A ((2008) ) The pentraxin serum amyloid P component is found in the male genital tract and attached to spermatozoa. Int J Androl 31: , 508–517. |
[43] | Kato M , Sung WK , Kato K , Goodman DS ((1985) ) Immunohistochemical studies on the localization of cellular retinol-binding protein in rat testis and epididymis. Biol Reprod 32: , 173–189. |
[44] | Darwin C ((1868) ) The variation of animals and plants under domestication. John Murray, London, UK. |
[45] | Sun J , Xia W , Cai R , Wang P , Huang R , Sun H , Tian S , Dong X , Wang S ((2016) ) Serum insulin degrading enzyme level and other factors in type 2 diabetic patients with mild cognitive impairment. Curr Alzheimer Res 13: , 1337–1345. |
[46] | Diamandis EP , Yousef GM , Soosaipillai AR , Bunting P ((2000) ) Human kallikrein 6 (zyme/protease M/neurosin): A new serum biomarker of ovarian carcinoma. Clin Biochem 33: , 579–583. |
[47] | Goedert M ((1999) ) Alzheimer’s disease. Pinning down phosphorylated tau. Nature 399: , 739–740. |
[48] | Vitek MP , Bhattacharya K , Glendening JM , Stopa E , Vlassara H , Bucala R , Manogue K , Cerami A ((1994) ) Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A 91: , 4766–4770. |
[49] | Queitsch C , Sangster TA , Lindquist S ((2002) ) Hsp90 as a capacitor of phenotypic variation. Nature 417: , 618–624. |
[50] | Rutherford SL , Lindquist S ((1998) ) Hsp90 as a capacitor for morphological evolution. Nature 396: , 336–342. |
[51] | Kaden D , Munter LM , Reif B , Multhaup G ((2012) ) The amyloid precursor protein and its homologues: Structural and functional aspects of native and pathogenic oligomerization. Eur J Cell Biol 91: , 234–239. |
[52] | Fujita M , Wei J , Nakai M , Masliah E , Hashimoto M ((2006) ) Chaperone and anti-chaperone: Two-faced synuclein as stimulator of synaptic evolution. Neuropathology 26: , 383–392. |
[53] | Ohtake H , Limprasert P , Fan Y , Onodera O , Kakita A , Takahashi H , Bonner LT , Tsuang DW , Murray IV , Lee VM , Trojanowski JQ , Ishikawa A , Idezuka J , Murata M , Toda T , Bird TD , Leverenz JB , Tsuji S , La Spada AR ((2004) ) Beta-synuclein gene alterations in dementia with Lewy bodies. Neurology 63: , 805–811. |
[54] | Fujita M , Sugama S , Sekiyama K , Sekigawa A , Tsukui T , Nakai M , Waragai M , Takenouchi T , Takamatsu Y , Wei J , Rockenstein E , Laspada AR , Masliah E , Inoue S , Hashimoto M ((2010) ) A beta-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat Commun 1: , 110. |
[55] | Taschenberger G , Toloe J , Tereshchenko J , Akerboom J , Wales P , Benz R , Becker S , Outeiro TF , Looger LL , Bahr M , Zweckstetter M , Kugler S ((2013) ) beta-synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann Neurol 74: , 109–118. |
[56] | Galvin JE , Uryu K , Lee VM , Trojanowski JQ ((1999) ) Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A 96: , 13450–13455. |
[57] | Surgucheva I , Sharov VS , Surguchov A ((2012) ) gamma-Synuclein: Seeding of alha-synuclein aggregation and transmission between cells. Biochemistry 51: , 4743–4754. |
[58] | Chen YC , Cheng CH , Chen GD , Hung CC , Yang CH , Hwang SP , Kawakami K , Wu BK , Huang CJ ((2009) ) Recapitulation of zebrafish sncga expression pattern and labeling the habenular complex in transgenic zebrafish using green fluorescent protein reporter gene. Dev Dyn 238: , 746–754. |
[59] | Ji H , Liu YE , Jia T , Wang M , Liu J , Xiao G , Joseph BK , Rosen C , Shi YE ((1997) ) Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res 57: , 759–764. |
[60] | Singh VK , Jia Z ((2008) ) Targeting synuclein-gamma to counteract drug resistance in cancer. Expert Opin Ther Targets 12: , 59–68. |
[61] | Pan ZZ , Bruening W , Giasson BI , Lee VM , Godwin AK ((2002) ) Gamma-synuclein promotes cancer cell survival and inhibits stress- and chemotherapy drug-induced apoptosis by modulating MAPK pathways. J Biol Chem 277: , 35050–35060. |
[62] | Wogulis M , Wright S , Cunningham D , Chilcote T , Powell K , Rydel RE ((2005) ) Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J Neurosci 25: , 1071–1080. |
[63] | Armstrong RA ((2006) ) Plaques and tangles and the pathogenesis of Alzheimer’s disease. Folia Neuropathol 44: , 1–11. |
[64] | Goedert M ((2004) ) Tau protein and neurodegeneration. Semin Cell Dev Biol 15: , 45–49. |
[65] | Sekiyama K , Takamatsu Y , Koike W , Waragai M , Takenouchi T , Sugama S , Hashimoto M ((2016) ) Insight into the dissociation of behavior from histology in Synucleinopathies and in related neurodegenerative diseases. J Alzheimers Dis 52: , 831–841. |
[66] | Oyanagi K , Makifuchi T , Ohtoh T , Chen KM , Gajdusek DC , Chase TN , Ikuta F ((1994) ) The neostriatum and nucleus accumbens in parkinsonism-dementia complex of Guam: A pathological comparison with Alzheimer’s disease and progressive supranuclear palsy. Acta Neuropathol 88: , 122–128. |
[67] | Kokubo Y , Kuzuhara S ((2004) ) Neurofibrillary tangles in ALS and Parkinsonism-dementia complex focus in Kii, Japan. Neurology 63: , 2399–2401. |
[68] | La Spada AR , Weydt P , Pineda VV ((2011) ) Huntington’s disease pathogenesis: Mechanisms and pathways. In Neurobiology of Huntington’s Disease: Applications to Drug Discovery, Lo DC, Hughes RE, eds. Boca Raton (FL). |
[69] | Paterson AD , Kennedy JL , Petronis A ((1996) ) Evidence for genetic anticipation in non-Mendelian diseases. Am J Hum Genet 59: , 264–268. |
[70] | Bassett AS , Honer WG ((1994) ) Evidence for anticipation in schizophrenia. Am J Hum Genet 54: , 864–870. |
[71] | Mukherjee A , Soto C ((2017) ) Prion-like protein aggregates and type 2 diabetes. Cold Spring Harb Perspect Med, doi: 10.1101/cshperspect.a024315 |
[72] | Silveira JR , Raymond GJ , Hughson AG , Race RE , Sim VL , Hayes SF , Caughey B ((2005) ) The most infectious prion protein particles. Nature 437: , 257–261. |
[73] | Partridge L , Barton NH ((2000) ) Evolving evolvability. Nature 407: , 457–458. |
[74] | Conway KA , Harper JD , Lansbury PT ((1998) ) Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med 4: , 1318–1320. |
[75] | Krohn M , Bracke A , Avchalumov Y , Schumacher T , Hofrichter J , Paarmann K , Frohlich C , Lange C , Bruning T , von Bohlen Und Halbach O , Pahnke J ((2015) ) Accumulation of murine amyloid-beta mimics early Alzheimer’s disease. Brain 138: , 2370–2382. |
[76] | Selkoe DJ ((2013) ) The therapeutics of Alzheimer’s disease: Where we stand and where we are heading. Ann Neurol 74: , 328–336. |
[77] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |


