
A Novel Therapeutic Approach to Treat Alzheimer’s Disease by Neurotrophic Support During the Period of Synaptic Compensation
Abstract
Alzheimer’s disease (AD), at present, is considered an incurable disease and a major dilemma with no drug to stop or slow down its progression. Drugs that are currently available in the market are able to only transiently improve the clinical symptoms. The repeated failures in developing an effective drug has led to the suggestion that the medical intervention was probably too late to be effective since the pathology starts many years before the appearance of the clinical symptoms. Probably, at the time of the appearance of clinical symptoms the brain has undergone major neuronal and synaptic loss. Because of the uncertainty on when to use a prevention therapy, especially targeting amyloid-β (Aβ) and tau pathologies, interventions that rely on the regenerative capacity of the brain such as the modulation of the inherent neurogenesis and neuronal plasticity represent a promising therapeutic strategy. Such an approach can act both at early as well as late stages of the disease and remove the barrier of the time of intervention. In this article, we review studies mainly from our laboratory that show the merit of early intervention during the synaptic and neuronal compensation period where the brain still has the capacity to self-repair by offering neurotrophic support in reversing cognitive impairment, neuronal and synaptic deficits, Aβ, and tau pathologies and decreasing mortality in a transgenic mouse model of AD.
INTRODUCTION
Alzheimer’s disease (AD) is a slow, progressive neurodegenerative disease defined by the presence of two hallmarks: β-amyloidosis and neurofibrillary degeneration mainly composed of amyloid-β and hyperphosphorylated tau, respectively. Synaptic deficit, neuronal loss, white matter loss, inflammation, and oxidative stress are other features of this disease [1]. Clinically, AD is defined by cognitive impairment and memory loss with recent memories lost first, followed by old memories when the disease progresses along with personality change. Despite being a major public health problem and dilemma in the US and worldwide, no effective disease-modifying therapy is, at present, available to treat AD. Currently, the only approved drugs to treat AD are five cholinomimetics and one NMDA receptor antagonist. These drugs are not able to slow or halt the progression of the disease; these drugs only temporarily relieve cognitive symptoms and their efficacy varies from person to person. They are usually administered as palliative therapy to slow down the deterioration of the quality of life for patients who are admitted in the nursing homes and receive dementia care.
AD is the most common cause of dementia. It accounts for 60–80% of all cases. Almost 50 million people worldwide suffer from AD and this number is projected to triple by 2050. In the US alone, AD affects around 5.4 million people and this number is projected to increase to 13.4 million by mid-century. Currently, in the US, a new case of AD is recorded every 66 s; by 2050, a new case would be recorded every 33 s, which means ∼1 million new cases per year if no treatment is developed. Between 2000–2013, deaths due to AD increased by 75% while deaths because of other diseases such as stroke, heart disease, and prostate cancer decreased by 23%, 14%, and 11%, respectively [2].
The repeated failure to develop a drug for AD has led to the common belief that the treatment was initiated too late to be effective since brain changes start at least 20 years before the appearance of the first clinical symptoms [2, 3]. This has led the field to focus on measures that can prevent AD at the very early stages. The timing of the treatment is considered a possible factor in the success of the development of new drugs since still reversible and less established pathological processes would be targeted [4]. This urges the development of new and effective diagnostic tools to detect AD in its very early stage [5]. More and more human and animal research show that during the early stages of the disease and before the appearance of the first clinical symptoms, the brain compensates for the damage and is able to maintain a reasonably normal function. Once certain neuronal and synaptic losses are reached, the brain can no longer compensate, resulting in memory loss and cognitive decline that become apparent and deteriorate when the disease progresses. Previous studies on AD patients showed that the brain attempts to repair itself by increasing neurogenesis and neuronal plasticity during the early stages of AD, but this attempt fails probably due to the lack of appropriate neurotrophic support [6–8]. In this article, we review studies recently published from our laboratory that show a novel therapeutic strategy in which we boost the neurotrophic environment of the diseased brain by providing neurotrophic support during the compensation period to help the brain’s self-repair attempts materialize into successful neurogenesis and synaptic plasticity and reversal of cognitive impairment.
ETHIOPAGONESIS OF AD AND RISK FACTORS
AD is a disease that has a multifactorial nature which apparently involves several ethiopagenic mechanisms, and it is accompanied by severe neuronal loss and synaptic deficit in the limbic and neocortical areas [9]. AD can be sporadic or familial. The sporadic form starts at >65 years and represents more than 95–99% of the AD cases. The exact causes of the sporadic form are still unknown and not yet understood. The remaining <5% of the AD cases are the early-onset familial form, which are genetically inherited through the mutation in one of three genes, the amyloid precursor protein (APP), presenilin1 (PS1), or presenilin2 (PS2) [10, 11]. Usually, the inherited form of the disease tends to be aggressive, has early onset as early as age ∼30 years [2], and follows a Mendelian pattern of inheritance [12]. Indeed, mutations in PS1 or APP are known to be associated with a complete penetrance which means that an individual who inherits a mutation in one of these genes is guaranteed to develop AD. While mutation in PS2 has a 95% penetrance and thus not every person who has the mutation develops the disease.
Age is a major risk factor for AD. Around 5% of those with AD are aged 65–74, while ∼45% are aged 75–84. However, AD is not a normal aging process and aging by itself does not cause AD [2]. This suggests that the age-associated biological processes may be implicated in the development of the disease [13]. Genetic susceptibility such as inheriting the APOE4 gene increases the risk of having AD. Indeed, people who inherit one copy of the APOE4 ɛ4 allele have a 3.5-fold higher risk of developing AD, while having two copies of the APOE4 ɛ4 allele increases the risk by 8–15-fold. The APOE4 ɛ4 is present in ∼14% of the general population and in 40–60% of all cases of AD as one or two copies [2]. The presence of each copy of the APOE ɛ4 allele lowers the age of onset of AD by 6–7 years in a dose-dependent manner [13]. However, the presence of the ɛ4 allele is not sufficient or necessary to develop AD [12]. While the presence of the APOE ɛ4 gene is considered a risk factor to develop AD, having the APOE ɛ2 allele which is present in ∼8.4% of the general population is considered to exert a protective effect against developing AD.
Several other factors are believed to increase the risk of developing AD such as having stroke, high blood pressure and increase in cholesterol in mid-life, type II diabetes, moderate and severe traumatic brain injury, amnestic mild cognitive impairment (aMCI), vitamin deficiency, smoking and cerebrovascular diseases such as hemorrhagic infarcts, small and large ischemic infarcts, and vasculopathies [13].
DIFFERENT CLINICAL STAGES OF AD
AD is believed to have three clinical stages.
The prodromal stage is considered to last for a relatively long period of one or two decades during which there are some subtle brain changes for people who are destined to have dementia but no appearance of clinical symptoms; AD in Down’s syndrome can have a prodromal stage of several decades. During this stage, there is a significant Aβ deposition, less significant tau aggregation, and minimal synaptic and neuronal loss [14]. This stage is considered to be perfect to start the primary prevention which is believed to delay the onset of the disease. However, due to the lack of the appropriate diagnostic tool such as a reliable biomarker or a genetic component it is very hard to identify when the disease starts.
The mild cognitive impairment stage (MCI) can last for a long period and it varies from person to person. It is characterized by the appearance of MCI and pathological changes that could be measurable by MRI, PET scans, or postmortem analysis. During this stage, neuronal loss, axonal loss, synaptic loss, and dysfunction reach beyond a certain threshold with further increase in tau pathology and Aβ42 deposition [14]. Usually this period is considered as transitional between normal aging and dementia [15]. There are two types of MCI: aMCI and non amnestic MCI; 10–15% of the aMCI progress to AD/dementia per annum. This stage is considered ideal for a secondary intervention in which one could delay the progression of the disease from its earliest symptoms to the full-blown disease.
AD/dementia stage: During this stage, the clinical symptoms become quite apparent with steady and progressive cognitive decline as well as the appearance of the neuropathological features such as Aβ plaques, neurofibrillary tangles (NFT), neurodegeneration, synaptic loss, neuroinflammation, and oxidative stress [16]. Medical intervention at this stage may be least effective since a massive synaptic and neuronal loss is already reached and it may or may not be reversed.
NEURONAL AND SYNAPTIC LOSS AND COGNITIVE IMPAIRMENT IN AD
Both histopathological hallmarks of AD, NFT and Aβ plaques, are also seen in non-demented aged individuals. The density and topographical localization of NFT are important parameters in histopathological diagnosis of AD [17]. In normal subjects, NFT are localized to some subcortical regions such as the medial temporal lobe while in AD they have a widespread presence in several neocortical areas. Furthermore, the increase in the density of NFT strongly correlates with the severity of cognitive impairment [17–21]. In an advanced stage of the disease, NFT tend to correlate both with the region in which neurons die and with the impaired cognitive domains. Indeed, in early stages (Braak stages III-IV) where NFT are localized to the areas of the medial temporal lobes, the memory component is affected; while at more advanced stages (Braak stages V-VI) when NFT spread to the neocortical areas, other cognitive domains such as executive functions, visuospatial domains, and speech become affected. Aβ plaques on the other hand are present in a high proportion of elderly individuals that are not demented. Neuritic plaques and diffuse plaques are two types of the Aβ plaques. Neuritic plaques are an important subtype of the Aβ plaques that are more associated with cognitive impairment than diffuse plaques. They are usually surrounded by degenerated axons and dendrites that contain aggregates of hyperphosphorylated tau. However, the presence of Aβ plaques alone is not sufficient to develop AD dementia. The distribution of Aβ plaques in AD patients starts in the neocortex and, as the disease advances, spreads to the hippocampus, entorhinal cortex, cingulate cortex, amygdala, basal ganglia, diencephalon, midbrain, medulla oblongata, and finally pons and cerebellum [21].
Although Aβ and tau pathologies are the two major histopathological hallmarks of AD, it has been proposed that the very early stages of the disease starts with synaptic dysfunction followed by neuronal loss and then Aβ and tau pathologies and cognitive impairment at an advanced stage [22]. Synaptic loss has been shown to be a consistent feature that differentiates demented from non-demented people and it correlates well with the severity of dementia [23–25]. It was also shown to correlate with other types of dementia and with the decline in cognitive performance in normal cognitive aging [26]. A decrease of 25–30% in the number of synapses was previously reported in frontal and temporal cortical biopsies and ∼15–35% loss in the number of synapses per neuron within 2–4 years of the onset of AD [27]. The major synaptic loss, however, happens in the hippocampus where it reaches 44% to 55% [28]. Synaptic loss is thought to start first in the entorhinal cortex with the dendrites undergoing degeneration since they make up to 90% of synaptic contacts. Synaptic loss was shown to not only happen in the degenerated neurons but also in the still surviving neurons, and the major synaptic loss happens very early in the AD process [27, 29]. The ratio of synapses to neurons decrease by 48% [30]. Since surviving neurons account for ∼38% of synaptic loss, it is believed that cognitive impairment happens not only because of the synaptic loss but also because of the impaired capacity of the surviving synapses.
Since in normal aging, brain mass loss/year is estimated to be ∼0.5% and in AD it is 5-fold higher, neuronal loss represents a major pathological component of AD [31]. Indeed, brain mass loss is estimated to be 200–400 g for 7–10 years of disease progression [32]. The hippocampus is the major area that undergoes severe neuronal loss with atrophy reaching up to 10% per year. More than 50% of the neurons in the hippocampus are lost and the loss increases with the severity of the disease. Thus, many studies suggest that the progression and the severity of the disease directly correlates with neuronal loss [33, 34] and synaptic dysfunction, especially the decrease in the presynaptic marker, synaptophysin, and with the increase in the number of NFT in the hippocampus, entorhinal cortex, and association cortices [23, 24, 35].
SYNAPTIC AND NEURONAL COMPENSATION DURING EARLY STAGES OF THE DISEASE AND THE WINDOW OF THERAPEUTIC INTERVENTION
In spite of the neuronal and synaptic loss during the progression of the disease, at the very early stages there is still some residual plasticity of the brain. Indeed, neurodegeneration and compensation often co-exist at the stage of aMCI which represents a transitional stage between normal cognitive aging and dementia [36]. Functional connectivity decreases between posterior cingulate cortex and regions with the default network such as superior frontal gyrus and middle frontal gyrus, and it is compensated by increased connectivity between posterior cingulate cortex, medial prefrontal cortex, and anterior cingulate cortex [37]. This compensation due to a large intracranial volume (ICV) seems to help the brain during aMCI and early clinical stages of AD, but this effect is no longer effective after a certain threshold of neuronal and synaptic loss is reached at an advanced stage of the disease. The compensation due to the ICV may be because large brain size is accompanied by an increase in the number of synapses, an increase in the synapse/neurons count, or better connectivity [36]. Cognitively active people also seem to be more protected and bear more pathological changes before the onset of dementia than less cognitively active people. However, at the onset of dementia, the rate of progression of AD in cognitively active people is fast and the pathological burden is high [38]. Synaptic and neuronal compensation during the very early stages of the disease does happen in early AD stages and in AD transgenic mice. In demented >90-year-old individuals, a decrease in the synaptophysin in the frontal cortex was found, while in cognitively impaired non-demented individuals, an increase in the expression of synaptophysin was seen [40]. This suggests that synaptic compensation counteracts for synaptic loss and accounts for the preserved cognition in the presence of extensive pathology. In the Rush Religious Orders Study, an increase in the level of synaptophysin was reported in the superior frontal cortex of MCI patients compared to normal individuals [39]. An increase in the level of synaptophysin in elderly people with extensive pathology and spared cognition was reported compared to patients diagnosed with AD and dementia [40, 41]. The increase in the level of synaptophysin does not necessarily mean an increase in the number of synapses but it can mean an increase in the synaptic size to keep the total synaptic area intact [25]. An increase in the level of synaptophysin and SNAP-23 in minimal to mild grades of dementia at Braak stage 3 and an increase in the expression of MAP2 and α-synuclein at Braak stage 4 in the neocortex of AD patients compared to controls were reported [6]. Furthermore, an increase in the level of PSD-95 and a decrease in the level of synaptophysin in AD cases compared to age-matched controls was found [42, 43]. Similarly, an increase in the number of glutamatergic buttons in the midfrontal gyrus in MCI patients was reported [44].
Synaptic compensation in animal models of AD was also reported. For example, in the APP K670N,M671L + PSlM146L double transgenic mice an increase in the synaptophysin and cholinergic buttons in the plaque proximity that decreases with the progression of the disease was found [45, 46]. Similarly an increase in the level of PSD-95 and GluR1 in spatial-reference memory impaired rats was reported [47]. Consistent with these studies, we found an increase in the level of different synaptic, dendritic, and neuronal markers following synaptic deficit in the 3xTg-AD mouse model of AD that harbors three different mutations APPSwe, P301L, and PS1 knock-in [48, 49]. Indeed, we found that at 12 weeks of age the 3xTg-AD mice show synaptic deficit that is compensated for from 13–16 weeks. We further found that the decrease in the level and expression of the synaptic markers was associated with spatial reference memory impairment and that the increase in their level due to synaptic compensation was associated with a partial rescue of spatial reference memory impairment in the Morris water maze in the learning phase but not in the retention phase. These findings led us to speculate that in the early stages of the disease the AD brain tries to repair itself by saving the intact synapses and increasing the expression of synaptic and dendritic markers. However, probably due to lack of appropriate neurotrophic support, the attempt to compensate for synaptic and neuronal loss and rescue cognitive impairment is unsuccessful [7].
BOOSTING THE NEUROTROPHIC ENVIRONMENT TO HELP THE BRAIN’S SELF-REPAIR ATTEMPT WITH A NEUROTROPHIC COMPOUND
We postulated that pharmacological treatment with a neurotrophic compound during the synaptic compensation period would be able to prevent synaptic and neuronal deficit inhibit Aβ and tau pathologies and rescue cognitive impairment. To test this hypothesis, we treated 3xTg-AD mice with an orally bioavailable neurotrophic compound P021 during the period of synaptic compensation.
P021 is a tetra-peptide derived from the most active region of the ciliary neurotrophic factor (CNTF). P021 is admantylated on its C-terminal to decrease its degradation by exopeptidases and increase its blood-brain barrier permeability. P021 acts by competitively inhibiting the Leukemia Inhibitory factor (LIF) signaling and by increasing the BDNF expression through increasing its transcription [50, 51]. Furthermore, P021 inhibits Aβ and tau pathologies by inhibiting the GSK3β through the activation of its phosphorylation on Ser-9 by the BDNF. GSK3β is a major tau kinase inhibition of which decreases tau’s hyperphosphorylation and the amyloidogenic processing of AβPP [52, 53].
Previously studies from our laboratory showed that neurotrophic compound P021 as well as its parent molecule Peptide 6 can increase neurogenesis and synaptic plasticity, improve cognitive performance of C57BL6 wild type mice, and rescue cognitive impairment in 3xTg-AD mouse model of AD, in a rat model of sporadic AD, in a rat model of cognitive aging, and in a mouse model of mild traumatic brain injury [50, 51, 54–59]. We further found that P021 was able to rescue tau and Aβ pathologies at an advanced stage of pathology in 3xTg-AD mice [51]. In a recent study we found that by providing P021 during the synaptic compensation period we could rescue synaptic deficit, increase neurogenesis, prevent cognitive impairment, and inhibit neurodegeneration as well as Aβ and tau pathologies at different stages of the disease [49, 60, 61]. We further found that chronic treatment with P021 for 18 months could dramatically decrease the mortality level in 3xTg-AD mice. These preventive effects with P021 were found with no apparent side effects such as a decrease in food intake or body weight. Our studies show that early intervention with a neurotrophic compound can prevent synaptic and neuronal loss, cognitive impairment, Aβ and tau pathologies, and decrease mortality in a transgenic mouse model of AD. The prevention of tau and Aβ pathologies by chronic treatment with P021 found by us in 3xTg-AD mice was most likely secondary to the enhancement of neuronal connectivity. Our studies on passive immunization with antibodies to the amino-terminal domain of tau showed inhibition of both tau and Aβ pathologies and reversal of cognitive impairment in 3xTg-AD mice [62]. We believe that the beneficial therapeutic effect of both P021 and tau immunization most likely involves enhancement of neuronal connectivity. While tau immunotherapy could be effective only after the occurrence of tau pathology, treatment with a neurotrophic molecule like P021 can be employed even before any tau or Aβ pathology as a primary prevention approach (Fig. 1).
Fig.1
A diagrammatic representation of AD neurofibrillary degeneration and beneficial therapeutic effect of neurotrophic compound P021 and immunization with antibodies to amino-terminal projection domain of tau. Dendrite and synaptic degeneration associated with formation of intraneuronal tau (in red) and Aβ (intraneuronal as black dots and extracellular as plaques) pathologies is a hallmark of AD. Increase in dendritogenesis and synaptogenesis (shown in green) can prevent loss of neuronal connectivity and thereby inhibit tau and Aβ pathologies, especially if the neurotrophic support such as P021is provided before any pathology. Clearance of tau pathology by passive immunization probably rescues neuronal connectivity deficit and consequently prevents Aβ pathology by inhibiting the amyloidogenic processing of AβPP. a.tau, anti tau.
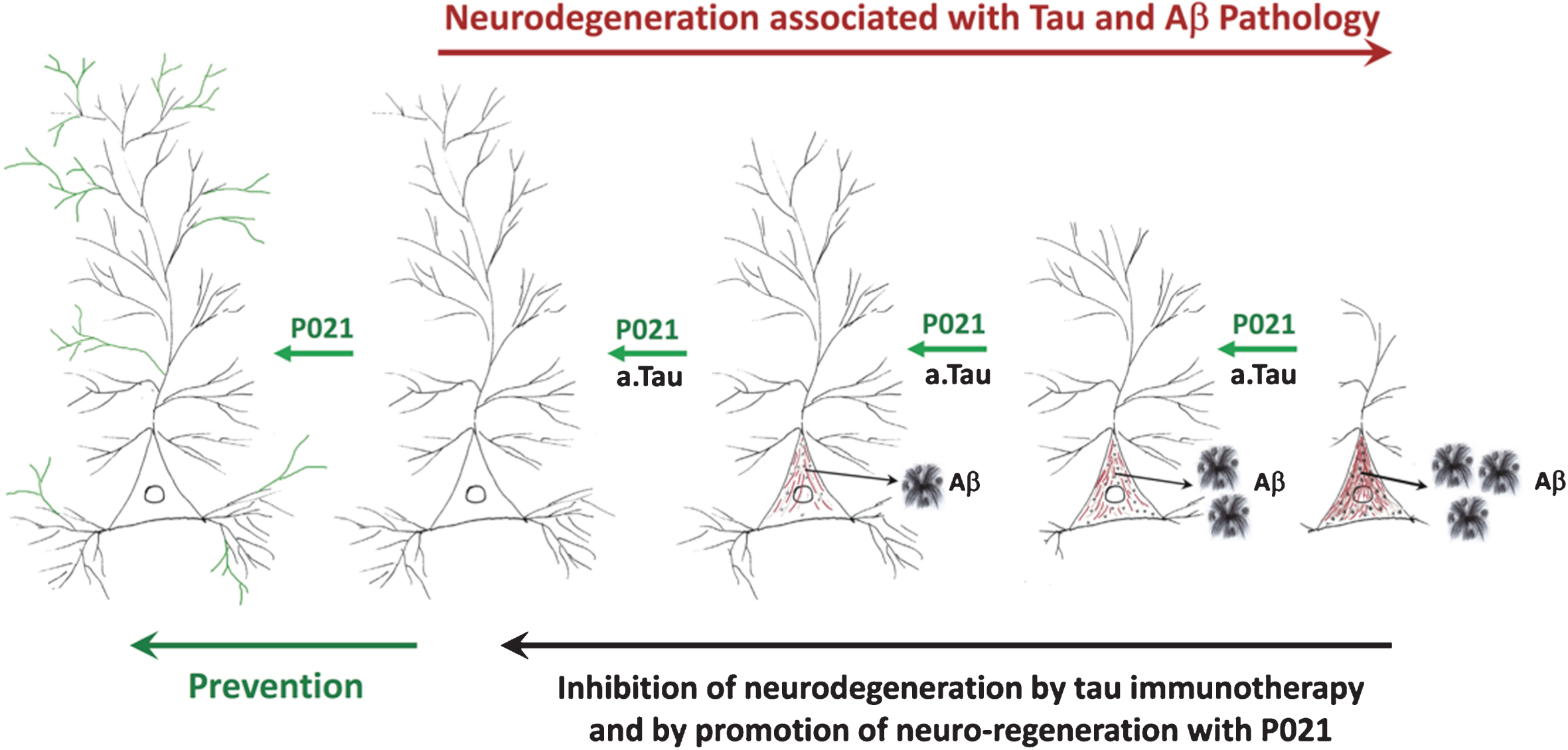
In conclusion, recent findings suggest that the timing of intervention to treat AD is very important in developing an effective drug that has a disease modifying effect. Starting an intervention, especially preceding or at a very early stage in the disease process during the synaptic and neuronal compensation period when the brain still has the capacity of self-repair, is a promising therapeutic approach. We hypothesize that the therapeutic beneficial effect of both tau immunotherapy and P021 treatment probably involves enhancement of neuronal connectivity. Removal of hyperphosphorylated tau by immunotherapy inhibits retrograde neurodegeneration and enables the affected neurons to form new functional connections. P021 treatment enhances dendritogenesis and synaptogenesis which prevents accumulation and hyperphosphorylation of tau.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0839r1).
REFERENCES
[1] | Querfurth HW , LaFerla FM ((2010) ) Alzheimer’s disease. N Engl J Med 362: , 329–344. |
[2] | Alzheimer’s Association ((2015) ) 2015 Alzheimer’s disease facts and figures. Alzheimers Dement 11: , 332–384. |
[3] | Sperling R , Mormino E , Johnson K ((2014) ) The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 84: , 608–622. |
[4] | Cavedo E , Habert MO , Lamari F , Gagliardi G , Lista S , Teichmann M , Bakardjian H , Hampel H , Dubois B ((2017) ) Preclinical Alzheimer’s disease: A systematic review of the cohorts underlying the concept. Alzheimers Dement 13,: , 454–467. |
[5] | Graham WV , Bonito-Oliva A , Sakmar TP ((2017) ) Update on Alzheimer’s disease therapy and prevention strategies. Annu Rev Med 68: , 413–430. |
[6] | Mukaetova-Ladinska EB , Garcia-Siera F , Hurt J , Gertz HJ , Xuereb JH , Hills R , Brayne C , Huppert FA , Paykel ES , McGee M , Jakes R , Honer WG , Harrington CR , Wischik CM ((2000) ) Staging of cytoskeletal and beta-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol 157: , 623–636. |
[7] | Li B , Yamamori H , Tatebayashi Y , Shafit-Zagardo B , Tanimukai H , Chen S , Iqbal K , Grundke-Iqbal I ((2008) ) Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol 67: , 78–84. |
[8] | Scheff SW , Price DA ((2006) ) Alzheimer’s disease-related alterations in synaptic density: Neocortex and hippocampus. J Alzheimers Dis 9: , 101–115. |
[9] | Iqbal K , Grundke-Iqbal I ((2010) ) Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement 6: , 420–424. |
[10] | Bird TD ((2008) ) Genetic aspects of Alzheimer disease. Genet Med 10: , 231–239. |
[11] | Guerreiro R , Hardy J ((2014) ) Genetics of Alzheimer’s disease. Neurotherapeutics 11: , 732–737. |
[12] | Reitz C , Mayeux R ((2014) ) Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 88,: , 640–651. |
[13] | Qiu C , Kivipelto M , von Strauss E ((2009) ) Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci 11: , 111–128. |
[14] | Tarawneh R , Holtzman DM ((2009) ) Critical issues for successful immunotherapy in Alzheimer’s disease: Developmentof biomarkers and methods for early detection and intervention. CNS Neurol Disord Drug Targets 8: , 144–159. |
[15] | Burns A , Zaudig M ((2002) ) Mild cognitive impairment in older people. Lancet 360: , 1963–1965. |
[16] | Feldman HH , Jacova C ((2006) ) Primary prevention and delay of onset of AD/dementia. Can J Neurol Sci 34: (Suppl 1), 84–90. |
[17] | Nelson PT , Alafuzoff I , Bigio EH , Bouras C , Braak H , Cairns NJ , Castellani RJ , Crain BJ , Davies P , Del Tredici K , Duyckaerts C , Frosch MP , Haroutunian V , Hof PR , Hulette CM , Hyman BT , Iwatsubo T , Jellinger KA , Jicha GA , Kövari E , Kukull WA , Leverenz JB , Love S , Mackenzie IR , Mann DM , Masliah E , McKee AC , Montine TJ , Morris JC , Schneider JA , Sonnen JA , Thal DR , Trojanowski JQ , Troncoso JC , Wisniewski T , Woltjer RL , Beach TG ((2012) ) Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol 71: , 362–381. |
[18] | Arriagada PV , Growdon JH , Hedley-Whyte ET , Hyman BT , Hedleywhyte ET , Hyman BT ((1992) ) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42: , 631–639. |
[19] | Duyckaerts C , Bennecib M , Grignon Y , Uchihara T , He Y , Piette F , Hauw JJ ((1997) ) Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging 18: , 267–273. |
[20] | Sabbagh MN , Cooper K , DeLange J , Stoehr JD , Thind K , Lahti T , Reisberg B , Sue L , Vedders L , Fleming SR , Beach TG ((2010) ) Functional, global and cognitive decline correlates to accumulation of Alzheimer’s pathology in MCI and AD. Curr Alzheimer Res 7: , 280–286. |
[21] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[22] | Selkoe DJ ((2002) ) Alzheimer’s disease is a synaptic failure. Science 298: , 789–791. |
[23] | DeKosky ST , Scheff SW ((1990) ) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27: , 457–464. |
[24] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[25] | Scheff SW , Price DA ((2003) ) Synaptic pathology in Alzheimer’s disease: A review of ultrastructural studies. Neurobiol Aging 24: , 1029–1046. |
[26] | Coleman P , Federoff H , Kurlan R ((2004) ) A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63: , 1155–1162. |
[27] | Davies CAA , Mann DMAM , Sumpter PQQ , Yates POO ((1987) ) A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci 78: , 151–164. |
[28] | Scheff SW , Price DA , Schmitt FA , Dekosky ST , Mufson EJ ((2007) ) Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68: , 1501–1508. |
[29] | Masliah E , Mallory M , Alford M , DeTeresa R , Hansen LA , McKeel DW , Morris JC ((2001) ) Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 56: , 127–129. |
[30] | Coleman PD , Yao PJ ((2003) ) Synaptic slaughter in Alzheimer’s disease. Neurobiol Aging 24: , 1023–1027. |
[31] | Fox NC , Freeborough PA , Rossor MN ((1996) ) Visualisation and quantification of rates of atrophy in Alzheimer’s disease. Lancet 348: , 94–97. |
[32] | Chan D , Fox NC , Jenkins R , Scahill RI , Crum WR , Rossor MN ((2001) ) Rates of global and regional cerebral atrophy inAD and frontotemporal dementia. Neurology 57: , 1756–1763. |
[33] | Gómez-Isla T , Hollister R , West H , Mui S , Growdon JH , Petersen RC , Parisi JE , Hyman BT ((1997) ) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41: , 17–24. |
[34] | Giannakopoulos P , Herrmann FR , Bussière T , Bouras C , Kövari E , Perl DP , Morrison JH , Gold G , Hof PR ((2003) ) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60: , 1495–1500. |
[35] | Ingelsson M , Fukumoto H , Newell KL , Growdon JH , Hedley-Whyte ET , Frosch MP , Albert MS , Hyman BT , Irizarry MC ((2004) ) Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology 62: , 925–931. |
[36] | Guo L-H , Alexopoulos P , Wagenpfeil S , Kurz A , Perneczky R ((2013) ) Brain size and the compensation of Alzheimer’s disease symptoms: A longitudinal cohort study. Alzheimers Dement 9: , 580–586. |
[37] | Wang Z , Liang P , Jia X , Jin G , Song H , Han Y , Lu J , Li K ((2012) ) The baseline and longitudinal changes of PCCconnectivity in mild cognitive impairment: A combined structure and resting-state fMRI study. PLoS One 7: , e36838. |
[38] | Wilson RS , Barnes LL , Aggarwal NT , Boyle PA , Hebert LE , Mendes De Leon CF , Evans DA ((2010) ) Cognitive activity and the cognitive morbidity of Alzheimer disease. Neurology 75: , 990–996. |
[39] | Counts SE , Nadeem M , Lad SP , Wuu J , Mufson EJ ((2006) ) Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol 65: , 592–601. |
[40] | Lue LF , Brachova L , Civin WH , Rogers J ((1996) ) Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. J Neuropathol Exp Neurol 55: , 1083–1088. |
[41] | Lue LF , Kuo YM , Roher AE , Brachova L , Shen Y , Sue L , Beach T , Kurth JH , Rydel RE , Rogers J ((1999) ) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 155: , 853–862. |
[42] | Leuba G , Walzer C , Vernay A , Carnal B , Kraftsik R , Piotton F , Marin P , Bouras C , Savioz A ((2008) ) Postsynaptic density protein PSD-95 expression in Alzheimer’s disease and okadaic acid induced neuritic retraction. Neurobiol Dis 30: , 408–419. |
[43] | Leuba G , Savioz A , Vernay A , Carnal B , Kraftsik R , Tardif E , Riederer I , Riederer BM ((2008) ) Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis 15: , 139–151. |
[44] | Bell KFS , Bennett DA , Cuello AC ((2007) ) Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci 27: , 10810–10817. |
[45] | Hu L , Wong T. P , Côté SL , Bell KFS , Cuello AC ((2003) ) The impact of Aβ-plaques on cortical cholinergic and non-cholinergic presynaptic boutons in Alzheimer’s disease-like transgenic mice. Neuroscience 121: , 421–432. |
[46] | Bell KFS , Kort GJL De , Steggerda S , Shigemoto R , Ribeiro-da-silva A , Cuello AC ((2003) ) Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci Lett 353: , 143–147. |
[47] | Nyffeler M , Zhang WN , Feldon J , Knuesel I ((2007) ) Differential expression of PSD proteins in age-related spatial learning impairments. Neurobiol Aging 28: , 143–155. |
[48] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron 39: , 409–421. |
[49] | Baazaoui Narjes , Iqbal K , Flory Michael IK ((2017) ) Synaptic compensation as a probable cause of prolonged mild cognitive impairment in Alzheimer’s disease: Implications from a transgenic mouse model of the disease. J Alzheimers Dis 57: , 1–17. |
[50] | Bolognin S , Buffelli M , Puoliväli J , Iqbal K ((2014) ) Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiol Aging 35: , 2134–2146. |
[51] | Kazim SF , Blanchard J , Dai C , Tung Y , Laferla FM , Iqbal I , Iqbal K ((2014) ) Disease modifying effect of chronic oral treatment with a neurotrophic peptidergic compound in a triple transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 71: , 110–130. |
[52] | Phiel CJ , Wilson CA , Lee VM-Y , Klein PS ((2003) ) GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423: , 435–439. |
[53] | Serenó L , Coma M , Rodríguez M , Sánchez-Ferrer P , Sánchez MB , Gich I , Agulló JM , Pérez M , Avila J , Guardia-Laguarta C , Clarimón J , Lleó A , Gómez-Isla T ((2009) ) A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis 35: , 359–367. |
[54] | Blanchard J , Chohan MO , Li B , Liu F , Iqbal K , Grundke-Iqbal I ((2010) ) Beneficial effect of a CNTF tetrapeptide on adult hippocampal neurogenesis, neuronal plasticity, and spatial memory in mice. J Alzheimers Dis 21: , 1185–1195. |
[55] | Li B , Wanka L , Blanchard J , Liu F , Chohan MO , Iqbal K , Grundke-Iqbal I ((2010) ) Neurotrophic peptides incorporating adamantane improve learning and memory, promote neurogenesis and synaptic plasticity in mice. FEBS Lett 584: , 3359–3365. |
[56] | Blanchard J , Wanka L , Tung Y-CC , Cárdenas-Aguayo MDC , Laferla FM , Iqbal K , Grundke-Iqbal I ((2010) ) Pharmacologic reversal of neurogenic and neuroplastic abnormalities and cognitive impairments without affecting Aβ and tau pathologies in 3xTg-AD mice. Acta Neuropathol 120: , 605–621. |
[57] | Chohan MO , Li B , Blanchard J , Tung Y-C , Heaney AT , Rabe A , Iqbal K , Grundke-Iqbal I ((2011) ) Enhancement of dentate gyrus neurogenesis, dendritic and synaptic plasticity and memory by a neurotrophic peptide. Neurobiol Aging 32: , 1420–1434. |
[58] | Bolognin S , Blanchard J , Wang X , Basurto-Islas G , Tung YC , Kohlbrenner E , Grundke-Iqbal I , Iqbal K ((2012) ) An experimental rat model of sporadic Alzheimer’s disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol 123: , 133–151. |
[59] | Chohan MO , Bragina O , Kazim SF , Statom G , Baazaoui N , Bragin D , Iqbal K , Nemoto E , Yonas H ((2015) ) Enhancement of neurogenesis and memory by a neurotrophic peptide in mild to moderate traumatic brain injury. Neurosurgery 76: , 201–214. |
[60] | Baazaoui N , Iqbal K ((2017) ) Prevention of dendritic and synaptic deficits and cognitive impairment with a neurotrophic compound. Alzheimers Res Ther 45: , 1–15. |
[61] | Baazaoui N , Iqbal K ((2017) ) Prevention of amyloid-β and tau pathologies, associated neurodegeneration, and cognitive deficit by early treatment with a neurotrophic compound. J Alzheimers Dis 58,: , 215–230. |
[62] | Dai CL , Chen X , Kazim SF , Liu F , Gong CX , Grundke-Iqbal I , Iqbal K ((2015) ) Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm 122: , 607–617. |


