
[18F]-Flutemetamol Uptake in Cortex and White Matter: Comparison with Cerebrospinal Fluid Biomarkers and [18F]-Fludeoxyglucose
Abstract
Flutemetamol (18F-Flut) is an [18F]-labelled amyloid PET tracer with increasing availability. The main objectives of this study were to investigate 1) cerebrospinal fluid (CSF) Aβ 1-42 (Aβ42) concentrations associated with regional 18F-Flut uptake, 2) associations between cortical 18F-Flut and [18F]-fludeoxyglucose (18F-FDG)-PET, and 3) the potential use of 18F-Flut in WM pathology. Cognitively impaired, nondemented subjects were recruited (n = 44). CSF was drawn, and 18F-Flut-PET, 18F-FDG-PET, and MRI performed. Our main findings were: 1) Different Alzheimer’s disease predilection areas showed increased 18F-Flut retention at different CSF Aβ42 concentrations (posterior regions were involved at higher concentrations). 2) There were strong negative correlations between regional cortical 18F-Flut and 18F-FDG uptake. 3) Increased 18F-Flut uptake were observed in multiple subcortical regions in amyloid positive subjects, including investigated reference regions. However, WM hyperintensity 18F-Flut standardized uptake value ratios (SUVr) were not significantly different, thus we cannot definitely conclude that the higher uptake in 18F-Flut(+) is due to amyloid deposition. In conclusion, our findings support clinical use of CSF Aβ42, putatively relate decreasing CSF Aβ42 concentrations to a sequence of regional amyloid deposition, and associate amyloid pathology to cortical hypometabolism. However, we cannot conclude that 18F-Flut-PET is a suitable marker for WM pathology due to high aberrant WM uptake.
INTRODUCTION
Aggregation and deposition of amyloid-β peptides (Aβ) into plaques is considered a key event in the evolution of Alzheimer’s disease (AD) [1]. There are two types of amyloid biomarkers: assays to measure cerebrospinal fluid (CSF) Aβ 1-42 (Aβ42) and PET tracers binding amyloid. Both reliably identify amyloid plaque deposition [2] and show high concordance in diagnosing AD [3, 4]. They are thus considered equal markers of brain amyloidosis [5], but discrepancies occur [4, 6], demonstrated by, for example, abnormal CSF Aβ42 and negative PET or vice versa. CSF Aβ42 becomes decreased (or pathological) before amyloid PET, as PET tracers bind strongly to fibrillar Aβ and less strongly to diffuse Aβ plaques [7]. Other possible explanations are inconsistencies in amyloid PET rating, incorrect handling or between-laboratory and batch-to-batch CSF assay variability, a known issue for these analyses [8]. Hence standardization is important for correct detection of incipient AD. We have insufficient knowledge about at which CSF Aβ42 concentrations amyloid PET becomes abnormal, and in which areas this happens first. CSF analyses are likely more available, and such knowledge may improve the diagnostic accuracy of CSF Aβ42 also to predict regional deposition at different CSF Aβ42 concentrations.
[11C] Pittsburgh compound B (11C-PiB) [9] is widely investigated, but 18F-labelled amyloid radiotracers (with increased availability due to longer half-life) have more recently been developed. Herein, we investigate [18F]-Flutemetamol (18F-Flut) [10] (a structural analogue to 11C-PiB). 11C-PiB and 18F-Flut are comparable with respect to cortical retention, but 18F-Flut typically presents with higher white matter (WM) uptake [11]. Possible explanations for the higher WM 18F-Flut uptake include non-specific uptake related to lipophilicity [12] in the high lipid content WM, and aberrant binding to the β-sheet structured myelin basic protein [13]. 18F-Flut clearance is also slower in WM compared to grey matter (GM) [14].
18F-Flut WM retention may pose challenges due to partial volume effects (PVE) (i.e., spill-over from areas of high uptake to adjacent areas of low uptake) [15]. It is not clear to which extent WM 18F-Flut uptake depends on pathology (e.g., WM amyloid load or pathology of the capillary bed) or is aberrant and unspecific, warranting further investigation of 18F-Flut for use in WM.
Applying PVE correction (PVC) enhances quantitative accuracy of 18F-Flut PET [15], and may also avoid bias in WM analyses. PVC may optimize PET images for studies of WM function and pathology, as we recently demonstrated with [18F]-fluorodeoxyglucose PET (FDG-PET) [16] (and also with diffusion tensor imaging [17]).
White matter hyperintensities of presumed vascular origin (WMHs) [18] (as visualized on T2-weighted MRI images) are thought to reflect underlying small-vessel cerebrovascular disease (CVD) [18]. There is strong evidence for an association between CVD and AD [19], but it is not certain whether there is a pathomechanistic interaction, or whether CVD is an independent process with manifestations like hippocampal atrophy [20] and synergistic effects on the clinical symptoms of AD. In both AD and CVD [21], there are microvascular alterations with capillary dysfunction, and the neurovascular unit is affected, impairing the blood-brain barrier and clearance of soluble substances [22, 23], and even amyloid deposits may be located at the outer basement membrane (cerebral amyloid angiopathy (CAA) type 1 [24]).
Establishing the potential contribution of amyloid pathology to vascular alterations (or vice versa) seen in AD may be of diagnostic, preventive and therapeutic value. To date, a handful studies have applied 11C-PiB to analyses of WM, showing reduced retention in WMHs compared to normal-appearing WM (NAWM) [25–27]. 11C-PiB is also shown to label vascular amyloid depositions [28, 29] as well as cortical deposits, a feature that may also be attributable to 18F-Flut given their structural similarities. WM 18F-Flut binding has been characterized [13], but to the best of our knowledge use of 18F-Flut in WMH has not been examined.
The objectives of the current study were therefore 1) to compare CSF Aβ42 and 18F-Flut uptake to evaluate the different Aβ concentrations at which regional 18F-Flut uptake becomes abnormal, 2) to investigate associations between cortical 18F-Flut and 18F-FDG uptake, and 3) to investigate the potential use of 18F-Flut in analyses of WM pathology. For comparative purposes, we analyzed 18F-Flut uptake in different reference regions, regions not prone to plaque deposition, and also compared subcortical retention in subjects classified as 18F-Flut positive and 18F-Flut negative to investigate potential contribution of amyloid dysmetabolism to WM pathology in AD.
MATERIAL AND METHODS
Subjects
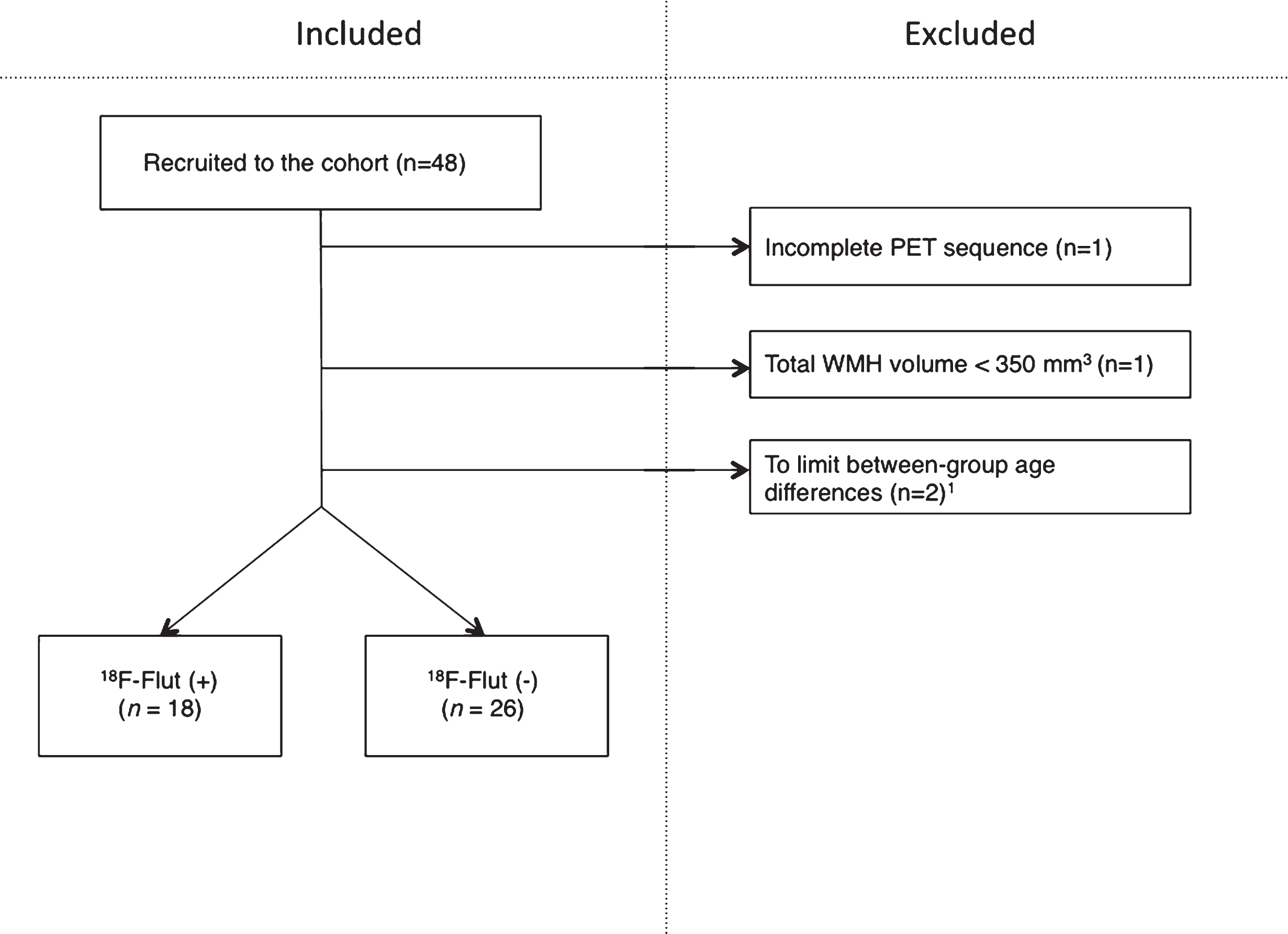
Forty-eight subjects were included from a longitudinal cohort followed at a university hospital from 2013 to 2016 (Fig. 1), of whom 29 are female. Inclusion criteria were age 40–80, complaints of cognitive decline relative to the previously normal state and Scandinavian first language. Exclusion criteria were established dementia, previous brain injury (including recognized stroke), neurodevelopmental disorder, and established serious psychiatric or somatic disorder, as well as drug use (including chemotherapy), which could significantly influence the cognitive function. All enrolled subjects had a standardized clinical assessment including physical examination, magnetic resonance imaging (MRI), lumbar puncture (n = 42), 18F-Flut PET (n = 48), and 18F-FDG PET (n = 34). All of the abovementioned procedures were performed within 3.5 months at the given assessment. The current data were collected either from time of inclusion (baseline) (n = 28), or at second assessment two years after inclusion (n = 20), given that they had not converted to dementia. The investigators were blinded to the subjects’ identities.
Fig.1
Inclusions and exclusions in the cohort. Two subjects of <55 years of age were excluded to limit between-group age differences. PET, positron emission tomography; WMH, white matter hyperintensities; 18F-Flut, 18F-Flutemetamol.
Physicians trained in diagnosing cognitive disorders established the final diagnoses in consensus conferences. Almdahl et al. have described the diagnostic criteria, including the cognitive test battery, in detail [30]. Mild cognitive impairment (MCI, n = 15) was defined according to the core criteria in the recommendations from the National Institute on Aging/Alzheimer’s Association (NIA/AA) [31]. Subjective cognitive decline (SCD) (n = 29) was defined based on the recommendations by the Subjective Cognitive Decline Initiative Working Group [32]. All forty-eight subjects had preserved functions of daily living, with Clinical Dementia Rating score≤0.5 [33], i.e., not demented. Two subjects were classified as preclinical AD stage 2 according to recommendations from the NIA/AA [34], based on one positive amyloid marker (positive 18F-Flut PET) and evidence of neuronal injury (pathological CSF total and/or phosphorylated tau). Two additional subjects were classified as preclinical AD stage 3 according the NIA/AA criteria, based on positive 18F-Flut PET, evidence of neurodegeneration (pathological CSF tau), and with subtle cognitive decline.
One subject was excluded from the current study due to an incomplete PET scan (one frame only). One subject was excluded due to total WMH volume <350 mm3, as we considered this as a minimum volume for robust analyses. Between-group age differences were limited by excluding subjects aged <50 years (n = 2). The remaining forty-four subjects were dichotomized according to cortical 18F-Flut PET uptake and classified as positive (18F-Flut(+)) or negative (18F-Flut(–)), as determined by visual interpretation by trained readers blinded to any clinical information and CSF Aβ42. The images were compared to a normal brain database supplied by the manufacturer of the tracer (CortexID, GE Healthcare, Chicago, IL, USA) and the subjects were classified as 18F-Flut(+) or 18F-Flut(–) according to the manufacturer’s guidelines.
Written informed consent was obtained from all participants prior to enrollment. The study was approved by the Regional Committee for Medical and Health Research Ethics, South East Norway (approval 2013/150) and conducted in accordance with the Helsinki Declaration.
MRI/PET Imaging acquisition
MRI was performed on a Philips Ingenia 3 Tesla system. A 3D T1-weighted turbo field echo sequence (TR/TE/TI/FA = 4.5 ms/2.2 ms/853 ms/8°, matrix = 256×213, 170 slices, thickness = 1.2 mm, in-plane resolution of 1 mm×1.2 mm) and a 3D axial fluid-attenuated inversion recovery (FLAIR) sequence (TR/TE/TI = 8000 ms/418 ms/2400 ms, slice thickness = 1.2 mm) were obtained.
18F-Flut and 18F-FDG PET were obtained from the same GE Discovery 690 PET/CT scanner on two separate days. Subjects fasted at least 6 hours in advance and blood glucose was routinely measured prior to 18F-FDG injection (subjects did not continue if blood glucose exceeded 8.0 mmol/l). For both the 18F-FDG and 18F-Flut scan sessions, a bolus injection of 185 MBq (5 mCi) was followed by rest before positioning the subject head-first supine in the scanner. Prior to PET acquisition, a low-dose CT scan for attenuation correction was acquired. PET scanning in 3D-mode started 45 minutes after injection of 18F-FDG and 90 minutes after 18F-Flut. PET data were acquired for 10 minutes for 18F-FDG (six frames) and for 20 minutes (four frames of five minutes) for 18F-Flut. Acquired data were corrected for random events, dead time, attenuation, scatter, and decay. PET volumes were reconstructed using an iterative algorithm (VUE Point FX SharpIR with six iterations, 24 subsets for 18F-FDG, four iterations, 16 subsets for 18F-Flut) and a post-reconstruction 3D Gaussian filter of 3 mm full-width half maximum was applied. The PET detector was cylindrical, 700 mm in the xy-plane and 153 mm in the z-plane, and there were 47 slices for both 18F-FDG and 18F-Flut. The matrix was 256×256 for 18F-FDG and 192×192 for 18F-Flut. Slice thickness was thus 3.27 mm for both 18F-FDG and 18F-Flut, original (xy plane) pixel size 3.27 mm×3.27 mm for 18F-FDG and 3.64 mm×3.64 mm for 18F-Flut.
Image processing
For the quantitative assessment, motion correction of the 18F-Flut PET was performed using frame-by-frame rigid registration, then the frames were summed to a single time-frame image and rigidly registered to the anatomical MRI volume using a 6-parameter rigid registration as implemented in the Spatial Parametrical Mapping (SPM 12, Wellcome Trust Centre for Neuroimaging, UCL, UK) toolbox.
Due to the relatively poor scanner resolution, PET images are susceptible to partial volume effects. Because these spill-over effects may impact the quantification of the regional uptake [15], we recently developed an algorithm to correct for partial volume effects [35]. This is a voxel-based algorithm that relies on MRI based segmentation of anatomical regions and accurate measurements of the effective point spread function of the PET imaging process. The PVC algorithm was applied to the 18F-Flut- and 18F-FDG PET images using an estimated PSF of 5.34×4.79×5.55 mm [36]. 18F-Flut- and 18F-FDG PET images and structural images were further co-registered to high-resolution 3D T1-weighted MRI scans for WMH segmentation and measurements. Registrations were manually inspected for accuracy.
An in-house developed object-based supervised machine-learning algorithm was used for automated segmentation and quantification of WMHs based on FLAIR image intensity and masks of tissue types [37]. For all segmentations, a single investigator (LFK) performed manual inspection of accuracy and edited when necessary. The reconstruction and segmentation of WM was performed with the FreeSurfer image analysis suite version 5.3.0 (http://surfer.nmr.mgh.harvard.edu/). This includes segmentation of the subcortical white-matter and deep grey-matter volumetric structures, and parcellation of the cortical surface. NAWM was defined by subtracting WMH from the total WM. Analyses and calculations were performed using The Oxford Centre for Functional MRI of the Brain (FMRIB) Software Library (FSL) version 5.0 [38, 39]. 18F-Flut and 18F-FDG standardized uptake value ratios (SUVr) were obtained by normalization to the cerebellar cortex. Prior to normalization, the cerebellar mask was eroded by 1 mm to avoid partial volume effects, inaccurate segmentation or co-registration. 18F-Flut and 18F-FDG uptake were analyzed in five pre-selected cortical regions of interest (ROIs), as defined by Desikan et al. [40], known to hold substantial amyloid plaques in AD: the precuneus and posterior cingulate combined, anterior cingulate, prefrontal, inferior parietal, and lateral temporal cortex [41]. These are also the regions used for clinical evaluation of 18F-Flut status according to the manufacturer’s guidelines [42]. We chose to only include early predilection sites for amyloid deposition in this subanalysis. We further calculated a composite SUVr by averaging the uptake in the above-mentioned regions. For comparative purposes, the 18F-Flut uptake was also analyzed in the hippocampus, parahippocampal gyrus, and entorhinal cortex. The 18F-Flut and 18F-FDG uptake for each region were averaged across the hemispheres, and the cerebellar cortex was used as the reference region.
Cerebrospinal fluid analyses
Lumbar puncture was performed consecutively after inclusion, between 8 a.m. and noon, at the L3/L4, L4/L5, or L5/S1 interspace. CSF was examined for the traditional CSF biomarkers Aβ42, T-tau, and P-tau and quantified with commercially available ELISAs (Fujirebio Europe, Gent, Belgium). The analyses were carried out in accordance with the manufacturers’ instructions at the national reference laboratory for these tests at the Department of Interdisciplinary Laboratory Medicine and Medical Biochemistry, Akershus University Hospital. The laboratory recommends the following cut-off values for abnormality (modified from Sjögren et al. [43]); t-tau >300 pg/ml for age <50 years, >450 pg/ml for age 50–69 years, and >500 pg/ml for age≥70 years, p-tau≥80 pg/ml and Aβ42 < 550 pg/ml. The national reference laboratory is part of the European multicenter project “Biomarkers for Alzheimer’s disease and Parkinson’s disease” (BIOMARKAPD), funded by EU Joint Programme-Neurodegenerative Disease Research (JPND), for standardization of analytical methods and laboratory procedures to increase its accuracy with regard to CSF biomarkers of neurodegenerative diseases.
Statistical analyses
Demographic and clinical features were assessed using chi-square tests for categorical data and independent-samples T-test or Mann-Whitney U test for continuous data. The WMH volume was logarithmically transformed prior to group-difference analyses.
Regional cortical 18F-Flut and CSF Aβ42
To determine the CSF Aβ42 concentrations associated with abnormal 18F-Flut uptake, slope and intercept values from regression equations representing associations between regional 18F-Flut and CSF Aβ42 were used. Abnormal 18F-Flut SUVr was defined as SUVr > 1.46, as previously published [44]. Receiver operating characteristic (ROC) curves were used to determine the sensitivity and specificity of the different CSF Aβ42 concentrations (as compared to 18F-Flut). All SUVr were PVE corrected. Linear regression was used to correct for age and sex, and the residuals were used for correlation analyses.
Associations between cortical 18F-Flut and 18F-FDG uptake
Spearman correlations were computed for assessment of associations between regional 18F-Flut SUVr and CSF Aβ42, as well as correlations between cortical 18F-Flut and 18F-FDG SUVr. All SUVr were PVE corrected.
Regional 18F-Flutemetamol retention and relevance to white matter pathology
We investigated 18F-Flut uptake prior to normalization to reference regions, exploring a putative pathology-specific increased retention in regions commonly used as reference for intensity normalization. Such standardized uptake values (SUV) were also analyzed in cerebral WM, WMHs, and NAWM for comparative purposes. SUV was calculated by dividing the mean regional radioactivity by the injected dose and body weight. SUVr were analyzed for cortical regions. Hippocampal, parahippocampal and entorhinal SUVr were analyzed for comparative purposes. Ratios were calculated by dividing the uptake for each region in 18F-Flut(+) by 18F-Flut(–), and Independent Samples T-tests and Mann-Whitney U tests were conducted to test for significance. For SUVr age and total WMH volume were included as covariates.
To analyze the potential use of 18F-Flut as a marker of WM pathology, 18F-Flut uptake in WMH and NAWM were determined prior to and after normalization to reference region, and group differences (i.e., 18F-Flut(+) versus 18F-Flut(–)) were assessed with Independent-Samples T-Tests. 18F-Flut WMHs retention was logarithmically transformed prior to the analyses. Age and total WMH volume were included as covariates for analyses of SUVr, and the ensuing residuals were normally distributed. Retention in WMHs and NAWM were compared using Paired-Samples T test. For assessment of the WMHs binding of 18F-Flut, we investigated associations between the 18F-FDG SUVr and 18F-Flut SUVr in WMHs using Spearman correlation. Linear regression was used to correct for age and total WMH volume, and the resulting residuals were used for the correlation analyses. In addition, Spearman correlations were used to determine associations between 18F-Flut SUVr in WMHs and cortex, as well as associations between 18F-FDG SUVr in WMHs and cortical 18F-Flut SUVr.
All tests were performed with significance level set at p < 0.05. Statistical analyses were performed with SPSS version 23 (SPSS Inc., Chicago, IL, USA).
RESULTS
Demographic and clinical data are listed in Table 1. There were no significant group differences in sex, age, WMH volume, or Mini-Mental State Examination score. By design, 18F-Flut (+) presented with significantly lower CSF Aβ42 and larger cortical SUVr of 18F-Flut when compared to 18F-Flut (–).
Table 1
Characteristics of the cohort
| Total study cohort (n = 44) | 18F-Flut (+) (n = 18) | 18F-Flut (–) (n = 26) | p-value (18F-Flut(+) versus 18F-Flut(–)) | |
| Age, y | 65.91 (6.88) | 68.17 (4.80) | 64.35 (7.72) | 0.070a |
| Women, n (%) | 25 (56.8) | 10 (55.56) | 15 (57.69) | 0.890b |
| MMSE, points | 28.53 (1.52) | 28.06 (1.89) | 28.85 (1.16) | 0.208c |
| CSF Aβ42 (pg/ml) | 873.57 (320.04) | 567.19 (167.31) | 1062.12 (232.67) | <0.001a |
| WMH volume (mm3) | 5850.50 (7235.81) | 7280.00 (9372.94) | 4860.85 (5274.92) | 0.448a,d |
Values are given in mean (standard deviation). Statistically significant differences shown in bold. aIndependent-samples T-test. bPearson Chi-square test. cMann-Whitney U test. dp-value for log transformed WMH volume adjusted for age. 18F-Flut, 18F-Flutemetamol; MMSE, Mini-Mental State Examination; CSF, cerebrospinal fluid; Aβ, amyloid β-peptide; WMH, white matter hyperintensities.
Regional cortical 18F-Flut and CSF Aβ42
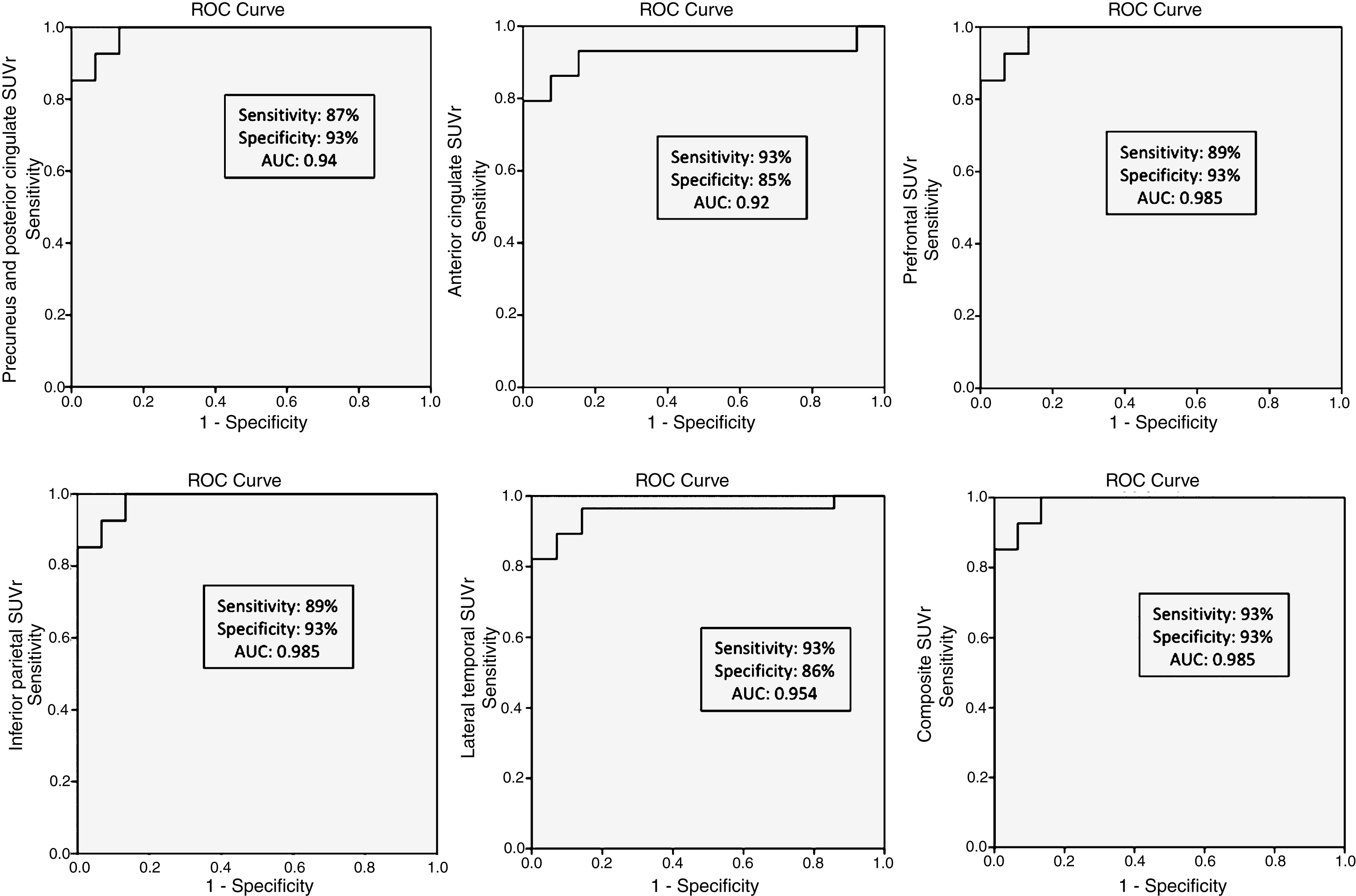
We compared regional cortical SUVr of 18F-Flut and CSF Aβ42. Figure 2 summarizes the results, and illustrates the CSF Aβ42 concentration (vertical dotted line) at which abnormal 18F-Flut SUVr (>1.46, horizontal dotted line) would be identified for each AD-specific region (also the composite region). Corresponding CSF Aβ42 levels ranged from 645 pg/ml in the anterior cingulate cortex to 762 pg/ml in the inferior parietal lobe, yielding sensitivities 87–93% and specificities 85–93% (Fig. 3). There were strong negative correlations between CSF Aβ42 and 18F-Flut SUVr for all regions; precuneus and posterior cingulate cortex (r = –0.85, p < 0.001), anterior cingulate cortex (r = –0.76, p < 0.001), prefrontal lobe (r = –0.68, p < 0.001), inferior parietal lobe (r = –0.79, p < 0.001) and lateral temporal lobe (r = –0.72, p < 0.001).
Fig.2
Associations between regional 18F-Flutemetamol SUVr and CSF Aβ42. Based on the visual interpretation by a trained reader, the subjects are marked as 18F-Flut(+) (green dots) or 18F-Flut(–) (blue dots). Using an SUVr cut-off of 1.46 yields different CSF Aβ42 cut-offs for each pre-selected region. SUVr, standardized uptake value ratios; CSF, cerebrospinal fluid; Aβ42, amyloid-β 1-42.
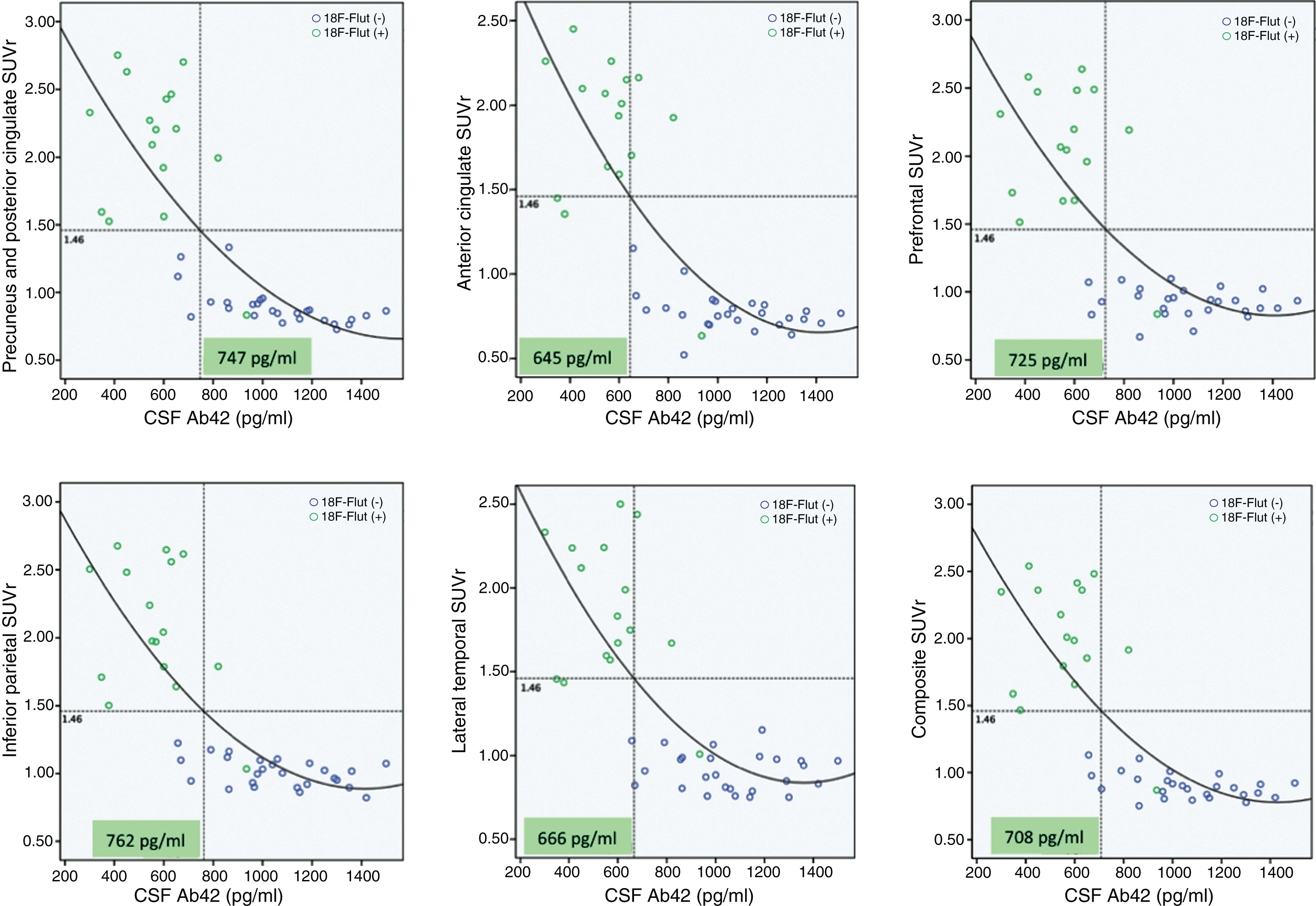
Fig.3
Receiver operating characteristic (ROC) curves for the accuracy of each CSF Aβ42 cut-off to predict regional 18F-Flutemetamol abnormality.
Associations between cortical 18F-Flut and 18F-FDG uptake
There were strong negative Spearman correlations between regional cortical 18F-Flut and 18F-FDG uptake in 18F-Flut(+) subjects in the precuneus and posterior cingulate (r = –0.797, p = 0.002), anterior cingulate cortex (r = –0.699, p = 0.011), inferior parietal lobe (r = –0.727, p = 0.007), lateral temporal lobe (r = –0.622, p = 0.031), as well as the prefrontal lobe (r = –0.811, p = 0.001). No significant correlations were observed in 18F-Flut(–) subjects.
Regional 18F-Flutemetamol retention and relevance to white matter pathology
18F-Flut SUVs (i.e., prior to intensity normalization against a reference region) were analyzed in several regions, in both PVE corrected and uncorrected images, primarily to investigate the validity of typical reference regions used for further analyses. We found numerically higher retention in all regions in subjects classified as 18F-Flut(+) compared to 18F-Flut(–). Based on the comparison of the SUV values between possible reference regions, we used the cerebellum cortex as reference region to calculate SUVr values. Before PVE correction, significant differences were also observed in subcortical WM and NAWM. Cortical ROIs were analyzed using SUVr. As expected, highly significant differences were observed in cortical regions (p < 0.001) when comparing 18F-Flut(+) and 18F-Flut(–). Results are summarized in Table 2.
Table 2
Regional 18F-Flutemetamol uptake in 18F-Flut(+) versus 18F-Flut(–)
| PVE corrected | PVE uncorrected | |||||||
| 18F-Flut + | 18F-Flut– | Ratio | p | 18F-Flut + | 18F-Flut – | Ratio | p | |
| A. SUV | ||||||||
| WMH | 767.72 (193.92) | 633.21 (173.71) | 1.21 | 0.015a | 700.66 (170.81) | 575.13 (159.05) | 1.22 | 0.019a |
| NAWM | 1028.62 (190.89) | 932.33 (240.24) | 1.10 | 0.112a | 978.49 (187.30) | 848.64 (224.09) | 1.15 | 0.035a |
| WM | 1020.95 (195.67) | 913.18 (242.43) | 1.12 | 0.081a | 918.69 (187.59) | 749.75 (211.26) | 1.23 | 0.007a |
| Cerebellum cortex | 429.59 (109.05) | 389.58 (142.53) | 1.10 | 0.143a | 429.13 (112.64) | 391.34 (147.88) | 1.10 | 0.169a |
| Cerebellum WM | 921.76 (192.45) | 830.06 (248.26) | 1.11 | 0.121a | 920.51 (192.29) | 828.68 (248.16) | 1.11 | 0.120a |
| Brainstem | 1025.93 (233.15) | 933.80 (305.42) | 1.10 | 0.157a | 1031.65 (232.99) | 938.33 (307.75) | 1.10 | 0.157a |
| B. SUVr | ||||||||
| Precun + post.cing. | 2.17 (0.54) | 0.89 (0.14) | 2.44 | <0.001b | 1.97 (0.31) | 1.23 (0.14) | 1.60 | <0.001b |
| Anterior cingulate | 1.94 (0.50) | 0.78 (0.12) | 2.49 | <0.001b | 1.88 (0.28) | 1.20 (0.13) | 1.57 | <0.001b |
| Prefrontal | 2.15 (0.53) | 0.92 (0.11) | 2.34 | <0.001b | 1.78 (0.28) | 1.13 (0.10) | 1.58 | <0.001b |
| Inferior parietal | 2.15 (0.50) | 1.01 (0.11) | 2.13 | <0.001b | 1.80 (0.27) | 1.21 (0.11) | 1.49 | <0.001b |
| Lateral temporal | 1.92 (0.43) | 0.91 (0.12) | 2.11 | <0.001b | 1.70 (0.23) | 1.17 (0.10) | 1.45 | <0.001b |
| Hippocampus | 1.07 (0.07) | 1.05 (0.06) | 1.02 | 0.274b | 1.25 (0.10) | 1.24 (0.12) | 1.01 | 0.526b |
| Parahippocampal | 1.33 (0.26) | 0.76 (0.09) | 1.75 | <0.001b | 1.35 (0.12) | 1.07 (0.10) | 1.26 | <0.001b |
| Entorhinal | 1.14 (0.24) | 0.90 (0.09) | 1.27 | 0.002b | 1.19 (0.14) | 1.04 (0.12) | 1.14 | 0.003b |
Values are given in standardized uptake value (SUV) in A and in standardized uptake value ratio (SUVr) in B. aCalculated using Independent-samples T test. bCalculated using Mann-Whitney U test. Ratio and p-values indicate 18F-Flut + versus 18F-Flut –. Statistically significant group differences are shown in bold. PVE, partial volume effects; WMH, white matter hyperintensities; NAWM, normal-appearing white matter; WM, white matter.
White matter hyperintensities. Before and after PVE corrections 18F-Flut SUVs were higher in WMHs in 18F-Flut(+) compared to 18F-Flut(–) (Table 2). No significant difference (p = 0.112) was observed in NAWM when comparing 18F-Flut(+) (mean = 1028.62, SD = 190.89) and 18F-Flut(–) (mean = 932.33, SD = 240.24). When normalizing against a reference region, corrected for age and total WMH volume, the group differences in WMHs were considerably reduced (p = 0.414; 18F-Flut(+) mean SUVr = 1.82, SD = 0.34; 18F-Flut(–) mean SUVr = 1.69, SD = 0.25) and not significant. No significance was observed in NAWM (p = 0.218; 18F-Flut(+) mean SUVr = 2.44, SD = 0.30; 18F-Flut(–) mean SUVr = 2.50, SD = 0.39). We found significantly reduced retention in WMHs compared to NAWM (Z = –5,78, p < 0,001), presenting with a reduction of 29.81%. We further assessed correlations between the 18F-Flut SUVr and 18F-FDG SUVr in WMHs and found strong positive correlations in 18F-Flut(+) subjects (r = 0.779, p = 0.003), while no significant correlation was found in 18F-Flut(–) subjects (r = 0.200, p = 0.371).
Associations between 18F-Flut SUVr in WMHs and GM, and between 18F-FDG SUVr in WMHs and cortical 18F-Flut SUVr were assessed (Table 3). 18F-FDG uptake in WMHs and cortical 18F-Flut SUVr in the AD-predilection cortical regions were negatively correlated. No significant correlations were found between 18F-Flut uptake in WMHs and cortex.
Table 3
Associations between 18F-Flutemetamol and 18F-FDG SUVr in WMHs and cortical 18F-Flutemetamol
| 18F-Flutemetamol in WMHs | 18F-FDG in WMHs | |
| 18F-Flutemetamol in | ||
| Precuneus + post.cing | r = 0.068, p = 0.663 | r = –0.359, p = 0.037 |
| Anterior cingulate | r = 0.046, p = 0.786 | r = –0.469, p = 0.005 |
| Prefrontal | r = –0.018, p = 0.908 | r = –0.396, p = 0.020 |
| Inferior parietal | r = 0.004, p = 0.978 | r = –0.422, p = 0.013 |
| Lateral temporal | r = –0.064, p = 0.681 | r = –0.358, p = 0.037 |
| Hippocampus | r = 0.009, p = 0.952 | r = 0.077, p = 0.666 |
| Parahippocampus | r = 0.139, p = 0.367 | r = –0.246, p = 0.161 |
| Entorhinal cortex | r = 0.086, p = 0.579 | r = –0.267, p = 0.127 |
Significance shown in bold. SUVr, standardized uptake value ratios; FDG, fluorodeoxyglucose; WMHs, white matter hyperintensities.
DISCUSSION
In this study, we combined three AD biomarkers, CSF Aβ42, 18F-Flut - and 18F-FDG PET, finding that CSF Aβ42 concentrations associated with abnormal 18F-Flut uptake differ between predilection areas for AD pathology, posterior areas demonstrating a pathological high retention at higher CSF Aβ42 levels. We showed strong negative correlations between cortical 18F-Flut and 18F-FDG uptake. To our knowledge, we are the first to evaluate the use of 18F-Flut in WMHs. We found generally higher 18F-Flut retention throughout the brain in 18F-Flut(+) subjects prior to normalization, including subcortical WM. We found significantly higher WMH 18F-Flut uptake in 18F-Flut(+) as compared to18F-Flut(–) without normalizing to a reference region, but after normalization the differences were noticeably reduced.
Brain amyloid pathology is a core diagnostic clinical criterion of AD, as evidenced by low CSF Aβ42 or positive amyloid PET. CSF biomarkers have shown similar diagnostic accuracy as both regional and composite PET measures [5]. CSF was recently demonstrated to reliably predict Aβ accumulation, where a CSF Aβ42 concentration of 647 pg/ml or less identified a positive composite 18F-Flut PET measure with high sensitivity and specificity [4]. However, regional differences in this aspect have not been investigated. In the current study, we find that regions known to harbor amyloid plaques in AD show abnormal 18F-Flut retention at different CSF Aβ42 concentrations (the inferior parietal lobe and posterior cingulate and precuneus present with abnormal 18F-Flut SUVr at the highest concentrations, followed by the prefrontal cortex). There were strong negative correlations between CSF Aβ42 and 18F-Flut SUVr in these regions, in line with previous findings [4], and our analyses yielded sensitivity and specificity around 90% for all investigated regions and CSF cut-offs (Fig. 3). The current findings are supported by the early involvement of these regions in the disease course [41] and local predilection for plaque deposition [45].
This gives additional valuable information for CSF as a clinical tool, also considering the 18F-Flut manufacturer’s guidelines stating that a positive 18F-Flut PET scan requires abnormal amyloid retention in at least one region [42]. This highlights the need for standardization of CSF biomarker analyses to define the optimal threshold for abnormal values. There is generally a large span in the CSF Aβ42 cut-offs and cut-offs reported in the current study are slightly higher than what Palmqvist et al. found [4]. This may result from the known between-laboratory variability in CSF Aβ42 analyses. There is a need for validation of these cut-offs to increase the diagnostic accuracy of CSF biomarkers, as CSF is an amenable clinical tool in many circumstances, and also to reduce the chances of false negatives.
Reduced cerebral glucose metabolism, as determined by 18F-FDG PET, is a well-established feature of AD [46]. Previous studies combining amyloid PET and 18F-FDG PET report conflicting results, some showing reduced glucose metabolism with increasing Aβ load [47, 48], while others do not [49, 50]. When associations were observed, they seemed to be region-specific, involving mostly posterior brain regions. This is consistent with our findings of significant cortical regional correlations, with the strongest correlations observed in the precuneus, posterior cingulate and inferior parietal lobe, and additional prefrontal regions. These predilection sites mimic early changes in 18F-Flut versus CSF (as described above). Significant associations were only found in 18F-Flut(+) subjects, which may not be surprising due to the by design low cortical uptake in 18F-Flut(–) subjects. However, it may also support previous hypotheses of toxic effects of Aβ on synaptic function in AD [47], as increased fibrillar Aβ (as is detected with amyloid PET) has been associated with higher levels of toxic oligomers [51].
We further investigated regional 18F-Flut retention (Table 2). As expected, significantly higher cortical uptake was observed in 18F-Flut(+) compared to 18F-Flut(–), using SUVr (Table 2 B). PET images are prone to PVE [52], primarily due to low spatial resolution, resulting in biased uptake measurements due to spill-over. For amyloid PET there may be an overestimation of cortical retention in healthy controls more than in AD due to relatively high WM retention, increasing the chances for false positives [53]. Therefore, correcting for PVE may improve statistical power [15]. This is illustrated by the higher cortical uptake in PVE uncorrected images compared to corrected images in 18F-Flut(–) subjects, likely due to spill-over from WM. Similarly, in 18F-Flut(+) subjects there may be a spill-over from GM to subcortical WM, illustrated by slightly larger ratios in PVE uncorrected compared to corrected images, causing a positive bias and overestimation of WM signal. This highlights the importance of such a correction, not only for accurate quantification of the cortical signal, but also for potential WM analyses.
SUV was used to investigate the validity of typical reference regions, for further analyses of WM (Table 2A). This was done because group differences in reference regions may potentially impact SUVr, especially in regions with more subtle differences in 18F-Flut uptake, e.g., WM. Higher SUV in 18F-Flut(+) was also observed in subcortical WM, with significant difference in WMHs, with and without PVE correction. Analyses with and without PVE correction were performed to investigate the potential impact of such correction also in WM analyses, as it is shown to increase the accuracy of analyses [15]. The high WM retention reported with 18F-Flut raises the possibility of 18F-Flut being a marker for WM pathology, though the binding characteristics of 18F-Flut in WM remain to be determined. We investigated subcortical retention in 18F-Flut(+) and 18F-Flut(–) to further explore the possible relationship between amyloid and WM pathology as this may improve the understanding of associations between CVD and AD. Investigating 18F-Flut retention in WMHs, we found significant group differences in WMH 18F-Flut uptake when comparing SUVs (i.e., prior to normalization) in 18F-Flut(+) and 18F-Flut(–), with the highest retention in 18F-Flut(+). The differences were noticeably reduced when normalizing the uptake to a reference region (SUVr) and no longer significant. There is generally a slightly higher 18F-Flut retention in subjects classified as 18F-Flut(+), and a possible explanation for the reduction in differences may be the observed higher aberrant uptake across the entire brain in 18F-Flut(+) subjects, including the typical reference regions cerebellum and brainstem in which specific binding is believed to be negligible. Furthermore, the findings must be interpreted with caution, as several factors are known to impact on SUV, and SUVr is considered the more precise and robust measure for amyloid PET.
High WM retention has been reported for several 18F-PET ligands [10, 54, 55]. The WM binding of 18F-Flut was recently investigated using spectral analysis and the authors concluded that WM binding was nonspecific, i.e., not related to amyloid binding [13]. This is in line with what has previously been reported with 18F-Florbetaben using kinetic model-based analyses [55]. This is supported by the current associations observed between 18F-Flut and 18F-FDG uptake in WMHs, as well as the lack of correlations between the 18F-Flut uptake in WMHs and cortex in the current study. Accumulating evidence links CVD and AD [19], and we have previously demonstrated impaired WMH microstructural integrity in Aβ positive subjects using diffusion tensor imaging [17]. Furthermore, we recently found reduced glucose metabolism in WMHs in the presence of amyloid dysmetabolism [16]. Increased fibrillar Aβ has been associated with higher levels of Aβ oligomers, suggested to be more neurotoxic and promote neurodegeneration [56]. Hence, the current findings of negative associations between the 18F-FDG uptake in WMHs and cortical 18F-Flut retention support our previous findings [16], connecting increasing Aβ pathology to glucose hypometabolism in WMHs.
Tracer tissue extraction and clearance is related to cerebral blood flow (CBF), and CBF decreases during AD progression [22]. WMHs are often considered to result from hypoperfusion [57], and previous studies using 11C-PiB found reduced retention in WMHs compared to NAWM [25–27]. This has in addition been reported with another 18F-PET ligand, 18F-Florbetaben [58], and we observed the same with 18F-Flut in the current study. Hence one might expect lower 18F-Flut uptake in WMHs in 18F-Flut(+). However, SUVr do not distinguish between the contributions of CBF and clearance, and clearance also depends on the capillary integrity, known to be reduced in AD [22]. In CAA type 1, as opposed to CAA type 2 (involving larger vessels), the capillaries are affected, reflected by Aβ deposition in the outer basement membrane [24]. Similar to Aβ plaques, these vascular changes present with surrounding microglia activation and inflammatory responses [59]. The vascular bed in AD further presents with several alterations. The microvascular distortion and capillary dysfunction affects the neurovascular unit, thereby affecting the blood-brain barrier function [60] causing impaired clearance of soluble substances [22, 23]. Given little or absent specific 18F-Flut amyloid binding in WM, one possible explanation for the higher 18F-Flut retention observed in WMHs in 18F-Flut(+) subjects might be vascular damage present in AD impairing tracer clearance. The lack of difference observed in NAWM raises the possibility that these alterations are confined to WMHs. For other 18F-ligands, white matter retention has also been hypothesized to result from reduced WM blood perfusion compared to the cortex and thus slower clearance [61]. However, due to the aberrant uptake observed 18F-Flut (+), we cannot conclude that 18F-Flut is a suitable marker for WM pathology and the current findings must be interpreted with caution. Replications in larger cohorts are warranted for further assessment of the use of 18F-Flut in WM pathology.
The cross-sectional study design may be a limitation when considering regional comparisons of cortical 18F-Flut retention and CSF Aβ42 in patients destined to develop dementia due to AD, and longitudinal studies should be conducted for further evaluations of the regional Aβ depositions related to CSF Aβ42 cut-offs.
In conclusion, although the study design and the relatively small sample size are limitations and replications in larger cohorts are warranted, this study gives further evidence for CSF Aβ42 as a clinical tool to predict regional cortical Aβ deposition. In addition, our findings support previous hypotheses of toxic effects of Aβ on synaptic function based on the strong correlations observed between cortical 18F-Flut and 18F-FDG uptake. Furthermore, we replicate our previous findings [16] linking amyloid dysmetabolism to glucose hypometabolism in WMHs. We also demonstrate generally higher 18F-Flut retention across the brain in subjects classified as 18F-Flut(+), including WM and WMHs, which may challenge the use of 18F-Flut for WM analyses. Hence replications in larger cohorts are warranted for further assessment of the use of 18F-Flut in WM pathology.
ACKNOWLEDGMENTS
Study funded by The Research Council of Norway (grant reference number 217780/H10); South-Eastern Norway Regional Health Authority (grant reference number 2013131); and EU-JPND via The Research Council of Norway (grant reference number 237250).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0582r2).
REFERENCES
[1] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[2] | Blennow K , Mattsson N , Scholl M , Hansson O , Zetterberg H ((2015) ) Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci 36: , 297–309. |
[3] | Weigand SD , Vemuri P , Wiste HJ , Senjem ML , Pankratz VS , Aisen PS , Weiner MW , Petersen RC , Shaw LM , Trojanowski JQ , Knopman DS , Jack CR Jr. ((2011) ) Transforming cerebrospinal fluid Abeta42 measures into calculated Pittsburgh Compound B units of brain Abeta amyloid. Alzheimers Dement 7: , 133–141. |
[4] | Palmqvist S , Zetterberg H , Blennow K , Vestberg S , Andreasson U , Brooks DJ , Owenius R , Hagerstrom D , Wollmer P , Minthon L , Hansson O ((2014) ) Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta-amyloid 42: A cross-validation study against amyloid positron emission tomography. JAMA Neurol 71: , 1282–1289. |
[5] | Palmqvist S , Zetterberg H , Mattsson N , Johansson P , Minthon L , Blennow K , Olsson M , Hansson O ((2015) ) Detailed comparison of amyloid PET and CSF biomarkers for identifying early Alzheimer disease. Neurology 85: , 1240–1249. |
[6] | Landau SM , Lu M , Joshi AD , Pontecorvo M , Mintun MA , Trojanowski JQ , Shaw LM , Jagust WJ ((2013) ) Comparing positron emission tomography imaging and cerebrospinal fluid measurements of beta-amyloid. Ann Neurol 74: , 826–836. |
[7] | Cairns NJ , Ikonomovic MD , Benzinger T , Storandt M , Fagan AM , Shah AR , Reinwald LT , Carter D , Felton A , Holtzman DM , Mintun MA , Klunk WE , Morris JC ((2009) ) Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: A case report. Arch Neurol 66: , 1557–1562. |
[8] | Mattsson N , Zetterberg H , Hansson O , Andreasen N , Parnetti L , Jonsson M , Herukka SK , van der Flier WM , Blankenstein MA , Ewers M , Rich K , Kaiser E , Verbeek M , Tsolaki M , Mulugeta E , Rosen E , Aarsland D , Visser PJ , Schroder J , Marcusson J , de Leon M , Hampel H , Scheltens P , Pirttila T , Wallin A , Jonhagen ME , Minthon L , Winblad B , Blennow K ((2009) ) CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302: , 385–393. |
[9] | Klunk WE , Engler H , Nordberg A , Wang Y , Blomqvist G , Holt DP , Bergstrom M , Savitcheva I , Huang GF , Estrada S , Ausen B , Debnath ML , Barletta J , Price JC , Sandell J , Lopresti BJ , Wall A , Koivisto P , Antoni G , Mathis CA , Langstrom B ((2004) ) Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol 55: , 306–319. |
[10] | Vandenberghe R , Van Laere K , Ivanoiu A , Salmon E , Bastin C , Triau E , Hasselbalch S , Law I , Andersen A , Korner A , Minthon L , Garraux G , Nelissen N , Bormans G , Buckley C , Owenius R , Thurfjell L , Farrar G , Brooks DJ ((2010) ) 18F-flutemetamol amyloid imaging in Alzheimer disease and mild cognitive impairment: A phase 2 trial. Ann Neurol 68: , 319–329. |
[11] | Landau SM , Thomas BA , Thurfjell L , Schmidt M , Margolin R , Mintun M , Pontecorvo M , Baker SL , Jagust WJ ((2014) ) Amyloid PET imaging in Alzheimer’s disease: A comparison of three radiotracers. Eur J Nucl Med Mol Imaging 41: , 1398–1407. |
[12] | Kepe V , Moghbel MC , Langstrom B , Zaidi H , Vinters HV , Huang SC , Satyamurthy N , Doudet D , Mishani E , Cohen RM , Hoilund-Carlsen PF , Alavi A , Barrio JR ((2013) ) Amyloid-beta positron emission tomography imaging probes: A critical review. J Alzheimers Dis 36: , 613–631. |
[13] | Heurling K , Buckley C , Vandenberghe R , Laere KV , Lubberink M ((2015) ) Separation of beta-amyloid binding and white matter uptake of (18)F-flutemetamol using spectral analysis. Am J Nucl Med Mol Imaging 5: , 515–526. |
[14] | Snellman A , Rokka J , Lopez-Picon FR , Eskola O , Wilson I , Farrar G , Scheinin M , Solin O , Rinne JO , Haaparanta-Solin M ((2012) ) Pharmacokinetics of [(1)(8)F]flutemetamol in wild-type rodents and its binding to beta amyloid deposits in a mouse model of Alzheimer’s disease. Eur J Nucl Med Mol Imaging 39: , 1784–1795. |
[15] | Thomas BA , Erlandsson K , Modat M , Thurfjell L , Vandenberghe R , Ourselin S , Hutton BF ((2011) ) The importance of appropriate partial volume correction for PET quantification in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 38: , 1104–1119. |
[16] | Kalheim LF , Selnes P , Bjornerud A , Coello C , Vegge K , Fladby T ((2016) ) Amyloid Dysmetabolism relates to reduced glucose uptake in white matter hyperintensities. Front Neurol 7: , 209. |
[17] | Kalheim LF , Bjornerud A , Fladby T , Vegge K , Selnes P ((2017) ) White matter hyperintensity microstructure in amyloid dysmetabolism. J Cereb Blood Flow Metab 37: , 356–365. |
[18] | Wardlaw JM , Smith EE , Biessels GJ , Cordonnier C , Fazekas F , Frayne R , Lindley RI , O’Brien JT , Barkhof F , Benavente OR , Black SE , Brayne C , Breteler M , Chabriat H , Decarli C , de Leeuw FE , Doubal F , Duering M , Fox NC , Greenberg S , Hachinski V , Kilimann I , Mok V , Oostenbrugge R , Pantoni L , Speck O , Stephan BC , Teipel S , Viswanathan A , Werring D , Chen C , Smith C , van Buchem M , Norrving B , Gorelick PB , Dichgans M ((2013) ) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: , 822–838. |
[19] | Iadecola C ((2013) ) The pathobiology of vascular dementia. Neuron 80: , 844–866. |
[20] | Selnes P , Grambaite R , Rincon M , Bjornerud A , Gjerstad L , Hessen E , Auning E , Johansen K , Almdahl IS , Due-Tonnessen P , Vegge K , Bjelke B , Fladby T (Metab) ((2015) ) Hippocampal complex atrophy in poststroke and mild cognitive impairment. J Cereb Blood Flow 35: , 1729–1737. |
[21] | Ostergaard L , Jespersen SN , Mouridsen K , Mikkelsen IK , Jonsdottir KY , Tietze A , Blicher JU , Aamand R , Hjort N , Iversen NK , Cai C , Hougaard KD , Simonsen CZ , Von Weitzel-Mudersbach P , Modrau B , Nagenthiraja K , Riisgaard Ribe L , Hansen MB , Bekke SL , Dahlman MG , Puig J , Pedraza S , Serena J , Cho TH , Siemonsen S , Thomalla G , Fiehler J , Nighoghossian N , Andersen G ((2013) ) The role of the cerebral capillaries in acute ischemic stroke: The extended penumbra model. J Cereb Blood Flow Metab 33: , 635–648. |
[22] | Ostergaard L , Aamand R , Gutierrez-Jimenez E , Ho YC , Blicher JU , Madsen SM , Nagenthiraja K , Dalby RB , Drasbek KR , Moller A , Braendgaard H , Mouridsen K , Jespersen SN , Jensen MS , West MJ ((2013) ) The capillary dysfunction hypothesis of Alzheimer’s disease. Neurobiol Aging 34: , 1018–1031. |
[23] | Iadecola C ((2010) ) The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 120: , 287–296. |
[24] | Thal DR , Ghebremedhin E , Rub U , Yamaguchi H , Del Tredici K , Braak H ((2002) ) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61: , 282–293. |
[25] | Stankoff B , Freeman L , Aigrot MS , Chardain A , Dolle F , Williams A , Galanaud D , Armand L , Lehericy S , Lubetzki C , Zalc B , Bottlaender M ((2011) ) Imaging central nervous system myelin by positron emission tomography in multiple sclerosis using [methyl-(1)(1)C]-2-(4’-methylaminophenyl)- 6-hydroxybenzothiazole. Ann Neurol 69: , 673–680. |
[26] | Goodheart AE , Tamburo E , Minhas D , Aizenstein HJ , McDade E , Snitz BE , Price JC , Mathis CA , Lopez OL , Klunk WE , Cohen AD ((2015) ) Reduced binding of Pittsburgh Compound-B in areas of white matter hyperintensities. Neuroimage Clin 9: , 479–483. |
[27] | Glodzik L , Rusinek H , Li J , Zhou C , Tsui W , Mosconi L , Li Y , Osorio R , Williams S , Randall C , Spector N , McHugh P , Murray J , Pirraglia E , Vallabhajolusa S , de Leon M ((2015) ) Reduced retention of Pittsburgh compound B in white matter lesions. Eur J Nucl Med Mol Imaging 42: , 97–102. |
[28] | Lockhart A , Lamb JR , Osredkar T , Sue LI , Joyce JN , Ye L , Libri V , Leppert D , Beach TG ((2007) ) PIB is a non-specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain 130: , 2607–2615. |
[29] | Johnson KA , Gregas M , Becker JA , Kinnecom C , Salat DH , Moran EK , Smith EE , Rosand J , Rentz DM , Klunk WE , Mathis CA , Price JC , Dekosky ST , Fischman AJ , Greenberg SM ((2007) ) Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 62: , 229–234. |
[30] | Almdahl IS , Lauridsen C , Selnes P , Kalheim LF , Coello C , Gajdzik B , Møller I , Wettergreen M , Grambaite R , Bjørnerud A , Bråthen G , Sando SB , White LR , Fladby T ((2017) ) Cerebrospinal fluid levels of amyloid beta 1-43 mirror 1-42 in relation to imaging biomarkers of Alzheimer’s disease. Front Aging Neurosci 9: , 9. |
[31] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[32] | Jessen F , Amariglio RE , van Boxtel M , Breteler M , Ceccaldi M , Chetelat G , Dubois B , Dufouil C , Ellis KA , van der Flier WM , Glodzik L , van Harten AC , de Leon MJ , McHugh P , Mielke MM , Molinuevo JL , Mosconi L , Osorio RS , Perrotin A , Petersen RC , Rabin LA , Rami L , Reisberg B , Rentz DM , Sachdev PS , de la Sayette V , Saykin AJ , Scheltens P , Shulman MB , Slavin MJ , Sperling RA , Stewart R , Uspenskaya O , Vellas B , Visser PJ , Wagner M ((2014) ) A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 10: , 844–852. |
[33] | Morris JC ((1997) ) Clinical dementia rating: A reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr 9: Suppl (1), 173–176; discussion 177–178. |
[34] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[35] | Coello C , Willoch F , Selnes P , Gjerstad L , Fladby T , Skretting A ((2013) ) Correction of partial volume effect in (18)F-FDG PET brain studies using coregistered MR volumes: Voxel based analysis of tracer uptake in the white matter. Neuroimage 72: , 183–192. |
[36] | Bettinardi V , Presotto L , Rapisarda E , Picchio M , Gianolli L , Gilardi MC ((2011) ) Physical performance of the new hybrid PETCT Discovery-690. Med Phys 38: , 5394–5411. |
[37] | Rincon M , Diaz-Lopez E , Selnes P , Vegge K , Altmann M , Fladby T , Bjornerud A ((2017) ) Improved automatic segmentation of white matter hyperintensities in mri Based on Multilevel Lesion Features. Neuroinformatics 15: , 231–245. |
[38] | Smith SM , Jenkinson M , Woolrich MW , Beckmann CF , Behrens TE , Johansen-Berg H , Bannister PR , De Luca M , Drobnjak I , Flitney DE , Niazy RK , Saunders J , Vickers J , Zhang Y , De Stefano N , Brady JM , Matthews PM ((2004) ) Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23: (Suppl 1), S208–S219. |
[39] | Woolrich MW , Jbabdi S , Patenaude B , Chappell M , Makni S , Behrens T , Beckmann C , Jenkinson M , Smith SM ((2009) ) Bayesian analysis of neuroimaging data in FSL. Neuroimage 45: , S173–S186. |
[40] | Desikan RS , Segonne F , Fischl B , Quinn BT , Dickerson BC , Blacker D , Buckner RL , Dale AM , Maguire RP , Hyman BT , Albert MS , Killiany RJ ((2006) ) An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31: , 968–980. |
[41] | Thal DR , Attems J , Ewers M ((2014) ) Spreading of amyloid, tau, and microvascular pathology in Alzheimer’s disease: Findings from neuropathological and neuroimaging studies. J Alzheimers Dis 42: (Suppl 4), S421–S429. |
[42] | ((2013) ) Vizamyl. (Prescribing information). GE Healthcare, Arlington Heights, IL. |
[43] | Sjogren M , Vanderstichele H , Agren H , Zachrisson O , Edsbagge M , Wikkelso C , Skoog I , Wallin A , Wahlund LO , Marcusson J , Nagga K , Andreasen N , Davidsson P , Vanmechelen E , Blennow K ((2001) ) Tau and Abeta42 in cerebrospinal fluid from healthy adults 21-93 years of age: Establishment of reference values. Clin Chem 47: , 1776–1781. |
[44] | Thurfjell L , Lilja J , Lundqvist R , Buckley C , Smith A , Vandenberghe R , Sherwin P ((2014) ) Automated quantification of 18F-flutemetamol PET activity for categorizing scans as negative or positive for brain amyloid: Concordance with visual image reads. J Nucl Med 55: , 1623–1628. |
[45] | Thal DR , Rub U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[46] | Mosconi L ((2005) ) Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging 32: , 486–510. |
[47] | Engler H , Forsberg A , Almkvist O , Blomquist G , Larsson E , Savitcheva I , Wall A , Ringheim A , Langstrom B , Nordberg A ((2006) ) Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 129: , 2856–2866. |
[48] | Cohen AD , Price JC , Weissfeld LA , James J , Rosario BL , Bi W , Nebes RD , Saxton JA , Snitz BE , Aizenstein HA , Wolk DA , Dekosky ST , Mathis CA , Klunk WE ((2009) ) Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: An example of brain reserve. J Neurosci 29: , 14770–14778. |
[49] | Li Y , Rinne JO , Mosconi L , Pirraglia E , Rusinek H , DeSanti S , Kemppainen N , Nagren K , Kim BC , Tsui W , de Leon MJ ((2008) ) Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging 35: , 2169–2181. |
[50] | Rabinovici GD , Furst AJ , Alkalay A , Racine CA , O’Neil JP , Janabi M , Baker SL , Agarwal N , Bonasera SJ , Mormino EC , Weiner MW , Gorno-Tempini ML , Rosen HJ , Miller BL , Jagust WJ ((2010) ) Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain 133: , 512–528. |
[51] | Esparza TJ , Zhao H , Cirrito JR , Cairns NJ , Bateman RJ , Holtzman DM , Brody DL ((2013) ) Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol 73: , 104–119. |
[52] | Hoffman EJ , Huang SC , Phelps ME ((1979) ) Quantitation in positron emission computed tomography: 1. Effect of object size. J Comput Assist Tomogr 3: , 299–308. |
[53] | Matsubara K , Ibaraki M , Shimada H , Ikoma Y , Suhara T , Kinoshita T , Itco H ((2016) ) Impact of spillover from white matter by partial volume effect on quantification of amyloid deposition with [11C]PiB PET. Neuroimage 143: , 316–324. |
[54] | Landau SM , Breault C , Joshi AD , Pontecorvo M , Mathis CA , Jagust WJ , Mintun MA ((2013) ) Amyloid-beta imaging with Pittsburgh compound B and florbetapir: Comparing radiotracers and quantification methods. J Nucl Med 54: , 70–77. |
[55] | Becker GA , Ichise M , Barthel H , Luthardt J , Patt M , Seese A , Schultze-Mosgau M , Rohde B , Gertz HJ , Reininger C , Sabri O ((2013) ) PET quantification of 18F-florbetaben binding to beta-amyloid deposits in human brains. J Nucl Med 54: , 723–731. |
[56] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[57] | Fernando MS , Simpson JE , Matthews F , Brayne C , Lewis CE , Barber R , Kalaria RN , Forster G , Esteves F , Wharton SB , Shaw PJ , O’Brien JT , Ince PG ((2006) ) White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 37: , 1391–1398. |
[58] | Matías-Guiu JA , Cabrera-Martín MN , Matías-Guiu J , Oreja-Guevara C , Riola-Parada C , Moreno-Ramos T , Arrazola J , Carreras JL ((2015) ) Amyloid PET imaging in multiple sclerosis: An (18)F-florbetaben study. BMC Neurology 15: , 243. |
[59] | Rozemuller AJ , van Gool WA , Eikelenboom P ((2005) ) The neuroinflammatory response in plaques and amyloid angiopathy in Alzheimer’s disease: Therapeutic implications. Curr Drug Targets CNS Neurol Disord 4: , 223–233. |
[60] | Zlokovic BV ((2011) ) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12: , 723–738. |
[61] | ((2013) ) Amyvid. (Prescribing information). Eli Lilly & Company, Indianapolis, IN. |


