
Evidence that the Human Innate Immune Peptide LL-37 may be a Binding Partner of Amyloid-β and Inhibitor of Fibril Assembly
Abstract
Background:
Identifying physiologically relevant binding partners of amyloid-β (Aβ) that modulate in vivo fibril formation may yield new insights into Alzheimer’s disease (AD) etiology. Human cathelicidin peptide, LL-37, is an innate immune effector and modulator, ubiquitous in human tissues and expressed in myriad cell types.
Objective:
We present in vitro experimental evidence and discuss findings supporting a novel hypothesis that LL-37 binds to Aβ42 and can modulate Aβ fibril formation.
Methods:
Specific interactions between LL-37 and Aβ (with Aβ in different aggregation states, assessed by capillary electrophoresis) were demonstrated by surface plasmon resonance imaging (SPRi). Morphological and structural changes were investigated by transmission electron microscopy (TEM) and circular dichroism (CD) spectroscopy. Neuroinflammatory and cytotoxic effects of LL-37 alone, Aβ42 alone, and LL-37/Aβ complexes were evaluated in human microglia and neuroblastoma cell lines (SH-SY5Y).
Results:
SPRi shows binding specificity between LL-37 and Aβ, while TEM shows that LL-37 inhibits Aβ42 fibril formation, particularly Aβ’s ability to form long, straight fibrils characteristic of AD. CD reveals that LL-37 prevents Aβ42 from adopting its typical β-type secondary structure. Microglia-mediated toxicities of LL-37 and Aβ42 to neurons are greatly attenuated when the two peptides are co-incubated prior to addition. We discuss the complementary biophysical characteristics and AD-related biological activities of these two peptides.
Conclusion:
Based on this body of evidence, we propose that LL-37 and Aβ42 may be natural binding partners, which implies that balanced (or unbalanced) spatiotemporal expression of the two peptides could impact AD initiation and progression.
INTRODUCTION
Alzheimer’s disease (AD) involves the assembly of amyloid-β (Aβ) peptides from soluble monomers into oligomers, fibrils, and plaques [1]. Studies of the spatiotemporal interplay between diffusible Aβ oligomers and fibrillar deposits as well as intracellular tau tangles are aimed at investigating drivers of neuronal dysfunction [2, 3]. However, root physiological causes of sporadic AD— most common—remain unspecified, preventing the development of effective therapies to prevent, halt, or reverse the disease [4].
Identification of physiologically relevant binding partners of Aβ that modulate fibril formation in vivo might yield new insights into causes of AD and help identify early instigators of Aβ accumulation and neurotoxic effects. A number of natural proteins that can bind to or interact with Aβ were identified by various assays [5]; however, their roles in sporadic AD are often challenging to assess. Recent, extensive in vitro and in vivo studies of Aβ interactions with the human Brichos domain, a molecular chaperone, exemplify how a discovery of this sort can yield intriguing mechanistic insights [6].
Numerous literature reports suggest that biophysical activities and signaling functions of Aβ peptides and LL-37, the only cathelicidin-derived innate immune system peptide found in humans, are related in vivo. For instance, the vitamin D receptor and retinoid X receptor (RXR) are both connected with AD, as well as with expression levels of LL-37. Vitamin D3 treatment was shown to reduce cerebral Aβ accumulation and improve cognition in a mouse model of AD [7], while RXR activation reduced neuronal loss and improved cognition in an aggressive mouse model of AD [8]. Expression levels of the CAMP gene that encodes hCAP-18, the precursor for LL-37 [9], are upregulated by activation of the vitamin D receptor [10], with an obligate involvement of RXR [11].
Another connection is the hypothesis that sporadic AD is essentially ‘type 3 diabetes’ occurring in brain tissue [12, 13]. In a recent study [14], it is shown that intraperitoneal administration of the murine cathelicidin peptide, CRAMP (which like LL-37 for humans, is unique in the mouse proteome) protects Non-Obese Diabetic (NOD) mice against the development of autoimmune diabetes. This treatment was motivated by the authors’ discovery that the genetic defect of NOD mice that creates susceptibility to autoimmune diabetes is a deficit in their ability to express cathelicidin; and this defect is more pronounced in female NOD mice, who have a higher incidence of disease as compared with males. In this animal model, immunomodulatory effects of the cathelicidin peptide, including effects on the phenotypes of white blood cells including macrophages, dendritic cells, T and B cells, reduced risks of inflammatory disease [14].
To date, most approaches to AD have relied on the supposition that pathological overexpression or hindered degradation of Aβ lays the primary foundation for disease [1]. What if chronic underexpression of another biomolecule, which normally opposes fibril formation, plays a key role? It is of course difficult for researchers to identify a systemic element that should be present, or perhaps should be better regulated, but is not.
Here we report a multifaceted approach to confirm and characterize, in vitro, both the interactions between Aβ and LL-37 and inhibitory effects of LL-37 on Aβ oligomer/fibril formation. We also demonstrate that LL-37 and Aβ42, both individually toxic and proinflammatory to neuroblastoma cell line SH-SY5Y via the stimulation of microglial production of inflammatory cytokines, lose most of their cytotoxicity to neurons if the two peptides are co-incubated prior to being added to the cell culture media.
The interactions between LL-37 and Aβ peptides were investigated by surface plasmon resonance imaging (SPRi), a recent evolution of traditional SPR, which couples the label-free monitoring of molecular interactions by scanning angle SPR measurements with simultaneous charge-coupled device (CCD)-based imaging of the whole surface for signal detection [15]. The multi-array SPRi configuration improves the overall accuracy of the study by allowing simultaneous detection of signals originating from both positive and negative control ligands immobilized on the same chip. In addition, the real-time imaging of the entire SPR-biochip allows for verification of the quality and the optical properties of different ligands after their immobilization on the chip surface. To evaluate the aggregation state and the presence of soluble oligomers in the Aβ samples used for SPRi, capillary electrophoresis (CE) analysis was carried out. The inhibitory effect on fibril formation was demonstrated through transmission electron microscopy (TEM) by investigating fibril formation in quasi-physiological conditions. Conformational analyses of Aβ42 peptide in solution, in the absence and presence of LL-37, were carried out by circular dichroism (CD) spectroscopy.
In this work, we demonstrate that Aβ and LL-37 bind to each other specifically, and that LL-37 inhibits the adoption by Aβ of ordered β-type secondary structure. We discuss a variety of different published findings that suggest a physiological interplay between, and potential co-regulation of these two peptides, as an aspect of human innate immunity that may affect the initiation and progression of AD-related pathology.
MATERIALS AND METHODS
Materials
Aβ40, Aβ42 free base, and 1% NH4OH were from AnaSpec (Fremont, CA). LL-37 was from Innovagen (Lund, Sweden). An unrelated peptide to serve as a binding study control (sequence SYSVQDQYQALLQQHAQYK) was kindly gifted by Dr. Alessandro Gori from the Peptide Synthesis Lab, ICRM, CNR, Milan. “Scrambled sequence” LL-37 peptide was from AnaSpec, and has the same amino acid composition as natural LL-37, but with the following sequence: GLKLRFEFSKIKGEFLKTPEVRFRDIKLKDNRISVQR. 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP), acetonitrile, sodium carbonate, sodium hydroxide as well as NaH2PO4 and Na2HPO4 were from Sigma-Aldrich (Stenheim, Germany). Buffer solutions were prepared daily using Millipore Direct-Q™-deionized water, filtered with 0.45 μm Millipore membrane filters (Bedford, MA) and degassed bysonication.
In SPRi and CE experiments, 1 mg/mL stock solutions of Aβ40 and unrelated peptide or scrambled-sequence LL-37 in PBS were diluted to the desired concentration. Aβ42 was prepared as described by Bartolini et al. [16]. Briefly, 0.5 mg of lyophilized Aβ42 powder was dissolved in HFIP (149 μM) and kept overnight at room temperature. After 12 h, the solution was aliquoted and the HFIP was left to evaporate overnight at room temperature. The Aβ42 film was redissolved in CH3CN/300 μM Na2CO3/250 mM NaOH (48.3:48.3:3.4, v/v/v). The obtained 500 μM Aβ42 solution was diluted to the desired concentration with 20 mM phosphate buffer, pH 7.4 (SPRi: 20 μM; CE: 100 μM).
For TEM and CD analysis, the samples were prepared as described previously [17], and analyzed at 50 and/or 100 μM concentrations. Briefly, to 1 mg lyophilized Aβ42 powder (AnaSpec) 80 μL of 1% NH4OH solution was added. After 1 min sonication, the solution was diluted with 20 mM phosphate buffer, pH 7.4, to the desired concentration, and divided into aliquots, which were freeze-dried and stored at –80°C. Each lyophilized peptide aliquot was resuspended in 20 mM phosphate buffer, pH 7.4 immediately before use. Samples were sonicated 3 min in an ultrasonic bath, to break down pre-formed aggregates and increase the peptide’s effective concentration [18]. For samples incubated with LL-37, lyophilized peptide was re-suspended in appropriately diluted LL-37 solutions, so as to keep the peptides at the desired molar concentrations and obtain proper Aβ42/LL-37 molar ratios. Samples were analyzed either immediately after solubilization, or after different lengths of incubation time, as noted in figure legends.
Surface plasmon resonance imaging
Bare gold SPRi-biochips (HORIBA Jobin Yvon SAS, France) were treated for 10 min with an Oxygen Plasma Generator (Harrick Plasma Cleaner), then coated using a solution of co-poly(DMA-NAS-MAPS) (MCP-2, Lucidant Polymers, Sunnyvale, CA) via surface immersion for 30 min (MCP-2 copolymer was dissolved at 1% w/v in aqueous 20% ammonium-sulfate). Coated SPRi-biochips were then washed with distilled water and dried at 80°C for 15 min. Residual reactive groups were then reacted with 50 mM ethanolamine/0.1M Tris pH 9.0 for 1 h, washed with water, and dried withnitrogen.
The SPRi-biochip surface coated with the copolymer was functionalized by spotting the following 20 μM solutions: 1) Aβ40 (t = 0 days); 2) Aβ42 (t = 0 days); 3) Aβ42 (t = 24 days, i.e., spotted after 24 days of incubation at room temperature); and 4) an unrelated peptide used as reference. These peptides were spotted in replicate (8 spots, around 190 μm diameter each) using a piezoelectric spotter (SciFLEXARAYER S5; Scienion) at 20°C and 50% humidity. After spotting, the SPRi-biochip was incubated overnight in a sealed chamber saturated with sodium chloride at room temperature.
The binding analysis was performed using a Horiba XelPleX SPRi imaging instrumentation (HORIBA Jobin Yvon SAS, France) after an overnight instrument equilibration with HBS-ET (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% Tween) used also as running buffer. Thereafter increasing concentrations of LL-37 peptide solution (1.0; 2.5; 5.0; 10; 25; 50; 100 μM in phosphate buffer 20 mM, pH 7.4) were injected onto the chip at 50 μL/min for 10 min, at 25°C, in order to reach a binding equilibrium for all the responses. Ovalbumin (Sigma-Aldrich, A5503) was used as the negative control sample. After each injection, the chip surface was regenerated by injecting 50 mM glycine at pH 2.0, 50 μL/min for 4 min, thus obtaining a complete regeneration of the chip surface without significant loss of binding capacity. The results were preprocessed using EzSuite (HORIBA Jobin Yvon SAS, France), then equilibrium-binding constants (KD and KA (1/ KD) values were calculated using Scrubbergen2 (licensed by HORIBA Jobin Yvon SAS)). Additional binding analysis experiments were performed using the same procedures and conditions, but by injecting a control scrambled-sequence LL-37 peptide.
Capillary electrophoresis
All CE experiments were carried out by slightly modifying the method reported by Brogi et al. [17]. Briefly, an Agilent Technologies 3D CE system with built-in diode-array detector (Waldbronn, Germany) was used and data were collected using Chemstation A.10.02 software. The fused-silica capillary (50 μm id, 360 μm od, total length 33 cm, effective length 24.5 cm) was from Polymicro Technologies (Phoenix, AZ). A new capillary was flushed at 1 bar with 1 M NaOH and water for 60 min each and background electrolyte (BGE, 80 mM sodium phosphate buffer, pH 7.4) for 90 min. The between-run rinsing cycle consisted of 50 mM SDS (1.5 min), water (1.5 min), and BGE (2 min). Sample injection (100 μM) was carried out by applying a pressure of 30 mbar for 3 s. Separations were carried out at 25°C and +12 kV (current 75–80 μA). The acquisition wavelengthwas 200 nm.
Transmission electron microscopy
For each TEM sample, a 10-μL droplet of suspension or solution was applied to a carbon-coated Formvar nickel grid (200 mesh) (Electron Microscopy Sciences, Washington, PA, USA). Each sample was allowed to sediment onto the carbon film for 15 min, then negative staining was performed with 10 μL of 2% w/v uranyl acetate solution (Electron Microscopy Sciences). After carefully draining off excess staining solution using filter paper, the specimen was transferred to a Philips CM12 TEM for examination, operating the TEM at 80 kV. Electron micrographs of negatively stained samples were photographed using Kodak film.
Circular dichroism
CD spectra were collected at 20°C using a Jasco-810 spectrophotometer and a quartz cuvette with a 0.1 cm path length. All experiments were performed with an Aβ42 concentration of 50 μM. Spectra were registered from 190 to 250 nm and run at a scan speed of 20 nm/min, with a time response of 2 s and data pitch of 0.2 nm. All spectra were baseline-corrected. Molar mean residue ellipticity [θ] is expressed in degrees cm2 dmol-1, and calculated as [θ] =θobs MWR/(10 l c), where θ is observed ellipticity in degrees; MWR, peptide mean residue molar weight; l, the optical path length in cm; and c, peptide concentration in g/mL.
SH-SY5Y cells – microglia co-culture
The human neuroblastoma SH-SY5Y cell line was a gift from Dr. R. Ross, Fordham University, NY. The cells were grown in DMEM/F12 medium containing 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and 100 IU/mL penicillin and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA) under humidified 5% CO2 and 95% air.
Human microglial cells were isolated from surgically resected temporal lobe tissue as described earlier [19]. Briefly, tissues were rinsed with a PBS solution and chopped into small pieces (<2 mm3) with a sterile scalpel. They were treated with 10 mL of a 0.25% trypsin solution at 37°C for 20 min. Subsequently DNase I (from bovine pancreas, Pharmacia Biotech, Baie d’Urfé, PQ, Canada) was added to reach a final concentration of 50 μg/mL. Tissues were incubated for an additional 10 min at 37°C. After centrifugation at 275 g for 10 min, the cell pellet was re-suspended in the serum-containing medium and passed through a 100 μm nylon cell strainer (Becton Dickinson, Franklin Lakes, NJ). The cell suspension was centrifuged again (275 g for 10 min) and re-suspended in 10 mL of DMEM/F12 medium with 10% FBS containing gentamicin (50 μg/mL), and plated onto tissue culture plates (Becton Dickinson) in a humidified 5% CO2, 95% air atmosphere at 37°C for 2 h. This achieved adherence of microglial cells. Cells were allowed to grow by replacing the medium once per week.
For estimating the purity of microglial cell cultures, aliquots of the cultures were placed on glass slides at 37°C for 48 h. The attached cells were then fixed with 4% paraformaldehyde for 1 h at 4°C, and made permeable with 0.1% Triton X-100 for 1 h at room temperature. After washing twice with PBS, the culture slides were treated with the monoclonal anti-GFAP antibody (1/4,000, DAKO) for astrocytic staining and polyclonal anti-Iba-1 antibody (1/500, Wako Chemicals, Richmond, VA) for microglial staining for 3 h at room temperature. The slides were then incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (Invitrogen, 1:500) and Alexa Fluor 546-conjugated goat anti-rabbit IgG antibody (Invitrogen, 1:500) in the dark for 3 h at room temperature to yield a positive red fluorescence. To visualize all cells, the slides were washed twice with PBS and counterstained with the nuclear dye DAPI (100 μg/mL, Sigma) to give a blue fluorescent color. Images were acquired using an Olympus BX51 microscope and a digital camera (Olympus DP71). Fluorescent images were co-localized with ImagePro software (Improvision Inc., Waltham, MA). The purity of microglia was more than 99% (1.93±0.54 astrocytes in 500 total cells in microglial culture, n = 30).
To achieve SH-SY5Y differentiation, the undifferentiated cells were treated for 4 days with 5 μM retinoic acid (RA) in DMEM/F12 medium containing 5% FBS, 100 IU/mL penicillin, and 100 μg/mL streptomycin [20]. The RA-containing medium was changed every two days. Differentiated SH-SY5Y cells demonstrated neurite extension, indicative of their differentiation [21].
Cell culture treatment with Aβ and LL-37 peptides
Human microglial cells (5×104 cells per well) or human neuroblastoma SH-SY5Y cells (5×105 cells per well) or their mixture were seeded into 24-well plates in 1 mL of DMEM/F12 medium containing 5% FBS. Aβ42 peptide (AnaSpec, Fremont, CA) was dissolved in mixture of 1% NH4OH and 0.1% Thioflavin S (Sigma, St. Louis, MO) in water to make 30 mM stock solution. Further dilution was made with water to reach 30 μM. Similar methods were used to prepare LL-37 peptide solutions (AnaSpec, Fremont, CA). Cultured cells were exposed to either 30 μM Aβ42 or 30 μM LL-37; or to a combined 1:1 equimolar mixture of the two peptides at 30 μM each. After incubation for 72 h cell free medium was collected to measure levels of proinflammatory cytokines such as TNFα and IL-6. For SH-SY5Y cell viability, MTT assays were performed [22].
SH-SY5Y-microglial cell viability assays
The viability of SH-SY5Y cells following incubation with glial cell supernatants was evaluated by MTT assays as previously described [22]. Briefly, the viability was determined by adding MTT to the cell cultures to reach a final concentration of 1 mg/mL. Following 1 h incubation at 37°C, the dark crystals formed were dissolved by adding a SDS/DMF extraction buffer (300 μL, 20% sodium dodecyl sulfate, 50% N, N-dimethylformamide, pH 4.7). Subsequently plates were incubated overnight at 37°C and optical densities at 570 nm were measured by transferring 100 μL aliquots to 96-well plates and using a plate reader with a corresponding filter. Data are presented as a percentage of the values obtained from cells incubated in fresh medium only.
Measurement of TNFα and IL-6 release
Cytokine levels were measured in cell-free supernatants after 72 h. Quantitation was performed with ELISA detection kits (Peprotech, NJ) following protocols described by the manufacturer.
Statistical data analysis for cell culture assays
The significance of differences between data sets was analyzed by one-way ANOVA tests. Multiple group comparisons were followed by a post-hoc Bonferroni test, e.g., cell viability in control condition versus cells treated with LL-37 alone; cell viability in control condition versus cells treated with Aβ42 alone; and viability in control condition versus cells treated with mixed LL-37 + Aβ42. P values are given in the figure legends.
RESULTS
SPRi analysis of LL-37 binding to immobilized Aβ and CE analysis
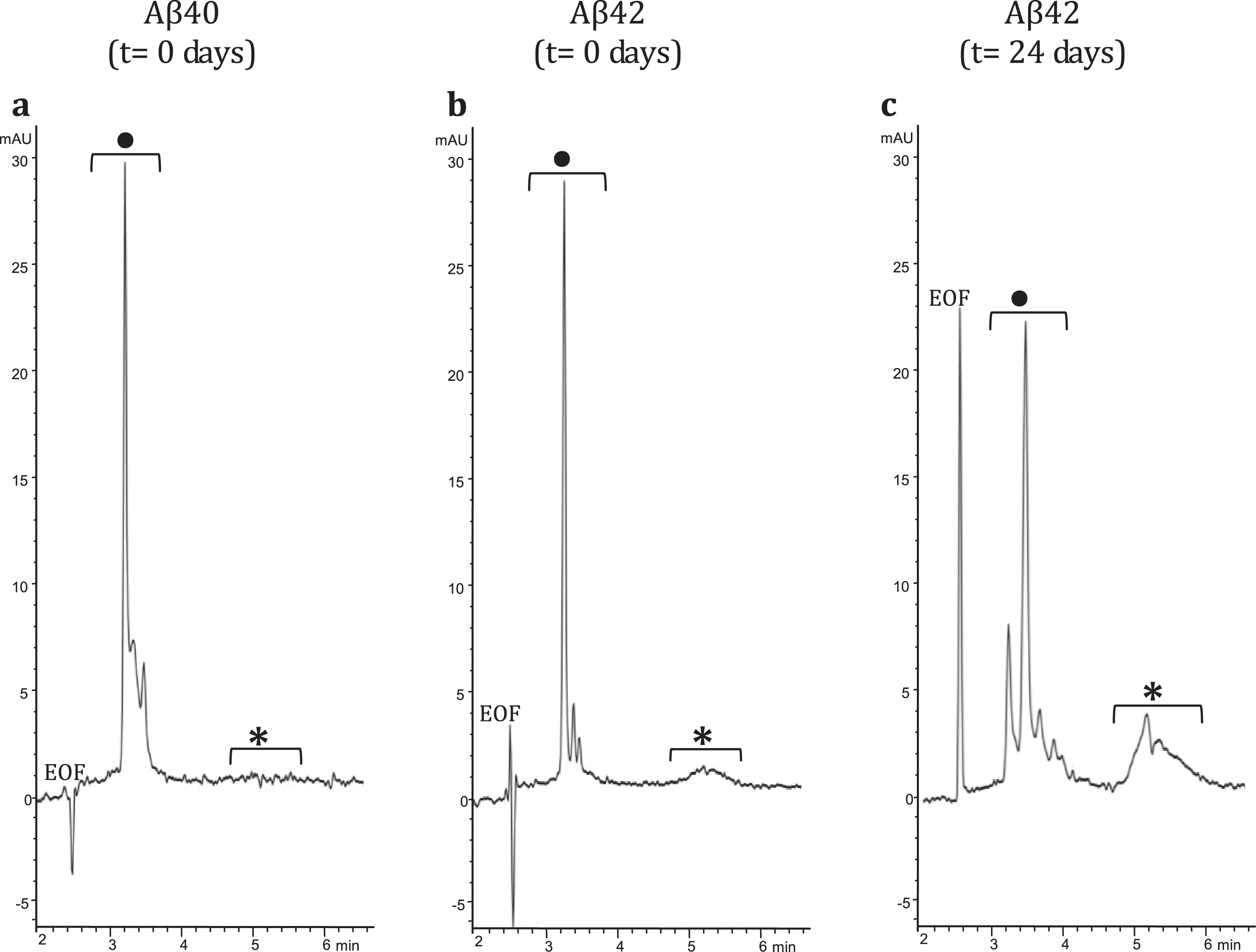
To demonstrate interaction between LL-37 and Aβ42, an SPRi biochip was functionalized with copoly(DMA-NAS-MAPS), a polydimethylacrylamide-based copolymer, widely used to immobilize biomolecules on microarray slides [23]. Three different solutions of 20 μM Aβ-peptides, in different states of aggregation, were spotted onto the SPRi-chip surface. The aggregation state and the presence of soluble oligomers were determined by CE analysis [17, 24]. Aβ40 (a) dissolved in PBS, Aβ42 (b) prepared as in [16], both solubilized immediately before spotting (t = 0 days), and Aβ42 (c) prepared as sample (b), but stored in the buffer for 24 days (t = 24 days) were spotted in different subarrays. Aβ42 was dissolved following the protocol detailed in the Material and Methods section. The solubilization buffer consisted of i) HFIP, a low-polarity solvent that stabilizes the α-helix and disrupts the interstrand hydrogen bonds of the β-sheets and ii) a mixture of acetonitrile/Na2CO3/NaOH. While acetonitrile stabilizes the unordered/α-helix structure, the basic pH increases the solubility of Aβ peptide. Overall, this procedure disfavors aggregation and provides a time window long enough to appreciate the prevailing formation of low-molecular weight (MW) oligomers at early stages (t = 0 days) and the building up of high-MW oligomers at late-stages (t = 24 days) of the in vitro fibrillogenesis process. Indeed, this was confirmed by analyzing in CE the three samples at 100 μM concentration using a method slightly modified from Sabella et al. [24]. Figure 1 shows CE electropherograms of the same Aβ peptide solutions spotted on the SPRi chip; consistent with what has been reported previously in literature [17, 24, 25], the fast-migrating group of peaks can be attributed to low MW-oligomers whereas the slow-migrating broad band corresponds to high MW-oligomers. In particular, in the Aβ42 sample, the high MW-oligomers build up over time, as evident in Fig. 1b and c.
Fig.1
CE electropherograms: a) Aβ40 (t = 0 days); b) Aβ42 (t = 0 days); and c) Aβ42 (t = 24 days). In the CE traces: •low MW-oligomers, *high MW-oligomers.
The interaction of Aβ in different aggregation states with LL-37 was investigated by SPRi. The CCD image of the chip surface, and the plasmonic curves acquired before the injection of LL-37 (Supplementary Figure 1), show the three Aβ forms conjugated to the surface. Specifically, the plasmonic curves of the different samples indicate that the sample containing a higher amount of Aβ42 high-MW oligomers (t = 24 days) binds to the surface more, compared to Aβ42 low-MW oligomers (t = 0 days) which, in turn, binds more than Aβ40 low-MW oligomers (t = 0 days). Differences in the immobilized mass could result from the increased MW of the samples in different aggregation states from low to high MW-oligomers [16, 17]. The sensograms taken after flowing a solution of LL-37 onto the SPRi chip for 10 min, revealed specific and reversible interactions with all the three Aβ forms (Fig. 2, top part). The injection of ovalbumin, a non-correlated protein, did not produce interaction signals. Analogously, the amount of LL-37 captured by non-correlated peptides (negative controls), spotted in parallel on the same SPRi chip, was significantly lower compared to that of positive signals. In a follow-up experiment, seven serial dilutions of LL-37, ranging from 1 to 100 μM, were injected to investigate the dependency of the SPR signal on ligand concentration for affinity studies (Supplementary Figure 2). An equilibrium analysis (steady-state analysis) was performed by fitting the SPRi response at the equilibrium state for each concentration (Fig. 2, bottom part). It is noteworthy that the system reached the equilibrium state at each LL-37 concentration and a complete regeneration of the SPRi chip surface was obtained after each injection. As expected, the maximal response value, or maximum binding capacity (Rmax), which is related to the maximum number of LL-37 peptides that can bind the Aβ peptides at a certain time, decreases in the following order: Aβ42 (t = 24 days) ⪢ Aβ42 (t = 0 days) > Aβ40 (t = 0 days) (Fig. 2, Table 1). This is in agreement with the amount of Aβ peptide immobilized on the SPRi chip, which increased with Aβ aggregation. To the contrary, as shown in Table 1, the affinity of LL-37 for the different Aβ forms shows the opposite trend, i.e., LL-37 binds to low-MW Aβ oligomers more strongly than to high-MW Aβ oligomers. The binding specificity of LL-37 was verified by performing the same equilibrium analysis with a control scrambled-sequence LL-37. For this experiment, it was difficult to extract adequate calibration curves (see Supplementary Figure 3), mostly due to non-specific and irregular interactions. For this reason, the binding affinities constants could not be calculated for experiments using scrambled LL-37.
Fig.2
Binding studies performed by SPRi. The SPRi-chip was functionalized with Aβ40 (t = 0 days) (a, d); Aβ42 (t = 0 days) (b, e); and Aβ42 (t = 24 days) (c, f). All the Aβ peptides were immobilized in replicate (n = 8) on the same SPRi chip at the same concentration (20 μM). SPRi reference-corrected responses related to LL-37 (10 μM) (black) and ovalbumin (10 μM) (gray) (negative control) flowed on the SPRi-chip functionalized with different Aβ forms (a, b, c). The three SPRi sensograms show the injection of running buffer (baseline) (1), the injection of the analyte (association phase) (2), and the subsequent injection of buffer (dissociation phase) (3). Calibration curve of LL-37 flowed onto different Aβ forms immobilized on the SPRi-chip Aβ40 (t = 0 days, d) Aβ42 (t = 0 days, e), Aβ42 (t = 24 days, f). The equilibrium binding constants (KA and KD) values were calculated using a nonlinear curve fit of the SPRi response at equilibrium (see Supplementary Figure 2).
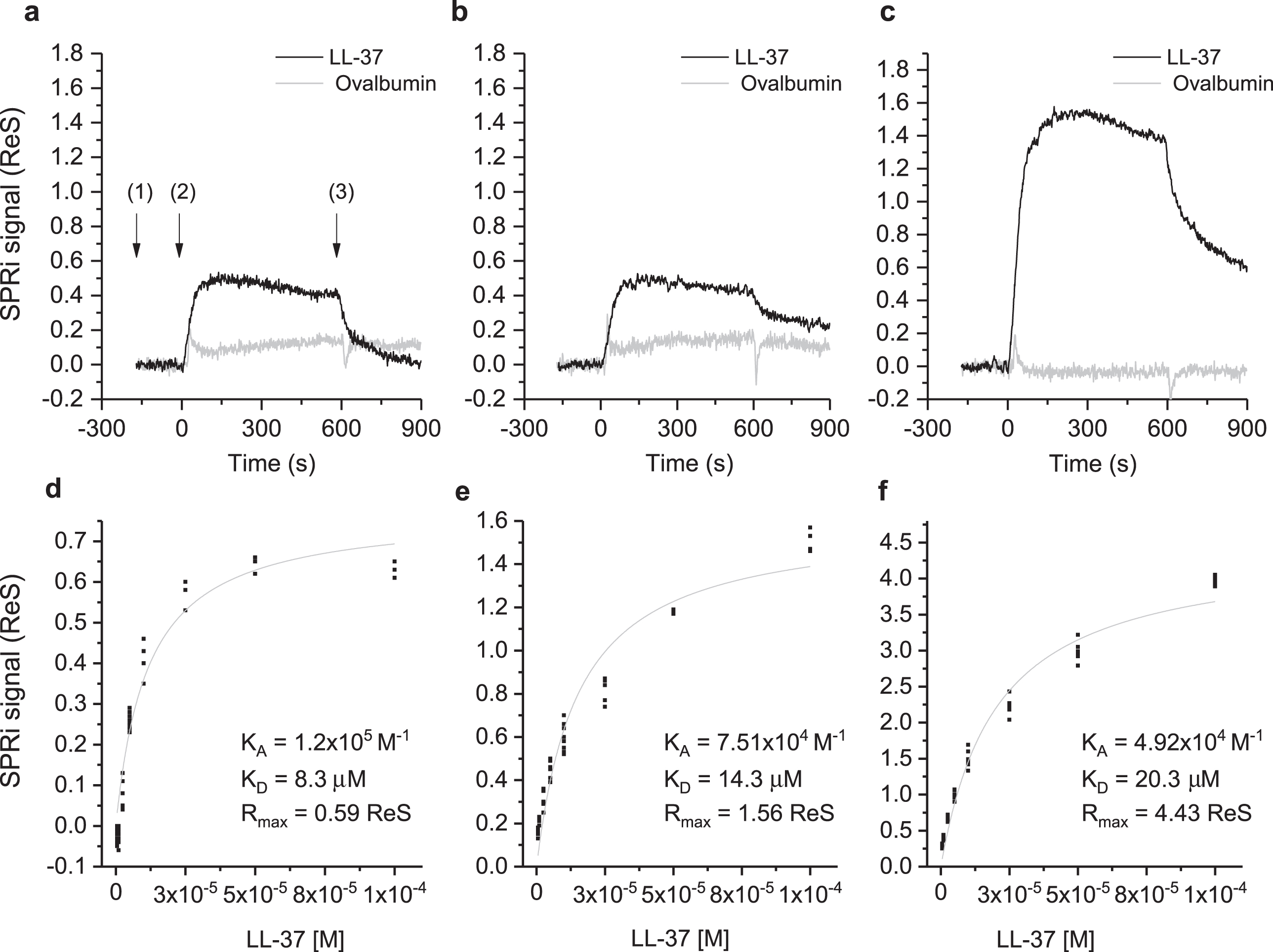
Table 1
Values of the maximal response (Rmax), (related to the absolute number of LL-37 molecules bound on the Aβ peptides at a certain time) and equilibrium binding constants (KA(1/KD) and KD), calculated using a nonlinear curve fit of the SPRi response at equilibrium
| Peptide | Rmax | KA (M-1) | KD (μM) |
| Aβ40 (t = 0 days) | 0.59 | 1.20×105 | 8.3 |
| Aβ42 (t = 0 days) | 1.56 | 7.51×104 | 13.3 |
| Aβ42 (t = 24 days) | 4.43 | 4.92×104 | 20.3 |
Transmission electron microscopy
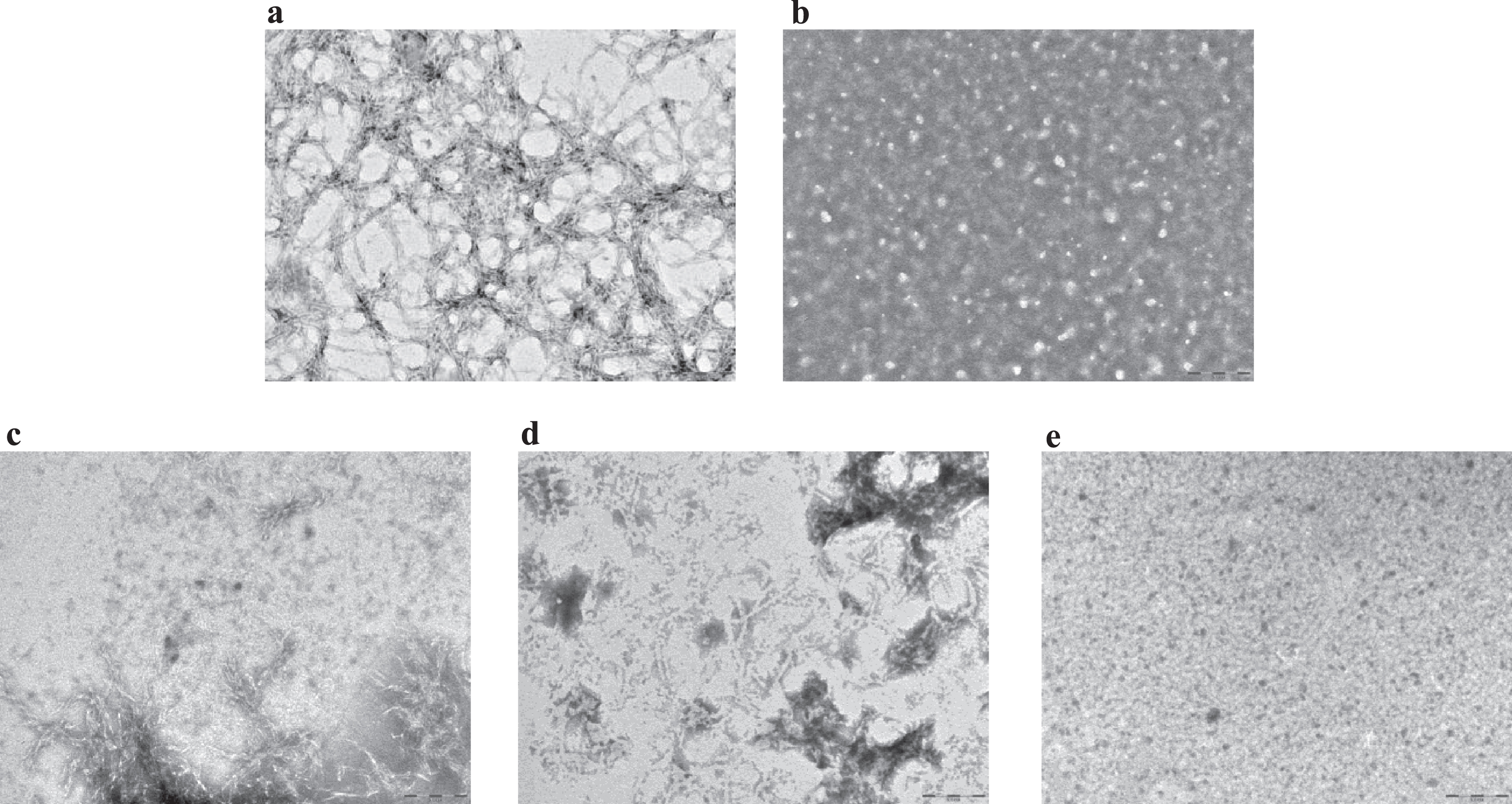
An inhibitory effect of LL-37 peptide binding on fibril formation was demonstrated by TEM. For these experiments (see Methods), samples were prepared according to a more aggregating protocol [17], to mimic quasi-physiological conditions and to better verify LL-37 anti-fibrillogenic activity. Aβ42 reproducibly forms a dense network of interpenetrating, μm-long, straight, unbranched filaments with a diameter of about 10 nm (Fig. 3a, n = 3), corresponding to known features of classic, mature amyloid fibrils [26]. This network of long, straight, interconnected fibrils covered the TEM plate uniformly. On the other hand, LL-37 alone in solution produced globular, amorphous aggregates (Fig. 3b, n = 3). To assess the influence of LL-37 on Aβ fibril formation, 50 μM Aβ42 was co-incubated with an equimolar amount of LL-37 peptide. Mixtures of Aβ42 and LL-37, prepared as described in Methods, were analyzed by TEM, either immediately after solubilization or after 3 or 9 days of incubation. In the equimolar solution of the two peptides at t = 0, shortened, less defined, more sparsely represented fibrils (as compared to Fig. 3a) were observed, in mixture with clumps of amorphous material (Fig. 3c, n = 2). These evolved into (as compared to Fig. 3a) sparser clumps of shorter and more curved fibrils at t = 3 days (Fig. 3d, n = 2), and then into globular amorphous aggregates at t = 9 days, with no visible fibrils present (Fig. 3e, n = 2). As reported in literature, molecules with a similar time-dependent antifibrillogenic effect are considered inhibitors of Aβ42 fibril formation [27, 28], thus from these data it can be deduced that LL-37 inhibits Aβ42 fibril formation.
Fig.3
Transmission electron microscopy images for (a) 50 μM Aβ42 at t = 0; (b) 100 μM LL-37 at t = 10 days; (c) equimolar mixtures of 50 μM Aβ42 and LL-37 at t = 0; (d) t = 3 days; (e) t = 9 days. Scale bar: 200 nm, magnification 60,000x.
Conformational analysis by circular dichroism spectroscopy
Conformational analyses of Aβ42 peptide in solution, in the absence and presence of LL-37, were carried out by CD spectroscopy. As in TEM analysis, spectra were recorded immediately after dissolving peptide according to [17] (Fig. 4a) and again after 24 h (Fig. 4b). We found that at t = 0, Aβ42 assumed an unordered conformation (Fig. 4a, blue line) whereas over 24 h, adoption of β-type conformations was observed (Fig. 4b, blue line), with a characteristic positive band at 195 nm and negative band at 215 nm. On the other hand, 50 μM LL-37 at t = 0, immediately adopted α-helical conformations and maintained this secondary structure over 24 h (Fig. 4a,b, red lines). Remarkably, in the presence of LL-37, the Aβ42 peptide in solution seemed to be prevented, to a large degree, from forming typical β structure (Fig. 4a,b, green lines).
Fig.4
Circular dichroism spectra of peptide solutions, recorded at (a) t = 0 min and (b) t = 24 hours. Blue line: 50 μM Aβ42; red line: 50 μM LL-37; green line: 1:1 mixture.
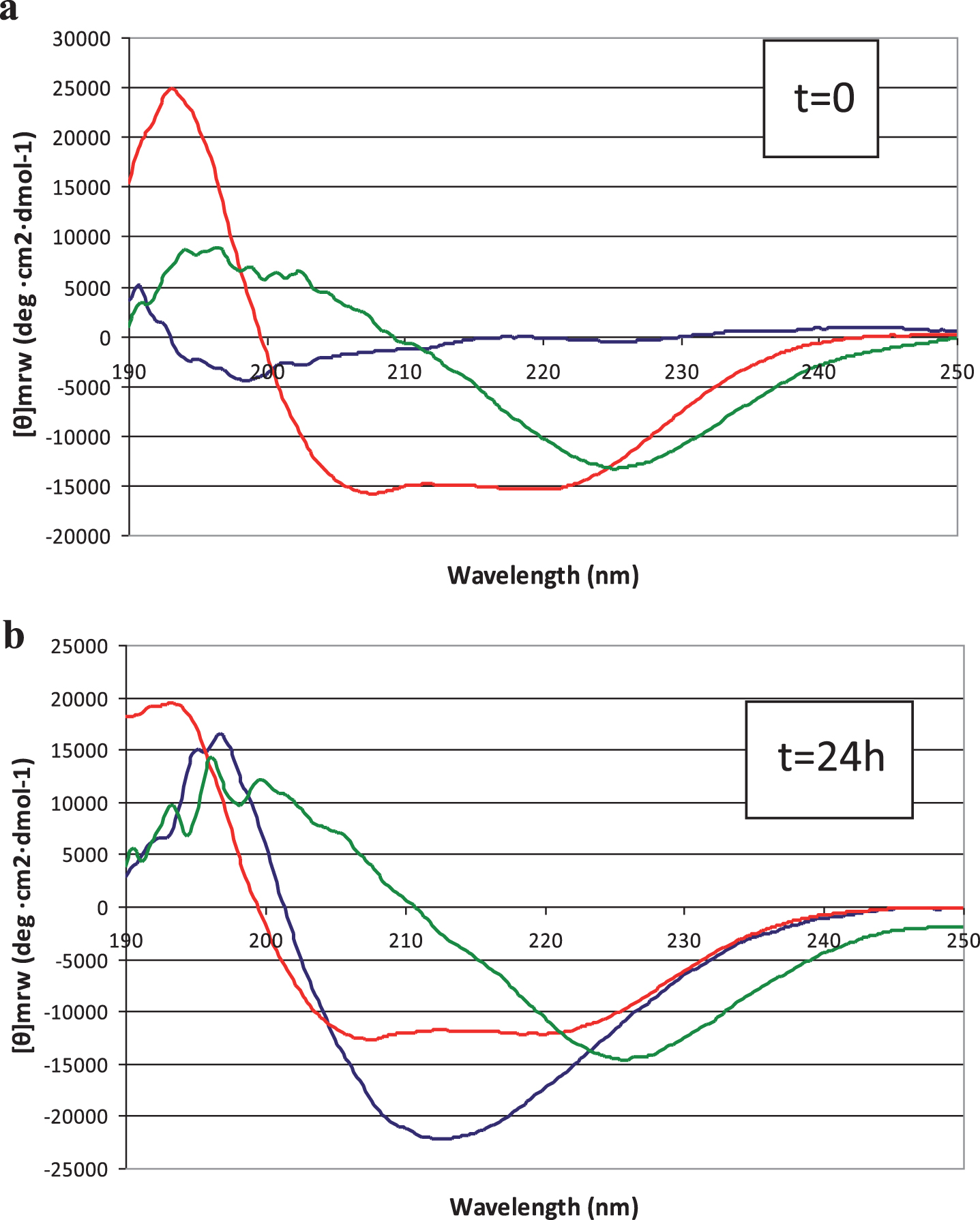
While CD is essentially an “averaging” spectroscopic method, the spectra of 1:1 mixed Aβ42 and LL-37 peptides do not show ordered β structure of the type that predominates in pure Aβ42 solutions. No time-dependence, within 24 h, of peptide secondary structure was observed in the mixed solutions, in contrast to the behavior of Aβ42 solutions. As discussed below, this interaction between the peptides makes sense, considering their opposite charges and mutually high and similar proportions of aromatic and aliphatic amino acids (Fig. 5). We can only conclude, definitively, that, in the presence of LL-37 we do not observe the typical CD spectrum of self-associated Aβ42.
Fig.5
Amino acid sequences: human cathelicidin peptide LL-37 (so named because it comprises 37 amino acids) and the human amyloid-β peptide, Aβ42.

Effects of Aβ42, LL-37, and Aβ42/LL-37 complexes on human microglia, SH-SY5Y neuronal cells, and microglia-mediated cytotoxicity to SH-SY5Y neuronal cells
We studied the effects of Aβ42 alone, LL-37 alone, and Aβ42/LL-37 complexes on SH-SY5Y neuroblastoma cells, human microglia, and on cytokine release from microglia that impact SH-SY5Y cells. Specifically, we exploited a simplified model that reproduces the cross-talk between the major players involved in neuronal damage by looking at the effect of cell-free supernatant from microglial cell culture, on cultured neuronal cells. Cultured cells were exposed to either Aβ42 alone or LL-37 alone (each at 30 μM concentration). After incubation for 72 h, MTT assays were performed and the cell-free medium was collected to measure levels of proinflammatory cytokines TNFα and IL-6. Note, as shown in the control experiments (Supplementary Figure 4), neither Aβ nor LL-37 peptide was toxic to SH-SY5Y cells alone, nor to human microglia alone, at any concentration.
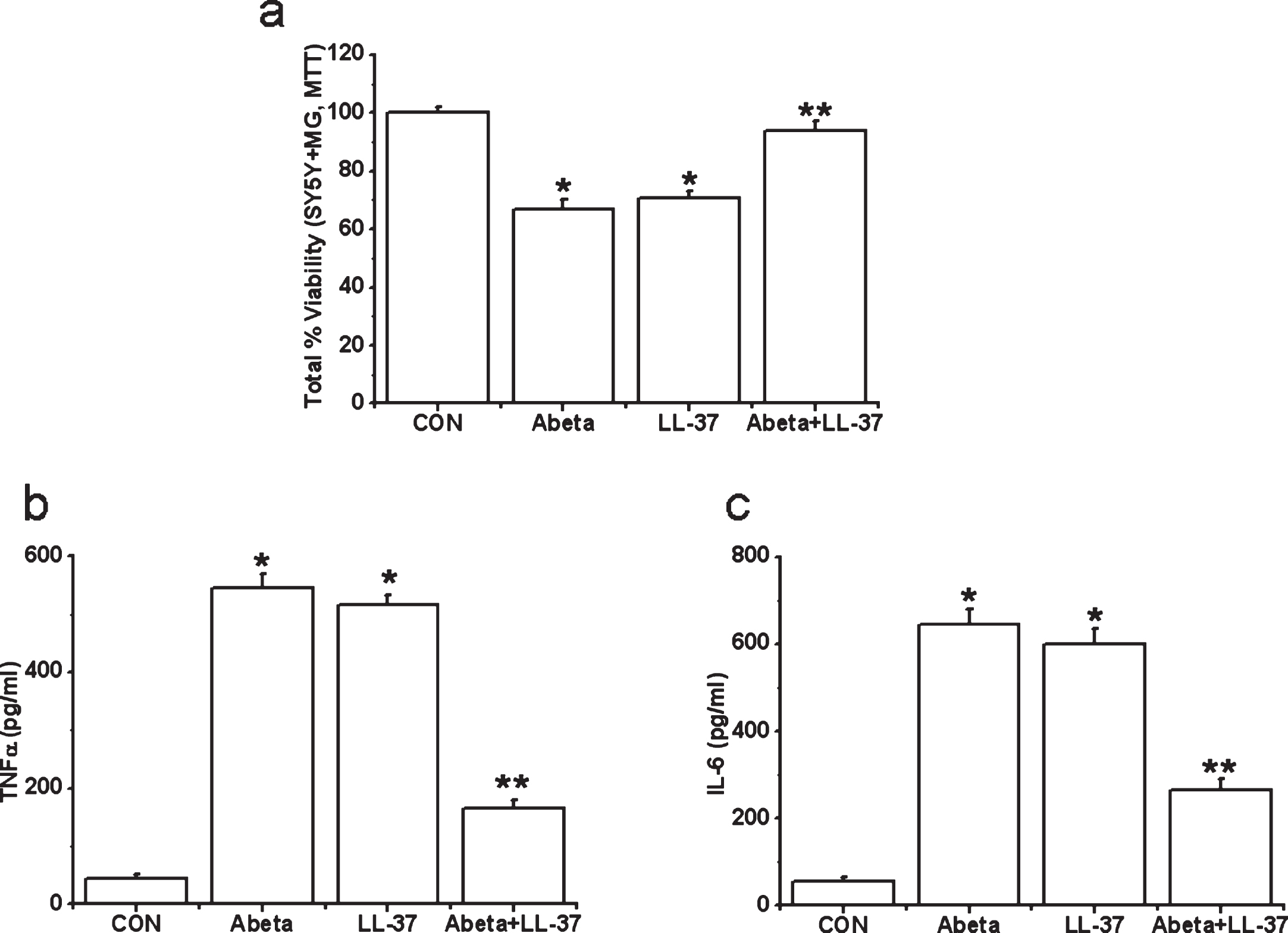
However, we find evidence for microglia-mediated neuroinflammation, for both peptides. Human microglia were exposed to either Aβ alone, LL-37 alone, or an equimolar LL-37/Aβ mixture for 2 days. Then cell-free supernatants from microglia cultures were transferred to differentiated SH-SY5Y cells. SH-SY5Y cell viability was measured after 72 h. We also measured TNFα and IL-6 levels in microglial cell-free supernatants after 2 days’ incubation with the peptides. Data are presented in Fig. 6a-c. Treatment of microglia with Aβ or LL-37 induced release of toxic microglial cytokines TNFα and IL-6 (Fig. 6b, c) and resulted in a reduction in SH-SY5Y cell viability (down to 70% after 3 days, Fig. 6a). However, when microglia were exposed to an equimolar Aβ:LL-37 mixture (30 μM each), the complexation of these two peptides reduced TNFα and IL-6 release, and attenuated SH-SY5Y cell viability loss by more than 90%, which is a major reduction in the deleterious impacts of either peptide, i.e., their complex is non-toxic.
Fig.6
Effects of treatment with Aβ, LL-37, or their mixture on the viability changes of SH-SY5Y cells induced by microglial-mediated toxicity in 72 h (a) and levels of cytokines, TNFα (b) and IL-6 (c) in microglial toxic supernatant. Values are mean±SEM, n = 4. One-way ANOVA was carried out to test significance. Multiple group comparisons were followed by a post-hoc Bonferroni test where necessary. *p < 0.01 for Aβ-treated cells and LL-37-exposed cells compared with control (CON) group and **p < 0.01 for Aβ-LL-37-treated groups compared with Aβ-treated cells and LL-37-exposed cells. Note that Aβ and LL-37 inhibit the microglial activation of each other.
DISCUSSION
Among the many inhibitors of Aβ aggregation and neurotoxicity that have been identified previously by various assays, or designed, synthesized, and/or selected [5, 6], no naturally occurring peptides that match Aβ in size were ever identified. The cathelicidin LL-37 is a pleiotropic, 37-residue innate immune effector that corresponds to residues 134–170 of the human cationic antimicrobial protein [9]. It is one of a kind in the human proteome and is expressed in many organs, including the brain [9, 29]. It fulfills myriad systemic roles as an immune system effector and modulator [30], links the endocrine and immune systems [31], and plays a central role in Vitamin D3-stimulated monocyte/macrophage autophagy [32].
This study aims at investigating a potential molecular binding interaction between Aβ and LL-37, as a step toward exploring a hypothesis that these two peptides may be natural binding partners. If so, dynamics of the spatiotemporal expression of LL-37 and Aβ could have major significance to human health. While there are myriad well-studied functions of LL-37 as an antimicrobial and antiviral weapon of immune cells [9, 30, 32], the physiological functions of Aβ are not yet fully elucidated [33]. Recently published work suggests that Aβ may itself be an antimicrobial and antiviral peptide [34–36].
Our study was initially inspired by an observation of a strong, apparent complementarity between the peptides’ amino acid (aa) sequences (Fig. 5). They are similar in size and molecular weight (Aβ: 39–42 aa, Aβ42 is 4493 g/mol; and LL-37:37 aa, 4514 g/mol). Aβ42 has a net negative charge of -3 (discounting the charge of histidines), while LL-37 has a positive charge of +6, providing for strong electrostatic attraction. Aβ has four aromatic residues (F or Y) with a binary “FF” motif within the KLVFF sequence known to be necessary for Aβ’s assembly [5], while LL-37 has four aromatic residues as well, and comprises the same “FF” motif within residues 4–8 (DFFRK). LL-37 has nine hydrophobic, aliphatic residues (L, V, I) while Aβ has eleven, again making them biophysically similar. And like Aβ, LL-37 associates into homo-oligomers when alone in solution [16, 37, 38], which remain helical in character, unlike Aβ which slowly self-assembles via adoption of β-type secondary structure when alone in solution. In our prior publication [29], we reported that the human cathelicidin precursor protein hCAP18 and its downstream cleavage product, the peptide LL-37, are both expressed in many organs of the human body, with the highest basic levels of LL-37 being expressed in the GI tract and the brain. Prior to that publication, LL-37’s expression and functional role in the human central nerve system had not been reported. Thus, LL-37 is known to be expressed in brain tissue in the human body, as is the Aβ peptide.
SPRi interaction study
The physical interaction and binding specificity between LL-37 and different Aβ forms was demonstrated by SPRi, taking advantage of the simultaneous monitoring of different interactions on the same chip enabled by the imaging configuration of the technique. CCD images and related plasmonic curves (Fig. 2) correlated to the amount of molecules adsorbed on the surface, showed an increasing immobilization density from low-MW oligomers to high-MW oligomers [16, 39]. While SPR measurements revealed higher absolute SPRi signals (Rmax) for Aβ with oligomeric features, the affinity for LL-37 was higher for the low-MW oligomeric forms present at the early stage of aggregation (i.e., Aβ40 (t = 0 days) and Aβ42 (t = 0 days)) [16, 24].
Several studies have reported the use of conventional SPR approaches to study Aβ potential interactors. Kai et al. reported the inhibition of Aβ fibril formation by tabersonine and demonstrated the binding between this small molecule and Aβ42 monomers and oligomers immobilized on the SPR chip surface [40]. Equilibrium binding studies yield KD values of 69 and 535 μM for Aβ42 monomers and oligomers, respectively. In a similar study, Barr et al. verified the inhibition of Aβ fibril formation by a 15-amino acid peptide [41]. SPR surfaces functionalized with monomeric, oligomeric, and fibrillar Aβ42 revealed a stronger affinity of the polypeptide toward the fibrillar form, although only a KD value of 11 μM was measured by injecting Aβ42 onto immobilized peptides and no affinity constant was given. Here we found kinetic constants in the range between 8 to 20 μM, similar to what is published for other Aβ fibrils inhibitors, as noted above. On the other hand, as mentioned earlier (and differently to what was previously shown) [40, 41], LL-37 shows higher affinity for low-MW Aβ oligomers. This strongly supports the ability of LL-37 to inhibit fibril formation by shifting the equilibrium toward smaller species of Aβ, according to the fibrillization mechanisms proposed by Cohen et al. [42] and previously studied in our laboratories [25, 39].
Effects of LL-37 and Aβ on cytotoxicity and inflammatory cytokine release
The results of the MTT assays showed that Aβ42 and LL-37 both induced microglial-mediated toxicity to SH-SY5Y cells. At the same time, the results of quantitative ELISA assays on IL-6 and TNFα showed that the levels of these proinflammatory markers released from microglia were markedly elevated compared with that of the untreated control group, when each peptide was added alone. However, the effects of each peptide on cell viability and inflammatory marker production were largely mitigated when the two peptides were co-incubated prior to being added to the culture medium. These data support the hypothesis that Aβ42 interacts with LL-37 as seen by the considerable attenuation of the toxicity of the peptides when they are in complexation.
Certain aspects of our results are not surprising, as previously we have shown that microglia, upon stimulation by LL-37, release pro-inflammatory factors that are toxic to human neurons [29]. Similarly, Aβ has been shown to activate microglia to produce neurotoxic factors [43]. However, an attenuation of the release of pro-inflammatory factors (IL-6 and TNFα) upon treatment of microglia with equimolar LL-37 and Aβ peptide mixtures is a novel finding, which lends support to a new hypothesis that these two peptides may be natural binding partners, which perhaps the body seeks to maintain in balance over the longer term (after an acute innate immune response). Unlike either peptide alone, which causes human microglia to release pro-inflammatory and neurotoxic cytokines, the complex of the two peptides lacks that neurotoxic effect.
Correlation of biophysical activities of Aβ-peptides and LL-37
No one has yet established a correlation between cathelicidin expression levels and AD. However, in addition to those mentioned in the introduction, other literature reports suggest that the biophysical activities and signaling functions of Aβ peptides and LL-37 are related in vivo. For instance, the Formyl-like Peptide Receptor 1 (FPRL1) is activated by both Aβ [44] and LL-37 [45]. This receptor is reportedly involved in inflammatory aspects of AD [46] via its effects on phagocyte responses [47]; and a decreased phagocyte clearance of central nervous system Aβ is a hallmark of AD [48]. As mentioned above, LL-37 itself is involved in monocyte and macrophage autophagy; its expression, stimulated by vitamin D3 [10], engenders enhanced autophagy [32]. Thus, a dearth of LL-37 generally would reduce phagocytic activity. Phenylbutyrate and vitamin D3 have both been shown to be neuroprotective in AD mouse models [7, 49], and these same two compounds are known cathelicidin inducers [50]. Finally, there is mounting evidence that innate immunity plays a larger role in AD than previously thought, and that Aβ’s normal function may be as an antimicrobial and antiviral peptide [34, 36, 51]. Kumar et al. showed that Aβ expression protects against fungal and bacterial infections in mouse, nematode, and cell culture models of AD [35]. And, part of the body’s natural response to infection is, of course, to upregulate cathelicidin [9].
Conclusions
This work demonstrates that these two peptides, the cathelicidin peptide LL-37 and Aβ, both ubiquitous in human tissues, interact in vitro with each other in different aggregation states, with affinity constants typical of protein-protein interactions and close to values reported in the literature for the binding of Aβ with optimized synthetic peptides. The addition of LL-37 to Aβ42, in a buffer that mimics quasi-physiological conditions, strongly inhibits fibril formation. The protective effect of LL-37’s presence against microglia-mediated Aβ42 toxicity to SH-SY5Y neuroblastoma cells is an important new finding, revealing that in combination the two peptides are 90% less pro-inflammatory than either peptide alone. Literature reports suggest that numerous biophysical activities and signaling functions of Aβ peptides and LL-37 are related in vivo. The in vitro data presented here constitute a starting point from which to investigate whether factors that affect cathelicidin gene CAMP regulation, which will in turn control the production of LL-37 in vivo, modulate Aβ aggregation and/or microglia-induced neuroinflammation.
DISCLOSURE STATEMENT
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0223r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-170223.
REFERENCES
[1] | Huang Y , Mucke L ((2012) ) Alzheimer mechanisms and therapeutic strategies. Cell 148: , 1204–1222. |
[2] | Goedert M ((2015) ) Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to asemsbled Abeta, tau, and alpha-synuclein. Science 349: , 1255555. |
[3] | Soejitno A , Tjan A , Purwata TE ((2015) ) Alzheimer’s disease: Lessons learned from amyloidocentric clinical trials. CNS Drugs 29: , 487–502. |
[4] | Andrieu S , Coley N , Lovestone S , Aisen PS , Vellas B ((2015) ) Prevention of sporadic Alzheimer’s disease: Lessons learned from clinical trials and future directions. Lancet Neurol 14: , 926–944. |
[5] | Stains CI , Mondal K , Ghosh I ((2007) ) Molecules that target beta-amyloid. Chem Med Chem 2: , 1674–1692. |
[6] | Cohen SI , Arosio P , Presto J , Kurudenkandy FR , Biverstal H , Dolfe L , Dunning C , Yang X , Frohm B , Vendruscolo M , Johansson J , Dobson CM , Fisahn A , Knowles TP , Linse S ((2015) ) A molecular chaperone breaks the catalytic cycle that generates toxic Abeta oligomers. Nat Struct Mol Biol 22: , 207–213. |
[7] | Durk MR , Han K , Chow EC , Ahrens R , Henderson JT , Fraser PE , Pang KS ((2014) ) 1alpha,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J Neurosci 34: , 7091–7101. |
[8] | Mariani MM , Malm T , Lamb R , Jay TR , Neilson L , Casali B , Medarametla L , Landreth GE ((2017) ) Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci Rep 7: , 42270. |
[9] | Burton MF , Steel PG ((2009) ) The chemistry and biology of LL-37. Nat Prod Rep 26: , 1572–1584. |
[10] | Gombart AF , Borregaard N , Koeffler HP ((2005) ) Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J 19: , 1067–1077. |
[11] | Cederlund A , Nylen F , Miraglia E , Bergman P , Gudmundsson GH , Agerberth B ((2014) ) Label-free quantitative mass spectrometry reveals novel pathways involved in LL-37 expression. J Innate Immun 6: , 365–376. |
[12] | de la Monte SM ((2014) ) Type 3 diabetes is sporadic Alzheimers disease: Mini-review. Eur Neuropsychopharmacol 24: , 1954–1960. |
[13] | de la Monte SM , Wands JR ((2008) ) Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2: , 1101–1113. |
[14] | Sun J , Furio L , Mecheri R , van der Does AM , Lundeberg E , Saveanu L , Chen Y , van Endert P , Agerberth B , Diana J ((2015) ) Pancreatic beta-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity 43: , 304–317. |
[15] | Scarano S , Mascini M , Turner AP , Minunni M ((2010) ) Surface plasmon resonance imaging for affinity-based biosensors. Biosens Bioelectron 25: , 957–966. |
[16] | Bartolini M , Naldi M , Fiori J , Valle F , Biscarini F , Nicolau DV , Andrisano V ((2011) ) Kinetic characterization of amyloid-beta 1-42 aggregation with a multimethodological approach. Anal Biochem 414: , 215–225. |
[17] | Brogi S , Butini S , Maramai S , Colombo R , Verga L , Lanni C , De Lorenzi E , Lamponi S , Andreassi M , Bartolini M , Andrisano V , Novellino E , Campiani G , Brindisi M , Gemma S ((2014) ) Disease-modifying anti-Alzheimer’s drugs: Inhibitors of human cholinesterases interfering with beta-amyloid aggregation. CNS Neurosci Ther 20: , 624–632. |
[18] | Stathopulos PB , Scholz GA , Hwang YM , Rumfeldt JA , Lepock JR , Meiering EM ((2004) ) Sonication of proteins causes formation of aggregates that resemble amyloid. Protein Sci 13: , 3017–3027. |
[19] | Lee M , Cho T , Jantaratnotai N , Wang YT , McGeer E , McGeer PL ((2010) ) Depletion of GSH in glial cells induces neurotoxicity: Relevance to aging and degenerative neurological diseases. FASEB J 24: , 2533–2545. |
[20] | Singh US , Pan J , Kao YL , Joshi S , Young KL , Baker KM ((2003) ) Tissue transglutaminase mediates activation of RhoA and MAP kinase pathways during retinoic acid-induced neuronal differentiation of SH-SY5Y cells. J Biol Chem 278: , 391–399. |
[21] | Lee M , McGeer E , McGeer PL ((2013) ) Neurotoxins released from interferon-gamma-stimulated human astrocytes. Neuroscience 229: , 164–175. |
[22] | Lee M , Kang Y , Suk K , Schwab C , Yu S , McGeer PL ((2011) ) Acidic fibroblast growth factor (FGF) potentiates glial-mediated neurotoxicity by activating FGFR2 IIIb protein. J Biol Chem 286: , 41230–41245. |
[23] | Cretich M , Bagnati M , Damin F , Sola L , Chiari M ((2011) ) Overcoming mass transport limitations to achieve femtomolar detection limits on silicon protein microarrays. Anal Biochem 418: , 164–166. |
[24] | Sabella S , Quaglia M , Lanni C , Racchi M , Govoni S , Caccialanza G , Calligaro A , Bellotti V , De Lorenzi E ((2004) ) Capillary electrophoresis studies on the aggregation process of beta-amyloid 1-42 and 1-40 peptides. Electrophoresis 25: , 3186–3194. |
[25] | Colombo R , Carotti A , Catto M , Racchi M , Lanni C , Verga L , Caccialanza G , De Lorenzi E ((2009) ) CE can identify small molecules that selectively target soluble oligomers of amyloid beta protein and display antifibrillogenic activity. Electrophoresis 30: , 1418–1429. |
[26] | Merlini G , Bellotti V ((2003) ) Molecular mechanisms of amyloidosis. N Engl J Med 349: , 583–596. |
[27] | Kokkoni N , Stott K , Amijee H , Mason JM , Doig AJ ((2006) ) N-Methylated peptide inhibitors of beta-amyloid aggregation and toxicity. Optimization of the inhibitor structure. Biochemistry 45: , 9906–9918. |
[28] | Kroth H , Ansaloni A , Varisco Y , Jan A , Sreenivasachary N , Rezaei-Ghaleh N , Giriens V , Lohmann S , Lopez-Deber MP , Adolfsson O , Pihlgren M , Paganetti P , Froestl W , Nagel-Steger L , Willbold D , Schrader T , Zweckstetter M , Pfeifer A , Lashuel HA , Muhs A ((2012) ) Discovery and structure activity relationship of small molecule inhibitors of toxic beta-amyloid-42 fibril formation. J Biol Chem 287: , 34786–34800. |
[29] | Lee M , Shi X , Barron AE , McGeer E , McGeer PL ((2015) ) Human antimicrobial peptide LL-37 induces glial-mediated neuroinflammation. Biochem Pharmacol 94: , 130–141. |
[30] | Mookherjee N , Hamill P , Gardy J , Blimkie D , Falsafi R , Chikatamarla A , Arenillas DJ , Doria S , Kollmann TR , Hancock RE ((2009) ) Systems biology evaluation of immune responses induced by human host defence peptide LL-37 in mononuclear cells. Mol Biosyst 5: , 483–496. |
[31] | Mayer ML , Hancock RE ((2010) ) Cathelicidins link the endocrine and immune systems. Cell Host Microbe 7: , 257–259. |
[32] | Yuk JM , Shin DM , Lee HM , Yang CS , Jin HS , Kim KK , Lee ZW , Lee SH , Kim JM , Jo EK ((2009) ) Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6: , 231–243. |
[33] | Pearson HA , Peers C ((2006) ) Physiological roles for amyloid beta peptides. J Physiol 575: , 5–10. |
[34] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[35] | Kumar DK , Choi SH , Washicosky KJ , Eimer WA , Tucker S , Ghofrani J , Lefkowitz A , McColl G , Goldstein LE , Tanzi RE , Moir RD ((2016) ) Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8: , 340ra372. |
[36] | Bourgade K , Garneau H , Giroux G , Le Page AY , Bocti C , Dupuis G , Frost EH , Fulop T Jr ((2015) ) beta-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 16: , 85–98. |
[37] | Oren Z , Lerman JC , Gudmundsson GH , Agerberth B , Shai Y ((1999) ) Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: Relevance to the molecular basis for its non-cell-selective activity. Biochem J 341: (Pt 3), 501–513. |
[38] | Xhindoli D , Morgera F , Zinth U , Rizzo R , Pacor S , Tossi A ((2015) ) New aspects of the structure and mode of action of the human cathelicidin LL-37 revealed by the intrinsic probe p-cyanophenylalanine. Biochem J 465: , 443–457. |
[39] | Butini S , Brindisi M , Brogi S , Maramai S , Guarino E , Panico A , Saxena A , Chauhan V , Colombo R , Verga L , De Lorenzi E , Bartolini M , Andrisano V , Novellino E , Campiani G , Gemma S ((2013) ) Multifunctional cholinesterase and amyloid Beta fibrillization modulators. Synthesis and biological investigation. ACS Med Chem Lett 4: , 1178–1182. |
[40] | Kai T , Zhang L , Wang X , Jing A , Zhao B , Yu X , Zheng J , Zhou F ((2015) ) Tabersonine inhibits amyloid fibril formation and cytotoxicity of Abeta(1-42). ACS Chem Neurosci 6: , 879–888. |
[41] | Barr RK , Verdile G , Wijaya LK , Morici M , Taddei K , Gupta VB , Pedrini S , Jin L , Nicolazzo JA , Knock E , Fraser PE , Martins RN ((2016) ) Validation and characterization of a novel peptide that binds monomeric and aggregated beta-amyloid and inhibits the formation of neurotoxic oligomers. J Biol Chem 291: , 547–559. |
[42] | Cohen SI , Linse S , Luheshi LM , Hellstrand E , White DA , Rajah L , Otzen DE , Vendruscolo M , Dobson CM , Knowles TP ((2013) ) Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 110: , 9758–9763. |
[43] | Block ML , Zecca L , Hong JS ((2007) ) Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat Rev Neurosci 8: , 57–69. |
[44] | Le Y , Gong W , Tiffany HL , Tumanov A , Nedospasov S , Shen W , Dunlop NM , Gao JL , Murphy PM , Oppenheim JJ , Wang JM ((2001) ) Amyloid (beta)42 activates a G-protein-coupled chemoattractant receptor, FPR-like-1. J Neurosci 21: , RC123. |
[45] | Yang D , Chen Q , Schmidt AP , Anderson GM , Wang JM , Wooters J , Oppenheim JJ , Chertov O ((2000) ) LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med 192: , 1069–1074. |
[46] | Cui Y , Le Y , Yazawa H , Gong W , Wang JM ((2002) ) Potential role of the formyl peptide receptor-like 1 (FPRL1) in inflammatory aspects of Alzheimer’s disease. J Leukoc Biol 72: , 628–635. |
[47] | Iribarren P , Zhou Y , Hu J , Le Y , Wang JM ((2005) ) Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease. Immunol Res 31: , 165–176. |
[48] | Mawuenyega KG , Sigurdson W , Ovod V , Munsell L , Kasten T , Morris JC , Yarasheski KE , Bateman RJ ((2010) ) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330: , 1774. |
[49] | Cuadrado-Tejedor M , Ricobaraza AL , Torrijo R , Franco R , Garcia-Osta A ((2013) ) Phenylbutyrate is a multifaceted drug that exerts neuroprotective effects and reverses the Alzheimer’s disease-like phenotype of a commonly used mouse model. Curr Pharm Des 19: , 5076–5084. |
[50] | Steinmann J , Halldorsson S , Agerberth B , Gudmundsson GH ((2009) ) Phenylbutyrate induces antimicrobial peptide expression. Antimicrob Agents Chemother 53: , 5127–5133. |
[51] | Vijaya Kumar DK , Moir RD ((2017) ) The emerging role of innate immunity in Alzheimer’s disease. Neuropsychopharmacology 42: , 362. |


