
Sembragiline in Moderate Alzheimer’s Disease: Results of a Randomized, Double-Blind, Placebo-Controlled Phase II Trial (MAyflOwer RoAD)
Abstract
Background: Sembragiline is a potent, selective, long-acting, and reversible MAO-B inhibitor developed as a potential treatment for Alzheimer’s disease (AD).
Objective: To evaluate the safety, tolerability, and efficacy of sembragiline in patients with moderate AD.
Methods: In this Phase II study (NCT01677754), 542 patients with moderate dementia (MMSE 13–20) on background acetylcholinesterase inhibitors with/without memantine were randomized (1:1:1) to sembragiline 1 mg, 5 mg, or placebo once daily orally for 52 weeks.
Results: No differences between treated groups and placebo in adverse events or in study completion. The primary endpoint, change from baseline in ADAS-Cog11, was not met. At Week 52, the difference between sembragiline and placebo in ADAS-Cog11 change from baseline was – 0.15 (p = 0.865) and 0.90 (p = 0.312) for 1 and 5 mg groups, respectively. Relative to placebo at Week 52 (but not at prior assessment times), the 1 mg and 5 mg sembragiline groups showed differences in ADCS-ADL of 2.64 (p = 0.051) and 1.89 (p = 0.160), respectively. A treatment effect in neuropsychiatric symptoms (as assessed by the difference between sembragiline and placebo on BEHAVE-AD-FW) was also seen at Week 52 only: – 2.80 (p = 0.014; 1 mg) and – 2.64 (p = 0.019; 5 mg), respectively. A post hoc subgroup analysis revealed greater treatment effects on behavior and functioning in patients with more severe baseline behavioral symptoms (above the median).
Conclusions: This study showed that sembragiline was well-tolerated in patients with moderate AD. The study missed its primary and secondary endpoints. Post hoc analyses suggested potential effect on neuropsychiatric symptoms and functioning in more behaviorally impaired study population at baseline.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by a progressive loss of cognitive function and deterioration of the performance of activities of daily living. Behavioral and neuropsychiatric symptoms (NPS), such as depression, agitation, aggression, apathy, delusions, and hallucinations, are also part of the disease, and can worsen over time, significantly contributing to disease burden [1].
Free-radical-induced oxidative stress is considered to be one of multiple factors contributing to AD pathogenesis [2], in part due to upregulation of the enzyme monoamine oxidase B (MAO-B) in astrocytes [3, 4]. MAO-B catalyzes the oxidative deamination of neurotransmitters including dopamine and norepinephrine, as well as neuromodulatory amines such as β-phenylethylamine [5]. Hydrogen peroxide is formed as a byproduct, and in the presence of trace amounts of metals, it can be converted into highly toxic hydroxyl radicals, which avidly react with polyunsaturated fats in neuronal membranes, initiating lipid peroxidation and cell death [5]. Consistent with this hypothesis, the amount of MAO-B in the human brain is reported to increase with age [6]. Notably, in patients with AD, brain MAO-B activity is further increased compared with age-matched controls [7] yet is unchanged in non-demented patients with neurodegeneration [8]. Furthermore, increases in brain MAO-B levels in the brains of patients with AD may also contribute to decreases in levels of neurotransmitters [7], leading to NPS [3].
A number of MAO-B inhibitors (MAO-Bi) have been studied as potential therapeutic compounds for AD and Parkinson’s disease. Selegiline and rasagiline have been used for more than 10 years in the treatment of Parkinson’s disease [9, 10], but neither has been approved for treatment in AD. Selegiline was tested in a 2-year study in patients with moderate-to-severe AD, and results suggested delayed deterioration in functioning and institutionalization [11]. However, lack of evidence for a clinically meaningful benefit prevented further development of the drug. Lazabemide, a selective MAO-Bi [12, 13], was tested as monotherapy in patients with mild-to-moderate AD in one exploratory placebo-controlled Phase II and two confirmatory pivotal placebo-controlled Phase III trials at doses of 50 and 100 mg twice daily. Both doses were selected based on brain MAO-B occupancy from a positron emission tomography (PET) study in humans. The pivotal Phase III studies provided consistent and statistically significant evidence of cognitive benefit relative to placebo, measured by Alzheimer’s Disease Assessment Scale-Cognitive Behavior 11-item Subscale (ADAS-Cog11), following 52 weeks of treatment with lazabemide [14]. One pivotal study showed statistically significant evidence supporting global clinical benefit of lazabemide treatment and an effect on behavior, measured by Alzheimer’s Disease Cooperative Study Clinician Global Impression of Change (ADCS-CGIC) and Neuropsychiatric Inventory, and the other pivotal study demonstrated overall benefit in functioning, measured by the Disability Assessment for Dementia and Functional Rating Scale (Roche data on file). Positive effect of lazabemide on apathy, measured by Neuropsychiatric Inventory-apathy subscore [15], was observed at end of treatment in a pooled post hoc analysis of the two pivotal trials. Although these studies supported the clinical benefit of MAO-Bi in the treatment of AD, the lazabemide program was not developed further, due to hepatotoxic liability. Also of note, the Phase III study, which enrolled patients in 1997, did not include cholinesterase inhibitors as concomitant medication.
Sembragiline, also referred to as RO4602522, RG1577, and EVT 302 in previous communications, was in development as a potent, selective, long-acting, and reversible inhibitor of MAO-B. It has a half-maximal inhibitory concentration for MAO-B of 5–6 nM and has approximately 600-fold selectivity for MAO-B, resulting in complete MAO-B inhibition without affecting MAO-A. In addition, it has no affinity to a wide variety of receptors, ion channels, or other enzymes distributed throughout the brain and other organs [16]. Experiments in animals indicate that sembragiline readily enters the brain and inhibits MAO-B enzymatic activity in a dose-dependent and reversible manner after oral administration. Administration of sembragiline in mice overexpressing MAO-B reduced oxidative stress and astrogliosis, and prevented loss of dopaminergic neurons in the substantia nigra (Borroni et al., J Pharm Exp Ther, in press), suggesting a potential role for sembragiline in modulating the neurodegenerative and neuroinflammatory processes that may be relevant to AD pathogenesis. In rats, administration of sembragiline also increased the production of neuromodulatory amines associated with motivation and cognition (Borroni et al., in preparation).
Sembragiline demonstrated a good safety profile in more than 450 healthy volunteers and patients with AD in Phase I and II studies, including a study of smoking cessation in over 400 subjects [17]. A PET study in elderly healthy volunteers and patients with AD has shown that daily treatment with 1 mg or 5 mg sembragiline resulted in near-maximal inhibition of brain MAO-B enzyme [18]. Based on observations from earlier clinical studies with lazabemide that suggest a greater effect of lazabemide in moderate AD patients, along with the support of preclinical mechanistic (Borroni et al., in preparation) and human PET data [19], we hypothesized that treatment with 1 or 5 mg sembragiline daily compared with placebo may result in slower worsening of cognition in patients with moderate AD dementia. Considering the mechanism of action and available data from selegiline and lazabemide suggesting that the expected effect is a delay of symptom deterioration rather than an improvement over baseline, the study enrolled patients with moderate AD (Mini-Mental State Examination (MMSE) score between 13 and 20). This moderate AD population is expected to show a higher rate of progression over 12 months compared with patients with mild AD. Therefore, the Phase II MAyflOwer RoAD study was conducted to evaluate the safety, tolerability, and efficacy of sembragiline in patients with moderate AD when administered on top of current standard background AD medication.
MATERIALS AND METHODS
Patients
Inclusion criteria for enrollment evaluated at screening comprised: age 50–90 years; diagnosis of probable AD, based on the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer’s Disease and Related Disorders Association and Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Text Revision (DSM-IV-TR) criteria; magnetic resonance imaging (MRI) supporting a diagnosis of AD; MMSE score 13–20; Modified Hachinski Ischemia Score of≤4; Cornell Scale for Depression in Dementia score≤13; treatment with acetylcholinesterase inhibitor (AChEI) alone or in combination with memantine for≥4 months, stabilized for≥3 months prior to screening; and a caregiver or some other identified responsible person having frequent contact with the patient (≥10 h per week).
Exclusion criteria comprised: uncontrollable behavioral symptoms, presence of a comorbid condition that might bias clinical or mental status assessments or put the patient at special risk; requirement for nursing home care in the absence of a reliable caregiver; non-AD pathology on brain MRI that may affect cognition; and recent (12 weeks) use of other MAO-Bi, including selegiline or rasagiline. Use of other psychotropic agents (e.g., antidepressants, antipsychotics, anxiolytics) was controlled with respect to dose and pharmacologic properties (details in Supplementary Material).
Study oversight
This study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. The protocol, Informed Consent Form, any information given to the patient and relevant supporting information were approved by the Independent Ethics Committee/Institutional Review Board prior to study initiation. Eligible patients (or their legal representatives) provided written informed consent prior to study inclusion. Separate caregiver consent was also required. The study is registered at ClinicalTrials.gov (NCT01677754).
Study design and treatment
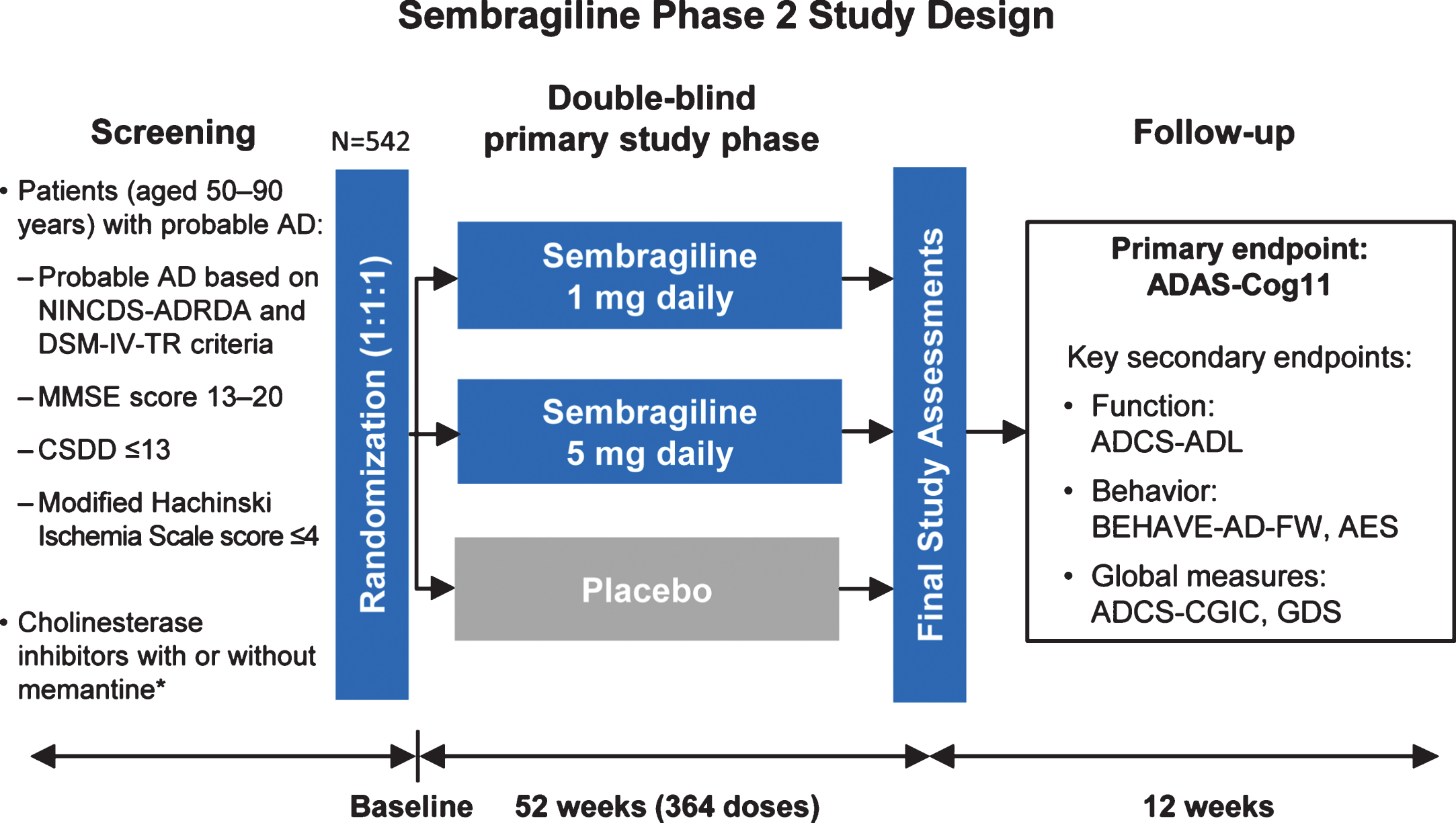
This Phase II trial was a prospective, multicenter, randomized, double-blind, parallel-group, placebo-controlled study to investigate the efficacy and safety of sembragiline in patients with moderate AD taking AChEI alone or in combination with memantine. Patients were recruited across more than 100 sites in 12 counties, and were stratified according to selective serotonin reuptake inhibitor use, memantine use and Apathy Evaluation Scale score (<42 versus≥42). Patients were randomized 1:1:1 to placebo, sembragiline 1 mg, or sembragiline 5 mg for a period of 52 weeks, after which treatment with study drug was discontinued and patients followed up for a further 12 weeks (Fig. 1). The study drug and placebo were administered as identical tablets to be taken orally once daily. The study remained blinded until all patients completed the follow-up period or subjects had been withdrawn.
Fig.1
MAyflOwer RoAD study design. *Background therapy stable for at least 4 months. AD, Alzheimer’s disease; ADAS-Cog11, Alzheimer’s Disease Assessment Scale-Cognitive Behavior 11-item Subscale; ADCS-ADL, Alzheimer’s Disease Cooperative Study-Activities of Daily Living; ADCS-CGIC, Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change; AES, Apathy Evaluation Scale; GDS, Global Deterioration Scale; BEHAVE-AD-FW, Behavioral Pathology in Alzheimer’s Disease Frequency-Weighted Severity Scale; CSDD, Cornell Scale for Depression in Dementia; MMSE, Mini-Mental State Examination; NINCDS-ADRDA, National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association.
Outcome measures
The primary outcome was mean change from baseline over time in ADAS-Cog11 scores. Secondary outcome measures included Alzheimer’s Disease Cooperative Study-Activities of Daily Living (ADCS-ADL) scale, Behavioral Pathology in Alzheimer’s disease Frequency-Weighted Severity Scale (BEHAVE-AD-FW) [20, 21], Apathy Evaluation Scale (AES-C: clinician-rated version) [22] and ADCS-CGIC scale [23]. Post hoc analysis was conducted comparing patient subgroups split at median baseline BEHAVE-AD-FW score. In an exploratory analysis, structural MRI was used to measure hippocampal volume, total brain volume, and ventricular volume at screening and after 52 weeks of treatment.
Safety monitoring
Safety and tolerability were assessed by physical, neurologic, and ophthalmologic examinations, vital signs, blood and urine safety tests, electrocardiography (ECG), Columbia Suicide Severity Rating Scale [24], and adverse event (AE) monitoring. AEs were defined as events that started or worsened in intensity between the first dose of study medication and 12 weeks after last dose. An independent Safety Monitoring Committee reviewed unblinded safety data throughout the trial to ensure patient safety.
Statistical analysis
The primary and secondary endpoints were analyzed using a mixed-effects model repeated measures analysis. The model included the baseline score, treatment groups, visit, and treatment-by-visit interaction as covariates. The repeated variable within a patient was visit week, and class variables included patient, treatment and visit week. The 95% confidence intervals (CIs) of treatment difference and nominal p-value without applying any adjustment for multiple comparisons are reported for each dose.
Analyses of hippocampal, whole-brain, and ventricular volumes, as well as voxel-based morphometry analyses, were tested for treatment-by-time interactions using repeated measures, general linear models using age, sex, and total intracranial volume as covariates, and were corrected for multiple comparisons. For regional volume analyses, an uncorrected p-value of p < 0.05 was applied. For exploratory voxel-wise analyses, a family-wise error corrected voxel-wise threshold of p < 0.05 was applied.
RESULTS
Subject disposition and baseline characteristics
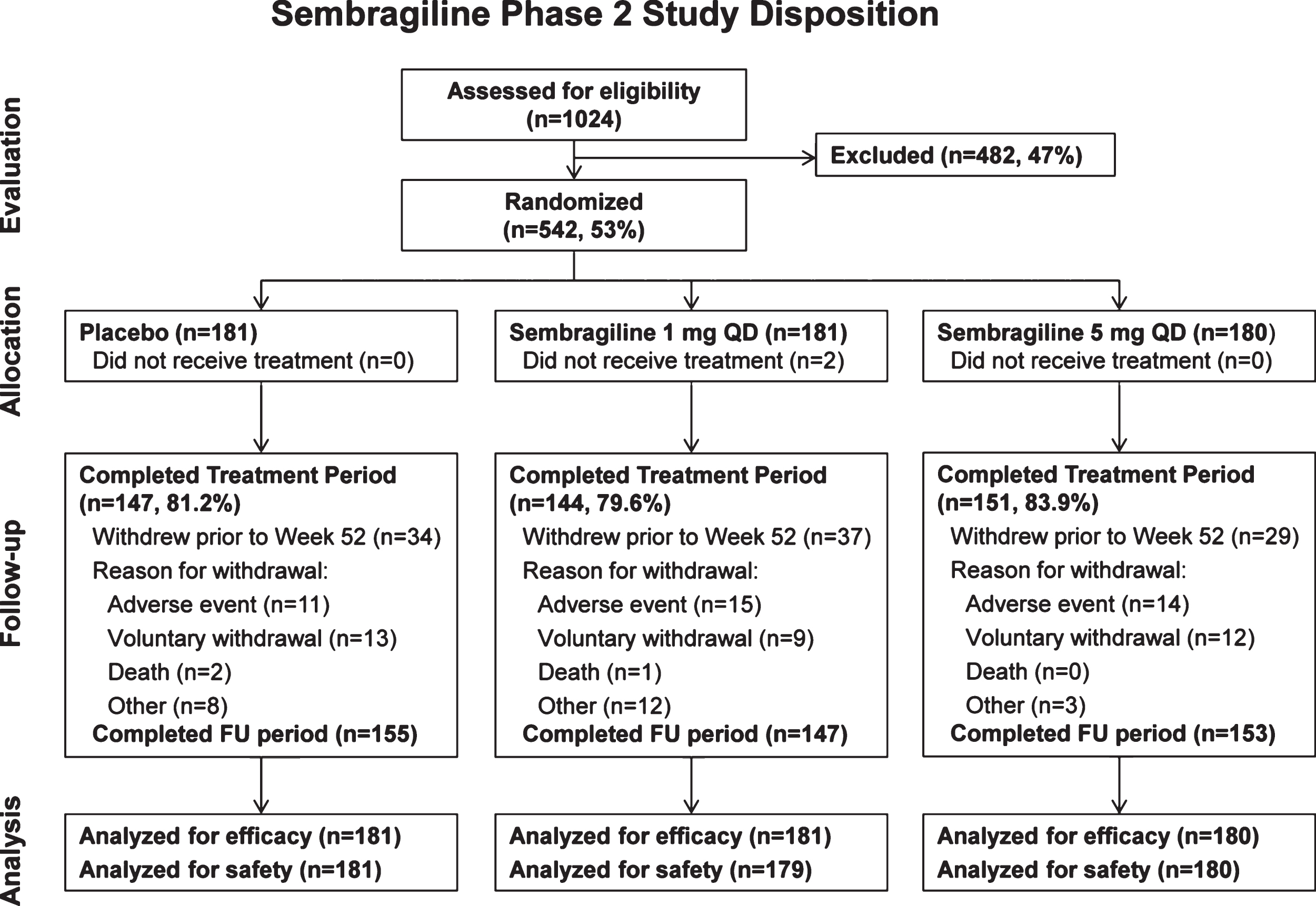
A total of 1,024 patients were screened from October 24, 2012 to February 6, 2014, and of these, 542 patients were randomized: 181 patients to placebo, 181 patients to sembragiline 1 mg, and 180 patients to sembragiline 5 mg (Fig. 2). The main reasons for screen failure were MMSE score (outside of 13–20) and ECG (QTcF >450 ms). Two patients in the sembragiline 1 mg group did not receive any study medication and therefore were not included in the safety population.
Fig.2
MAyflOwer RoAD study disposition. FU, follow-up; QD, once a day.
For the intent-to-treat population, the mean age at baseline was similar across the three treatment groups. There were fewer male patients in the 5 mg group than in the 1 mg and placebo groups. No relevant differences were observed among treatment groups for other demographic variables, time from AD diagnosis to study entry or first AD symptoms (Table 1). Background AD therapy was generally similar across the three treatment groups. Although use of antipsychotics at baseline was very low in general, it was reported for a greater proportion of patients in the 5 mg group (20 patients, 11.1%) than in the 1 mg (5 patients, 2.8%) and placebo groups (10 patients, 5.5%).
Table 1
Subject baseline demographics, AD characteristics, and concomitant medications
| Placebo | Sembragiline | Sembragiline | |
| (n = 181) | 1 mg QD (n = 181) | 5 mg QD (n = 180) | |
| Age, years* | 73.8 (8.3, 51–91) | 72.8 (9.0, 51–89) | 72.5 (9.5, 51–90) |
| Sex | |||
| Male | 77 (42.5%) | 68 (37.6 %) | 57 (31.7%) |
| Female | 104 (57.5%) | 113 (62.4%) | 123 (68.3%) |
| Time from AD diagnosis, months* | 26.9 (20.8, 2.8–114.4) | 30.0 (23.2, 0.3–114.3) | 28.7 (23.1, 1.7–133.9) |
| APOE4 status | |||
| Non-carrier | 74 (42.5%) | 73 (44.5%) | 67 (40.1%) |
| Carrier | 100 (57.5%) | 91 (55.5%) | 100 (59.9%) |
| Concomitant medications | |||
| Acetylcholinesterase inhibitors alone | 141 (77.9%) | 139 (76.8%) | 144 (80.0%) |
| Acetylcholinesterase inhibitors + memantine | 40 (22.1%) | 42 (23.2%) | 36 (20.0%) |
| Antipsychotics | 10 (5.5%) | 5 (2.8%) | 20 (11.1%) |
*Mean (SD, range). QD, once-daily; APOE, Apolipoprotein E.
There were no clinically meaningful differences between the three treatment groups in any of the baseline cognitive, behavioral or functioning scores (Table 2).
Table 2
Baseline cognitive, behavioral, and functional scores
| Placebo | Sembragiline | Sembragiline | |
| (n = 181) | 1 mg QD (n = 181) | 5 mg QD (n = 180) | |
| ADAS-Cog11 | 26.6 (9.6, 9.7–52.0) | 27.3 (9.3, 10.3–50.3) | 27.4 (9.1, 6.7–54.3) |
| ADCS-ADL | 55.5 (13.5, 16–77) | 54.8 (13.2, 17–78) | 54.2 (13.87, 16–77) |
| MMSE | 17.5 (3.00, 9–28) | 16.6 (3.04, 10–27) | 17.0 (2.85, 8–25) |
| BEHAVE-AD-FW | 6.7 (8.74, 0–40) | 7.9 (9.89, 0–52) | 7.9 (10.00, 0–60) |
| AES | 42.8 (10.97, 18–68) | 43.1 (10.82, 18–69) | 42.8 (11.01, 18–72) |
Mean (SD, range). ADAS-Cog11, Alzheimer’s Disease Assessment Scale-Cognitive Behavior 11-item Subscale; ADCS-ADL, Alzheimer’s Disease Cooperative Study-Activities of Daily Living); AES, Apathy Evaluation Scale; BEHAVE-AD-FW, Behavioral Pathology in Alzheimer’s Disease Frequency-Weighted Severity Scale; MMSE, Mini-Mental State Examination. QD, once-daily.
Efficacy
For change from baseline in ADAS-Cog11 at 52 weeks, no statistically significant difference was seen in the 1 mg or 5 mg groups compared with placebo (Fig. 3), and the least-squares (LS) mean change from baseline was similar for both dose groups compared with placebo (a – 0.15 mean difference for 1 mg favoring treatment [95% CI: – 1.90, 1.60; p = 0.865, ES = –0.02] and a 0.90 mean difference for 5 mg favoring placebo [95% CI: – 0.84, 2.63; p = 0.312, ES = 0.12]). There were also no significant differences observed between treatment and placebo in change from baseline in ADAS-Cog11 at any other time points analyzed (Weeks 12, 24, or 36).
The LS mean change from baseline in ADCS-ADL at Week 52 favored treatment (but not significantly), where the mean difference between sembragiline and placebo groups was 2.64 (95% CI: – 0.01, 5.30; p = 0.051; ES = 0.22) for the 1 mg group and 1.89 (95% CI: – 0.75, 4.53; p = 0.160; ES = 0.16) for the 5 mg group (Fig. 4A). No significant differences between treatment and placebo in change from baseline in ADCS-ADL were observed at any other time points analyzed (Weeks 12,24, or 36).
Fig.3
Change of cognitive function (primary endpoint) in moderate AD patients treated with sembragiline and placebo. Mean change from baseline to Week 52 in ADAS-Cog11 in total study population. Error bars represent SEM. ADAS-Cog11, Alzheimer’s Disease Assessment Scale-Cognitive Behavior 11-item Subscale; SEM, standard error of the mean.
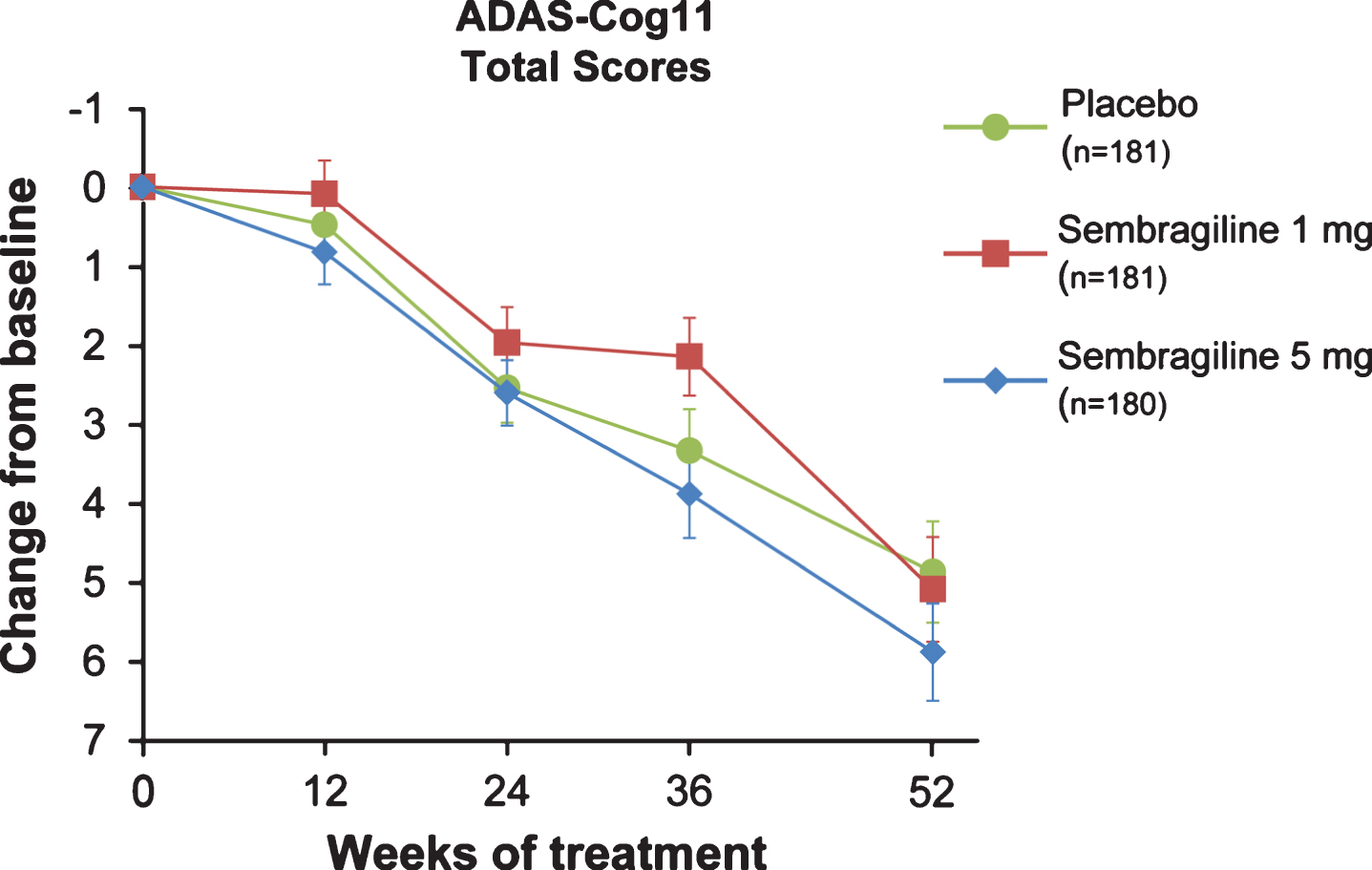
Fig.4
Change from baseline in (A) ADCS-ADL, (B) BEHAVE-AD-FW, and (C) ADCS-CGIC (secondary endpoint) by treatment group. Change from baseline to Week 52 in mean scores in total study population. Error bars represent SEM. ADCS-ADL, Alzheimer’s Disease Cooperative Study-Activities of Daily Living; ADCS-CGIC, Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change; BEHAVE-AD- FW, Behavioral Pathology in Alzheimer’s Disease Frequency-Weighted Severity Scale; SEM, standard error of the mean.
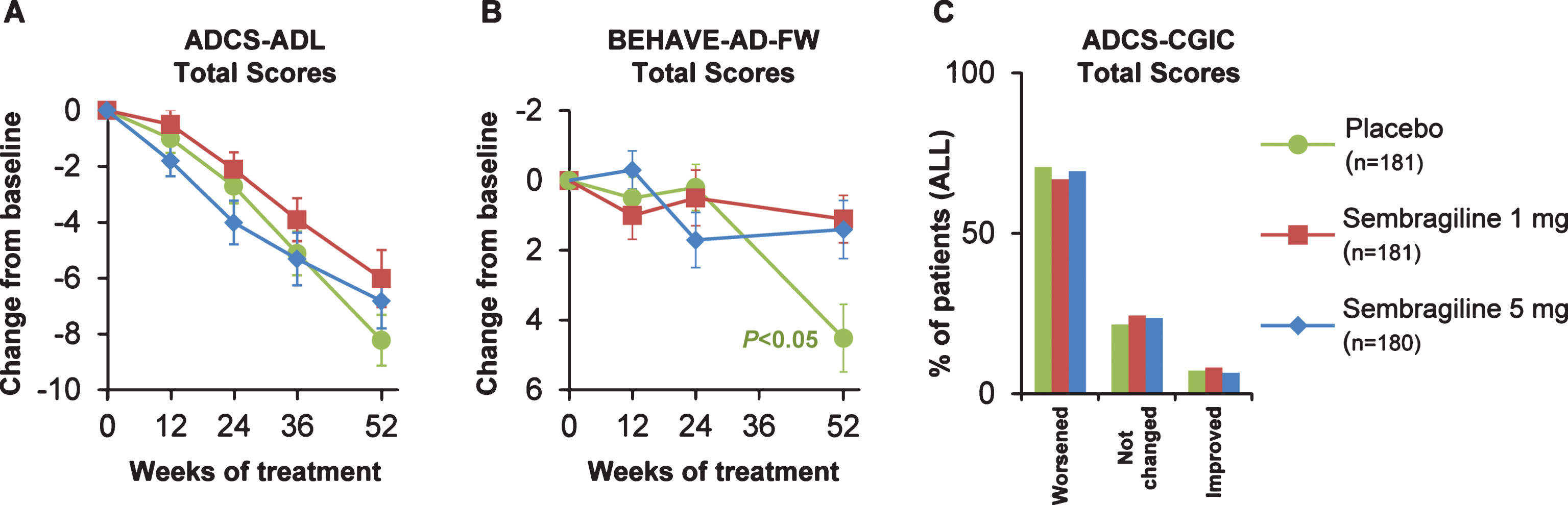
Baseline BEHAVE-AD-FW total scores were similar but low across the treatment groups (Table 2). At Week 52, there was less development of behavioral symptoms compared with placebo in the 1 mg and 5 mg groups, with a mean difference of – 2.80 (95% CI: – 5.02, – 0.57; p = 0.014; ES = 0.28) and – 2.64 (95% CI: – 4.85, – 0.43; p = 0.019; ES = 0.27), respectively (Fig. 4B). This treatment effect was observed in the context of greater worsening in a small subset of patients in the placebo group between Weeks 24 and 52, and no significant differences between groups were observed at Weeks 12 and 24. Furthermore, a large number of patients had a zero value— indicative of a floor effect. Thus, assumptions of the pre-defined statistical analysis model may be invalidated and the p-values and confidence intervals should be interpreted with caution.
Analysis of BEHAVE-AD-FW individual domains showed a potential trend toward a treatment effect in the paranoid and delusional domain at Week 52 in both the 1 mg and 5 mg dose groups (p = 0.051 and p = 0.011, respectively) (Supplementary Figure 1A). Consistent effects were not observed in other domains. At Week 52, no difference in the overall proportion of patients with BEHAVE-AD-FW emerging symptoms was observed between either treatment groups or placebo.
Only a small proportion of patients in any of the groups showed improvement on the ADCS-CGIC at either Week 24 (<15.0%) or at Week 52 (<10.0%). The majority of patients experienced no change or minimal-to-moderate worsening at both time points, with no significant differences at either time point for the 1 mg and 5 mg groups compared with placebo (Fig. 4C).
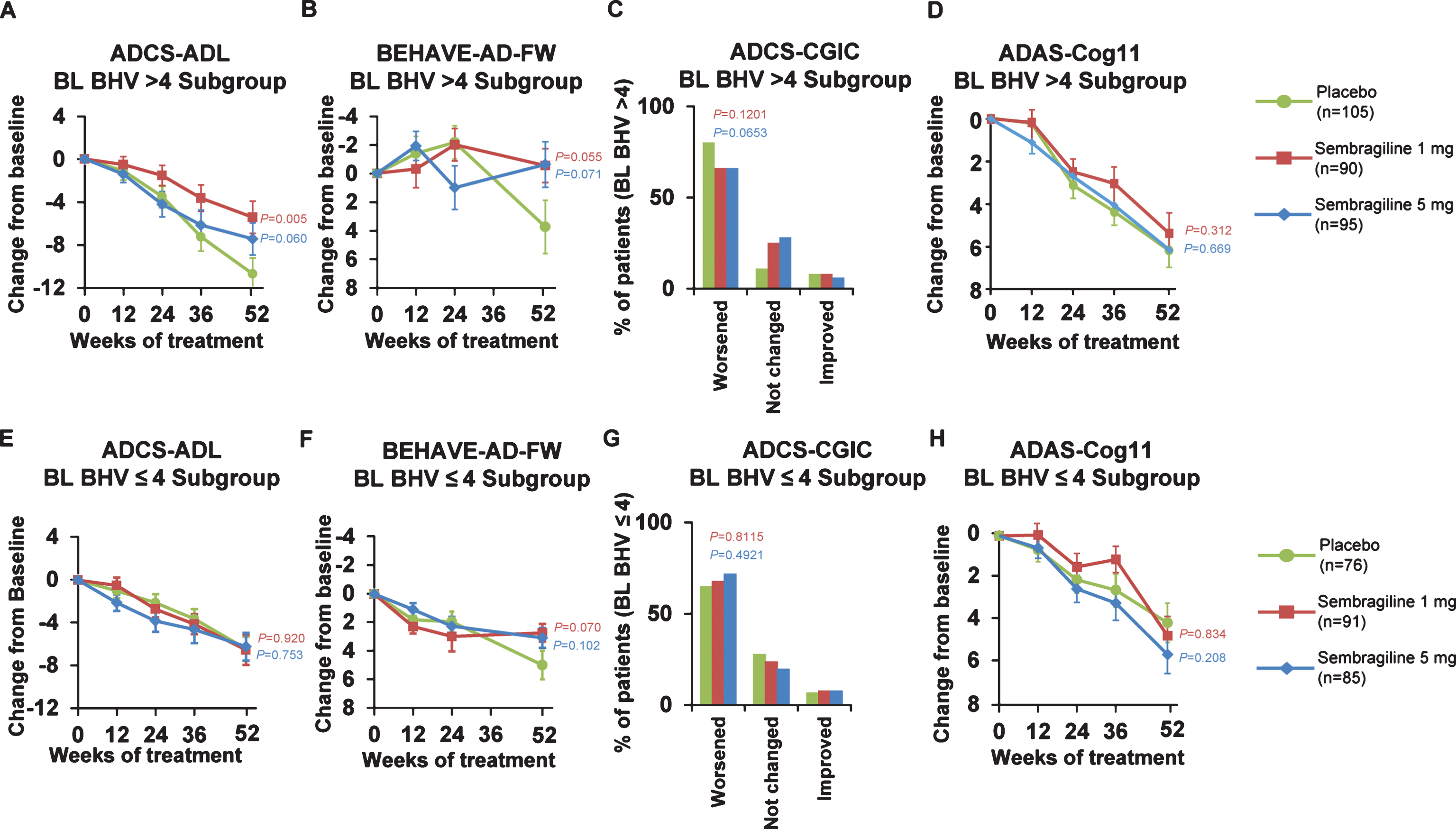
The AES total scores increased throughout the study with mean (±SD) change from baseline at Week 52 of: 4.3 (±8.92) for the 1 mg group, 4.5 (±9.77) for the 5 mg group and 4.2 (±9.60) for the placebo group (difference from placebo was not significant). Similarly, there were no differences in GDS at Week 52 in the 1 mg and 5 mg groups compared with the placebo group. The finding of a treatment effect in behavior for this relatively behaviorally unimpaired patient population prompted a post hoc subgroup analysis to be performed in the more behaviorally impaired half of the patient population— those with BEHAVE-AD-FW above the median (>4) at baseline: 105 patients in placebo, 90 in sembragiline 1 mg, and 95 in sembragiline 5 mg groups (Fig. 5). In this subgroup, baseline characteristics were well balanced between treatment groups. The LS mean change in ADCS-ADL total score from baseline to Week 52 for both dose groups favored treatment, with a mean difference relative to placebo of 5.91 (p = 0.005; ES = 0.48) and 3.94 (p = 0.060; ES = 0.32) for the 1 mg and 5 mg groups, respectively (Fig. 5A). The LS mean change in BEHAVE-AD-FW from baseline to Week 52 for both dose groups favored treatment, with a mean difference relative to placebo of 4.04 (p = 0.055; ES = 0.33) and 3.82 (p = 0.071; ES = 0.31) for the 1 mg and 5 mg groups, respectively (Fig. 5B). BEHAVE-AD-FW individual domains showed a potential trend toward a treatment effect in two domains for both 1 mg and 5 mg dose groups: activity disturbances (p = 0.098 and p = 0.12, respectively) and aggressiveness (p = 0.076 and p = 0.072, respectively). In the ADCS-CGIC, a smaller proportion of patients in the 1 mg (66%) and 5 mg (66%) sembragiline groups showed worsening relative to placebo (80%) but this difference was not significant (Fig. 5C). In contrast, those with BEHAVE-AD-FW below or equal to the median (≤4) at baseline— 76 patients in the placebo group, 91 in the sembragiline 1 mg group, and 85 in the sembragiline 5 mg group (Fig. 5)—showed no treatment effects across ADCS-ADL (Fig. 5E), BEHAVE-AD-FW (Fig. 5F), or ADCS-CGIC (Fig. 5G). Treatment effect was not observed using the ADAS-Cog11 in either subgroup (Fig. 5D, H).
Fig.5
Post hoc subgroup analyses in subpopulations differing in behavioral impairment at baseline (exploratory endpoints). Change from baseline to Week 52 in mean ADCS-ADL, BEHAVE-AD-FW, ADCS-CGIC and ADAS-Cog11 scores in post hoc analyses in study subpopulations (A-D: more impaired, BHV >4 at baseline; E-H: less impaired BHV≤4 at baseline). Error bars represent SEM. ADAS-Cog11, Alzheimer’s Disease Assessment Scale-Cognitive Behavior 11-item Subscale; ADCS-ADL, Alzheimer’s Disease Cooperative Study-Activities of Daily Living; ADCS-CGIC, Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change; BEHAVE-AD-FW, Behavioral Pathology in Alzheimer’s Disease Frequency-Weighted Severity Scale; BL BHV, baseline BEHAVE-AD-FW; SEM, standard error of the mean.
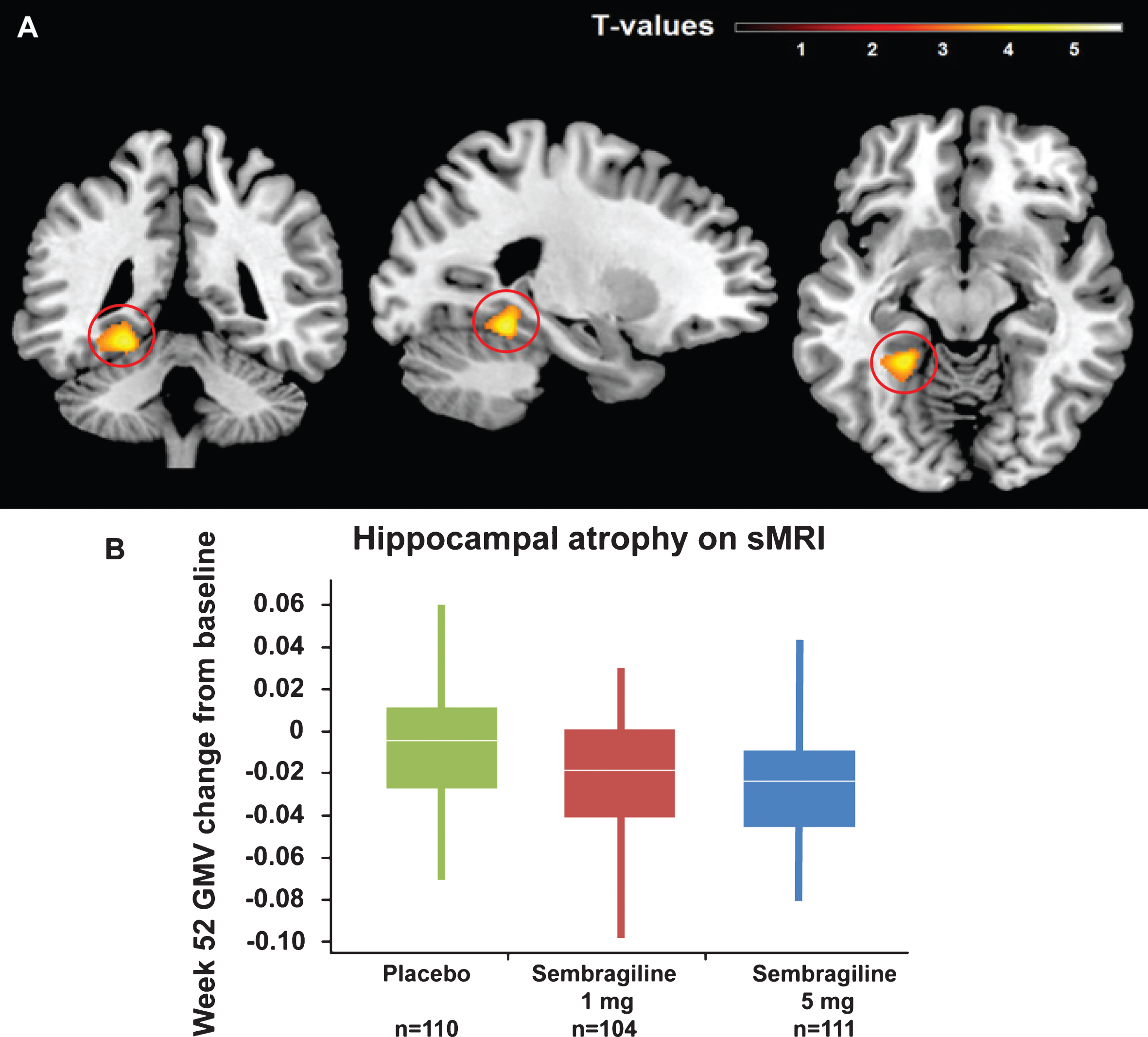
As a pre-specified exploratory analysis to assess potential treatment effects on brain atrophy progression, changes in brain volumes between baseline and Week 52 were evaluated by structural MRI. In region-of-interest analyses, no significant treatment-by-time interactions were observed for hippocampal (F[2,638] = 0.03, p = 0.968), ventricular (F[2,638] = 0.03, p = 0.972), and total (F[2,638] = 0.06, p = 0.938) brain volumes. In voxel-wise analyses, a significantly faster decrease in left posterior hippocampal gray matter volume was observed for treated patients when comparing pooled treatment groups with the placebo cohort [p = 0.031, family-wise error corrected (Fig. 6)]. In post hoc direct group comparisons, this effect was also significant in high dose group versus placebo but not for the low dose or between both doses.
Fig.6
Change in hippocampal volume at Week 52 by sMRI. A) Representative MRI image depicting regions of significant treatment-associated decline observed in voxel-based morphometry analyses (p < 0.001 used for visualization). B) Decline in GMV of posterior hippocampal region by treatment group. Mean peak voxel change from baseline (Montreal Neurological Institute space coordinate: – 24, – 45, – 14). There were no baseline differences in hippocampal volume (data not shown).
Safety
Sembragiline was generally well tolerated in patients with moderate AD. There were no meaningful differences between sembragiline and placebo in the proportion of subjects experiencing one or more severe AE, serious adverse event (SAE), AE with an outcome of death, or AE leading to treatment discontinuation (Table 3). There also did not appear to be any AEs that were drug- or dose-related. The majority of AEs were assessed as either mild or moderate in intensity. Overall, 100 patients (18.5%) discontinued study medication during the treatment period, including 37 patients (20.4%) in the 1 mg group, 29 patients (16.1%) in the 5 mg group, and 34 patients (18.8%) in the placebo group. The primary reported reasons for treatment discontinuation for the greatest proportion of patients were “adverse event” and “voluntary withdrawal.”
Table 3
Safety evaluation
| Placebo | Sembragiline | Sembragiline | |
| (n = 181) | 1 mg QD (n = 179) | 5 mg QD (n = 180) | |
| At least one AE* | 141 (77.9) | 142 (79.3) | 137 (76.1) |
| At least one treatment-related AE† | 50 (27.6) | 44 (24.6) | 56 (31.1) |
| At least one AE resulting in study discontinuation | 13 (7.2) | 15 (8.4) | 14 (7.8) |
| At least one severe AE‡ | 20 (11.0) | 15 (8.4) | 11 (6.1) |
| At least one SAE | 23 (12.7) | 18 (10.1) | 19 (10.6) |
| Deaths | 3 (1.7) | 3 (1.7) | 2 (1.1) |
Number of patients, n (%). AE, adverse event; QD, once a day; SAE, serious adverse event. *AEs, as defined by investigators, started or worsened in intensity on or after the first dose of study medication and with onset up to and including 12 weeks from the last dose of study medication. †Related = relationship to study medication reported as “remote”, “possible”, “probable”, or missing. ‡Severe = most extreme intensity reported as “severe” or missing.
The most common AEs overall were cataracts, diarrhea, headache, urinary tract infections, and nasopharyngitis, which had similar incidence for all three treatment groups. SAEs reported for≥2 patients within a treatment group were fall and pneumonia (placebo), urinary tract infection (1 mg sembragiline), and hip fracture (5 mg sembragiline). Deaths occurred in three patients receiving 1 mg sembragiline, two patients receiving 5 mg, and three patients receiving placebo. The SAEs and causes of death appeared to be due to underlying medical conditions such as cerebrovascular disease, cardiovascular disease, injuries, or cancer, and were not considered to be due to study drug. There also did not appear to be any drug- or dose-related AEs or changes in laboratory findings, vital signs, or ECG.
DISCUSSION
This Phase II study demonstrated that treatment with sembragiline 1 mg and 5 mg in conjunction with background AD therapy in patients with moderate AD, although safe and well tolerated, did not affect cognition, as assessed by ADAS-Cog11, during 52 weeks of treatment. Similarly, no significant benefit in function assessed by the ADCS-ADL was found. A treatment effect in NPS, as assessed by the BEHAVE-AD-FW, was suggested in both 1 mg and 5 mg groups at Week 52. This effect was likely driven by a greater worsening in a small subset of patients in the placebo group between Weeks 24 and 52, as no significant differences between groups were observed at Weeks 12 and 24. The large number of patients with a zero value indicates a floor effect, and thus assumptions of the pre-defined statistical analysis model may be invalid, and p-values provided should be interpreted with caution. Nevertheless, this finding, in the context of low baseline BEHAVE-AD-FW score across the study, prompted a post hoc subgroup analysis in a more behaviorally impaired study population (BEHAVE-AD-FW>4 at baseline), which then suggested treatment effects in behavior, functioning, and global measures. MRI findings indicated no slowing of atrophy rates throughout treatment, but exploratory voxel-wise analyses suggested a stronger decrease in posterior hippocampal gray matter volume in the treatment groups relative to placebo. No new safety signals were found, and number of AEs, treatment discontinuations, and deaths did not differ between treatment and placebo groups, demonstrating a favorable safety profile.
A number of MAO-Bis have previously shown initial promise as therapeutic agents for AD, including selegiline and lazabemide. These programs, however, were not developed further due to lack of evidence for clinically meaningful benefit [25] and to hepatotoxicity (Roche data on file). The MAyflOwer RoAD study was initiated to further explore the therapeutic benefits of MAO-B inhibition as adjunctive therapy to current standard of care, using sembragiline— a potent, selective, and long-acting inhibitor of MAO-B that is chemically unrelated to lazabemide [14], at doses confirmed by PET studies to achieve near-complete MAO-B enzyme occupancy (Sturm et al., submitted).
In this trial, although cognitive function, as measured by ADAS-Cog11, declined over 52 weeks as expected for a moderate patient population taking AChEI (with or without memantine) [26], treatment with sembragiline did not provide any additional benefit on cognition at any time point measured. The absence of sembragiline efficacy with regards to cognition contrasted with results of the lazabemide trials. Importantly, the lazabemide trials started in 1997, and were therefore monotherapy trials without background treatments. In contrast, in the MAyflOwer RoAD study, sembragiline was administered on a background of standard of care, and it is possible that any subtle treatment effect (if present) would be more difficult to show in this setting.
The lack of a cognitive effect in moderate AD is consistent with a selegiline study in moderate-to-severe AD that showed no effect on ADAS-Cog11 or MMSE score when administered as monotherapy or in combination with alpha-tocopherol [11]. Evidence of a long-term benefit of selegiline on cognition has not been demonstrated; rather, a treatment effect has been observed only in 4–6- or 8–17-week studies [25]. Together, these studies suggest that MAO-Bis, when administered on top of background therapy in patients with moderate AD, do not provide cognitive benefit.
Apathy, or lack of motivation and loss of interest in previously enjoyed activities, is a common, but under-recognized, change in behavior in AD. Of relevance to MAO-B inhibition, evidence for dopamine dysfunction in brain areas appears to be an important correlate of apathy in AD [27]. However, neither sembragiline dose group showed treatment effects on apathy at Week 52. These data were unexpected, since improvement in apathy had been observed in lazabemide trials and MAO-B inhibition is believed to modulate dopaminergic transmission, thus sembragiline would be expected to improve apathy and other behavioral functions (Roche data on file, [7, 28]). Apathy was not evaluated in the 2-year selegiline study [11].
In this study, a greater decrease in hippocampal volume was observed in the treatment groups relative to placebo but was not associated with a greater cognitive decline in ADAS-Cog11. Although the cause for this finding is unknown, increased rates of brain atrophy in treatment arms unassociated with cognitive deterioration have been reported in immunotherapy trials targeting amyloid removal [29–31]. The findings with immunotherapies and sembragiline are consistent with a phenomenon known as “pseudoatrophy”, which is thought to be caused by reduction in the number and volume of inflammatory cells, particularly glial cells [32, 33]. Given that hypertrophic activated astrocytes surrounding amyloid plaques express high levels of MAO-B, the hippocampal atrophy observed in sembragiline-treated patients may be due to reduction of astrocytosis from MAO-B inhibition. In animal models, MAO-B overexpression-induced astrocyte activation can be blocked by selegiline [34] and sembragiline (Borroni et al., in preparation), supporting this hypothesis. Further studies are required to test this hypothesis and to better understand the role of MAO-B in neuroinflammation.
The results of this Phase II study indicate that sembragiline is safe and well tolerated in patients with moderate AD over a 1-year period. While efficacy in cognition, functioning, or global outcomes was absent, treatment with sembragiline adjunctive to current standard of care showed a numerical reduction in the worsening of NPS relative to placebo at Week 52. As the trial was not designed to investigate this potential benefit of sembragiline on NPS, validity of this effect is difficult to interpret. A small number of pharmacologic treatments with different mechanisms of action have been recently investigated for the treatment of NPS with modest results, limited efficacy, and safety concerns [35]. A clear unmet need for management of NPS in dementia exists. There are currently no approved medications for NPS in AD, with the exception of risperidone, which is licensed in some regions with a narrow indication for short-term treatment. Several agents currently used for the management of NPS carry serious safety liabilities. Moreover, NPS in AD are associated with poor prognosis and more rapid progression to severe dementia, leading to increased mortality and institutionalization [36], and have the greatest impact on patient and caregiver quality of life and associated costs of the disease [37, 38]. Therefore, broadening the understanding of the pathogenesis of NPS in AD and exploring the potential role of MAO-Bis in development as treatment options for NPS will be particularly meaningful for the AD community.
ACKNOWLEDGMENTS
The authors would like to thank Kirsten Taylor for her input regarding the structural MRI interpretation. We are also grateful to the MayflOwer RoAD investigators, caregivers, patients and families who contributed to this trial (NCT01677754).
The study is sponsored by F. Hoffmann-La Roche Ltd, which was responsible for the administration or conduct of many of the study procedures. Some administrative or clinical roles were given to external organizations including QLAB, Quintiles, Nuvisan, S-Clinica, Medavante, and NeuroRx. Support for third-party writing assistance for this manuscript, furnished by Judy Wang, PhD and Athena Lemesiou, PhD of MediTech Media, was provided byF. Hoffmann-La Roche.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1309r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-161309.
REFERENCES
[1] | Alzheimer’s Disease International ((2013) ) World Alzheimer’s report, http://www.alz.co.uk/research/WorldAlzheimerReport2013.pdf 2013, July, 2014. |
[2] | Markesbery WR ((1997) ) Oxidative stress hypothesis in Alzheimer’sdisease. Free Radic Biol Med 23: , 134–147. |
[3] | Saura J , Luque JM , Cesura AM , Da Prada M , Chan-Palay V , Huber G , Loffler J , Richards JG ((1994) ) Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience 62: , 15–30. |
[4] | Gulyas B , Pavlova E , Kasa P , Gulya K , Bakota L , Varszegi S , Keller E , Horvath MC , Nag S , Hermecz I , Magyar K , Halldin C ((2011) ) Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem Int 58: , 60–68. |
[5] | Riederer P , Danielczyk W , Grunblatt E ((2004) ) Monoamine oxidase-B inhibition in Alzheimer’s disease. Neurotoxicology 25: , 271–277. |
[6] | Saura J , Andres N , Andrade C , Ojuel J , Eriksson K , Mahy N ((1997) ) Biphasic and region-specific MAO-B response to aging in normal human brain. Neurobiol Aging 18: , 497–507. |
[7] | Reinikainen KJ , Paljarvi L , Halonen T , Malminen O , Kosma VM , Laakso M , Riekkinen PJ ((1988) ) Dopaminergic system and monoamine oxidase-B activity in Alzheimer’s disease. Neurobiol Aging 9: , 245–252. |
[8] | Zellner M , Baureder M , Rappold E , Bugert P , Kotzailias N , Babeluk R , Baumgartner R , Attems J , Gerner C , Jellinger K , Roth E , Oehler R , Umlauf E ((2012) ) Comparative platelet proteome analysis reveals an increase of monoamine oxidase-B protein expression in Alzheimer’s disease but not in non-demented Parkinson’s disease patients. J Proteomics 75: , 2080–2092. |
[9] | Valeant Pharamaceuticals North America, LLC. ((2014) ) ZELAPARâ (selegiline HCl) Orally Disintegrating Tablets [prescribing information]. |
[10] | Teva Neuroscience I ((2014) ) AZILECTâ (rasagilinemesylate) [prescribing information] |
[11] | Sano M , Ernesto C , Thomas RG , Klauber MR , Schafer K , Grundman M , Woodbury P , Growdon J , Cotman CW , Pfeiffer E , Schneider LS , Thal LJ ((1997) ) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med 336: , 1216–1222. |
[12] | Henriot S , Kuhn C , Kettler R , Da Prada M ((1994) ) Lazabemide (Ro 19-6327), a reversible and highly sensitive MAO-B inhibitor: Preclinical and clinical findings. J Neural Transm Suppl 41: , 321–325. |
[13] | Cesura AM , Borroni E , Gottowik J , Kuhn C , Malherbe P , Martin J , Richards JG ((1999) ) Lazabemide for the treatment of Alzheimer’s disease: Rationale and therapeutic perspectives. Adv Neurol 80: , 521–528. |
[14] | Magni G , Meibach RC ((1999) ) Lazabemide for the long-term treatment of Alzheimer’s disease. Eur Neuropsychopharmacol 9: , 142. |
[15] | Cummings JL , Mega M , Gray K , Rosenberg-Thompson S , Carusi DA , Gornbein J ((1994) ) The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 44: , 2308–2314. |
[16] | Borroni E , Wyler R , Messer J , Nave S , Cesura A ((2013) ) Preclinical characterization of R04602522, a novel, selective, and orally active monoamine oxidase type B inhibitor for the treatment of Alzheimer’s disease. Alzheimers Dement 9: , 818. |
[17] | Berlin I , Hunneyball IM , Greiling D , Jones SP , Fuder H , Stahl HD ((2012) ) A selective reversible monoamine oxidase B inhibitor in smoking cessation: Effects on its own and in association with transdermal nicotine patch. Psychopharmacology (Berl) 223: , 89–98. |
[18] | Ricci B , Sturm S , Seneca N , Jamois C , Nave S , Ejduk Z , Stenkrona P , Varrone A , Forsberg A , Fazio P , Halldin C , Al-Tawil NA , Akenine U , Andreasen N ((2013) ) Brain MAO-B inhibition in healthy elderly and people with Alzheimer’s disease after oral administration of RO4602522. Alzheimers Dement 9: , 667. |
[19] | Sturm S , Forsberg A , Nave S , Stenkrona P , Seneca N , Varrone A , Comley RA , Fazio P , Jamois C , Nakao R , Ejduk Z , Al-Tawil N , Akenine U , Halldin C , Andreasen N , Ricci B ((2017) ) Positron emission tomography measurement of brain MAO-B inhibition in patients with Alzheimer’s disease and elderly controls after oral administration of sembragiline. Eur J Nucl Med Mol Imaging 44: , 382–391. |
[20] | Monteiro IM , Boksay I , Auer SR , Torossian C , Ferris SH , Reisberg B ((2001) ) Addition of a frequency-weighted score to the Behavioral Pathology in Alzheimer’s Disease Rating Scale: The BEHAVE-AD-FW: Methodology and reliability. Eur Psychiatry 16: (Suppl 1), 5s–24s. |
[21] | Reisberg B , Monteiro I , Torossian C , Auer S , Shulman MB , Ghimire S , Boksay I , Guillo BenArous F , Osorio R , Vengassery A , Imran S , Shaker H , Noor S , Naqvi S , Kenowsky S , Xu J ((2014) ) The BEHAVE-AD assessment system: A perspective, a commentary on new findings, and a historical review. Dement Geriatr Cogn Disord 38: , 89–146. |
[22] | Marin RS , Biedrzycki RC , Firinciogullari S ((1991) ) Reliability and validity of the Apathy Evaluation Scale. Psychiatry Res 38: , 143–162. |
[23] | Olin JT , Schneider LS , Doody RS , Clark CM , Ferris SH , Morris JC , Reisberg B , Schmitt FA ((1996) ) Clinical evaluation of global change in Alzheimer’s disease: Identifying consensus. J Geriatr Psychiatry Neurol 9: , 176–180. |
[24] | Posner K , Brown GK , Stanley B , Brent DA , Yershova KV , Oquendo MA , Currier GW , Melvin GA , Greenhill L , Shen S , Mann JJ ((2011) ) The Columbia-Suicide Severity Rating Scale: Initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168: , 1266–1277. |
[25] | Birks J , Flicker L ((2003) ) Selegiline for Alzheimer’s disease. Cochrane Database Syst Rev (1), CD000442. |
[26] | Ito K , Ahadieh S , Corrigan B , French J , Fullerton T , Tensfeldt T , Alzheimer’s Disease Working Group ((2010) ) Disease progression meta-analysis model in Alzheimer’s disease. Alzheimers Dement 6: , 39–53. |
[27] | Mitchell RA , Herrmann N , Lanctot KL ((2011) ) The role of dopamine in symptoms and treatment of apathy in Alzheimer’s disease. CNS Neurosci Ther 17: , 411–427. |
[28] | Berry MD ((2007) ) The potential of trace amines and their receptors for treating neurological and psychiatric diseases. Rev Recent Clin Trials 2: , 3–19. |
[29] | Novak G , Fox N , Clegg S , Nielsen C , Einstein S , Lu Y , Tudor IC , Gregg K , Di J , Collins P , Wyman BT , Yuen E , Grundman M , Brashear HR , Liu E ((2015) ) Changes in brain volume with bapineuzumab in mildto moderate Alzheimer’s disease. J Alzheimers Dis 49: , 1123–1134. |
[30] | Fox NC , Black RS , Gilman S , Rossor MN , Griffith SG , Jenkins L , Koller M , AN1792(QS-21)-201 Study ((2005) ) Effects of Abetaimmunization (AN1792) on MRI measures of cerebral volume inAlzheimer disease. Neurology 64: , 1563–1572. |
[31] | Salloway S , Sperling R , Fox NC , Blennow K , Klunk W , Raskind M , Sabbagh M , Honig LS , Porsteinsson AP , Ferris S , Reichert M , Ketter N , Nejadnik B , Guenzler V , Miloslavsky M , Wang D , Lu Y , Lull J , Tudor IC , Liu E , Grundman M , Yuen E , Black R , Brashear HR ((2014) ) Two Phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 322–333. |
[32] | De Stefano N , Arnold DL ((2015) ) Towards a better understanding of pseudoatrophy in the brain of multiple sclerosis patients. Mult Scler 21: , 675–676. |
[33] | Sastre-Garriga J , Tur C , Pareto D , Vidal-Jordana A , Auger C , Rio J , Huerga E , Tintore M , Rovira A , Montalban X ((2015) ) Brain atrophy in natalizumab-treated patients: A 3-year follow-up. Mult Scler 21: , 749–756. |
[34] | Mallajosyula JK , Kaur D , Chinta SJ , Rajagopalan S , Rane A , Nicholls DG , Di Monte DA , Macarthur H , Andersen JK ((2008) ) MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS One 3: , e1616. |
[35] | Seitz DP , Gill SS , Herrmann N , Brisbin S , Rapoport MJ , Rines J , Wilson K , Le Clair K , Conn DK ((2013) ) Pharmacological treatments for neuropsychiatric symptoms of dementia in long-term care: A systematic review. Int Psychogeriatr 25: , 185–203. |
[36] | Peters ME , Schwartz S , Han D , Rabins PV , Steinberg M , Tschanz JT , Lyketsos CG ((2015) ) Neuropsychiatric symptoms as predictors of progression to severe Alzheimer’s dementia and death: The Cache County Dementia Progression Study. Am J Psychiatry 172: , 460–465. |
[37] | Lyketsos CG , Carrillo MC , Ryan JM , Khachaturian AS , Trzepacz P , Amatniek J , Cedarbaum J , Brashear R , Miller DS ((2011) ) Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement 7: , 532–539. |
[38] | Alzheimer’s disease International ((2009) ) World Alzheimer’s Report, http://www.alz.co.uk/research/files/WorldAlzheimerReport.pdf 2009, July, 2014. |


