ABCA1 Deficiency Affects Basal Cognitive Deficits and Dendritic Density in Mice
Abstract
ATP-binding cassette transporter A1 (ABCA1) mediates cholesterol efflux to lipid-free apolipoproteins and regulates the generation of high density lipoproteins. Previously, we have shown that lack of Abca1 significantly increases amyloid deposition and cognitive deficits in Alzheimer’s disease model mice expressing human amyloid-β protein precursor (APP). The goal of this study was to determine if ABCA1 plays a role in memory deficits caused by amyloid-β (Aβ) oligomers and examine neurite architecture of pyramidal hippocampal neurons. Our results confirm previous findings that Abca1 deficiency significantly impairs spatial memory acquisition and retention in the Morris water maze and long-term memory in novel object recognition of APP transgenic mice at a stage of early amyloid pathology. Neither test demonstrated a significant difference between Abca1ko and wild-type (WT) mice. We also examined the effect of intra-hippocampal infused Aβ oligomers on cognitive performance of Abca1ko mice, compared to control infusion of scrambled Aβ peptide. Age-matched WT mice undergoing the same infusions were also used as controls. In this model system, we found a statistically significant difference between WT and Abca1ko mice infused with scrambled Aβ, suggesting that Abca1ko mice are vulnerable to the effect of mild stresses. Moreover, examination of neurite architecture in the hippocampi revealed a significant decrease in neurite length, number of neurite segments, and branches in Abca1ko mice when compared to WT mice. We conclude that mice lacking ABCA1 have basal cognitive deficits that prevent them from coping with additional stressors, which is in part due to impairment of neurite morphology in the hippocampus.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by cognitive decline, amyloid-β (Aβ) plaques and tangles of hyperphosphorylated tau. Accumulation of soluble extracellular high molecular weight oligomeric Aβ species in the brain is considered a major risk factor for the onset and progression of cognitive deficits associated with AD. Inheritance of the ɛ4 allele of apolipoprotein E (APOE) is the major genetic risk factor for late-onset AD [1]. Carriers of APOE4 allele have increased amyloid plaque load and an earlier onset of the disease [2]. In general, human amyloid-β protein precursor (APP) transgenic mice recapitulate AD amyloid pathology, at least in regard to amyloid pathology and cognitive deficits [3–5].
ATP binding cassette transporter A1 (ABCA1) regulates cholesterol and phospholipids efflux from cells to lipid-poor apolipoprotein A-I (ApoA-I) and ApoE and controls the generation of high density lipoproteins (HDL) (reviewed in [6]). Previous data from our laboratory and other groups have demonstrated significantly increased amyloid plaques in different APP transgenic mice lacking Abca1 [7–10] associated with a significantly decreased ApoE protein level in brain. In contrast, Abca1 overexpression in PDAPP mice decreases amyloid burden [11]. We have reported that mice expressing human APOE4 were more susceptible to Abca1 haplo-deficiency than APOE3 mice [12], suggesting that the Abca1 genotype can interact with other genetic risk factors to worsen the AD phenotype.
Recently, using APP/PS1dE9 (in this study referred to as APP mice) transgenic mouse model crossed to Abca1ko mice, we demonstrated an increased level of Aβ oligomers in APP/Abca1ko mice. We also observed that the dendritic complexity in the CA1 region of the hippocampus, but not in CA2 region, was significantly impaired in APP/Abca1ko mice [13]. An unexpected finding was that lack of ABCA1 affected the performance of Abca1ko mice in contextual fear conditioning paradigm similarly in APP transgenic and wild-type (WT) mice [13]. Since different behavior paradigms test discrete domains of the learning process, we undertook this study to determine if ABCA1 plays a role in memory deficits caused by Aβ oligomers and how this correlates to the neurite morphology of Abca1ko mice.
MATERIAL AND METHODS
Transgenic mice
All experiments followed NIH guidelines for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Data was collected and analyzed for all experiments where researchers were blinded to genotypes and/or treatments described below. Abca1het mice were bred to Abca1het mice to yield Abca1ko, Abca1het, and Abca1wt littermates. All mice used in this study were purchased from Jackson Laboratory and bred onto a C57BL/6J background for greater than 11 generations. APP/PS1ΔE9 (B6.Cg-Tg(APPswe, PSEN1ΔE9)85Dbo/Mmjax; referred to as APP) transgenic mice, also purchased from Jackson Laboratory, were crossbred to Abca1het mice to generate APP/Abca1ko, APP/Abca1het, APP/Abca1wt and corresponding non-APP expressing littermates. Non-APP expressing –Abca1wt littermates are referred here in as WT mice. The progeny were identified by PCR. All reagents purchased through Fisher Scientific unless otherwise stated.
Cannula implantation
To examine the effects of Aβ oligomers on memory, mice were infused with Aβ oligomers directly into the hippocampus through guide cannulas. Following anesthesia with isoflurane, the head was shaven and sterilized with two separate iodine – alcohol washes. A 50% mixture of bupivacaine and lidocaine were applied to the surgical site and ophthalmic ointment applied to the eyes. The head was leveled in a stereotaxic frame and an incision made exposing the dorsal aspect of the skull. Two holes were drilled into the skull (coordinates: P = 2.46 mm, L =±1.50 mm) and 26-gauge guide cannulas (Plastics One) were lowered into the dorsal part of the hippocampi to a depth of 1.30 mm. Cannulas were fixed to the skull with acrylic dental cement attached to two bone anchoring screws and the surgical opening sutured closed. Animals were allowed to recover for 8 days before behavioral testing started.
Aβ oligomer preparation
Under a fume hood, 1 mg of Aβ42 peptide (American Peptide Company) was dissolved in ice cold 1,1,1,3,3,3-Hexafluoro-2-Propanol (HFIP, Fluka) to obtain a 1 mM solution then vortex for few seconds. The solution was quickly aliquoted into 3 polypropylene vials and dried with a gentle stream of N2 to obtain a clear peptide film in the bottom of the vials. Prior to use, one film was re-suspended in anhydrous DMSO to form a 5 mM solution, sonicated in water bath for 10 min and diluted 200X with sterile phosphate-buffered saline (PBS). Aβ samples were left at room temperature for 24 h to form oligomer complexes and stored at –20°C until use. The same concentration of scrambled Aβ (AnaSpec) was dissolved in vehicle and utilized as a negative control.
Aβ oligomer characterization
Western blot
To examine Aβ oligomers on western blotting, proteins were resolved on 4–12% Bis-Tris gels (Invitrogen) and transferred onto nitrocellulose membranes. These membranes were probed with 6E10 antibody (1 : 1000; Signet) and immunoreactive signals visualized using enhanced chemiluminescence.
Dot blot
For detection of prefibrillar Aβ oligomers, 1 μg of protein was spotted on a nitrocellulose membrane and probed with A11 antibody (1 : 2000, Invitrogen). The membranes were probed with anti-rabbit secondary antibody, and immunoreactive signals visualized using enhanced chemiluminescence. The exact same number of samples was spotted on additional dot blots and probed with 6E10 antibody and Bradford reagent for normalization.
Electron microscopy
Imaging was performed as before [14]. Briefly, time zero and 24 h incubated Aβ oligomer aliquots were placed on freshly glow-discharged carbon-coated grids (Electron Microscopy Sciences) and incubated for 1 min. Excess sample was removed with filter paper and sample grid was then washed with deionized water, stained with 1% uranyl acetate (w/v) solution for 3 s, and blotted dry. Grids were imaged using Tecnai T12 microscope (FEI Co.) operating at 120 kV and 30,000×magnification and equipped with an ultrascan 1000 CCD camera (Gatan) with post-column magnification of 1.4×.
Behavioral testing
Morris water maze
Behavioral tests to assess spatial navigational learning and memory retention were performed with a modified version of the Morris water maze (MWM) as before [15]. Briefly, in a circular pool of water (diameter 122 cm, height 51 cm, temperature 21±1°C), we determined the ability of mice to form a spatial representation between a safe but invisible platform (10 cm diameter; submerged 1 cm below the water surface) and visual cues surrounding the pool. Animals were handled for 2 min for two days prior to testing and received a habituation trial, during which they were allowed to explore the pool of water without the platform present for 1 min. Beginning the next day, they received four daily hidden platform training (acquisition) trials with 5 min inter-trial interval over five consecutive days. Animals were allowed 60 s to locate the platform and rest there for 20 s. Mice that failed to find the platform were lead to the platform by the experimenter and allowed to rest there for 20 s. 24 h following the last acquisition trial, a single 60 s probe trial was administered to assess spatial memory retention. For the probe trial, animals were returned to the maze as during training but with no platform present. Performance was recorded with AnyMaze video tracking (Stoelting Co.) during all phases. During the acquisition trials, escape latency and path length were subsequently used to analyze and compare the performance between different genotypes. In addition, the swimming speed during the acquisition phase was analyzed (this was used to evaluate the locomotor activity). The latency to reach the target quadrant and time spent in the target quadrant were recorded and analyzed during the probe trials.
Novel object recognition
Changes in long term memory were assessed utilizing a novel object recognition paradigm as before [16]. On day 1 of testing, mice were acclimated to the behavioral arena (40 cm × 40 cm×30 cm tall white plastic box) for 5 min. For training, 24 h following acclimation mice were placed into the center of the arena with two similar objects and allowed to explore the objects for 30 total visits but no longer than 10 min. The two identical objects were made of weighted plastic to prevent movement and located in the southeast and northwest quadrant, spaced equidistant from the arena walls. This training trial was completed twice, separated by a 10-min interval. 24 h following the last training trial, one object was replaced with a novel object; the object replaced was alternated for each mouse to avoid a side preference, and mice were again placed into the arena and allowed to explore the objects for 30 total visits or 10 min. An exploratory visit was defined as the mouse sniffing, climbing on, or touching an object or within 1 cm while facing an object. Testing was recorded with automated tracking system (AnyMaze; Stoelting Co.) and the arena was cleaned with 70% alcohol between trials to eliminate olfactory cues. An increased percentage of visits exploring the novel object (number of novel object visits/total visits×100) was considered an index for improved long term memory retention in this task.
Radial arm water maze
Two day radial arm water maze (RWM) was used to measure the ability of mice to form a spatial relationship between a safe, but hidden, platform and visual cues surrounding the maze [12]. The RWM consisted of six arms (20 cm wide, 40 cm long, 8 cm high walls above the water) and a central area (30 cm diameter), filled with water (temperature, 21±1°C) to a level 1 cm above the hidden platform (10 cm diameter). All animals were handled for 2 min for 2 consecutive days before behavioral testing and 1 day before testing allowed to explore the water maze without the platform present. Acquisition testing was performed over 2 consecutive days with mice trained in groups of five or six. Each day, a mouse received two 6 trial blocks and a final 3 trial block (total of 15 trials per day) with a 30-min rest between blocks. 30 min prior to each training block, animals were infused with the Aβ oligomer or scrambled Aβ. Briefly, the dummy cannulas were removed and infusion cannulas, attached to microsyringe pump by polyethylene tubing, were placed in the guide cannula. Aβ oligomer or scrambled Aβ (final volume of 1 μl per hemisphere) was infused over 1 min, the cannulas left in place for 1 min to allow for diffusion of sample, and finally dummy cannulas replaced. Each animal received three infusions of Aβ oligomer or scrambled Aβ prior to each of the 3 training blocks (2–6 trial blocks and 1–3 trial blocks).
During day 1 of training, a visible platform (flag projecting 6 cm from the platform) was used during trials 1, 3, 5, 7, 9, and 11 to define the rule of a safe platform. All other trials consisted of animals finding the location of a hidden platform. Animals were allowed 60 s to find the platform and 20 s to rest on it. Mice that failed to find the platform were led there by the experimenter and allowed to rest there for 20 s. All animals in a group completed the trial before proceeding, providing a 5 min inter-trial interval. The start location was changed for each trial and the platform location was changed between groups. Performance was recorded with an automated tracking system (AnyMaze; Stoelting Co.) during training. During the acquisition phase, total number of incorrect arm entries and time errors were combined for the overall performance of an animal during a trial. An incorrect arm entry was defined as the entry of 50% of the animal’s body into an arm that did not contain the hidden platform. A time error was defined as the failure of an animal to enter an arm after 15 s elapsed. For the 15 daily trials, performance during 3 consecutive trials was averaged into a block (total of 5 blocks per day). During the open pool task of training, speed and latency to the platform were used to compare the performance between genotypes.
Animal tissue processing
Mice were anesthetized with Avertin (250 mg/kg of body weight, i.p.) and blood was drawn from the heart. The mice were perfused transcardially with 25 ml of cold 0.1 m PBS, pH 7.4. One hemisphere was drop fixed in 4% phosphate-buffered paraformaldehyde at 4°C for 48 h before storage in 30% sucrose.
Neurite morphology quantification
Neurite morphometry analysis was performed as described before [13]. Coronal brain sections (30 μm thickness) were obtained with a sliding microtome. Sections were stored in cryoprotectant at –20°C until staining commenced. For immunofluorescence labeling, brain tissue sections were rinsed 3 times in PBS for 10 min and incubated with 1% Triton X-100 (permeabilizing reagent) in PBS solution for 5 h at 4°C. The tissue was washed 3 times in PBS for 10 min each and blocked with 10% serum and 0.3% Triton X-100 in PBS solution for 30 min at room temperature (RT). The sections were incubated for 72 h at 4°C with microtubule-associated protein 2 (MAP2) primary antibody (1 : 2000; Millipore), a cytoskeletal protein that binds to tubulin and stabilizes microtubules and is essential for the development and maintenance of neuronal morphology. Tissue was rinsed 3 times in PBS for 10 min each to remove unreacted primary antibodies. Tissue sections were incubated with Cy3-conjugated anti-sheep antibody (1 : 500; Jackson-ImmunoResearch) secondary antibody for 2 hr at RT. Sections were washed 2 times in PBS for 10 min and counterstained with H33342 nuclear reagent (1 : 3000; Sigma-Aldrich) for 5 min at RT. Sections were washed 3 times in PBS for 10 min each and then, were mounted onto plus-coated slides and coverslipped using gelvatol mounting media. The FilamentTracer module of Imaris (Bitplane), which facilitates 3D neuron reconstruction, was utilized to determine neuronal patterning and connections of hippocampal brain sections. The MAP2 (Cy3) channel was used to quantify total neurite length, the number of segments, and the number of branches in the CA1 and CA2 regions of the hippocampus. 5 sections per animal, starting at the beginning of the dentate gyrus, and every 300 μm were used. For neurite analysis, 4-5 confocal images (considered replicates) were acquired for the CA1 region while 3-4 confocal pictures were captured for the CA2 region. Confocal fluorescence micrographs were obtained using a 60x lens magnification at very high resolution (100 μs exposure). For unbiased examination, the size and the length of the neurites were the only parameters that required manual introduction. For parity, image assessment must use identical grid dimensions. Neurite length was normalized to the number of MAP2 neurons.
Statistical analysis
Data are reported as means±SEM. Statistical differences between mean scores during acquisition phase of training in the MWM were analyzed with two-way ANOVA and t-test was used to determine difference for each trial day. One-way ANOVA followed by Tukey’s post hoc analysis for multiple comparisons was used to analyze the MWM probe trial data. Performance in RWM was assessed with two-way ANOVA Repeated measure (using Genotype or Infusion Group and Trial as sources of variation) followed by Tukey’s post hoc analysis. Significant differences between groups for the rest of the data were determined by t-test. All statistical analyses were performed in GraphPad Prism, version 6.0, and differences were considered significant where p < 0.05.
RESULTS
Abca1 deficiency significantly impairs spatial memory in APP transgenic mice
Recently we showed that lack of Abca1 increases amyloid load and worsens the performance of APP expressing mice in the contextual fear conditioning test [13]. The contextual fear condition paradigm assesses hippocampal-dependent associative learning to unconditional stimulus such as electric shock [5]. To test if ABCA1 deficiency affects other types of memory, such as spatial learning and retention and long-term memory, we employed MWM and the novel object recognition test, respectively. We used 7-month-old APP mice that express wild type Abca1 (APP/Abca1wt) or Abca1ko (APP/Abca1ko) and their non-APP transgenic littermates (WT and Abca1ko). As illustrated in Fig. 1A and B, during the acquisition phase, APP mice with intact Abca1 performed similarly to WT mice (no significant main effect of genotype); however, there was a statistical difference on the last day of testing (p < 0.05) suggesting impaired acquisition. In contrast, APP/Abca1ko mice performed much worse than Abca1ko (significant main effect of genotype, p < 0.001), particularly on the last days of MWM. This suggests that the lack of ABCA1 impairs spatial acquisition in mice expressing human APP. In the MWM probe trial, APP/Abca1ko mice demonstrated decreased memory retention exemplified by a longer latency time to enter the target zone compared to APP/Abca1wt and non-transgenic Abca1ko mice (Fig. 1C). APP/Abca1ko mice also spent the shortest time in the target quadrant compared to the other genotypes; however, the difference with Abca1ko was not significant (Fig. 1D). These changes in performance were not due to swim speeds as there was no difference in the swim speeds of the assessed genotypes.
To confirm that the memory impairment is not restricted only to MWM, the mice were also tested using the novel object recognition paradigm as before [16]. Once more, APP/Abca1ko mice demonstrated the worst performance (Fig. 1E, p < 0.05 compared are APP/Abca1ko and APP/Abca1wt), with APP/Abca1ko mice exploring the novel object far less than the other genotypes.
The behavior experiments presented on Fig. 1 demonstrate that ABCA1 deficiency exaggerates cognitive deficits in APP transgenic mice that could be a result of either increased endogenously formed Aβ oligomers or amyloid plaques present in APP/Abca1ko mice.
Characterization of Aβ oligomers utilized in this study
It has been reported that Aβ oligomers affect synaptic plasticity and cognitive function [17]. Recently we have shown that amyloid load and A11-positive Aβ oligomers are significantly increased in 7-month-old APP/Abca1ko mice in comparison with APP mice expressing Abca1wt [13]. To determine how the acute infusion of Aβ oligomers into the hippocampus affects the memory of WT and Abca1ko mice, we used Aβ42 peptide and employed several methods to characterize the Aβ oligomers. Fig. 2A shows SDS PAGE followed by western blotting with anti-Aβ 6E10 antibody. As visible, at time 0, Aβ existed only as monomers and low MW oligomers such as dimers, trimers, and tetramers. Upon oligomerization, following 24 h incubation, there was an increase of higher MW oligomers such as 9-mer, 12-mer etc.; however, we were unable to detect oligomers with MW higher than 100 kDa. For comparison, on the right of Fig. 2A is shown western blotting of Aβ fibrils. Aβ oligomers were also confirmed using conformation specific A11 antibody. As seen on Fig. 2B, when the dot blot was probed with A11, there was an increase in the intensity of oligomeric Aβ from time 0 to 48 h incubation. Finally, on Fig. 2C are shown electron micrographs of Aβ at time 0 and 24 h incubation. At time 0 (Fig. 2C-a), disaggregated Aβ42 is present as a monomer.After incubation for 24 h at room temperature, Aβ42 was converted into rod-shaped oligomers with diameter of approximately 10 nm and length between 20–50 nm (Fig. 2C-b).
Infusion of Aβ oligomers into hippocampus differentially affects cognitive deficits in WT and Abca1ko mice
To test the effect of the oligomers shown on Fig. 2, on cognition we infused WT and Abca1ko mice, which do not express human APP transgene, with the 24 h oligomeric Aβ preparation. Young 7-month-old mice were implanted with cannulas into both hippocampi (see picture on Fig. 3A) followed by a recovery period of 8 days before behavioral testing. Aβ42 oligomers were infused bilaterally into the hippocampus 30 min prior to each of the RWM training blocks (2–6 trial blocks and 1–3 trial blocks) for a total of 3 infusions. RWM paradigm tests reference memory [5] and were used as a more demanding task, compared to MWM, to detect relatively subtle changes in cognitive function. Control mice were infused with scrambled Aβ. In addition, mice were compared to naïve mice that were not subjected to surgery. As shown on Fig. 3B, in WT mice Aβ42 oligomers significantly affected cognitive performance as compared to the control mice infused with scrambled Aβ (compare purple circles to grey squares). In contrast, there was no statistical difference between naïve (no surgery) and mice infused with scrambled Aβ (Fig. 3B, compare grey squares to black triangles). This experiment suggests that surgical procedures followed by the infusion of scrambled peptide do not affect the memory of WT mice.
We next examined how Aβ oligomers infused into the hippocampus would impact cognitive performance of age-matched Abca1ko mice and compared them to controls of Abca1ko and WT mice infused with scrambled Aβ. Surprisingly, we found a statistically significant difference between WT and Abca1ko infused with control scrambled Aβ (see Fig. 3C, compare black to grey squares) suggesting that Abca1ko mice are vulnerable to the effect of stress/trauma induced by surgery and infusion of scrambled Aβ. Due to the worsened performance of the control Abca1ko mice (injected with scrambled Aβ), there was no statistical difference between their behavior and Abca1ko mice infused with Aβ oligomers (compare black squares to red circles). Our conclusion is that Abca1ko mice have basal cognitive deficits that prevent them from coping with additional stressors that do not impact performance of normal healthy WT mice.
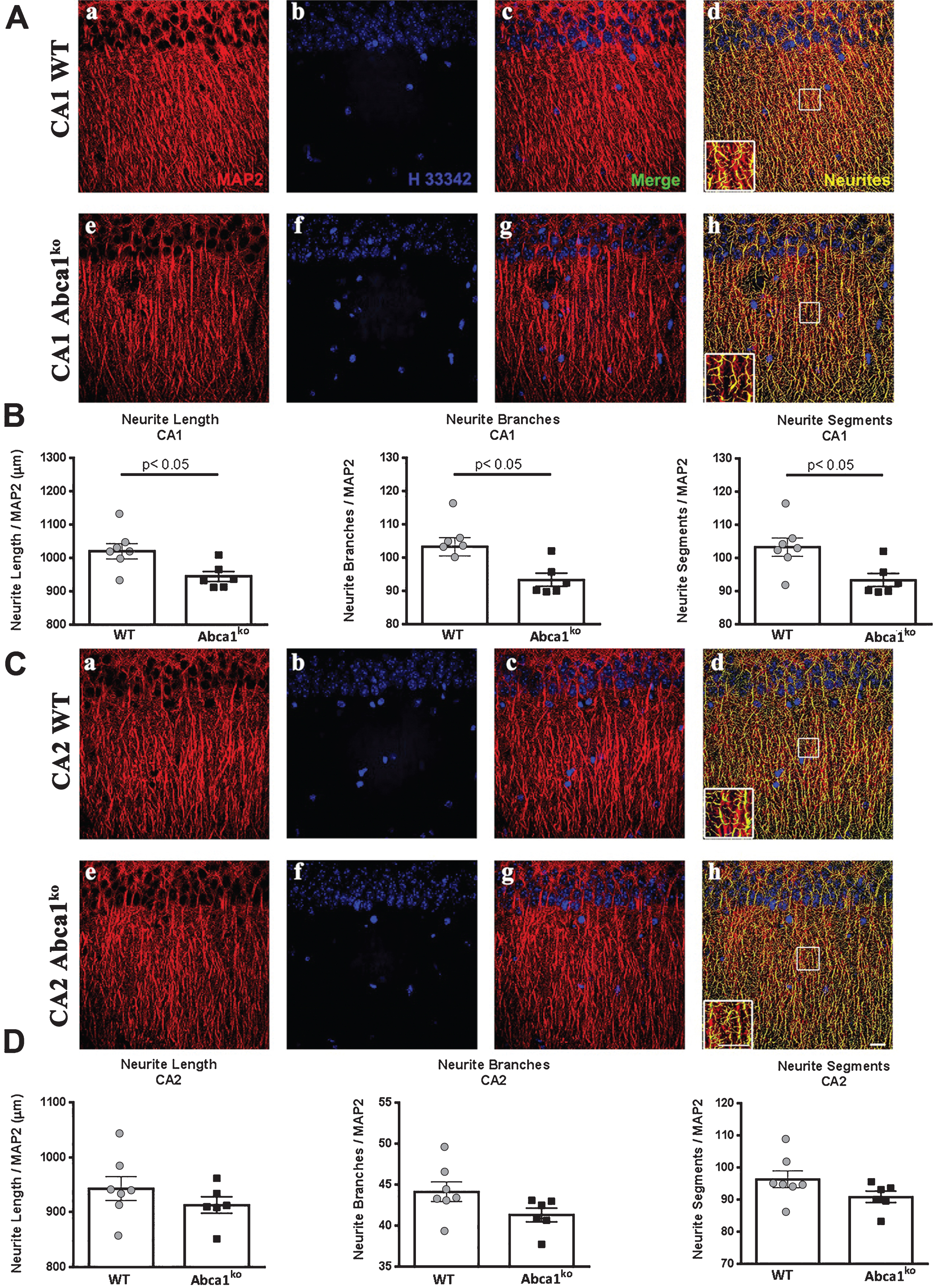
Deficiency of Abca1 significantly impairs neurite morphology in CA1 but not in CA2 region of the hippocampus
Recently we have demonstrated that Abca1 as well as Apoe deficiency significantly affects dendrite architecture in human APP transgenic mice [13]. The behavior tests shown on Fig. 2 prompted us to examine the dendritic parameters of Abca1ko mice that do not express human APP transgene. To test the effect of Abca1 deficiency we used 7-month-old Abca1ko mice and compared them to WT mice (the same age as in Fig. 2). For neurite morphometry of pyramidal hippocampal neurons, we used MAP2 stained brain sections and performed analysis in two different regions (CA1 and CA2) of medial hippocampal sections. Since neurites form complex tree-like structures, 3D image reconstruction is necessary to obtain accurate measurements. Examination of neurites in the CA1 region (Fig. 4A) revealed a significant decrease in neurite length, number of neurite segments and number of branches (Fig. 4A, B; p < 0.05) in Abca1ko mice when compared to WT. These changes in neurite architecture were restricted to the CA1 region of the hippocampus as we did not observe any changes in the CA2 region when comparing Abca1ko to WT mice (Fig. 4C, D). The data were normalized to the number of neurons and there was no significant change in the numbers of counted nuclei (not shown) when comparing Abca1ko mice and WT mice in either region of the hippocampus. Our data demonstrate that the genetically engineered deletion of Abca1 disrupts neurite morphology in the hippocampus, and this effect is specific for CA1 region.
DISCUSSION
In this study, we examined the effect of global deletion of Abca1 on cognitive performance and neurite morphology. We also determined the potential impact the deletion of Abca1 has on cognitive impairment induced by oligomeric Aβ. In regards to cognitive performance and in correlation to the level of soluble Aβ oligomers, we have previously shown that old APP23 mice with one copy of Abca1 performed significantly worse than mice with intact Abca1 in a MWM paradigm [10]. Furthermore, we have reported that simultaneous deletion of Apoe and Apoa1 in the AD mouse model used in the current study, significantly aggravated memory impairment, similarly to deletion of Abca1, revealed in the contextual fear conditioning. Consistent with the behavior data, APP expressing mice with double –Apoe and Apoa1, or Abca1 knockout, both demonstrated significant impairment in dendrite morphology compared to APP/WT mice. Herein, we confirmed the behavior deficits caused by Abca1 deletion in APP/PS1ΔE9 mice at an early stage of amyloid pathology utilizing two behavioral tests: MWM and novel object recognition.
Accumulation of soluble extracellular high molecular weight oligomeric Aβ species in brain is considered pathogenic for onset and progression of cognitive deficits associated with AD. Studies have found that soluble oligomers of Aβ rapidly and potently inhibit long-term potentiation [18]. Aβ oligomers also activate glial/neuronal stress kinases and increase the production and release of nitric oxide, superoxide, and other mediators [19]. It has been shown that Aβ oligomers negatively impact neuronal viability [20, 21] and synaptic plasticity [17]. We have previously shown that cognitive performance can be correlated to levels of soluble Aβ oligomers [10], and here we wanted to demonstrate that ABCA1 influences the effects of Aβ oligomers on cognitive performance. In this study, we utilized mice which expressed endogenous APP only and determined the effects of Aβ oligomers without the context of amyloid pathology. While we have been able to demonstrate impairments in cognitive performance in both WT and Abca1ko mice following infusion of Aβ oligomers into the hippocampi, we found a statistically significant difference between WT and Abca1ko mice infused with control scrambled Aβ peptide, suggesting that Abca1ko mice have basal deficits that prevent them from coping with additional stressors that do not affect performance of a normal healthy WT mouse. A possible explanation of the results could be, Abca1ko mice are, generally, unable to cope with additional stressors ultimately resulting in diminished cognitive performance. However, since ABCA1 regulates cholesterol and phospholipids efflux from cells to lipid-poor ApoE and controls the generation of HDL-like particles in brain, the overall impact could be a result of insufficient supply of cholesterol to neurons, emphasizing the regulatory role of ABCA1-ApoE axis on cholesterol homeostasis and promotion of synaptogenesis. While our study has not been designed to answer all these questions, the overall consequence of deficient, properly lipidated ApoE could explain reduced number of synapses [22], negative effects on reactive sprouting response following lesions in entorhinal cortex [23], and decreased MAP2-positive staining of neurons in the amygdala [24]. While evidence exists that adult neurons can synthesize cholesterol [25, 26], they cannot produce cholesterol efficiently, depend on an external source, and rely on exogenously supplied lipids especially during periods of increased demands such as repair following additional stressors [27, 28]. Yet, another plausible explanation could be, ApoE as the main carrier of cholesterol and phospholipids in the brain, in conditions of nonfunctional ABCA1 not only inefficiently delivers cholesterol and lipids to neurons to support growth and connectivity, but indirectly influences transcriptional activity of genes, and thus the expression of proteins critical for synaptogenesis and cognitive performance. The exact molecular mechanism by which ApoE influences neuronal structure and complexity remains unresolved, however.
In conclusion, our study demonstrates behavior deficits caused by Abca1 deletion in APP/PS1ΔE9 mouse model at an early stage of amyloid pathology. The basal deficits of Abca1ko, manifested by diminished cognitive performance, prevent them from coping with additional stressors, which is in part due to the impairment of neurite morphology in the hippocampus. The results of this study emphasize the important role of ABCA1 in brain cholesterol homeostasis and its ability to cope with external insults: environmental, diet-induced, physical trauma, or disease associated neuropathology.
ACKNOWLEDGMENTS
This work was supported by NIH Grants R01AG037481, R01AG037919, R01ES024233, and K01AG044490.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1056r1).
REFERENCES
[1] | Saunders AM , Roses AD , Pericak-Vance MA , Dole KC , Strittmatter WJ , Schmechel DE , Szymanski MH , McCown N , Manwaring MG , Schmader K , Breinter JCS , Goldgaber D , Benson MD , Goldfarb L , Brown WT ((1993) ) Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer’s disease and in other amyloid-forming diseases. Lancet 342: , 710–711. |
[2] | Kanekiyo T , Xu H , Bu G ((2014) ) ApoE and Aβ in Alzheimer’s disease: Accidental encounters or partners? Neuron 81: , 740–754. |
[3] | Webster SJ , Bachstetter AD , Nelson PT , Schmitt FA , Van Eldik LJ ((2014) ) Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet 5: , 88. |
[4] | Bilkei-Gorzo A ((2014) ) Genetic mouse models of brain ageing and Alzheimer’s disease. Pharmacol Ther 142: , 244–257. |
[5] | Puzzo D , Lee L , Palmeri A , Calabrese G , Arancio O ((2014) ) Behavioral assays with mouse models of Alzheimer’s disease: Practical considerations and guidelines. Biochem Pharmacol 88: , 450–467. |
[6] | Koldamova R , Fitz NF , Lefterov I ((2014) ) ATP-binding cassette transporter A1: From metabolism to neurodegeneration. Neurobiol Dis 72: (Pt A), 13–21. |
[7] | Hirsch-Reinshagen V , Maia LF , Burgess BL , Blain JF , Naus KE , McIsaac SA , Parkinson PF , Chan JY , Tansley GH , Hayden MR , Poirier J , Van Nostrand W , Wellington CL ((2005) ) The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J Biol Chem 280: , 43243–43256. |
[8] | Wahrle SE , Jiang H , Parsadanian M , Hartman RE , Bales KR , Paul SM , Holtzman DM ((2005) ) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem 280: , 43236–43242. |
[9] | Koldamova R , Staufenbiel M , Lefterov I ((2005) ) Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem 280: , 43224–43235. |
[10] | Lefterov I , Fitz NF , Cronican A , Lefterov P , Staufenbiel M , Koldamova R ((2009) ) Memory deficits in APP23/Abca1+/- mice correlate with the level of Aβ oligomers. ASN Neuro 1: , pii: e00006. |
[11] | Wahrle SE , Jiang H , Parsadanian M , Kim J , Li A , Knoten A , Jain S , Hirsch-Reinshagen V , Wellington CL , Bales KR , Paul SM , Holtzman DM ((2008) ) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 118: , 671–682. |
[12] | Fitz NF , Cronican AA , Saleem M , Fauq AH , Chapman R , Lefterov I , Koldamova R ((2012) ) Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci 32: , 13125–13136. |
[13] | Fitz NF , Tapias V , Cronican AA , Castranio EL , Saleem M , Carter AY , Lefterova M , Lefterov I , Koldamova R ((2015) ) Opposing effects of Apoe/Apoa1 double deletion on amyloid-β pathology and cognitive performance in APP mice. Brain 138: (Pt 12), 3699–3715. |
[14] | Lefterov I , Fitz NF , Cronican AA , Fogg A , Lefterov P , Kodali R , Wetzel R , Koldamova R ((2010) ) Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem 285: , 36945–36957. |
[15] | Fitz NF , Cronican A , Pham T , Fogg A , Fauq AH , Chapman R , Lefterov I , Koldamova R ((2010) ) Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci 30: , 6862–6872. |
[16] | Fitz NF , Cronican AA , Lefterov I , Koldamova R ((2013) ) Comment on “ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models”. Science 340: , 924–c. |
[17] | Tu S , Okamoto S , Lipton SA , Xu H ((2014) ) Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener 9: , 48. |
[18] | Wang Q , Walsh DM , Rowan MJ , Selkoe DJ , Anwyl R ((2004) ) Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24: , 3370–3378. |
[19] | Rowan MJ , Klyubin I , Wang Q , Anwyl R ((2004) ) Mechanisms of the inhibitory effects of amyloid beta-protein on synaptic plasticity. Exp Gerontol 39: , 1661–1667. |
[20] | Dahlgren KN , Manelli AM , Stine WB Jr , Baker LK , Krafft GA , LaDu MJ ((2002) ) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 277: , 32046–32053. |
[21] | Hoshi M , Sato M , Matsumoto S , Noguchi A , Yasutake K , Yoshida N , Sato K ((2003) ) Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc Natl Acad Sci U S A 100: , 6370–6375. |
[22] | Diaz-Cintra S , Yong A , Aguilar A , Bi X , Lynch G , Ribak CE ((2004) ) Ultrastructural analysis of hippocampal pyramidal neurons from apolipoprotein E-deficient mice treated with a cathepsin inhibitor. J Neurocytol 33: , 37–48. |
[23] | Champagne D , Rochford J , Poirier J ((2005) ) Effect of apolipoprotein E deficiency on reactive sprouting in the dentate gyrus of the hippocampus following entorhinal cortex lesion: Role of the astroglial response. Exp Neurol 194: , 31–42. |
[24] | Robertson J , Curley J , Kaye J , Quinn J , Pfankuch T , Raber J ((2005) ) apoE isoforms and measures of anxiety in probable AD patients and Apoe-/- mice. Neurobiol Aging 26: , 637–643. |
[25] | Dietschy JM ((2009) ) Central nervous system: Cholesterol turnover, brain development and neurodegeneration. Biol Chem 390: , 287–293. |
[26] | Huang Y , Mahley RW ((2014) ) Apolipoprotein E: Structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis 72: (Pt A), 3–12. |
[27] | Nieweg K , Schaller H , Pfrieger FW ((2009) ) Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem 109: , 125–134. |
[28] | Pfrieger FW , Barres BA ((1997) ) Synaptic efficacy enhanced by glial cells in vitro. Science 277: , 1684–1687. |
Figures and Tables
Fig.1
Abca1 deficiency negatively affects memory of APP transgenic mice. A-D) MWM results from 7-month-old APP mice expressing mouse Abca1 (APP/Abca1wt; n = 16) or Abca1ko (APP/Abca1ko; n = 14). Age- and gender-matched non-APP expressing Abca1wt (n = 14) or Abca1ko (n = 16) littermates were used as controls. Analysis is by two-way ANOVA to determine interaction and main effect of genotype or training, and t-test to determine genotype differences in performance within trial days. A, B) Training phase of MWM with each bar representing performance from all trials for a training day. A) Escape latency to the hidden platform: APP/Abca1wt versus WT, no interaction and no main effect of genotype. APP/Abca1ko versus Abca1ko, no interaction and significant main effect of genotype; F(1,140) = 22.26, p < 0.001. B) Path length to the hidden platform: APP/Abca1wt versus WT, no interaction and no main effect of genotype. APP/Abca1ko versus Abca1ko, no interaction and significant main effect of genotype; F(1,140) = 21.09, p < 0.001. For A and B, **p < 0.01, *p < 0.05 versus non-APP expressing controls by t-test. C and D represents probe trial of MWM performed 24 h following last training trial. Analysis is by one-way ANOVA followed by Tukey’s post-test (shown on the graph). C) Latency to reach the target quadrant of the hidden platform. p < 0.0001. D) Time spent in the target quadrant of the hidden platform. p < 0.05. E) Novel object recognition was performed on a different group of 7-month-old mice of the same genotypes. Shown is percent novel object recognition as indicated in the Methods. Analysis is by one-way ANOVA followed by Tukey’s post-test. n = 8–12 male and female mice per group.
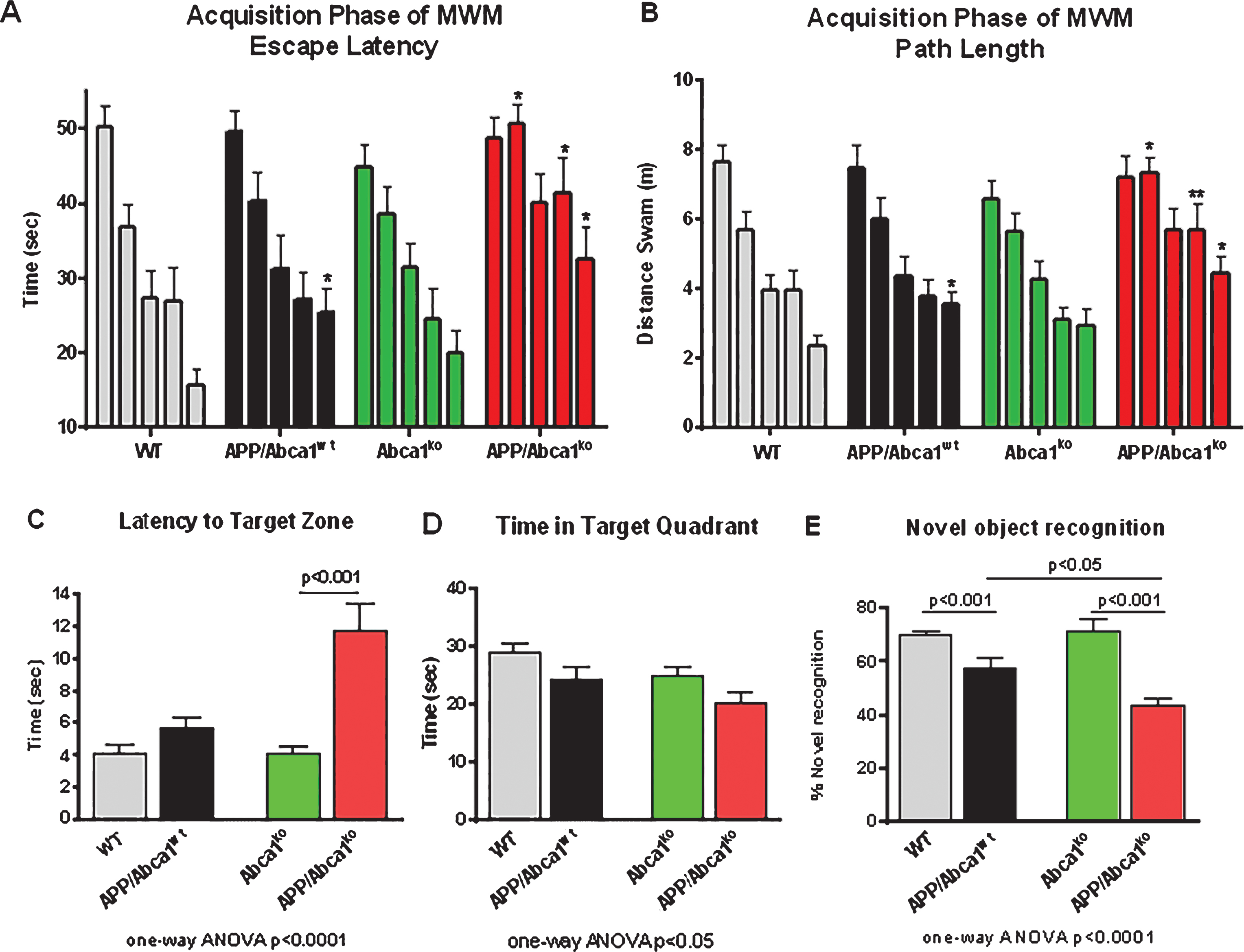
Fig.2
Characterization of Aβ oligomer. Disaggregated Aβ42 was incubated at room temperature for 24 h as indicated in the Methods. A) SDS PAGE of Aβ42 at time 0 and 24 h after the start of incubation. Western blotting was performed with anti-Aβ antibody, 6E10. On the left are shown molecular weight markers. Aβ oligomers migrate between 40 and 90 kDa marker. Western blot of Aβ fibrils is shown for comparison. B) Aβ oligomers were identified by dot blotting performed with A11 antibody. Notice with the dot blot an increase in Aβ oligomers with increased incubation time. C) Electron micrograph of Aβ42 at time 0 (a) and 24 h after the start of incubation (b).
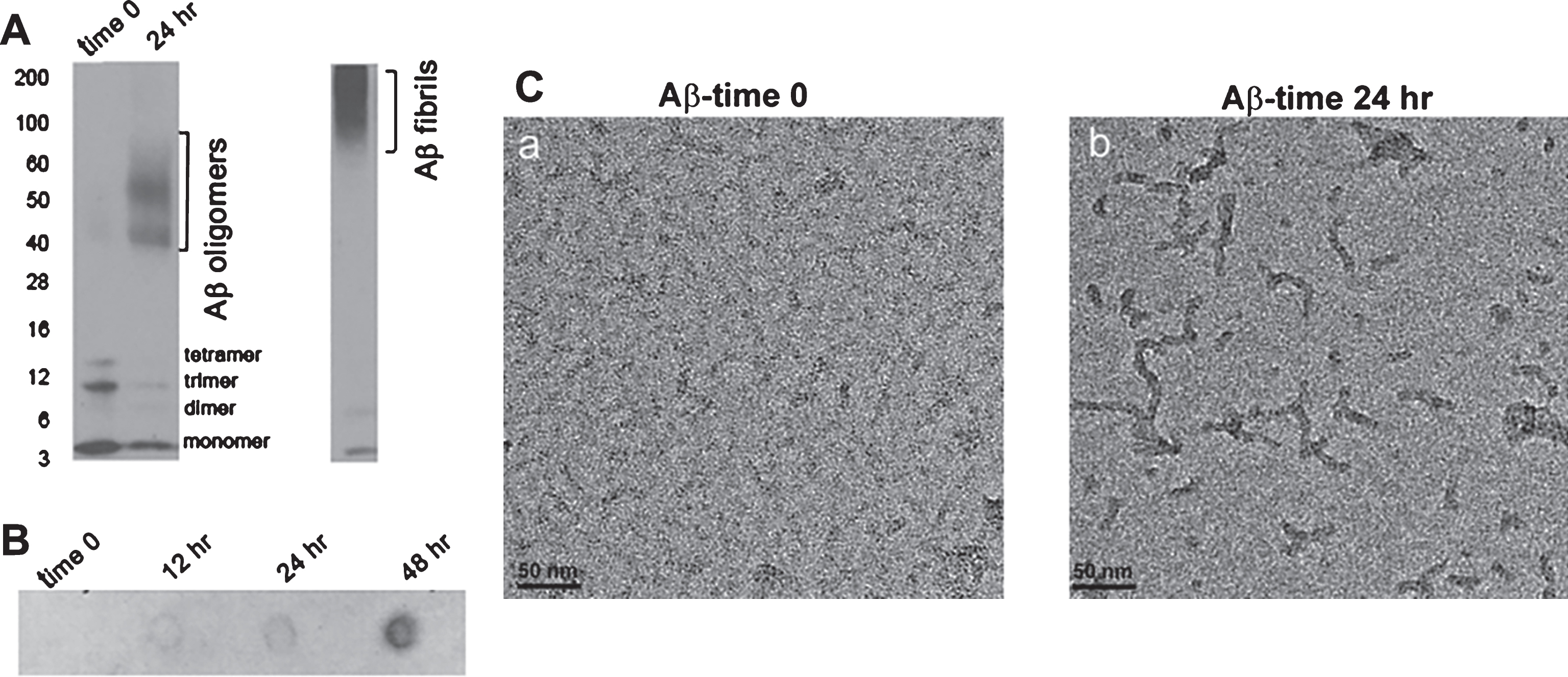
Fig.3
Infusion of Aβ oligomers into the hippocampus differentially affects cognitive performance in WT and Abca1ko mice. Aβ42 oligomers (characterized on Fig. 2) were infused into the hippocampus of 7-month-old WT or Abca1ko mice and cognitive performance was evaluated by Radial arm water maze (RWM) paradigm. Control mice were infused with scrambled Aβ. As additional control, we used WT naïve mice without surgery. A) Represents the location of cannulas implanted into the hippocampus. B) Cognitive performance of WT mice is significantly affected by Aβ42 oligomers but not by scrambled Aβ. Analysis by two-way repeated measures ANOVA shows no interaction between training and treatment; however there was a significant main effect of treatment (Aβ42 oligomers); F(2,279) = 17.84, p < 0.0001 and trial block (trial block); F(9,279) = 7.99, p < 0.0001. Post-test for multiple comparisons demonstrates the difference between mice infused with Aβ42 oligomers and scrambled Aβ (compare purple circles to grey squares): ***p < 0.001; **p < 0.01; *p < 0.05. n = 11 –12 mice per group. C) There is a significant difference in RWM performance between WT and Abca1ko mice infused with scrambled Aβ. In contrast there is no significant difference in RWM performance between Abca1ko mice infused with Aβ42 oligomers and scrambled Aβ. Analysis by two-way repeated measures ANOVA shows no interaction between training and treatment; but a significant main effect of treatment; F(2,90) = 5.56, p = 0.0238 and training F(9,90) = 6.29, p < 0.0001. Post-test for multiple comparisons demonstrates the difference between WT and Abca1ko mice infused with scrambled Aβ (compare black to grey squares): *p < 0.05. n = 4 –5 mice per group.
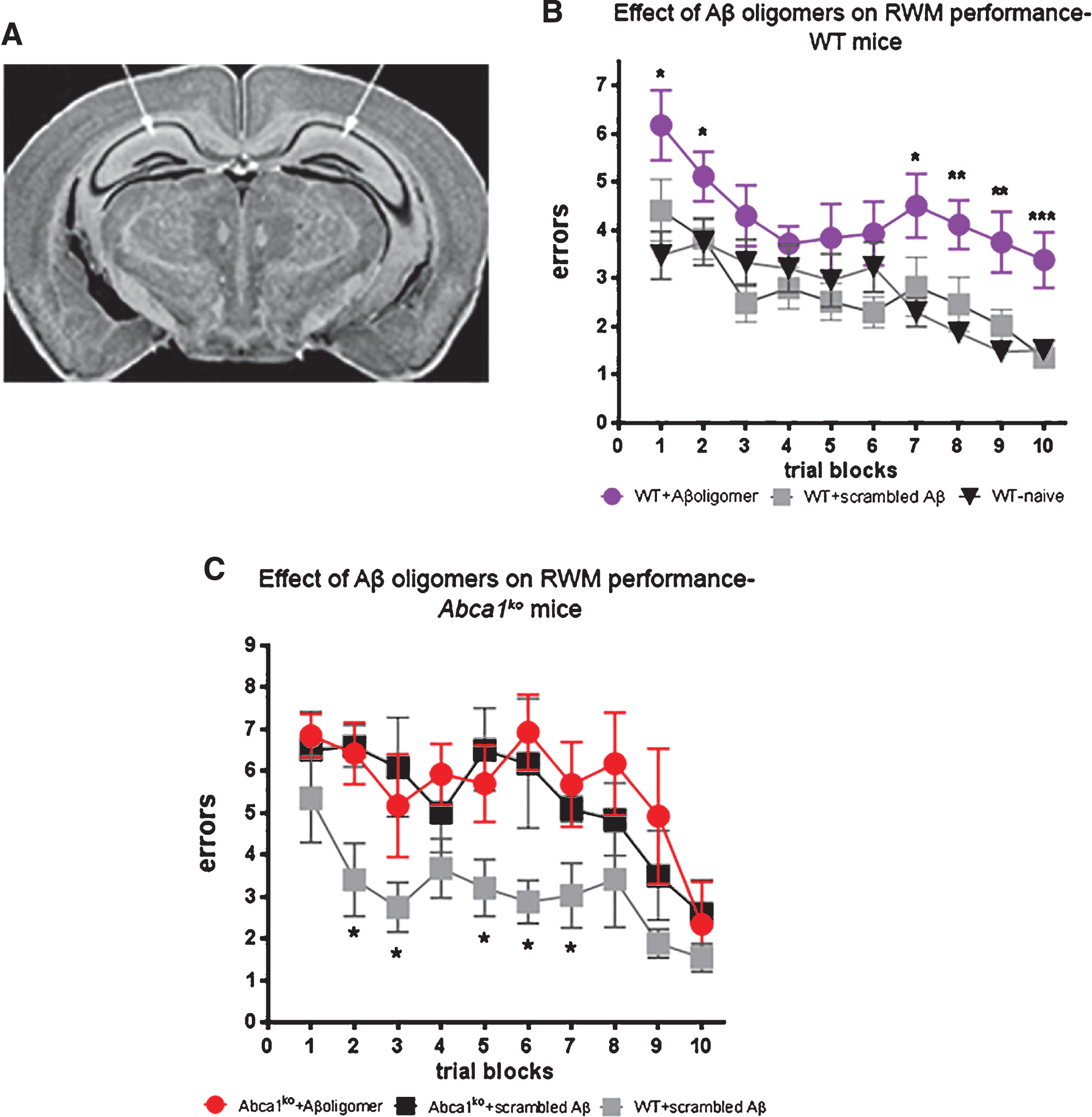
Fig.4
Lack of Abca1 significantly affects dendritic architecture in CA1 but not CA2 region of the hippocampus. MAP2 and DAPI staining were used for dendritic tree reconstruction in the hippocampal CA1 and CA2 regions of WT and Abca1ko mice. Data were analyzed from a total of four images (60X confocal imaging) from each of the four sections for each mouse (n = 5–7 mice per group; 16 images per mouse). The total dendritic length, branch points and segments were quantified using Imaris filament tracing macros and were normalized to the total DAPI positive nuclei of the CA1 and CA2 regions. Analysis by t-test. A) Representative images for CA1 region (a –MAP2 staining in red, b – DAPI staining in blue, c – Merged MAP2 and DAPI staining, d – Imaris neurite tracings in yellow). B) Quantification of neurite length, segments, and branches. C) Representative images for CA2 region (scale bar = 100 μM). D) Quantification of neurite length, segments, and branches in the CA2 region. Note that lack of Abca1 significantly affects dendritic architecture in CA1 but not CA2 region of the hippocampus.