
Localization and Processing of the Amyloid-β Protein Precursor in Mitochondria-Associated Membranes
Abstract
Alteration of mitochondria-associated membranes (MAMs) has been proposed to contribute to the pathogenesis of Alzheimer’s disease (AD). We studied herein the subcellular distribution, the processing, and the protein interactome of the amyloid-β protein precursor (AβPP) and its proteolytic products in MAMs. We reveal that AβPP and its catabolites are present in MAMs in cellular models overexpressing wild type AβPP or AβPP harboring the double Swedish or London familial AD mutations, and in brains of transgenic mice model of AD. Furthermore, we evidenced that both β- and γ-secretases are present and harbor AβPP processing activities in MAMs. Interestingly, cells overexpressing APPswe show increased ER-mitochondria contact sites. We also document increased neutral lipid accumulation linked to Aβ production and reversed by inhibiting β- or γ-secretases. Using a proteomic approach, we show that AβPP and its catabolites interact with key proteins of MAMs controlling mitochondria and ER functions. These data highlight the role of AβPP processing and proteomic interactome in MAMs deregulation taking place in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative pathology characterized by the progressive decline of cognitive functions. The accumulation of intracellular aggregates of tau protein in the neurofibrillary tangles and of extracellular aggregates of a set of polypeptides called amyloid-β peptides (Aβ) in the senile plaques [1] are the major histological hallmarks of AD. Aβ peptides are generated from a large transmembrane protein, the amyloid-β protein precursor (AβPP) through its sequential cleavage by the β-secretase generating the C-terminal fragment (CTF) of 99 amino acids (C99), and by the γ-secretase releasing Aβ peptide, and the APP intracellular domain (AICD) [2]. Cleavage of AβPP by α-secretase precludes the formation of Aβ peptides and leads to the generation of another CTF of 83 amino acids (C83) [3, 4].
Aβ peptides are generated extracellularly, but also within intracellular organelles such as the endoplasmic reticulum (ER) and mitochondria. AβPP is folded and modified in the ER and transported through the Golgi complex to the plasma membrane. It was proposed that Aβ is generated in the ER lumen as a result of deficits in axonal transport [5]. Aβ oligomers (considered to be the most toxic forms of Aβ) accumulate in the ER in hippocampal neurons and thereby, likely lead to cell death in transgenic mice expressing AβPP mutant [6]. Association of AβPP with mitochondria was also reported by several groups [7–9]. Aβ was shown to accumulate into mitochondria prior to extracellular plaques and in a time-dependent manner, leading to mitochondrial damage [10–12]. In addition, different components of the γ-secretase complex have been located in different subcellular compartments, including the ER and mitochondria [9, 13–16].
Physical association between the ER and mitochondria, also known as mitochondria-associated membranes (MAMs), play important roles in various cellular housekeeping functions such as phospholipids-, glucose-, cholesterol-, and fatty-acid- metabolism, as well as calcium signaling [17, 18]. The hypothesis stating that AD could be a disorder linked to MAMs alterations started to emerge only in the last 6 years [19–21]. In this study, we show that AβPP and its catabolites accumulate in MAMs and increase ER-mitochondria contact sites. We also evidence that both β-secretase and γ-secretase complex are present and may harbor enzymatic activities in MAMs. Of most importance, we document increased neutral lipid accumulation in cellular models of AD, which was reversed by inhibiting β- or γ-secretases. A proteomic approach revealed the protein interactome of AβPP and its catabolites with key proteins of MAMs controlling mitochondria and ER functions.
MATERIAL AND METHODS
Cells
Human SH-SY5Y neuroblastoma cells (CRL-2266, ATCC) stably expressing pcDNA3.1 (control), wild type human AβPP (APPwt), or human AβPP harboring the double Swedish mutations (APPswe: APPKM670/671NL) constructs were generated as already described [22]. Polyclonal stable lines were maintained in DMEM containing 10% FBS in the presence of 400 μg geneticin (Gibco).
Mock-transfected or APP695LDN-expressing CHO cells were obtained by stable transfection of pcDNA4 empty vector (Control), wild type hAPP695 cDNA or harboring the London mutation (APPLDN: APPV642I) and subcloned in pcDNA4 vector. Single clones were maintained in DMEM containing 10% FBS, sodium hypoxanthine-thymidine supplement, and 300 μM proline as already described [23].
Cells were treated overnight (20 h) with β- or γ-secretase inhibitors. γ-secretase inhibitor ELND006 was used at 5 μM final concentration and vehicle (methylcellulose/polysorbate 80; Sigma-Aldrich) was used as control [24, 25]. β-secretase inhibitor (Eli Lilly’s inhibitor LY288672 [26], synthesized by Elan Pharmaceutical) was used at 30 μM final concentration prepared in DMSO. Vehicle was used as control since no difference was observed as compared to cells treated with DMSO.
Mice
We used brains isolated from double-transgenic APP23xPS45 mice (B6-Tg hAPP751 K670 N/M671 L×PS1 G384A mThy1). These mice were obtained through Material Transfer Management from Novartis. To generate APP23xPS45 mice, we crossed APP23 transgenic line overexpressing hAβPP with the Swedish double mutation at positions 670/671 [hAPP751: K670 N/M671L], with PS45 transgenic line overexpressing human PS1 carrying G384A mutation [hPS1: G384A] [27]. Both lines are driven by a Thy-1 promoter and were developed in C57BL/6 genetic background. Control wild type (WT) mice were from the same genetic background.
Ethics statements
In vivo experiments were performed in accordance with relevant guidelines and regulations established by the European community council (Directive of November 24, 1986), and approved by Nice University Animal care and use Committee, and the National Council on animal care of the Ministry of Health (Project n°: NCE/2013-152). Mice were housed in SPF animal facility.
Cellular fractionation
Cell fractionation was performed using SH-SY5Y and CHO cell lines, and brains dissected from WT, APP23, PS45, and APP23xPS45 mice using an optimized protocol to isolate subcellular fractions from tissues and cells [28]. Cells (≈109) were harvested, washed by centrifugation at 500×g for 5 min with PBS. Cells or brains (n = 5 for each mice group) were resuspended in homogenization buffer (225 mM mannitol, 75 mM sucrose, 30 mM Tris-HCl pH 7.4, 0.1 mM EGTA) and gently disrupted by Dounce homogenization at 4°C. The homogenate was centrifuged twice at 600×g for 5 min to remove nuclei and unbroken cells, and then the supernatant was centrifuged at 10 300×g for 10 min to pellet crude mitochondria (Mc). The resultant supernatant was centrifuged at 20 000×g for 30 min at 4°C. The pellet consists of lysosomal and plasma membrane fractions. Further centrifugation of the obtained supernatant at 100 000×g for 90 min (70-Ti rotor; Beckman, France) at 4°C results in the isolation of ER (pellet) and cytosolic fraction (supernatant). The crude mitochondrial (Mc) fraction, resuspended in MAM isolation buffer (250 mM mannitol, 5 mM HEPES pH 7.4 and 0.5 mM EGTA), was subjected to Percoll gradient centrifugation (Percoll medium: 225 mM mannitol, 25 mM HEPES pH 7.4, 1 mM EGTA, and 30% vol/vol Percoll) in a 10-ml polycarbonate ultracentrifuge tube. After centrifugation at 95,000 ×g for 30 min (SW40 rotor; Beckman, France), a dense band containing purified mitochondria (Mp) was recovered approximately 3/4 down of the tube, resuspended in MAM isolation buffer and washed by centrifugation at 6300×g for 10 min to remove the Percoll and finally resuspended in small volume of MAM isolation buffer. The MAMs, containing the structural contacts between mitochondria and ER, collected from the Percoll gradient as a diffuse white band located above the mitochondria, were diluted in MAM isolation buffer and centrifuged at 6300×g for 10 min; then the supernatant was further centrifuged at 100 000×g for 90 min (70-Ti rotor, Beckman) to pellet the MAMs fraction.
SDS-PAGE analysis
To detect Aβ peptide, protein extracts (40 μg) were incubated with 70% formic acid (Sigma) and Speed Vac evaporated for 40 min. The pellets were dissolved in 1 M Tris pH 10.8, 25 mM Betaine and diluted in 2x Tris-Tricine loading buffer (125 mM Tris-HCl pH 8.45, 2% SDS, 20% Glycerol, 0.001% Bromophenol blue, and 5% β-mercaptoethanol). Proteins were resolved by 16.5% Tris-Tricine SDS-PAGE, transferred onto PVDF membranes, and incubated overnight with specific antibodies as specified in legends. All the other proteins were resolved by Tris-Glycine SDS-PAGE following standard procedures.
Antibodies
Aβ, C99, and full length AβPP were detected using 6E10 antibody (Covance) recognizing 1-16 residues of Aβ. AβPP C-terminal fragments (AβPP CTF: C99 and C83), were detected using AβPP C-terminal antibody (Sigma Aldrich) recognizing 676–695 residues of AβPP. Other antibodies directed toward the following proteins were as follow: Cytochrome C oxidase subunit II (Cox), and RyR (Thermo scientific); VDAC-1 (Calbiochem); HSP60, Calreticulin, and GRP75 (Santa Cruz); Chapronin 10 (Stressgen, Biotech); Nicastrin, Tom20, α-Tubulin, and β-Actin (Sigma Aldrich), SERCA2b (clone IID8), and Aph1 (Thermo Scientific Pierce Products); BACE-1 and Cytp450 (Abcam); PS1-Nter is a generous gift from Gopal Thinakaran.
Detection of Aβ by ELISA
The concentrations of Aβ40 and Aβ42 were measured in subcellular fractions by using the respective ELISA kits (Invitrogen) following the manufacturer’s instructions. Briefly, 40 μg of each fraction was treated with 5 M guanidine, 50 mM HCl solution for 3-4 h at room temperature and stored at –20°C overnight. Supernatant obtained after centrifugation at 16,000 rpm for 20 min at 4°C, were diluted at >1/10 in D-PBS-BSA 5% and used for ELISA.
In vitro α-secretase assay
α-secretase activity was monitored as already described [29]. Briefly, 20 μg of each subcellular fraction were incubated in 90 μl final volume of Tris buffer (10 mM, pH 7.5) containing α-secretase substrate (JMV2770, 100 μM) in the absence or presence of the metal chelator o-phenanthroline (100 μM) reported to block metalloproteases. α-secretase activity corresponds to the α-secretase inhibitor-sensitive fluorescence recorded at 320 and 420 nm as excitation and emission wavelengths, respectively.
In vitro β-secretase assay
β-secretase activity was monitored as already described [22]. Briefly, 20 μg of each subcellular fraction were incubated in a final volume of acetate buffer (25 mM, pH4.5, 100 μl) containing BACE-1 substrate [(7-methoxycoumarin-4-yl)-acetyl-SEVNLDAEFRK (2,4-dinitrophenyl)-RRNH2; 10 μm; R&D Systems] in the absence or presence of β-secretase inhibitor I (50 μm; PromoCell). BACE-1 activity corresponds to the β-secretase inhibitor I-sensitive fluorescence recorded at 320 and 420 nm as excitation and emission wavelengths, respectively.
In vitro γ-secretase assay
In vitro γ-secretase assay was assessed as already described [30]. Twenty μg of each subcellular fraction were resuspended in solubilization buffer (150 mM sodium citrate pH6.4 containing 3-[(3-cholamydopropyl) dimethylammonio]-2-hydroxy-1-propanesulfonate 1% (v/v)) supplemented with protease inhibitor mixture. All steps were performed at 4°C. Solubilized membranes were diluted once with sodium citrate buffer (150 mM pH 6.4), and with reaction buffer (150 mM sodium citrate pH 6.4, 20 mM dithiothreitol, 0.2 mg/ml BSA, 1 mg/ml egg phosphatidyl choline and 50 μg/mL recombinant C100-FLAG). The resulting reaction mix were then either incubated over constant agitation for 16 h at 37°C or stored at 4°C (negative controls). Samples were then supplemented with 2x Tris-Tricine loading buffer, boiled for 5 min and subjected to 16.5% Tris-Tricine SDS-PAGE.
Immunofluorescence analysis
Cells grown on 25 mm round coverslips were fixed in PFA 4% solution for 10 min at room temperature. Cells were permeabilized with triton 0.5%, and non-specific binding sites were blocked for 1 h with BSA (5%). Cells were then incubated at 4°C overnight with primary antibodies diluted in BSA (5%). After 3 washes, coverslips were incubated with secondary antibodies [fluorescent Alexa Fluor antibodies, Alexa 488- and Alexa 594- or Alexa 633-conjugated (Invitrogen; 1:1000)] at room temperature during 1 h. Immunofluorescence images were acquired on Leica SP5 confocal microscope using excitation filters 488, 594, and 633 nm. Images were analyzed using ImageJ.
High resolution imaging
To visualize mitochondria and ER organelle colocalization, we used GFP construct specifically targeted to the ER. Mitochondria were labeled by using 10 nM Mitotraker® Deep Red dye. Z-series images acquired on Leica SP5 microscope were deconvolved. ER and mitochondria colocalization was calculated as the number of voxels (volume pixels) occupied by both signals (i.e., erGFP and Mitotraker Deep Red) over all voxels occupied by the mitochondria (i.e. Mitotraker® Deep Red signal) in thresholded images [31]. The spectral proprieties of these two fluorochromes allow specific identification of the two compartments and quantification of contact sites.
Electron microscopy
Cells were fixed in situ with 2.5% glutaraldehyde in 0.1 M phosphate buffer at room temperature (RT) for 20 min. Samples were rinsed and then post-fixed with 1% osmium tetroxide and 1% potassium ferrocyanide in 0.1 M cacodylate buffer for 1 h at RT to enhance the staining of cytoplasmic membranes. Cells were rinsed with distilled water, embedded in epoxy resin, sectioned, and examined with a Philips CM12 transmission electron microscope equipped with an Olympus SIS CCD camera. Images were acquired randomly by a blinded investigator. We counted the total number of mitochondria (n > 150) in contact or not with ER in more than 10 different micrographs obtained from two different experiments. Data are presented as a percentage of mitochondria in contact with ER.
Neutral lipid staining
Cells grown on 25 mm round coverslips were fixed in PFA 4% solution for 20 min at room temperature. Fixed cells were gently rinsed with PBS 3 times to remove residual formaldehyde. Cells were stained with 1X LipidTOX™ prepared in PBS for 30 min at room temperature. Coverslips were then processed for image acquisition on Leica SP5 confocal microscope using excitation filters 488. Images were thresholded, converted to mask and analyzed using Particle analysis ImageJ plugin.
Oligomeric Aβ preparation
Human synthetic Aβ1–42 (Bachem AG, Switzerland) was suspended in hexafluoroisopropanol, HFIP; Sigma–Aldrich) to 1 mM. Peptide samples were vortexed to obtain a homogenous solution, aliquoted into microfuge tubes, and lyophilized overnight under the hood. The Aβ1–42 peptide films were stored desiccated at –20°C until further processed. To form Aβ oligomers, peptide films were resuspended to 5 mM in DMSO, diluted in ice cold PBS. Aggregation was allowed to proceed for 24 h at 4 °C before the peptide solution was centrifuged at 14000×g for 10 min at 4°C. Supernatant containing oligomeric Aβ was then used at ≈5 μM.
Immunoprecipitation and in-gel digestion
After preclearing with protein A-agarose (Sigma-Aldrich) (1 h, 4°C), MAM fractions (40 μg) isolated from control CHO cells and overexpressing APPLDN were immunoprecipitated using 6E10 antibody (5 μg). Immunoprecipitates were recovered by overnight incubation at 4°C, and then incubated with protein A-agarose for 3 h at 4°C. Beads were washed three times with CHRIS buffer (50 mM Tris pH 8, 10% glycerol, 200 mM NaCl, 0.5% Nonidet p-40, and 0.1 mM EDTA) supplemented with protease inhibitors (Complete, Roche diagnostics) and once with PBS. Beads were then resupended in Laemli 2x buffer, heated at 95°C, and then run on Tris-Glycine PAGE. Gels were stained using Imperial Protein Stain (Thermo scientific), a ready-to-use colorimetric stain formulated with Coomassie dye.
Gel pieces containing proteins were excised and distained by adding 100 μL of H2O/ACN solvent (1/1). After 10 min incubation with vortexing the liquid was discarded. This procedure was repeated 2 times. Gel piece were then rinsed (15 min) with acetonitrile and dried under vacuum. Gel pieces were reswelled in 50 μL of 20 mM dithiothreitol (DTT) in NH4HCO3 100 mM, incubated for 30 min at 56°C, and next cooled down to RT. The DTT solution was replaced with 50 μL of 55 mM of iodoacetamide in 100 mM of NH4HCO3. After 15 min incubation in the dark at room temperature, the solution was discarded and gel pieces were washed by adding successively i) 100 μL of H2O/ACN (1/1), repeated 2 times and ii) 100 μL of acetonitrile. Next gel pieces were reswelled in 50 μL of 50 mM NH4HCO3 buffer containing 10 ng/μL of trypsin (modified porcine trypsin sequence grade, Promega) incubated for 1 h at 4°C. Then the solution was removed and replaced by 50 μL of 50 mM NH4HCO3 buffer (without trypsin), and incubated 18 h at 37°C. After trypsin digestion the solution was transferred into an Eppendorf tube and tryptic peptides were extracted with i) 50 μL of 1% AF (acid formic) in water (10 min at RT) and ii) 50 μL acetonitrile (10 min at RT). Peptides extracts were pooled, concentrated under vacuum and solubilized in 15 μL of 0.1% TFA (trifluoroacetic acid) in water.
NanoHPLC-MALDI-TOF/TOF analysis
Peptide separation was carried out using a nanoHPLC offline (ultimate 3000, Thermo Fisher Scientific) coupled with a MALDI-TOF/TOF mass spectrometer (4800 plus, Applied Biosystems). Peptides solution was concentrated on a μ-Precolumn Cartridge Acclaim PepMap 100 C18 (i.d. 5 mm, 5 μm, 100Å, Thermo Fisher Scientific) at a flow rate of 20 μL/min and using solvent containing H2O/ACN/TFA 98% /2% /0.04%. Next peptides separation was performed on a 75 μm i.d.×150 mm (3 μm, 100 Å) Acclaim PepMap 100 C18 column (Thermo Fisher Scientific) at a flow rate of 200 nL/min and with detection at 214 nm. Solvent systems were: (A) 100% water, 0.05% TFA, (B) 100% acetonitrile, 0.04% TFA. The following gradient was used t = 0 min 100% A; t = 3 min 100% A; t = 63 min, 80% B; t = 64 min, 100% B; t = 69 min 100% B (temperature was regulatedat 30°C).
For offline nanoHPLC-MALDI-TOF/TOF-MS and MS/MS analyses, fractions were collected on an Opti-TOF LC/MALDI target (123×81 mm, Applied Biosystems) and fractionation was done using the Probot fractionation robot (DIONEX, LC Packings). Matrix solution (α-cyano-4-hydroxycinnamic acid, 2.5 mg/mL in 50% water, 50% acetonitrile, 0.1% TFA solution) and nanoHPLC fractions were mixed (in rate 4:1, matrix:fractions) and collected every 20 s (208 fractions were collected per run).
MS spectra were recorded automatically in a mass range of 700–4000 Da resulting from 200 laser shots of constant intensity. Data were collected using 4000 series Explorer (Applied Biosystems) allowing for an automatic selection of peptide masses for subsequent MS/MS experiments. Each MS/MS spectra acquired using 1000 laser shots were further processed using 4000 series Explorer. Finally all raw data were transferred into ProteinPilot software (Applied Biosystems, MDS Analytical Technologies) and proteins identification was processed using ParagonTM Algorithm. Our analyses took in consideration only peptides with confidence index 95%. This experiment was done twice.
Statistical analyses
Results are reported from at least three different experiments or as indicated. Statistical analyses were done using Student’s t-test, or ANOVA one-way and Tukey post-test or Newman-Keuls Multicomparison post-test. p value <0.05 was considered significant.
RESULTS
AβPP and its proteolytic products are present in MAMs fraction of in vitro and in vivo study models of AD
We first aimed at establishing by subcellular fractionation, the distribution of AβPP and its proteolytic catabolites in human SH-SY5Y neuroblastoma cells stably expressing pcDNA3.1 (control) or human AβPP harboring the double Swedish mutations (APPswe: APPKM670/671NL) constructs. We already reported that SH-SY5Y cells expressing APPswe harbor increased AβPP processing yielding the production of AβPP CTFs fragments (C99 and C83) and of Aβ peptides [22].
We investigated the presence of AβPP and its proteolytic catabolites in MAMs. Although several techniques are available to isolate mitochondria, only few are specifically tuned to the isolation of MAMs, containing unique regions of ER membranes attached to the outer mitochondrial membrane and mitochondria without contamination from other organelles (i.e., pure mitochondria). We used an optimized protocol to isolate these fractions from tissues and cells [28]. Subcellular fractionation procedure was validated by confirming the expression of different proteins in cellular compartments where they were known to be enriched, namely in the ER (SERCA2b, calreticulin (CRT)), in mitochondria (cytochrome oxidase subunit II (Cox), and voltage dependent anion channel 1 (VDAC1)), and in MAMs (Glucose regulated protein 75 (GRP75)) (Fig. 1A, B). Fraction enrichment was further demonstrated by the loss of actin signal in pure mitochondria (Mp), ER, and MAMS fractions (Fig. 1A, B). We observed AβPP and Aβ expressions in the whole homogenate (homog), in crude mitochondria (Mc), in pure mitochondria (Mp), and in ER fractions (Fig. 1B). Interestingly, we noticed the presence of full-length AβPP and of Aβ in MAMs fraction of APPswe expressing cells (Fig. 1B). Full-length AβPP was also observed in MAMs fraction of control cells, thus indicating that both endogenous and overexpressed AβPP are present in MAMs. However, only a slight Aβ signal was noticed in control cells. This may be related to low expression level of AβPP in control cells.
We then investigated AβPP subcellular localization in a second cellular model expressing the London APP mutation (APPLDN: APPV642I). In fact, while the Swedish mutation increases the rate of β-cleavage and is proposed to alter the trafficking of AβPP [32], the “London” mutation increases the relative amount of the more toxic Aβ42 produced by γ-cleavage, but is not proposed to alter trafficking [33]; Furthermore, It was already reported that CHO cells expressing APPLDN yield high levels of intracellular and secreted Aβ oligomers [23, 34], and several lines of evidence concord to demonstrate that at least part of the AD-related neurodegenerative process could be due to Aβ oligomers [34]. Thus, we carried out similar analyses on control and APPLDN cells (Supplementary Figure 1A). We revealed the presence of full length AβPP, and its C-terminal fragments (C99 and C83), and of Aβ in the whole homogenate, and in Mc, ER, and MAMs fractions of CHO cells expressing APPLDN (Supplementary Figure 1A). Of most interest, we also revealed supplementary bands (Supplementary Figure 1A, see (*) symbol) likely corresponding to dimeric and low molecular oligomeric forms of Aβ in the Mc and MAMs fractions of CHO cells expressing APPLDN (Supplementary Figure 1A). In this model, we also observed the presence of full-length AβPP in MAMs fraction of control cells. These first set of data show that AβPP, CTFs, and Aβ are present in MAMs in two in vitro models of AD.
We then examined whether AβPP and Aβ could also be present in MAMs, in vivo. We measured expressions of AβPP and its catabolites C99, C83, and Aβ in subcellular fractions prepared from brains isolated from WT and transgenic mice expressing human AβPP harboring the double Swedish mutations (APPswe: APP23), human presenilin 1 (PS1) (component of the γ-secretase complex) mutation (PS45: G384A), or both (APP23xPS45). Cytochrome p450 (Cytp450, an ER resident protein (Fig. 1A) was indeed enriched in the ER fraction (Fig. 1C). Chapronin 10 (Cpn10), a mitochondrial protein (Fig. 1A) was detected in the homogenate and Mc, and largely enriched in Mp fraction (Fig. 1C). Both APP23 and APP23xPS45 showed AβPP, its C-terminal fragments (C99 and C83), and Aβ expression in MAMs fraction (Fig. 1C). Interestingly, we also noticed the presence of C83 but to a lesser extent than C99 in WT and PS45 mice (Fig. 1C). The increased C99 fragment in APP23 and APP23xPS45 is likely linked to the overexpression of the Swedish mutation known to increase the rate of β-cleavage [32]. Finally specific ELISA approaches allowed unraveling both Aβ1 - 40 and Aβ1 - 42 peptides in MAMs fraction of APP23 (Fig. 1D). We also unraveled a slightly elevated Aβ40/Aβ42 ratio in Mc, ER, and MAMs as compared to homogenate and Mp fractions. Overall, this set of data indicates that AβPP and its catabolites are present in MAMs in vivo (Fig. 1C, D) in agreement with the in vitro data (Fig. 1B and Supplementary Figure 1).
Immunolocalization of AβPP and its catabolites in the ER, mitochondria, and MAMs
We then analyzed subcellular localization of AβPP and its catabolites by immunofluorescence in SH-SY5Y cells expressing APPswe. We investigated the extent of AβPP, and its catabolites co-localization with the ER and mitochondria by using AβPP C-ter antibody (recognizing full length AβPP, C-terminal fragments (C83 and C99), and AβPP intracellular domain (AICD)), or 6E10 antibody (recognizing full length AβPP, C99, and Aβ peptides) concomitantly with specific antibodies recognizing proteins localized in the ER (the ryanodine receptor (RyR) and Cytp450), or in the mitochondria (Tom20 and Cox). Representative images show the co-localization (yellow signal) of AβPP and its catabolites with both the ER (Fig. 2A, C), and mitochondria (Fig. 2B, D) markers. Similar results were obtained in CHO cells expressing APPLDN using AβPP C-ter antibody (Supplementary Figure 1B, C).
We then examined AβPP and its catabolites colocalization within ER-mitochondria contact sites. SH-SY5Y cells expressing APPswe were immunolabeled concomitantly with AβPP C-ter, Tom20, and the ER-resident protein calreticulin (CRT) antibodies (Fig. 2E). Merge, high magnificence images (Fig. 2E, merge and inset), and super imposable line scan traces (Fig. 2F) (corresponding to the red, green, and blue fluorescence signals along the red line in the inset) demonstrate co-localization of AβPP and CTFs with both ER and mitochondria. These data are in agreement with our data obtained by subcellular fractionation and further support the presence of AβPP and its metabolites in MAMs in cellular models overexpressing both the Swedish and the London mutations.
Full length AβPP as revealed by using specific antibody recognizing AβPP N-terminal epitope (APP N-ter) shows also slight co-localization with the ER (Supplementary Figure 3A) and mitochondria (Supplementary Figure 3B).
Using immunofluorescence and imaging, we also examined the subcellular distribution of wild type AβPP in both SH-SY5Y and CHO cells. We show the immunolocalization of WT AβPP and its metabolites in the ER (colocalization with SERCA2b) (Supplementary Figures 3A and 4A), mitochondria (colocalization with mitochondrial HSP60) (Supplementary Figures 3B and 4B), and MAMs (Supplementary Figures 3C, D and 4C, D) in both SH-SY5Y(Supplementary Figure 3) and CHO (Supplementary Figure 4) cells overexpressing wild type AβPP. The results reveal that like the Swedish and London mutated AβPP, wild type AβPP harbor partial colocalization with the ER, mitochondria, and MAMs. These data strengthen our conclusion regarding the subcellular localization and proteolytic processing of AβPP in MAMs.
β- and γ-secretases are present and show in vitro activities in MAMs fraction
We next revealed by subcellular fractionation of APP23 transgenic mice brains the presence of β-secretase (BACE-1) and of components of the γ-secretase complex (PS1, Aph1, and nicastrin) in MAMs (Fig. 3A). In vitro γ-secretase activity assay allowed us to reveal the hydrolysis of recombinant C100 fragment at 37°C and not at 4°C (used as negative control) that yield Aβ production in the homogenate, Mc, Mp, ER, and MAMs fractions isolated from brains of APP23 mice (Fig. 3B). We did not notice a difference in γ-secretase activity between WT and APP23 mice (Supplementary Figure 5A). Similarly, γ-secretase activity was also observed in MAMs fraction isolated from CHO cells expressing APPLDN (Supplementary Figure 5B). In agreement with BACE-1 expression in APP23 MAMs (Fig. 3A), we detect β-secretase activity in MAMs fraction in both WT and APP23 mice fractions (Fig. 3C), and in control CHO cells and expressing APPLDN (Supplementary Figure 5C). Related to the presence of C83 (the α-secretase-derived CTF) in vivo (Fig. 1C), we also measured α-secretase activity in subcellular fractions isolated form APP23 transgenic mice. We indeed could observe in vitro α-secretase activity in the homogenate, Mc, ER, and MAMs. However, the in vitro α-secretase activity was lower in MAMs as compared to the other fractions (Supplementary Figure 5D). All over, and according to already published data [9, 13, 16, 20], we revealed the presence and activity of γ-secretase in MAMs fraction. We also provide for the first time evidences showing that BACE-1 is present in MAMs fraction and that BACE-1 and α-secretase may harbor enzymatic activities in this domain.
Increased ER and mitochondria contacts in SH-SY5Y neuroblastoma APPswe cell model
Increased ER-mitochondria colocalization has been reported in human-derived fibroblasts isolated from familial and sporadic AD patients and in mouse embryonic fibroblasts isolated from either PS1 or PS1 and PS2 knockout mice [21]. This was also observed in cells overexpressing familial AD PS2 mutant [35]. We examined herein ER-mitochondria contact sites in live cells overexpressing APPswe (Fig. 4A). For live cells imaging, the ER was visualized by transfecting GFP construct targeted to the ER (ER GFP). Mitochondria were labeled by using Mitotracker Deep red dye as already described [31]. We noticed that APPswe expressing cells exhibit a significant increased co-localization of the ER and mitochondria as compared to control cells (evidenced by an increase of mitochondria volume in contact with the ER versus total mitochondria volume) (Fig. 4A). Accordingly, we also revealed increased mitochondria in contact with the ER by electron microscopy in cells overexpressing APPswe as compared to controls (Fig. 4B).
These data demonstrate that AβPP mutation/overexpression and most likely its proteolytic products lead to increased ER-mitochondria contact sites.
The increased ER-mitochondria contacts in SH-SY5Y neuroblastoma APPswe cell model may depend on mitochondrial or ER shape and volume. Supplementary experiments are necessary to fully study this issue.
Overexpression of AβPP familial mutations increases neutral lipids accumulation
MAM have the proprieties of an intracellular lipid raft [21]. This micro-domain is also the site of phospholipid synthesis and transport between the ER and mitochondria [36]. We explored the potential lipid dysfunction in relation with AβPP and its catabolites presence in the MAM and increased ER and mitochondria contacts. In particular, lipid droplets are found in close proximity with the ER and mitochondria [37, 38].
We used LipidTox GreenTM dye to stain and quantify neutral lipid accumulation (major components of lipid droplets). Whereas the lipidTox stain in control CHO cells reveal few positive droplets, the CHO cells expressing APPLDN contained numerous lipidTox positive droplets (∼2-fold over control cells) (Fig. 5A, B). Similar results were also observed in SH-SY5Y neuroblastoma cells expressing APPswe as compared to its respective control cells (Supplementary Figure 6A, B). Importantly, the increase in lipid droplets in CHO cells expressing APPLDN was reversed by inhibition of β- or γ-secretases. As expected, both inhibitors block Aβ production (Supplementary Figure 6C). However, while the inhibitor of β-secretase strongly reduces C99 fragment production, inhibitor of γ-secretase enhances the accumulation of this fragment (Supplementary Figure 6C). Based on these results, we may postulate that lipid droplets accumulation in our study models is likely linked to the production of Aβ peptides instead of CTFs.
We further demonstrate the specificity of our data regarding the reduction of neutral lipid accumulation obtained with β- and γ-secretase inhibitors observed in CHO cells expressing the London AβPP mutant. First, we analyzed neutral lipid accumulation in naïve SH-SY5Y cells upon the application of exogenous oligomeric Aβ (Fig. 5C, 5D). We could indeed show that the treatment with exogenous oligomeric Aβ leads to increased neutral lipid accumulation (Fig. 5C, D). However, since our Aβ preparation contains both monomeric and oligomeric forms of Aβ (Fig. 5E), we cannot conclude whether neutral lipids accumulation is linked to either monomeric or oligomeric Aβ (Fig. 5E). Second, we analyzed neural lipid accumulation in mouse embryonic fibroblasts (MEF) isolated from wild type (expressing endogenous AβPP) (WT MEF), or invalidated for AβPP and its family member APLP2 (referred to as APP-TKO MEF) [39]. We established that knock out of AβPP did not affect neutral lipid accumulation (Supplementary Figure 6D). Importantly, we did not notice an effect of both β- and γ-secretase inhibitors neither in WT MEF, nor in APP-TKO MEF cells (Supplementary Figure 6D).
All over, these data demonstrates that neutral lipid accumulation is likely linked to AβPP overexpression and processing and to Aβ production.
Protein interactome of AβPP and its catabolites in MAMs
Since we showed that AβPP is present and is processed in MAMs likely contributing to increased ER-mitochondria contact sites, we hypothesized that AβPP and its catabolites may thus interact with proteins localized in this micro-domain. We used immunoprecipitation combined to proteomic approach to reveal AβPP and its catabolites protein interactome in MAMs. By using 6E10 antibody, we expected to immunoprecipitate MAMs proteins interacting directly or indirectly with AβPP holoprotein, C99, and/or Aβ peptides. To exclude false positives due to adsorption of MAMs proteins with IgG beads, we incubated MAMs fractions isolated from CHO cells expressing pcDNA4 (Control) or APPLDN with IgG beads alone (Supplementary Figure 7). We validated our experimental procedure by revealing AβPP peptides (accession number: P12023) in MAMs fraction isolated from APPLDN CHO cells but not in CHO control cells, thus providing a positive control of our experimental protocol (Supplementary Figure 8).
We specifically immunoprecipitated 135 proteins from CHO APPLDN MAM fraction (obtained in 6E10 immunoprecipitate conditions, but not in samples incubated with IgG beads alone) (Supplementary Figure 7). In the analyses, we excluded IgG and ribosomal proteins. Among immunoprecipitated proteins from CHO APPLDN MAMs fraction, 39 were also identified in CHO control MAMs fraction. The common proteins identified in both samples may reflect MAMs proteins interacting with full length AβPP rather than those interacting with C99 and Aβ peptides. Indeed, the amount of AβPP catabolites was clearly lower in control as compared to APPLDN MAMs fraction (Supplementary Figure 1).
We analyzed the subcellular localization and biological functions of the 96 unique proteins obtained in CHO APPLDN MAMs fraction by using gene onthology (GO) annotation and UniProt (Universal Protein Resource) database. In this study, we exclude from the analysis proteins showing nuclear, extracellular or microtubule localization. We then determined that 38 proteins are resident in mitochondria (44%); 23 proteins have ER localization (27%), 12 proteins are mainly cytosolic (14%), 7 proteins are found in lysosomes, Golgi apparatus, and endosomes (8%), and 6 proteins are associated with the plasma membrane (7%) (Supplementary Figure 7). It is important to note that mitochondria and ER comprise 71% of the total immunoprecipitated proteins. However, proteins associated with other organelles should not be discounted as contaminants since the plasma membrane, Golgi apparatus, and lysosomes all have tight association with the ER or mitochondria and may execute fundamental cellular functions in these contact microdomains [40, 41].
We then analyzed the types of function of the proteins identified. Of most interest, the identified molecular functions (Supplementary Figure 7) are mostly attributable to MAMs functions [41, 42]. We focused on the four most significant biological processes: 1) mitochondrial function and transport (n = 23) (Supplementary Figure 7 and Table 1); 2) protein synthesis, maturation, folding, processing, and degradation (n = 18) (Supplementary Figure 7 and Table 2); 3) vesicle formation and trafficking (n = 12) (Supplementary Figure 7 and Table 3); and 4) fatty-acid metabolism (n = 10) (Supplementary Figure 7 and Table 4). The remaining pathways correspond to proteins implicated in mitochondria and ER structure (n = 3), apoptosis (n = 2), cellular signaling (n = 7), and other metabolic pathways (n = 4) (Supplementary Figure 7 andTable 1).
Protein interactome related to mitochondrial function
The identified mitochondrial proteins (n = 23) are implicated in mitochondrial respiratory chain complexes (n = 9), mitochondrial transport (n = 6), and mitochondrial metabolism (n = 8) (Supplementary Figure 7 and Table 1). Interestingly, 6 out of 10 isolated mitochondrial respiratory chain proteins are components or chaperone proteins of the mitochondrial complex I. We also identified Cytochrome b-c1 complex subunit1 (component of the mitochondrial respiratory chain complex III), and two components of the mitochondrial ATP synthase complex (Supplementary Figure 7 and Table 1). The protein interactome of AβPP and its catabolites related to mitochondrial function include also proteins implicated in mitochondrial metabolism (e.g., Acyl-CoA, and coenzyme Q biosynthesis) (Table 1). In accordance with already reported data, our approach allowed us to identify proteins implicated in mitochondrial transport (e.g., TIM, and TOM complexes) [8, 43, 44] (Supplementary Figure 7 and Table 1). This result could be considered as a positive control validating our experimentalprocedure.
Protein interactome related to protein synthesis, maturation, folding, processing, and degradation
We identified different proteins implicated in ER protein maturation (e.g., glycosylation and signal peptidase complex), protein folding (e.g., inhibition of protein aggregation of misfolded proteins, protein quality control), and protein processing and degradation (e.g., peptidases, proteases) (Supplementary Figure 7 and Table 2). The protein interactome includes also chaperone proteins inhibiting the aggregation of misfolded proteins (e.g., the protein disulfide-isomerase A4, the protein disulfide-isomerase A6, and the peptidyl-prolyl cis-isomerase FKBP11), or implicated in the ER quality control (e.g., the protein sel-1 homolog 1, and the DnaJ homolog subfamily B member 11). Interestingly, we also identified proteins implicated in mitochondrial protein translation, and the mitochondrial presequence protease involved in the degradation of mitochondrial transit peptides (Supplementary Figure 7 and Table 2).
Protein interactome related to vesicle formation and trafficking
In addition to the mitochondria, the ER is associated with different subcellular domains that execute divers function such as vesicular traffic [40]. As a matter of fact, we identified a class of AβPP protein interactome (n = 12) including proteins implicated in vesicle formation and trafficking (Supplementary Figure 7 and Table 3). These include Ras-related family proteins (Rab-10, Rab18, and Rab1b) implicated in ER-Golgi transport and vesicular traffic, and Praf2 (PRAF1 family protein 2) known to interacts with numerous Rab GTPases thus assisting in the packaging of Rab proteins into vesicles for the transport to target compartments [45, 46]. We also revealed other proteins implicated in vesicular transport namely the transmembrane emp24 domain containing proteins (Tmed2, Tmed10, and Tmed1) and Arl8b (an Arf-like GTPase being a critical regulator of cargo delivery to lysosomes). We also revealed in the protein interactome Flotillin 1, known to be a scaffolding protein within caveolar membranes and to interact with lipid rafts [47, 48].
Protein interactome related to fatty-acid metabolism
We classified a fourth cellular pathway including proteins (n = 10) implicated in fatty-acid metabolism (Supplementary Figure 7 and Table 4). This includes proteins implicated in fatty-acid elongation or modification (Hsd17b12, Acot1, Far1), fatty-acid beta-oxidation (Acaa2, Hadhb, and Acad9), sterol and cholesterol biosynthesis (Nsdh1, Cyp51a1, Lss), and sphingolipid metabolism (Sptlc1). Based on GO annotation, these proteins are mostly localized in the ER (Hsd17b12, Nsdhl, Sptlc1, Cyp51a1, and Lss), or in the mitochondria (Acot1, Acaa2, Far1, and Hadhb,and Acad9). Nsdhl may be also localized in lipid droplets (Table 4).
All over, these analyses identify potential new AβPP and its catabolites interactors in MAMs. However, a dedicated study is necessary to confirm specific direct or indirect interactions of AβPP or its catabolites with these proteins and their role in MAMs alteration in AD.
DISCUSSION
We used both biochemical and imaging approaches to demonstrate the presence of AβPP and its catabolites in MAMs in in vitro and in in vivo AD-like models. It is well established that AβPP is synthesized in the ER [49]. Recent studies reported that AβPP is also associated with mitochondria in AD affected neurons [50, 51]. This was further supported by the presence of AβPP catabolites (i.e., Aβ, C99, and AICD) in the mitochondria [9, 52–55]. We report in this study AβPP localization, and processing in MAMs. These results were obtained in two distinct cellular systems (SH-SY5Y and CHO cells) expressing wild type AβPP or harboring two different familial mutations (APPswe and APPLDN) thus demonstrating that the observed presence and processing of WT or mutated AβPP in MAMs is not linked to a specific cell type and not to a specific familial AβPP mutation. This may also reveal that the subcellular localization and proteolytic processing of AβPP in MAMs was not linked to AD familial cases but may also be observed in sporadic cases of AD. These data also suggest that even if trafficking of AβPP may vary according to the mutation, transit through MAMs compartment where AβPP undergo cleavage is a common denominator independent of the nature of the AβPP species.
We also revealed the presence and activity of α-, β-, and γ-secretases in the MAMs. γ-secretase was shown to exert its activity in the plasma membrane but also in other intracellular compartments such as the trans-Golgi network, lysososmes, pure mitochondria fraction, and MAMs [16, 20, 56]. However, it is important to emphasize that even if γ-secretase complex has been found in mitochondria [16], the putative generation of intra-mitochondrial Aβ and C99 remains puzzling, given the C-terminus-out orientation of AβPP in the outer mitochondrial membrane. A hypothetical model has been proposed stating that Aβ may be produced at the interface between the ER and mitochondria and then transferred to the mitochondria via the TOM import machinery [9, 44]. Another gap to envision the production of Aβ in mitochondria was the demonstration of the presence of active β-secretase (BACE-1) in mitochondria or in MAMs. Importantly, we report herein and for the first time the presence of BACE-1 in crude mitochondria and MAMs fractions. Current knowledge on AβPP and BACE-1 trafficking indicates that plasma membrane and endosomes are likely the major sites of BACE-1-mediated cleavage of AβPP [57, 58]. Mature BACE-1 localizes largely within plasma membrane cholesterol-rich lipid rafts [57, 58], and replacing the BACE-1 transmembrane domain with a glycosylphosphatidylinositol anchor extensively targets BACE-1 to lipid rafts and substantially increases Aβ production [59]. In addition, various types of lipids stimulate BACE-1 activity [60]. Interestingly, a recent study revealed that MAMs behave as a detergent-resistant lipid raft-like domain, consistent with the presence and activity of β- and γ-secretase in rafts [21, 61–63]. In accordance with the presence of α-CTF fragment in MAMs, we also could measure an in vitro α-secretase activity in this compartment in mice brains (Supplementary Figure 5D). Other experiments are still needed to further characterize expression and subcellular distribution and trafficking of α-secretase in vitro and in vivo. We must take into account that both β- and γ-secretases harbor optimal activities in acidic compartments and that the pH of the ER is almost neutral (pH 7.2), and that of mitochondria matrix is markedly alkaline (pH 8) [64]. Thus, it remains not clear how these enzymes may function in the ER, mitochondria, and MAMs compartments in intact cells. One may postulate that AβPP, or at least its C-terminal fragments (C99/C83), and BACE-1 are transported to the ER-mitochondria contact sites through a mechanism implicating retrograde vesicular trafficking. Indeed, we identified different proteins related to vesicle formation and trafficking in the protein interactome data of MAMs fraction isolated from CHO cells expressing APPLDN (Table 3). Dedicated experiments are necessary to confirm this hypothesis and to further explore the molecular mechanisms underlying BACE-1 localization and in situ function in MAMs.
We report an increased ER-mitochondria contact sites in SH-SY5Y cells overexpressing APPswe. Accordingly, an increased ER-mitochondria connectivity was detected in human fibroblasts isolated from individuals with familial AD mutations as well as sporadic AD cases, and in MEFs isolated from mice simple or double knockout for PS1 or PS2 [21]. Similar results were obtained in neuroblastoma cells expressing familial AD-linked PS2 mutations [35]. Interestingly, increased ER-mitochondria connectivity was also reported in a transgenic AD mouse model expressing mutant tau protein [65], and most recently, in cells exposed to apolipoprotein E (ApoE4), a major risk factor for developing sporadic AD [66]. These results and our data may lead to consider MAMs structural alteration as a common denominator underlying the pathogenesis of AD associated to both sporadic and APP/PS-linked familial AD mutations. In agreement with these data, the proteomic approach revealed potential AβPP interactors implicated in the regulation of mitochondrial and ER structure (Dnajc11 and atlastin 3 respectively) (Supplementary Table 1) [67–69]. Accordingly, postmortem analysis of human AD brain and those of AD mouse models showed altered expression levels of PACS2 and Sigma1R, thus further establishing deregulated MAMs as a hallmark of AD [70]. Moreover, it is interesting to note that AβPP processing relies on intact ER-mitochondria crosstalk [21]. Thus, cells deficient of MFN2 show diminished MAM function in terms of lipid metabolism and reduced γ-secretase activity by approximately 50% [21]. In turn, exposing hippocampal neurons or neuroblastoma cells to Aβ enhances ER-mitochondria contacts [70, 71]. These observations are in favor of a vicious cycle where AβPP is processed in MAMs, and its derivative catabolites enhance ER-mitochondria communication and function.
We analyzed a second aspect of MAMs function in AD by combining immunoprecipitation and proteomic mass spectrometric analysis to identify molecules that associate with AβPP and its proteolytic products in MAMs. This approach allows the unbiased and extensive exploration of all possible direct or indirect protein interactors. Nevertheless, we may consider some limitations of the approach used here namely the difficulty to identify interactors of lower expression. This approach failed also to reveal components of γ-secretase and/or β-secretase in MAM proteome analyses. This may be explained by the limitation of immunoprecipitation approach and/or to instable interaction of AβPP and its cleaving enzymes. Although, the strength of our data is supported by the identification of AβPP interactors described in previous studies using distinct experimental approaches (i.e., TIM/TOM complex) [8, 43, 44]. Furthermore, we also revealed new protein partners belonging to cellular pathways known to participate to AD pathogenesis. Interestingly, six out of nine of AβPP and its catabolites interactome related to mitochondrial respiratory chain complexes are components or chaperone of complex I. Previous studies have revealed that Aβ may cause both a selective defect in complex I activity associated with an increase of intracellular reactive oxygen species [72, 73]. We also unraveled an interaction of AβPP and or its catabolites with the mitochondrial ATP synthase (complex V). Interestingly this enzyme shows reduced activity in AD affectedbrains [74].
We revealed the interaction of AβPP and its catabolites with several proteins implicated in protein synthesis, maturation, folding, processing, and degradation. Several studies have suggested that altered maturation, processing, and degradation of AβPP holoprotein may drive the availability of AβPP for Aβ generation [75–78]. It was also suggested that different PPIases regulate Aβ production, or the toxicity associated with Aβ [79]. Concerning mitochondria, we showed that AβPP and its catabolites may directly or indirectly interact with mitochondrial presequence protease (PreP, GN: pitrm), a pitrilysin metallopeptidase 1 implicated in the degradation of mitochondrial transit peptides after their cleavage. PreP has been shown to degrade various Aβ peptides thus clearing mitochondrial (Aβ) [80, 81]. Accordingly, increased neuronal PreP activity attenuates neuroinflammation and improves mitochondrial and synaptic function in AD mice model [82, 83].
Finally, we delineate in this study the potential interaction of AβPP and its catabolites with different proteins related to fatty-acid metabolism. Accordingly, we show increased neutral lipids accumulation in both neuroblastoma and CHO cells expressing APPswe or APPLDN. Neutral lipids accumulation was reported in different AD study models including PS-mutant MEFs, PS double knockout MEFs, fibroblasts from familial AD patients [21], and more recently fibroblasts exposed to ApoE4 conditioned media [66]. We also revealed that neutral lipids accumulation in our study models is likely linked to Aβ peptides rather to C99 fragment. A more dedicated study is necessary to investigate the molecular mechanisms underlying lipid droplets accumulation in AD. In particular, lipid droplets are found in close proximity with the ER and mitochondria [37, 38], and the functions of these interactions are still largely unknown. It is, however, noticeable that proteolytic cleavage of AβPP is tightly regulated by several lipids such as cholesterol and sphingolipids. In turn, Aβ as well as other AβPP processing products play an essential role in regulating lipid homeostasis [84]. Moreover, in vitro studies have shown that fatty acids may affect Aβ secretion [85].
In summary, our data extends the previous studies suggesting MAMs as an important contributor to AD pathogenesis. Maintenance and tight regulation of MAM structure and function could be envisioned as potential therapeutic strategies for AD.
ACKNOWLEDGMENTS
This work was supported by CNRS (FC), INSERM (PPB), and LECMA (Ligue Européenne Contre la Maladie d’Alzheimer) (MC). This work has been developed and supported through the LABEX (excellence laboratory, program investment for the future) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to Alzheimer’s disease) (FC), and the University Hospital Federation (FHU OncoAge). We acknowledge fellow support from Bright Focus foundation to DDP. PP is grateful to Camilla degli Scrovegni for continuous support. This research was supported by grants from the Polish National Science Centre (UMO-2011/01/M/NZ3/02128), Iuventus Plus (UMO-0531/IP1/2011/71) to MRW and JMS.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0953).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-160953.
REFERENCES
[1] | Taylor JP , Hardy J , Fischbeck KH ((2002) ) Toxic proteins in neurodegenerative disease. Science 296: , 1991–1995. |
[2] | Pardossi-Piquard R , Checler F ((2012) ) The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem 120: (Suppl 1), 109–124. |
[3] | Checler F ((1995) ) Processing of the beta-amyloid precursor protein and its regulation in Alzheimer’s disease. J Neurochem 65: , 1431–1444. |
[4] | Shen J , Kelleher RJ 3rd ((2007) ) The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104: , 403–409. |
[5] | Muresan V , Muresan Z ((2012) ) A persistent stress response to impeded al transport leads to accumulation of amyloid-beta in the endoplasmic reticulum, and is a probable cause of sporadic Alzheimer’s disease. Neurodegener Dis 10: , 60–63 axon. |
[6] | Umeda T , Tomiyama T , Sakama N , Tanaka S , Lambert MP , Klein WL , Mori H ((2011) ) Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. J Neurosci Res 89: , 1031–1042. |
[7] | Yamaguchi H , Yamazaki T , Ishiguro K , Shoji M , Nakazato Y , Hirai S ((1992) ) Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol 85: , 15–22. |
[8] | Anandatheerthavarada HK , Biswas G , Robin MA , Avadhani NG ((2003) ) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 161: , 41–54. |
[9] | Pavlov PF , Wiehager B , Sakai J , Frykman S , Behbahani H , Winblad B , Ankarcrona M ((2011) ) Mitochondrial gamma-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein. FASEB J 25: , 78–88. |
[10] | Gouras GK , Tsai J , Naslund J , Vincent B , Edgar M , Checler F , Greenfield JP , Haroutunian V , Buxbaum JD , Xu H , Greengard P , Relkin NR ((2000) ) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: , 15–20. |
[11] | Mungarro-Menchaca X , Ferrera P , Moran J , Arias C ((2002) ) beta-Amyloid peptide induces ultrastructural changes in synaptosomes and potentiates mitochondrial dysfunction in the presence of ryanodine. J Neurosci Res 68: , 89–96. |
[12] | Manczak M , Anekonda TS , Henson E , Park BS , Quinn J , Reddy PH ((2006) ) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: , 1437–1449. |
[13] | Ankarcrona M , Hultenby K ((2002) ) Presenilin-1 is located in rat mitochondria. Biochem Biophys Res Commun 295: , 766–770. |
[14] | Cook DG , Sung JC , Golde TE , Felsenstein KM , Wojczyk BS , Tanzi RE , Trojanowski JQ , Lee VM , Doms RW ((1996) ) Expression and analysis of presenilin 1 in a human neuronal system: Localization in cell bodies and dendrites. Proc Natl Acad Sci U S A 93: , 9223–9228. |
[15] | Annaert WG , Levesque L , Craessaerts K , Dierinck I , Snellings G , Westaway D , George-Hyslop PS , Cordell B , Fraser P , De Strooper B ((1999) ) Presenilin 1 controls gamma-secretase processing of amyloid precursor protein in pre-golgi compartments of hippocampal neurons. J Cell Biol 147: , 277–294. |
[16] | Hansson CA , Frykman S , Farmery MR , Tjernberg LO , Nilsberth C , Pursglove SE , Ito A , Winblad B , Cowburn RF , Thyberg J , Ankarcrona M ((2004) ) Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem 279: , 51654–51660. |
[17] | Hayashi T , Rizzuto R , Hajnoczky G , Su TP ((2009) ) MAM: More than just a housekeeper. Trends Cell Biol 19: , 81–88. |
[18] | Giorgi C , Missiroli S , Patergnani S , Duszynski J , Wieckowski MR , Pinton P ((2015) ) Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal 22: , 995–1019. |
[19] | Schon EA , Area-Gomez E ((2010) ) Is Alzheimer’s disease a disorder of mitochondria-associated membranes?S281-S. J Alzheimers Dis 2: (20 Suppl), 292–. |
[20] | Area-Gomez E , de Groof AJ , Boldogh I , Bird TD , Gibson GE , Koehler CM , Yu WH , Duff KE , Yaffe MP , Pon LA , Schon EA ((2009) ) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175: , 1810–1816. |
[21] | Area-Gomez E , Del Carmen Lara Castillo M , Tambini MD , Guardia-Laguarta C , de Groof AJ , Madra M , Ikenouchi J , Umeda M , Bird TD , Sturley SL , Schon EA ((2012) ) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31: , 4106–4123. |
[22] | Oules B , Del Prete D , Greco B , Zhang X , Lauritzen I , Sevalle J , Moreno S , Paterlini-Brechot P , Trebak M , Checler F , Benfenati F , Chami M ((2012) ) Ryanodine receptor blockade reduces amyloid-beta load and memory impairments in Tg2576 mouse model of Alzheimer disease. J Neurosci 32: , 11820–11834. |
[23] | Guillot-Sestier MV , Sunyach C , Ferreira ST , Marzolo MP , Bauer C , Thevenet A , Checler F ((2012) ) alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J Biol Chem 287: , 5021–5032. |
[24] | Probst G , Aubele DL , Bowers S , Dressen D , Garofalo AW , Hom RK , Konradi AW , Marugg JL , Mattson MN , Neitzel ML , Semko CM , Sham HL , Smith J , Sun M , Truong AP , Ye XM , Xu YZ , Dappen MS , Jagodzinski JJ , Keim PS , Peterson B , Latimer LH , Quincy D , Wu J , Goldbach E , Ness DK , Quinn KP , Sauer JM , Wong K , Zhang H , Zmolek W , Brigham EF , Kholodenko D , Hu K , Kwong GT , Lee M , Liao A , Motter RN , Sacayon P , Santiago P , Willits C , Bard F , Bova MP , Hemphill SS , Nguyen L , Ruslim L , Tanaka K , Tanaka P , Wallace W , Yednock TA , Basi GS ((2013) ) Discovery of (R)-4-cyclopropyl-7,8-difluoro-5-(4-(trifluoromethyl)phenylsulfonyl)-4,5-dihydro- 1H-pyr-azolo[4,3-c]quinoline (ELND006) and (R)-4-cyclopropyl-8-fluoro-5-(6-(trifluoromethyl)pyridin-3-ylsulfonyl)-4,5-dihydr o-2H-pyrazolo[4,3-c]quinoline (ELND007): Metabolically stable gamma-secretase Inhibitors that selectively inhibit the production of amyloid-beta over Notch. J Med Chem 56: , 5261–5274. |
[25] | Lauritzen I , Pardossi-Piquard R , Bauer C , Brigham E , Abraham JD , Ranaldi S , Fraser P , St-George-Hyslop P , Le Thuc O , Espin V , Chami L , Dunys J , Checler F ((2012) ) The beta-secretase-derived c-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci 32: , 16243–16255. |
[26] | May PC , Dean RA , Lowe SL , Martenyi F , Sheehan SM , Boggs LN , Monk SA , Mathes BM , Mergott DJ , Watson BM , Stout SL , Timm DE , Smith Labell E , Gonzales CR , Nakano M , Jhee SS , Yen M , Ereshefsky L , Lindstrom TD , Calligaro DO , Cocke PJ , Greg Hall D , Friedrich S , Citron M , Audia JE ((2011) ) Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci 31: , 16507–16516. |
[27] | Busche MA , Eichhoff G , Adelsberger H , Abramowski D , Wiederhold KH , Haass C , Staufenbiel M , Konnerth A , Garaschuk O ((2008) ) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321: , 1686–1689. |
[28] | Wieckowski MR , Giorgi C , Lebiedzinska M , Duszynski J , Pinton P ((2009) ) Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc 4: , 1582–1590. |
[29] | Cisse MA , Gandreuil C , Hernandez JF , Martinez J , Checler F , Vincent B ((2006) ) Design and characterization of a novel cellular prion-derived quenched fluorimetric substrate of alpha-secretase. Biochem Biophys Res Commun 347: , 254–260. |
[30] | Sevalle J , Amoyel A , Robert P , Fournie-Zaluski MC , Roques B , Checler F ((2009) ) Aminopeptidase A contributes to the N-terminal truncation of amyloid beta-peptide. J Neurochem 109: , 248–256. |
[31] | Chami M , Oules B , Szabadkai G , Tacine R , Rizzuto R , Paterlini-Brechot P ((2008) ) Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell 32: , 641–651. |
[32] | Sinha S , Lieberburg I ((1999) ) Cellular mechanisms of beta-amyloid production and secretion. Proc Natl Acad SciU S A 96: , 11049–11053. |
[33] | Goate AM ((1998) ) Monogenetic determinants of Alzheimer’s disease: APP mutations. Cell Mol Life Sci 54: , 897–901. |
[34] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: , 535–539. |
[35] | Zampese E , Fasolato C , Kipanyula MJ , Bortolozzi M , Pozzan T , Pizzo P ((2011) ) Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+cross-talk. Proc Natl Acad Sci U S A 108: , 2777–2782. |
[36] | Vance JE ((2015) ) Phospholipid synthesis and transport in mammalian cells. Traffic 16: , 1–18. |
[37] | Goodman JM ((2008) ) The gregarious lipid droplet. J Biol Chem 283: , 28005–28009. |
[38] | Murphy S , Martin S , Parton RG ((2009) ) Lipid droplet-organelle interactions; sharing the fats. Biochim Biophys Acta 1791: , 441–447. |
[39] | Heber S , Herms J , Gajic V , Hainfellner J , Aguzzi A , Rulicke T , von Kretzschmar H , von Koch C , Sisodia S , Tremml P , Lipp HP , Wolfer DP , Muller U ((2000) ) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20: , 7951–7963. |
[40] | Lynes EM , Simmen T ((2011) ) Urban planning of the endoplasmic reticulum (ER): How diverse mechanisms segregate the many functions of the ER. Biochim Biophys Acta 1813: , 1893–1905. |
[41] | Poston CN , Krishnan SC , Bazemore-Walker CR ((2013) ) In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteomics 79: , 219–230. |
[42] | Grimm S ((2012) ) The ER-mitochondria interface: The social network of cell death. Biochim Biophys Acta 1823: , 327–334. |
[43] | Devi L , Prabhu BM , Galati DF , Avadhani NG , Anandatheerthavarada HK ((2006) ) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26: , 9057–9068. |
[44] | Hansson Petersen CA , Alikhani N , Behbahani H , Wiehager B , Pavlov PF , Alafuzoff I , Leinonen V , Ito A , Winblad B , Glaser E , Ankarcrona M ((2008) ) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105: , 13145–13150. |
[45] | Abdul-Ghani M , Gougeon PY , Prosser DC , Da-Silva LF , Ngsee JK ((2001) ) PRA isoforms are targeted to distinct membrane compartments. J Biol Chem 276: , 6225–6233. |
[46] | Figueroa C , Taylor J , Vojtek AB ((2001) ) Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J Biol Chem 276: , 28219–28225. |
[47] | Rajendran L , Knobloch M , Geiger KD , Dienel S , Nitsch R , Simons K , Konietzko U ((2007) ) Increased Abeta production leads to intracellular accumulation of Abeta in flotillin-1-positive endosomes. Neurodegener Dis 4: , 164–170. |
[48] | Rajendran L , Le Lay S , Illges H ((2007) ) Raft association and lipid droplet targeting of flotillins are independent of caveolin. Biol Chem 388: , 307–314. |
[49] | Placido AI , Pereira CM , Duarte AI , Candeias E , Correia SC , Santos RX , Carvalho C , Cardoso S , Oliveira CR , Moreira PI ((2014) ) The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: Implications for Alzheimer’s disease. Biochim Biophys Acta 1842: , 1444–1453. |
[50] | Anandatheerthavarada HK , Devi L ((2007) ) Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist 13: , 626–638. |
[51] | Anandatheerthavarada HK , Devi L ((2007) ) Mitochondrial translocation of amyloid precursor protein and its cleaved products: Relevance to mitochondrial dysfunction in Alzheimer’s disease. Rev Neurosci 18: , 343–354. |
[52] | Chen JX , Yan SS ((2010) ) Role of mitochondrial amyloid-beta in Alzheimer’s disease.S569-S. J Alzheimers Dis 20: (Suppl 2), 578–. |
[53] | Pagani L , Eckert A ((2011) ) Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011: , 925050–. |
[54] | Devi L , Ohno M ((2012) ) Mitochondrial dysfunction and accumulation of the beta-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis 45: , 417–424. |
[55] | Schreiner B , Hedskog L , Wiehager B , Ankarcrona M ((2015) ) Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis 43: , 369–374. |
[56] | Pasternak SH , Bagshaw RD , Guiral M , Zhang S , Ackerley CA , Pak BJ , Callahan JW , Mahuran DJ ((2003) ) Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem 278: , 26687–26694. |
[57] | Riddell DR , Christie G , Hussain I , Dingwall C ((2001) ) Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol 11: , 1288–1293. |
[58] | Ehehalt R , Keller P , Haass C , Thiele C , Simons K ((2003) ) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: , 113–123. |
[59] | Kalvodova L , Kahya N , Schwille P , Ehehalt R , Verkade P , Drechsel D , Simons K ((2005) ) Lipids as modulators of proteolytic activity of BACE: Involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. J Biol Chem 280: , 36815–36823. |
[60] | Puglielli L , Ellis BC , Saunders AJ , Kovacs DM ((2003) ) Ceramide stabilizes beta-site amyloid precursor protein-cleng enzyme 1 and promotes amyloid beta-peptide biogenesis. J Biol Chem 278: , 19777–19783 avi. |
[61] | Marquer C , Devauges V , Cossec JC , Liot G , Lecart S , Saudou F , Duyckaerts C , Leveque-Fort S , Potier MC ((2011) ) Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J 25: , 1295–1305. |
[62] | Cordy JM , Hussain I , Dingwall C , Hooper NM , Turner AJ ((2003) ) Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 100: , 11735–11740. |
[63] | Vetrivel KS , Cheng H , Lin W , Sakurai T , Li T , Nukina N , Wong PC , Xu H , Thinakaran G ((2004) ) Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem 279: , 44945–44954. |
[64] | Casey JR , Grinstein S , Orlowski J ((2010) ) Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11: , 50–61. |
[65] | Perreault S , Bousquet O , Lauzon M , Paiement J , Leclerc N ((2009) ) Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L tau. J Neuropathol Exp Neurol 68: , 503–514. |
[66] | Tambini MD , Pera M , Kanter E , Yang H , Guardia-Laguarta C , Holtzman D , Sulzer D , Area-Gomez E , Schon EA ((2016) ) ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep 17: , 27–36. |
[67] | Huynen MA , Muhlmeister M , Gotthardt K , Guerrero-Castillo S , Brandt U ((2016) ) Evolution and structural organization of the mitochondrial contact site (MICOS) complex and the mitochondrial intermembrane space bridging (MIB) complex. Biochim Biophys Acta 1863: , 91–101. |
[68] | Barlowe C ((2009) ) Atlasin GTPases shape up ER networks. Dev Cell 17: , 157–158. |
[69] | Orso G , Pendin D , Liu S , Tosetto J , Moss TJ , Faust JE , Micaroni M , Egorova A , Martinuzzi A , McNew JA , Daga A ((2009) ) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460: , 978–983. |
[70] | Hedskog L , Pinho CM , Filadi R , Ronnback A , Hertwig L , Wiehager B , Larssen P , Gellhaar S , Sandebring A , Westerlund M , Graff C , Winblad B , Galter D , Behbahani H , Pizzo P , Glaser E , Ankarcrona M ((2013) ) Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci U S A 110: , 7916–7921. |
[71] | Ferreira IL , Ferreiro E , Schmidt J , Cardoso JM , Pereira CM , Carvalho AL , Oliveira CR , Rego AC ((2015) ) Abeta and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol Aging 36: , 680–692. |
[72] | Vargas T , Ugalde C , Spuch C , Antequera D , Moran MJ , Martin MA , Ferrer I , Bermejo-Pareja F , Carro E ((2010) ) Abeta accumulation in choroid plexus is associated with mitochondrial-induced apoptosis. Neurobiol Aging 31: , 1569–1581. |
[73] | Bobba A , Amadoro G , Valenti D , Corsetti V , Lassandro R , Atlante A ((2013) ) Mitochondrial respiratory chain Complexes I and IV are impaired by beta-amyloid via direct interaction and through Complex I-dependent ROS production, respectively. Mitochondrion 13: , 298–311. |
[74] | Terni B , Boada J , Portero-Otin M , Pamplona R , Ferrer I ((2010) ) Mitochondrial ATP-synthase in the entorhinal cortex is a target of oxidative stress at stages I/II of Alzheimer’s disease pathology. Brain Pathol 20: , 222–233. |
[75] | Huttunen HJ , Guenette SY , Peach C , Greco C , Xia W , Kim DY , Barren C , Tanzi RE , Kovacs DM ((2007) ) HtrA2 regulates beta-amyloid precursor protein (APP) metabolism through endoplasmic reticulum-associated degradation. J Biol Chem 282: , 28285–28295. |
[76] | Yang Y , Turner RS , Gaut JR ((1998) ) The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem 273: , 25552–25555. |
[77] | Chen Q , Kimura H , Schubert D ((2002) ) A novel mechanism for the regulation of amyloid precursor protein metabolism. J Cell Biol 158: , 79–89. |
[78] | Bunnell WL , Pham HV , Glabe CG ((1998) ) gamma-secretase cleavage is distinct from endoplasmic reticulum degradation of the transmembrane domain of the amyloid precursor protein. J Biol Chem 273: , 31947–31955. |
[79] | Pastorino L , Sun A , Lu PJ , Zhou XZ , Balastik M , Finn G , Wulf G , Lim J , Li SH , Li X , Xia W , Nicholson LK , Lu KP ((2006) ) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440: , 528–534. |
[80] | Falkevall A , Alikhani N , Bhushan S , Pavlov PF , Busch K , Johnson KA , Eneqvist T , Tjernberg L , Ankarcrona M , Glaser E ((2006) ) Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem 281: , 29096–29104. |
[81] | Alikhani N , Guo L , Yan S , Du H , Pinho CM , Chen JX , Glaser E , Yan SS ((2011) ) Decreased proteolytic activity of the mitochondrial amyloid-beta degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J Alzheimers Dis 27: , 75–87. |
[82] | Fang D , Wang Y , Zhang Z , Du H , Yan S , Sun Q , Zhong C , Wu L , Vangavaragu JR , Hu G , Guo L , Rabinowitz M , Glaser E , Arancio O , Sosunov AA , McKhann GM , Chen JX , Yan SS ((2015) ) Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum Mol Genet 24: , 5198–5210. |
[83] | Matic I , Strobbe D , Frison M , Campanella M ((2015) ) Controlled and impaired mitochondrial quality in neurons: Molecular physiology and prospective pharmacology. Pharmacol Res 99: , 410–424. |
[84] | Grimm MO , Rothhaar TL , Hartmann T ((2012) ) The role of APP proteolytic processing in lipid metabolism. Exp Brain Res 217: , 365–375. |
[85] | Amtul Z , Uhrig M , Rozmahel RF , Beyreuther K ((2011) ) Structural insight into the differential effects of omega-3 and omega-6 fatty acids on the production of Abeta peptides and amyloid plaques. J Biol Chem 286: , 6100–6107. |
Figures and Tables
Fig.1
AβPP and its catabolites are present in MAMs fraction. A) Scheme representing ER-mitochondria contact sites and the molecular markers of ER and mitochondria used. MRC, mitochondrial respiratory chain. B, C) Representative SDS-PAGE of subcellular fractions isolated from: (B) SH-SY5Y neuroblastoma cells expressing pcDNA3.1 empty vector (Control) or hAPP695 harboring double Swedish mutations (APPKM670/671NL: APPswe), and (C) brains of WT mice, PS45, APP23, and APP23xPS45 transgenic mice. We load 40 μg (B) or 20 μg (C) of total homogenate (homog), Crude mitochondria (Mc), pure mitochondria (Mp), the endoplasmic reticulum (ER), and MAMs fractions. AβPP, C99, and Aβ were detected by 6E10 antibody. AβPP C-terminal fragments (C99 and C83) were detected by AβPP C-ter antibody. SERCA2b, calreticulin (CRT), and Cytochrome p450 (Cytp450) were used as loading controls for ER fraction. Cox, VDAC1, and Chapronin 10 (Cpn10) were used as loading controls for Mc and Mp fractions. GRP75 was used as loading controls for MAMs fraction. Actin and Tubulin were used as loading controls. Data are representative of 3 independent experiments for (B), and were obtained from 5 animals per group of mice and repeated twice for (C). D) ELISA of Aβ40 and Aβ42 done on subcellular fractions isolated from APP23 transgenic mice. Graph represents Mean±S.E.M from three experiments and expressed in pg per μg of proteins. *p value <0.05, **p value <0.01, ***p value <0.001 calculated using ANOVA one way and Newman-Keuls Multicomparison post-test.
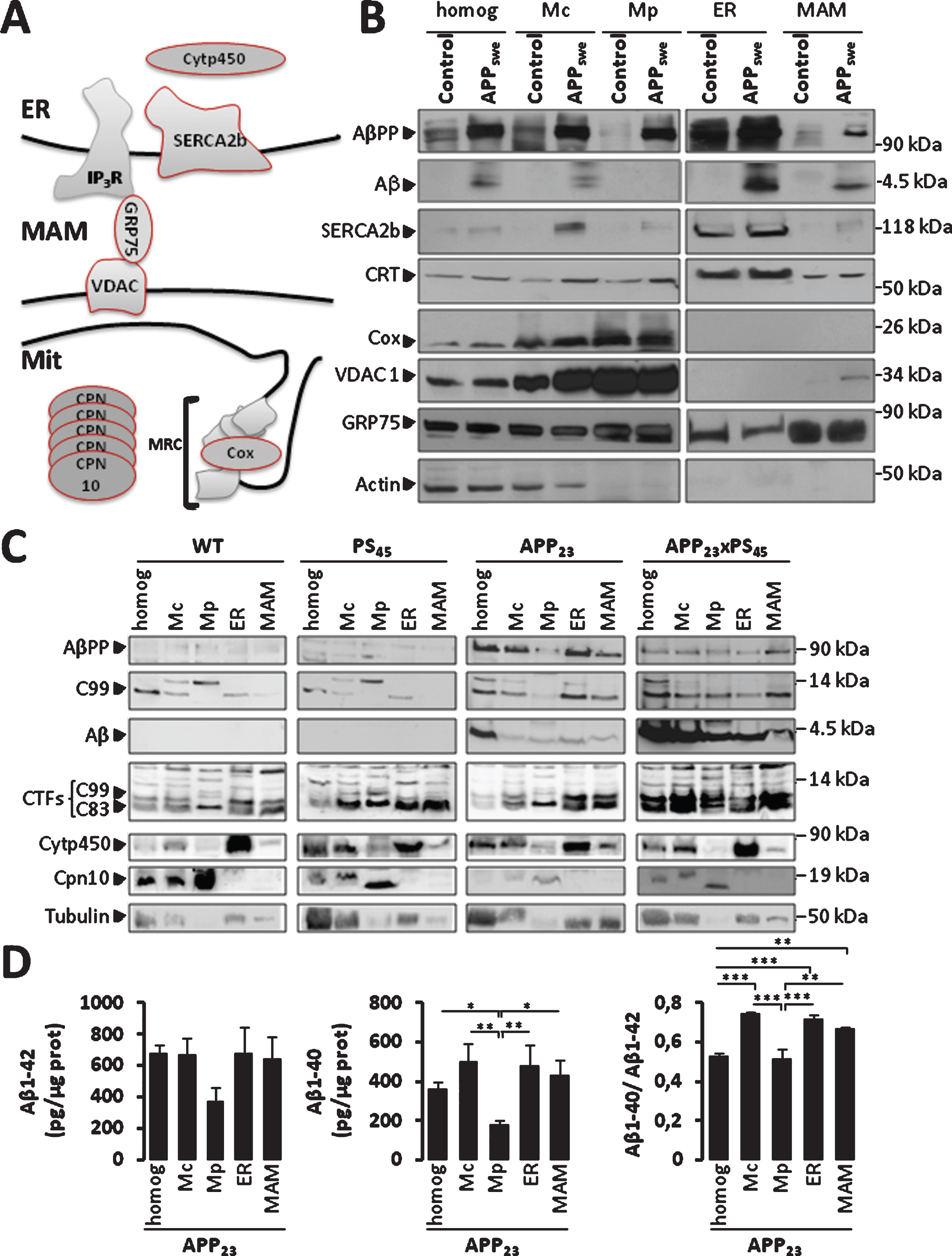
Fig.2
AβPP and its catabolites colocalize with ER and mitochondria. A, B) Immunostaining of SH-SY5Y APPswe cells with AβPP C-ter antibody recognizing total AβPP and CTF (C99 and C83). RyR was used to stain the ER (A). Tom20 was used to stain mitochondria (B). C, D) Immunostaining of SH-SY5Y APPswe cells with 6E10 antibody recognizing total AβPP, C99, and Aβ peptides. Cytp450 was used to stain the ER (C). Cox was used to stain mitochondria (D). A-D) Merge images show green and red signals and were used to show the colocalization of AβPP and its catabolites with the ER or mitochondria (yellow signal). Scale bars represent 10 μm. E) Immunostaining of SH-SY5Y APPswe cells with AβPP C-ter, Tom20, and calreticulin (CRT) antibodies. Merge images show overlay of green, red and blue signals, where colocalization is depicted in white (see merge image and magnified overlay). Scale bar represents 10 μm. F) RGB (Red, Green, Blue) profile plots of a representative line scan (red line in inset) shows the maximum intensity of Red, Green, and Blue plots observed at the same distance, and demonstrating the colocalization of Red (Tom20), Green (C-ter), and Blue (CRT) signals.
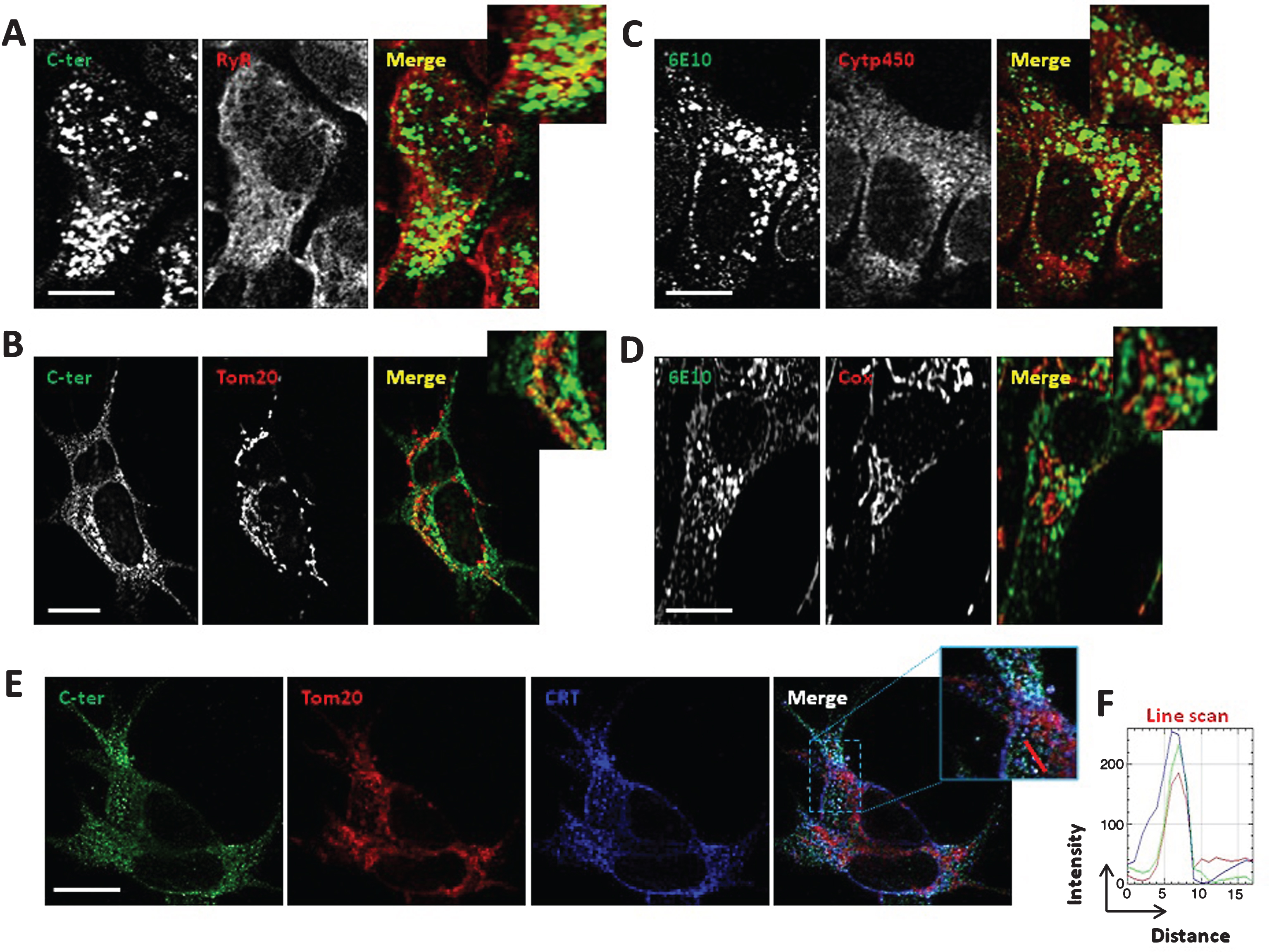
Fig.3
β-secretase and γ-secretase complex are present and active in MAMs fraction in mice brains. A) Representative SDS-PAGE of subcellular fractions isolated from APP23 transgenic mice as in Fig. 1 showing the expression of γ-seceretase complex (cleaved Presenilin 1 N-terminal fragment (PS1-NT); Anterior Pharynx-Defective 1 (Aph1); and Nicastrin (NCT)) and of β-secretase (BACE-1). B) Cell-free Aβ production from recombinant C100 peptide performed at 37°C or 4°C in the presence of subcellular fractions isolated from APP23 transgenic mice. C100 and Aβ were detected using 6E10 antibody. C) In vitro β-secretase specific activity in WT and APP23 mice brain fractions. The graph represents β-secretase specific activity obtained by subtracting residual activity in the presence of β-secretase inhibitor from total β-secretase activity and is expressed in fluorescent units (FU)/hour (h)/mg of proteins of each fraction. Data are presented as Mean±S.E.M. obtained from 3 independent experiments. Differences are statistically non-significant using ANOVA one-way and Newman-Keuls Multicomparison post-test.
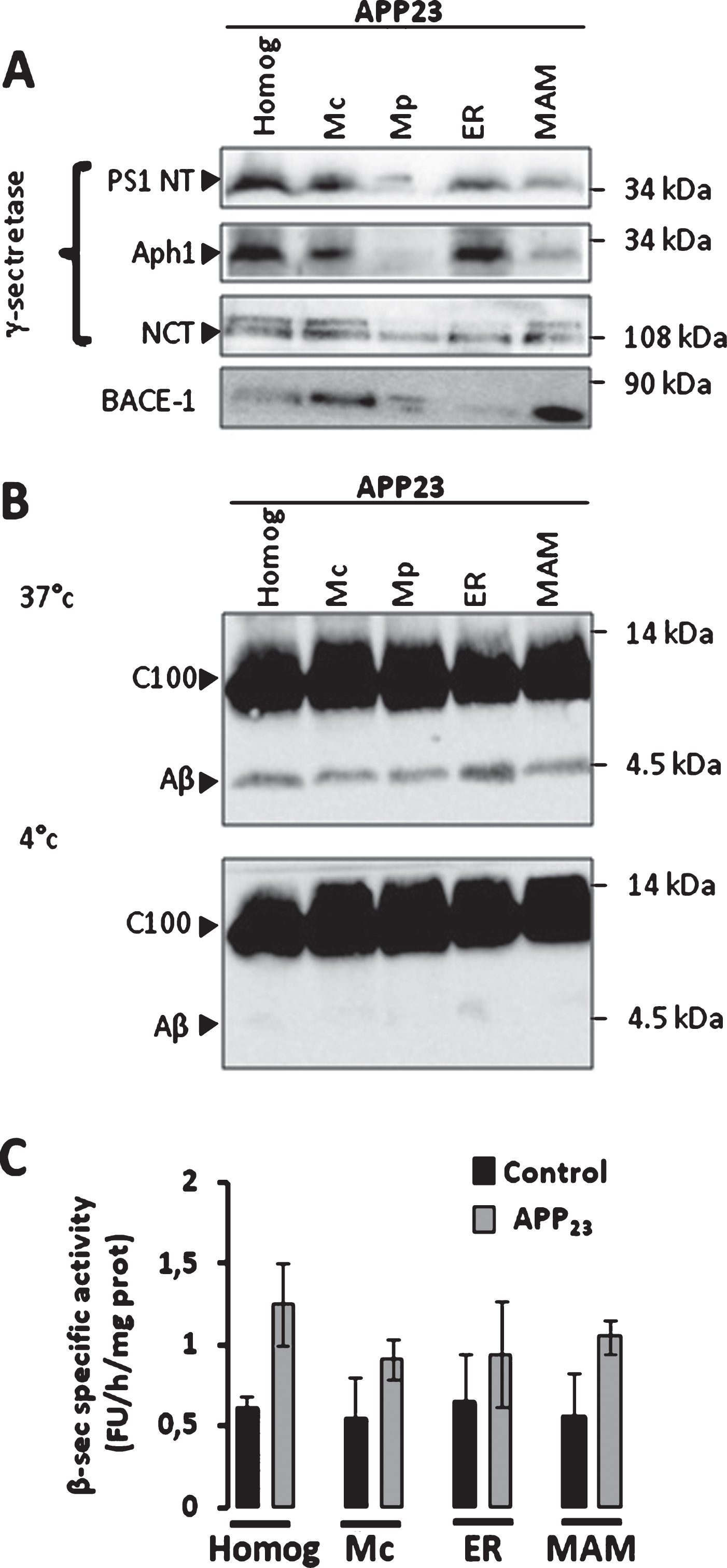
Fig.4
Increased ER-mitochondria contact sites in SH-SY5Y cells expressing APPswe. A) Quantitative analyses of the colocalization of ER and mitochondria in SH-SY5Y live cells expressing pcDNA3.1 [Control (n = 4) or APPswe (n = 7)]. ER is visualized by transfecting ER GFP (green). Mitochondria are stained with Mitotracker Deep red dye (red). Merge images show overlay of green and red signals. Insets (magnified overlay) showing ER and mitochondria colocalization depicted in yellow. The graph represents the quantification of ER and mitochondria colocalization presented as percentage of total mitochondrial volume (% mean±S.E.M.). **p value < 0.01 using Student’s t test. Scale bars represent 5 μm. B) Representative electron microscopy micrographs of control and APPswe expressing cells. Red arrows show mitochondria in contacts with ER. High magnificence of ER-mitochondria contacts is shown for APPswe cells. Data are presented as % of mitochondria in contacts with ER. Quantification was obtained from a total number of mitochondria in control (n = 195) and in APPswe expressing cells (n = 166). Scale bars represent 2 μm.
![Increased ER-mitochondria contact sites in
SH-SY5Y cells expressing APPswe. A) Quantitative analyses of the colocalization of ER and
mitochondria in SH-SY5Y live cells expressing pcDNA3.1 [Control (n = 4) or APPswe
(n = 7)]. ER is visualized by transfecting ER GFP (green). Mitochondria are stained with Mitotracker
Deep red dye (red). Merge images show overlay of green and red signals. Insets (magnified overlay) showing ER and
mitochondria colocalization depicted in yellow. The graph represents the quantification of ER and mitochondria
colocalization presented as percentage of total mitochondrial volume (% mean±S.E.M.). **p
value < 0.01 using Student’s t test. Scale bars represent 5 μm. B) Representative electron
microscopy micrographs of control and APPswe expressing cells. Red arrows show mitochondria in
contacts with ER. High magnificence of ER-mitochondria contacts is shown for APPswe cells. Data are
presented as % of mitochondria in contacts with ER. Quantification was obtained from a total number of
mitochondria in control (n = 195) and in APPswe expressing cells (n = 166). Scale
bars represent 2 μm.](https://content.iospress.com:443/media/jad/2017/55-4/jad-55-4-jad160953/jad-55-jad160953-g004.jpg)
Fig.5
Increased neutral lipid droplets accumulation in CHO cells expressing APPLDN. A) Representative images showing neutral lipids staining in CHO control cells or expressing APPLDN. Cells were treated with vehicle or with β- or γ-secretase inhibitors for 20 h. Scale bars represent 25 μm. B) The graph shows the quantification of lipid droplets (mean±S.E.M. number of puncta/cell). The number of analyzed cells is indicated for each condition and was obtained from at least three independent experiments. *p value < 0.05, and **p value < 0.01 using ANOVA one way and Tukey post-test. C) Representative images showing neutral lipids staining in SH-SY5Y cells non treated or treated with 5 μM oligomeric Aβ (oAβ) preparation for 20 h. Scale bars represent 10 μm. D) The graph shows the quantification of lipid droplets (mean±S.E.M. number of puncta/cell). The number of analyzed cells is indicated for each condition and was obtained from two independent experiments. *p value < 0.05, using Student t-test. E) Representative SDS-PAGE of oligomeric Aβ (3 μM and 6 μM) preparation.
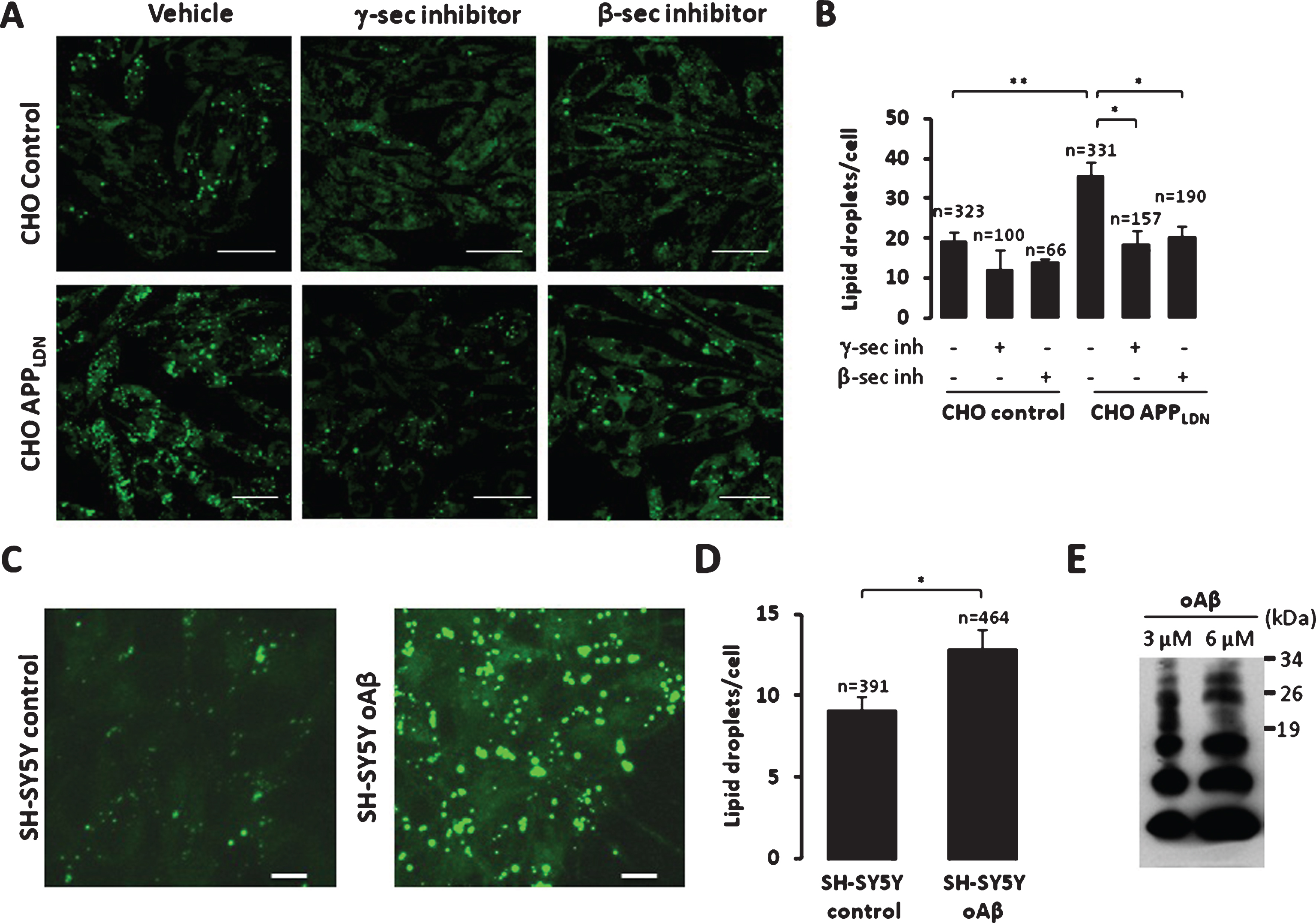
Table 1
Classification of protein interactome of the AβPP and its catabolites related to mitochondrial function
| UniProt ID | Name(a) | Peptides (95%)(b) | Function |
| Mitochondrial respiratory chain complexes | |||
| Q9CZ13 | Cytochrome b-c1 complex subunit 1, mitochondrial GN = Uqcrc1 | 3 | Component of complex III or cytochrome b-c1 complex |
| Q8BGH2(*) | Sorting and assembly machinery component 50 homolog GN = Samm50 | 2 | Maintenance of the structure of mitochondrial cristae and the proper assembly of the MRC complexes |
| Q91VD9 | NADH-ubiquinone oxidoreductase 75 kDa subunit, mitochondrial GN = Ndufs1 | 1 | Core subunit of the mitochondrial Complex I |
| Q91WD5 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 2, mitochondrial GN = Ndufs2 | 1 | Core subunit of the mitochondrial Complex I |
| Q91YT0 | NADH dehydrogenase [ubiquinone] flavoprotein 1, mitochondrial GN = Ndufv1 | 1 | Core subunit of the mitochondrial Complex I |
| Q9CQN1 | Heat shock protein 75 kDa, mitochondrial GN = Trap1 | 1 | Chaperone protein involved in maintaining mitochondrial function and polarization, likely through stabilization of mitochondrial complex I |
| Q9CWX2 | Complex I intermediate-associated protein 30, mitochondrial GN = Ndufaf1 | 1 | Chaperone protein involved in the assembly of the mitochondrial complex I |
| P97450 | ATP synthase-coupling factor 6, mitochondrial GN = Atp5j | 1 | Mitochondrial F1F0 ATP synthase orComplex V |
| Q9CQQ7 | ATP synthase subunit b, mitochondrial GN = Atp5f1 | 1 | Mitochondrial F1F0 ATP synthase orComplex V |
| Mitochondrial transport | |||
| Q9Z0V7 | Mitochondrial import inner membrane translocase subunit Tim17-B GN = Timm17b | 1 | Essential component of the TIM23 complex |
| Q8BGH2(*) | Sorting and assembly machinery component 50 homolog GN = Samm50 | 2 | Assembly of TOM40 into the TOM complex |
| Q9CPQ3 | Mitochondrial import receptor subunit TOM22 homolog GN = Tomm22 | 1 | Central receptor component of the translocase of the TOM complex |
| Q9QZD8 | Mitochondrial dicarboxylate carrier GN = Slc25a10 | 1 | Translocation of malonate, malate and succinate in exchange for phosphate, sulfate, sulfite or thiosulfate |
| Q9Z2Z6 | Mitochondrial carnitine/acylcarnitine carrier protein OS = Mus musculus GN = Slc25a20 | 1 | Transport of acylcarnitines of different length across the mitochondrial inner membrane |
| Q9CQX2 | Cytochrome b5 type B GN = Cyb5b | 1 | Electron carrier for several membrane bound oxygenases |
| Mitochondrial metabolism | |||
| Q9JHI5 | Isovaleryl-CoA dehydrogenase, mitochondrial GN = Ivd | 1 | Synthesis of (S)-3-hydroxy-3-methylglutaryl-CoA from 3-isovaleryl-CoA |
| Q9WUM5 | Succinyl-CoA ligase [ADP/GDP-forming] subunit alpha, mitochondrial GN = Suclg1 | 1 | ATP- or GTP-dependent ligation of succinate and CoA to form succinyl-CoA |
| Q9R112 | Sulfide:quinone oxidoreductase, mitochondrial GN = Sqrdl | 1 | Oxidation of hydrogen sulfide with the help of a quinone, such as ubiquinone |
| Q9D2G2 | Dihydrolipoyllysine-residue succinyltransferase component of 2-oxoglutarate dehydrogenase complex, mitochondrial GN = Dlst | 1 | Conversion of 2-oxoglutarate to succinyl-CoA and CO2 |
| Q60936 | Chaperone activity of bc1 complex-like, mitochondrial GN = Adck3 | 1 | Biosynthesis of coenzyme Q (ubiquinone) |
| Q64521 | Glycerol-3-phosphate dehydrogenase, mitochondrial GN = Gpd2 | 1 | Belongs to the FAD-dependent glycerol-3-phosphate dehydrogenase family |
| Q99KI0 | Aconitate hydratase, mitochondrial GN = Aco2 | 1 | Isomerization of citrate to isocitrate via cis-aconitate |
| Q9D051 | Pyruvate dehydrogenase E1 component subunit beta, mitochondrial GN = Pdhb | 1 | Conversion of pyruvate to acetyl-CoA and CO2 |
UniProtID, protein accession number in Swiss-Prot/Trembl. (a)Protein name given by UniProt database. (b)Number of peptides matching the protein with a confidence limit of 95%. (*)A protein that have several functions. GN, gene name.
Table 2
Classification of protein interactome of the AβPP and its catabolites related to protein synthesis, maturation, folding, processing and degradation
| UniProt ID | Name(a) | Peptides (95%)(b) | Function |
| Protein synthesis | |||
| Q8K0D5 | Elongation factor G, mitochondrial GN = Gfm1 | 2 | Mitochondrial GTPase that catalyzes the GTP-dependent ribosomal translocation step during translation elongation. |
| Q8R2Y8 | Peptidyl-tRNA hydrolase 2, mitochondrial GN = Ptrh2 | 2 | The natural substrate for this enzyme may be peptidyl-tRNAs which drop off the ribosome during protein synthesis. |
| Protein maturation or modification | |||
| Q9DBG6 | Dolichyl-diphosphooligosaccharide–protein glycosyltransferase subunit 2 GN = Rpn2 | 1 | Involved in the pathway of protein glycosylation |
| P46978 | Dolichyl-diphosphooligosaccharide–protein glycosyltransferase subunit STT3A GN = Stt3a | 1 | Involved in the pathway of protein glycosylation |
| Q9R0P6 | Signal peptidase complex catalytic subunit SEC11A GN = Sec11a | 1 | Component of the microsomal signal peptidase complex. |
| Q9CXY9 | GPI-anchor transamidase GN = Pigk | 1 | Involved in the pathway glycosylphosphatidylinositol-anchor biosynthesis |
| Protein folding | |||
| P08003 | Protein disulfide-isomerase A4 GN = Pdia4 | 2 | Chaperone that inhibits aggregation of misfolded proteins. |
| Q922R8 | Protein disulfide-isomerase A6 GN = Pdia6 | 2 | Chaperone that inhibits aggregation of misfolded proteins. |
| Q9D1M7 | Peptidyl-prolyl cis-trans isomerase FKBP11 GN = Fkbp11 | 2 | PPIases accelerate the folding of proteins during protein synthesis. |
| Q9Z2G6 | Protein sel-1 homolog 1 GN = Sel1l | 2 | May play a role in Notch signaling. May be involved in the ER quality control |
| P24369 | Peptidyl-prolyl cis-trans isomerase B GN = Ppib | 1 | PPIases accelerate the folding of proteins |
| Q99KV1 | DnaJ homolog subfamily B member 11 GN = Dnajb11 | 1 | Co-chaperone for HSPA5. Binds directly to both unfolded proteins that are substrates for ERAD |
| Protein processing and degradation | |||
| Q8K411 | Presequence protease, mitochondrial GN = Pitrm1 | 1 | ATP-independent protease that degrades mitochondrial transit peptides after their cleavage |
| Q6I6G8 | E3 ubiquitin-protein ligase HECW2 GN = Hecw2 | 1 | Involved in the pathway of protein ubiquitination, |
| O70481 | E3 ubiquitin-protein ligase UBR1 GN = Ubr1 | 1 | E3 ubiquitin-protein ligase which is a component of the N-end rule pathway |
| O88696 | Putative ATP-dependent Clp protease proteolytic subunit, mitochondrial GN = Clpp | 1 | Protease component of the Clp complex that cleaves peptides and various proteins in an ATP-dependent process |
| Q9QXV0 | ProSAAS GN = Pcsk1n | 1 | Serine-type endopeptidase inhibitor activity |
| Q9CYN2 | Signal peptidase complex subunit 2 GN = Spcs2 | 1 | Component of the microsomal signal peptidase complex |
UniProtID, protein accession number in Swiss-Prot/Trembl. (a)Protein name given by UniProt database. (b)Number of unique peptides matching the protein with a confidence limit of 95%. GN, gene name.
Table 3
Classification of protein interactome of the AβPP and its catabolites related to vesicle formation and trafficking
| UniProt ID | Name(a) | Peptides (95%)(b) | Function |
| Q3V009 | Transmembrane emp24 domain-containing protein 1 GN = Tmed1 | 1 | Potential role in vesicular protein trafficking, mainly in the early secretory pathway |
| Q9R0Q3 | Transmembrane emp24 domain-containing protein 2 GN = Tmed2 | 2 | Involved in vesicular protein trafficking (early secretory pathway and post-Golgi membranes) |
| Q9D1D4 | Transmembrane emp24 domain-containing protein 10 GN = Tmed10 | 1 | Involved in vesicular protein trafficking |
| O08917 | Flotillin-1 GN = Flot1 | 1 | May act as a scaffolding protein within caveolar membranes, functionally participating in formation of caveolae or caveolae-like vesicles |
| O55100 | Synaptogyrin-1 GN = Syngr1 | 1 | Involved in the regulation of short-term and long-term synaptic plasticity |
| P35293 | Ras-related protein Rab-18 GN = Rab18 | 1 | Plays a role in apical endocytosis/recycling |
| P61027 | Ras-related protein Rab-10 GN = Rab10 | 2 | Regulator of intracellular membrane trafficking, may play a role in ER dynamics and morphology |
| Q9D1G1 | Ras-related protein Rab-1B GN = Rab1b | 1 | Regulates vesicular transport between the ER and Golgi compartments. Plays a role in the initial events of the autophagic vacuole development |
| Q61548 | Clathrin coat assembly protein AP180 GN = Snap91 | 1 | Components of the adaptor complexes which link clathrin to receptors in coated vesicles |
| Q9CQW2 | ADP-ribosylation factor-like protein 8B GN = Arl8b | 1 | May play a role in lysosomes motility |
| Q9EQH3 | Vacuolar protein sorting-associated protein 35 GN = Vps35 | 1 | Component of the retromer cargo-selective complex (CSC). |
| Q9JIG8 | PRA1 family protein 2 GN = Praf2 | 1 | May be involved in ER/Golgi transport and vesicular traffic |
UniProtID, protein accession number in Swiss-Prot/Trembl. (a)Protein name given by UniProt database. (b)Number of unique peptides matching the protein with a confidence limit of 95%. GN, gene name.
Table 4
Classification of protein intercatome of the AβPP and its catabolites related to fatty-acid metabolism
| UniProt ID | Name(a) | Peptides (95%)(b) | Function |
| O70503 | Estradiol 17-beta-dehydrogenase 12 GN = Hsd17b12 | 2 | Catalyzes the second of the four reactions of the long-chain fatty acids elongation cycle |
| Q9R1J0 | Sterol-4-alpha-carboxylate 3-dehydrogenase, decarboxylating GN = Nsdhl | 1 | Involved in the sequential removal of two C-4 methyl groups in post-squalene cholesterol biosynthesis |
| O35704 | Serine palmitoyltransferase 1 GN = Sptlc1 | 1 | Involved in the pathway sphingolipid metabolism |
| O55137 | Acyl-coenzyme A thioesterase 1 GN = Acot1 | 1 | Catalyze the hydrolysis of acyl-CoAs to the free fatty acid and coenzyme A (CoASH) |
| Q8BWT1 | 3-ketoacyl-CoA thiolase, mitochondrial GN = Acaa2 | 1 | Involved in the pathway fatty acid beta oxidation |
| Q8K0C4 | Lanosterol 14-alpha demethylase GN = Cyp51a1 | 1 | Catalyzes C14-demethylation of lanosterol |
| Q8JZN5 | Acyl-CoA dehydrogenase family member 9, mitochondrial GN = Acad9 | 2 | Mitochondrial complex I assembly (By similarity) |
| Q922J9 | Fatty acyl-CoA reductase 1 GN = Far1 | 1 | Catalyzes the reduction of saturated fatty acyl-CoA with chain length C16 or C18 to fatty alcohols |
| Q99JY0 | Trifunctional enzyme subunit beta, mitochondrial GN = Hadhb | 1 | Involved in the pathway fatty acid beta-oxidation |
| Q8BLN5 | Lanosterol synthase GN = Lss | 1 | Catalyzes the cyclization of (S)-2,3 oxidosqualene to lanosterol |
UniProtID, protein accession number in Swiss-Prot/Trembl. (a)Protein name given by UniProt database. (b)Number of unique peptides matching the protein with a confidence limit of 95%. GN, gene name.


