Tocotrienol-Rich Fraction Modulates Amyloid Pathology and Improves Cognitive Function in AβPP/PS1 Mice
Abstract
Alzheimer’s disease (AD) is the most common cause of dementia. The cardinal neuropathological characteristic of AD is the accumulation of amyloid-β (Aβ) into extracellular plaques that ultimately disrupt neuronal function and lead to neurodegeneration. One possible therapeutic strategy therefore is to prevent Aβ aggregation. Previous studies have suggested that vitamin E analogs slow AD progression in humans. In the present study, we investigated the effects of the tocotrienol-rich fraction (TRF), a mixture of vitamin E analogs from palm oil, on amyloid pathology in vitro and in vivo. TRF treatment dose-dependently inhibited the formation of Aβ fibrils and Aβ oligomers in vitro. Moreover, daily TRF supplementation to AβPPswe/PS1dE9 double transgenic mice for 10 months attenuated Aβ immunoreactive depositions and thioflavin-S-positive fibrillar type plaques in the brain, and eventually improved cognitive function in the novel object recognition test compared with control AβPPswe/PS1dE9 mice. The present result indicates that TRF reduced amyloid pathology and improved cognitive functions, and suggests that TRF is a potential therapeutic agent for AD.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder that invariably progresses from early mild cognitive impairments such as memory loss to severe impairments across multiple cognitive domains (dementia) at advance stage. The neuropathological hallmarks of AD are the accumulation of senile plaques and neurofibrillary tangles in the cerebral cortex and select subcortical regions [1, 2]. According to the amyloid cascade hypothesis, accumulation of amyloid-β (Aβ) due to an imbalance between production and clearance in the brain is the primary event driving AD pathogenesis [3]. The hypothesis is supported by molecular genetics showing that mutations in the amyloid precursor protein and presenilin genes accelerate Aβ production in familial AD [4–6]. Abnormal Aβ aggregation and hyperphosphorylation of the intermediate filament protein tau induce progressive synaptic and neuronal dysfunction, ultimately impairing cognitive performance. Therefore, one possible therapeutic approach to combat AD is by treating the disease with a compound that could modulate Aβ aggregation thus reducing its cytotoxiceffect [1, 7].
Vitamin E is composed of eight analogs: α-, β-, δ-, and γ-tocopherol and α-, β-, δ-, and γ-tocotrienol. Tocopherols are abundant in the leaves and seeds of most dicots, whereas tocotrienols are rich in the seeds of monocots, two broad groups of flowering plants distinguished by stem and leaf morphology [8]. Palm oil from the Elais guineensis tree is among the most abundant natural sources of vitamin E with the major constituent is 70% tocotrienols while another 30% is tocopherols [9, 10]. Tocotrienol-rich fraction (TRF) extracted from palm oil consists of mainly tocotrienols family. Numerous studies have demonstrated the beneficial effects of vitamin E family in preventing degenerative diseases such as antioxidant, anti-inflammatory, anti-aging, and anti-cancer [11–17], and there is emerging evidence that vitamin E can also slow the progression of AD [18–22]. The incidences of mild cognitive impairment and AD are associated with low plasma levels of tocopherols and tocotrienols as reported by Mangialasche et al. [23]. They also suggested a combination of vitamin E analogs linked to reduce risk of AD at advanced age [24].
The α-tocotrienol is more potent neuroprotectant than α-tocopherol at nanomolar concentrations [25]. Furthermore, TRF exerted neuroprotective effect by reducing DNA damage and apoptosis induced by Aβ and glutamate in cell culture [26, 27]. While treatment with α-tocopherol reduced Aβ levels and deposition in an AD mouse model [28], no previous study has examined such effects using TRF. Moreover, the mechanisms for reduced Aβ by vitamin E are still unknown. Overall, these findings demonstrated various neuropathological effects of vitamin E as individual analog or a mixture in in vitro and in vivo experiments. In the present study, we investigated direct interactions between Aβ and TRF in vitro and the effects of TRF supplementation on AD-like pathogenesis and age-related cognitive decline in the AβPPswe/PS1dE9 double-transgenic AD mouse model.
MATERIALS AND METHODS
Preparation of TRF and PO
An in vitro assay was conducted to study the effects of TRF on Aβ42 aggregation. The TRF and palm oil stripped of vitamin E (PO) were purchased from Sime Darby Foods and Beverages Sdn Bhd (Selangor, Malaysia) and Malaysian Palm Oil Board (MPOB) respectively. The TRF used consists of α-tocotrienol (196.0 mg/g), β-tocotrienol (24.0 mg/g), γ-tocotrienol (255.0 mg/g), δ-tocotrienol (75.0 mg/g), and α-tocopherol (168.0 mg/g), while vitamin E was not detected in the PO as mentioned by Nesaretnam, et al. [29]. The TRF was prepared for experimental application according to a previous study with modification [30]. Briefly, TRF or PO stock at 5% v/v was dissolved in 100% ethanol and stored at –30°C. Prior to use, 60 μL of 5% v/v TRF or PO stock was incubated with 45 μL of phosphate-buffered saline (PBS) at 37°C overnight. To obtain 1% v/v TRF or PO, 40 μL of 100% ethanol and 55 μL of PBS were added to the TRF or PO mixture, and then 100 μL of a mixture containing 100% ethanol and PBS [1:1] was added. Further dilutions were carried out using PBS to obtain concentrations of 0.001%, 0.01%, and 0.1% v/v TRF or PO.
Preparation of Aβ42 aggregates
Human Aβ1-42 peptide (Aβ42, Peptide Institute, Osaka, Japan) was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at 1 mM [31]. The mixture was then incubated at 37°C for 1 h, aliquoted into microcentrifuge tubes and evaporated for 30 min in an evaporation chamber. The peptide residue was stored at –30°C until use. Prior to use, this HFIP-treated Aβ42 was dissolved in dimethyl sulfoxide (DMSO; Wako, Osaka, Japan) to 0.5 mM, vortexed, and further diluted to 20 μM with PBS. To examine the effects of TRF and PO on Aβ42 aggregation, the Aβ42 solution was mixed with TRF or PO to a final concentration of 10 μM Aβ42 plus 0.001%, 0.01%, or 0.1% v/v TRF or PO. As a control, PBS containing 5% ethanol was added to the Aβ42 solution. The mixture was incubated at 37°C for 6, 24, and 48 h and then analyzed by gel electrophoresis, thioflavin T assay and cell viabilityassay.
Gel electrophoresis and western blotting
After incubation, samples were diluted in 4× sample buffer (62.5 mM Tris, pH 6.8, 1% sodium dodecyl sulfate (SDS), 25% glycerol) in PBS without heating to concentration of 2.25 μM. Ten-microliter of samples were loaded into 10% –20% precast polyacrylamide gels (SuperSep Ace; Wako) for electrophoresis (20 mA/gel for 65 min). After electrophoresis, samples were transferred onto polyvinylidene difluoride (PVDF) membranes (Immobilon-P, Merck Millipore). Membranes were washed with Tris-buffered saline (25 mM Tris-HCl, pH 7.4, 0.9% NaCl) containing 0.1% Tween 20 (TBS-T) for 10 min, blocked with 5% skim milk in TBS-T for 60 min, washed three times with TBS-T (7 min/wash), and incubated with mouse monoclonal anti-Aβ antibody clone 6E10 (1:2000; Covance, Princeton, NJ, USA) in TBS-T at 4°C overnight. After three washes with TBS-T (7 min/wash), immunolabeled membranes were incubated with HRP-conjugated anti-mouse IgG antibody (1:20000; Jackson ImmunoResearch, West Grove, PA, USA) in TBS-T containing 0.5% skim milk for 60 min at room temperature (RT). The membranes were then washed three times with TBS-T, and labeling was visualized by chemiluminescence (SuperSignal West Pico; Thermo Scientific, Rockford, IL, USA) using a lumino image analyzer (ImageQuant LAS 4000 mini; GE Healthcare Life Sciences, Japan).
Thioflavin T (ThT) fluorescence assay
A stock solution was prepared by dissolving ThT (AnaSpec, Fremont, CA, USA) in DMSO at 5 mM. A 20 μL sample of Aβ42 incubated with TRF, PO, or PBS (containing 5% ethanol) was periodically added to 180 μL of 5 μM ThT dissolved in PBS and the fluorescence measured in 96-well black plates (Greiner Bio-one, Frickenhausen, Germany) using an Infinite M200 microplate reader (Tecan, Grödig, Austria) at excitation and emission wavelengths of 450 nm and 482 nm, respectively. As a control, ThT was mixed with PBS containing only TRF or PO. The Aβ42-specific fluorescence was calculated by subtracting the fluorescence intensity of the control solution. Each measurement was performed in duplicate. Data represent the means of three independent experiments.
Photo-induced cross-linking of unmodified protein (PICUP) assay
The PICUP assay was performed according to a previous study [32]. Briefly, a mixture of 50 μM Aβ1-40 or Aβ1-42 peptides, 2 mM tris(2,2′-bipyridyl)dichlororuthenium(II) and 40 mM ammonium persulfate was mixed with TRF or PO at 1:1 ratio. Immediately, the mixture was cross-linked by irradiation to visible light for 1 s and then the reaction was quenched by addition of Tricine sample buffer and dithiothreitol. The samples were subjected to SDS-polyacrylamide gel electrophoresis under reducing conditions with 15–20% Tricine gel (SuperSep Ace; Wako) and the protein was visualized by silver staining with a SilverQuest Staining Kit (Invitrogen, California, USA).
Cell viability assay
Human neuroblastoma cell line SH-SY5Y (ATCC, Manassas, Virginia, USA) was cultured in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 (Nacalai Tesque, Kyoto, Japan) containing 10% (v/v) fetal bovine serum in a humidified atmosphere of 5% CO2/95% air at 37°C.
Ten-micromolar Aβ42 was incubated at 37°C with 0.001%, 0.01%, and 0.1% v/v TRF or PO as described in the method above. The vehicle was served as a control containing 5% ethanol. For treatment, the mixtures were dissolved in DMEM/Ham’s F-12 (Nacalai Tesque) to final concentration of 0.3 μM Aβ42 and 0.00003%, 0.0003%, 0.003% v/v TRF or PO. The final concentration of vehicle was 0.15% ethanol in medium.
Cell viability was assessed by the MTT assay with a MTT Cell Count Kit (Nacalai Tesque). The assay was based on a reduction of tetrazolium salt (MTT; 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) by dehydrogenase in viable cells into a purple formazan product. Therefore, high amount of formazan formed reflects an increase in number of living cells. The cells used in this study was seeded at 5.0×104 cells per well in a 96-well microplate and incubated at 37°C in humidified atmosphere for 24 h. After incubation, the culture medium was replaced with 100 μl of new medium with or without Aβ and TRF or PO. Ten-microliter of MTT solution was added to each well after 20 h of incubation at 37°C in humidified atmosphere and further incubated for another 4 h. Then, 100 μl of solubilization solution was added to the wells and incubated at 37°C in humidified atmosphere overnight. The absorbance was measured at 570 nm with a reference wavelength of 700 nm using an Infinite M200 microplate reader (Tecan). Each measurement was performed in triplicate. Data represent the means of three independent experiments.
Animals
AβPPswe/PS1dE9 double transgenic mice (AβPP/PS1), expressing a chimeric mouse/human amyloid protein precursor (Mo/HuAPP695swe) and a mutant human presenilin 1 with deletion at exon-9 (PS1-dE9), were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Mice were maintained by breeding heterozygous females with wild-type (WT) males, and offspring were genotyped by polymerase chain reaction. Only heterozygous and WT male mice were used in this study. All mice were housed at 23°C under a 12-h/12-h light/dark cycle (lights on from 8:00–20:00) with ad libitum access to food and water. Experiments were conducted with approval of the Animal Care and Use Committee of Shiga University of Medical Science.
Supplementation
The TRF was dissolved at 12 mg/mL in PO. Five-month-old AβPP/PS1 mice were divided into three groups. Two groups received daily water (control, 5 mL/kg body weight) or PO as a vehicle (5 mL/kg body weight) and the other received daily TRF (60 mg/kg body weight) by oral administration. All mice received daily supplementation from 5 to 15 months of age.
Novel object recognition test
The novel object recognition test was conducted according to a previous study with modification [33]. A circular home-built chamber was made with 38 cm in diameter, with a 22-cm high wall. The test was conducted over four consecutive days and consisted of four phases: pre-habituation, habituation, training, and testing. Wild-type mice (WT; n = 13) and AβPP/PS1 mice that received water (Cont; n = 9), TRF (n = 11), or PO (n = 10) were kept in the testing room at least 30 min before the test for acclimation. On day 1, pre-habituation was conducted by allowing each mouse to freely explore the empty chamber for 5 min. On days 2 and 3, mice were habituated in the empty chamber for 20 min. On day 4, each mouse was given a training trial followed by a testing trial. During the training trial, two identical objects were placed in opposite positions of the chamber at the same distance from the wall, and the mouse was allowed to explore the objects for 10 min before return to the cage. In the test trial conducted 1 h later, the mouse was placed again into the same chamber, but one of the identical objects had been replaced with a novel object. The mouse was allowed to freely explore the objects for 5 min before return to its cage. The objects used in this study were of different shapes and colors but identical in size and were fixed to the chamber floor to prevent movement. All objects and the chamber were cleaned with 70% ethanol after each trial.
Novel object recognition was measured by the ratio of novel object exploration time to total exploration time (familiar plus novel) or the recognition index. Exploration time was defined as the time the mouse spent within 2 cm of the object sniffing with nose directed toward the object or directly pawing or biting the object. Sitting or standing on the object was not recognized as exploration. To exclude environmental factor that may contribute to location preference independent of object familiarity, the ratio of exploration times of the two identical objects was also measured during the training trial. The formula for calculating location preference in the training trial and the recognition index in the testing trial are as follows:
Location preference = Time exploring one of the identical objects/Total time exploring both identical objects×100%
Recognition index = Time exploring novel object/(Time exploring novel object + Time exploringfamiliar object)×100%
Brain tissue preparation
Four mice from each group (water, PO, TRF) were used for biochemical analysis and histochemical staining. The rest of the mice were employed for other experiments. Mice were sacrificed at 15 months of age by cervical dislocation and brain tissue prepared as described previously [31]. In brief, the brain was removed and divided into two hemispheres. One hemisphere was fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (PB, pH 7.4) for 24 h and then cryoprotected in 15% sucrose dissolved in 0.1 M PB containing 0.1% sodium azide for at least 2 days. Coronal sections were cut at 20-μm thickness using a cryostat and kept in 15% sucrose until used for immunohistochemistry or thioflavin-S staining (below). The other hemisphere was kept at –80°C until preparation of homogenate for biochemical measures of Aβ40, Aβ42, and Aβ oligomers. The frozen brains were cut into 1-mm thick coronal sections. To measure Aβ40 and Aβ42 levels, the hippocampus and cortex from the two brain sections were dissected. To measure Aβ oligomers, the hippocampus and cortex were combined because Aβ oligomers could not be detected in separate 1:3 dilutions of these regions (data not shown). Thus, we decided to use a lower dilution (1:1), which required a higher amount of brain tissue. The combination of the hippocampus and cortex enabled the detection of Aβ oligomers using an ELISA kit. Samples of the hippocampus, cortex, and the combined hippocampus and cortex were homogenized in cold TBS (1 mL/150-mg wet weight) containing protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). Homogenized tissue was sonicated and centrifuged at 104,300× g for 60 min at 4°C. Supernatant was collected as soluble Aβ (TBS fraction) and pellet was then resuspended with 70% formic acid in water, sonicated, and centrifuged at 104,300× g for 60 min at 4°C. The recovered supernatant was neutralized with 1 M Tris base and stored at –80°C as insoluble Aβ (FA fraction). Protein concentration was determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules,CA, USA).
Immunohistochemical and thioflavin-S staining
All staining was performed using free-floating coronal brain sections fixed in 4% paraformaldehyde in 0.1 M PB. The brain sections were washed with 0.1 M PBS containing 0.3% Triton X-100 (PBS-T, pH 7.4) and then incubated in 0.3% hydrogen peroxide containing 0.1% sodium azide in PBS-T for 20 min at RT to eliminate endogenous peroxidase activity. After incubation, the sections were washed three times with PBS-T (10 min/wash) and blocked with 2% bovine serum albumin (BSA) in PBS-T for 30 min at RT. Blocked sections were then incubated with rabbit polyclonal antibodies against the N-terminal of human Aβ (1:500; Immuno-Biological Laboratories) or ionized-calcium binding adapter molecule 1 (Iba1, 1:5000; Wako) in PBS-T containing 0.2% BSA overnight at 4°C. After several washes with PBS-T, immunolabeled sections were incubated with biotinylated anti-rabbit IgG antibody (1:1000; Vector Laboratories, Burlingame, CA, USA) for 60 min at RT. Following several washes with PBS-T, sections were incubated with avidin–biotin complex solution (Vectastain ABC Elite kit, 1:3000; Vector Laboratories) for 60 min at RT, washed several times with PBS-T, and the immunostaining visualized by reaction with 3,3′-diaminobenzidine (Dojindo Laboratories, Kumamoto, Japan) and nickel ammonium. The sections were then mounted onto glass slides, dehydrated in an increasing ethanol series, and cover slipped with Entellan mounting medium (Merck Millipore). Images were captured using a DP27 digital camera (Olympus, Tokyo, Japan).
Thioflavin-S staining was performed according to a previous study [34]. Briefly, the brain sections from all supplemented groups were mounted onto glass slides, dehydrated with 70% and 80% ethanol for 1 min each and stained with freshly made and filtered 1% thioflavin-S (Chroma-Gesellschaft Schmid, Kongen, Germany) dissolved in 80% ethanol for 15 min in the dark. As a control, slices were subjected to the same staining protocol but omitting thioflavin-S. The slides were then washed with 80% and 70% ethanol for 1 min each, rinsed twice in distilled water, mounted with Immuno-Mount (Thermo, Pittsburgh, PA, USA), cover slipped, and dried for at least 2 h in the dark. The slides were examined with an upright Olympus BX61 microscope (Olympus, Japan) equipped with an FITC filter set (U-MWB; band-pass excitation 450–480 nm, dichroic mirror 505 nm, long-pass emission peak 515 nm).
Enzyme-linked immunosorbent assay
Levels of Aβ40 and Aβ42 in the cortex and hippocampus were determined using an enzyme-linked immunosorbent assay (ELISA) kit (Wako) according to the manufacturer’s protocol. Briefly, 100 μL of homogenate was pipetted onto the primary antibody-coated microplate and incubated overnight at 4°C. Homogenate was then removed from wells and the plates rinsed five times with washing solution. Then, a 100-μL secondary antibody solution was added to each well. Plates were sealed and incubated at 4°C for 1 h for Aβ42 detection and 2 h for Aβ40 detection. After five rinses with washing solution, chromogen was added to each well, and blue color was developed in the dark. The reaction was terminated by addition of 100 μL stop solution, and absorbance was measured at 450 nm using an Infinite M200 microplate reader (Tecan). The levels of Aβ42 and Aβ40 were calculated using standard curves.
The oligomeric form of Aβ peptide was measured with ELISA kit from Biosensis (Thebarton, Australia). A 100-μL TBS fraction was added to the pre-coated microplate well and incubated for 24 h at 4°C. After washed for five times with washing solution, 100 μL of detection antibody was added to each well and the plate was incubated for 1 h on a shaker (140 rpm) at room temperature. After five rinses, streptavidin-HRP conjugate was added into each well and the plate was incubated for 30 min on a shaker at room temperature. Then, a 100-μL TMB solution was added to the well after five rinses and incubated for 8 min at room temperature in dark. The reaction was stopped by addition of 100 μL stop solution into each well. The absorbance was measured at 450 nm using an Infinite M200 microplate reader (Tecan). The levels of Aβ oligomers were calculated using a standard curve.
Evaluation of plaques
Aβ- and Iba-1-positive areas in the cortex and hippocampus were viewed with an Olympus BX50 microscope (Olympus, Tokyo, Japan) and images captured with a digital camera (DP27, Olympus). Images were analyzed using ImageJ 1.48 V (National Institutes of Health; USA). First, images were transformed to eight-bit grayscale. The threshold was set to 110, and the percent immunoreactive area was obtained. Four mice from each group were subjected to immunohistochemical staining, and for each mouse, five brain sections wereanalyzed.
Thioflavin-S-positive plaques were evaluated according to previous studies with modification [35, 36]. Images were captured at four different locations within the cortex and hippocampus at ×20 magnification. The images were transformed to eight-bit grayscale format using ImageJ. The calibration was set at 2.58 (distance in pixels), 1.00 (known distance), 1.0 (pixel aspect ratio), and μm (unit length) and “global” was activated. The threshold was set by comparison with the original images because of differences in background staining across sections. The number of plaques was counted setting the size to 100–10000 μm2, in a defined area of 0.2 mm2. Four mice from each group were used for thioflavin-S staining, and two brain sections were analyzed per mouse.
Data analysis
Statistical analysis was performed using Graphpad Prism 6 (GraphPad Software; La Jolla, CA, USA). Data are presented as mean±standard error of mean (S.E.M.). Group mean was compared by one-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test for multiple comparisons.
RESULTS
In vitro Aβ42 fibrillogenesis was inhibited by TRF
We investigated direct interactions between Aβ and TRF in in vitro experiments. Firstly, SDS-polyacrylamide gel electrophoresis was carried out to study the effect of TRF on the profiles of Aβ42 aggregates. Incubation of Aβ42 with vehicle (PBS containing 5% ethanol) at 37°C for 24 h resulted in a large amount of high molecular weight Aβ42 aggregate at the top of the gel and little “smear” down the length of the gel, indicating that high molecular weight Aβ42 aggregate was too large to enter the gel (Fig. 1A). A similar gel staining pattern was observed for Aβ42 incubated with 0.1% v/v PO. In contrast, a darker smear and more intense bands of low molecular weight Aβ42 aggregates, which are small enough to enter the gel, were observed in gels loaded with Aβ42 incubated in 0.1% v/v TRF. We further examined the dose-dependent effect of TRF on this disruption of high molecular weight Aβ42 fibril formation. Indeed, as the concentration of TRF was increased from 0.001% to 0.01% and 0.1%, the intensity of lower molecular weight fibrils increased, and trimers and tetramers appeared. These results strongly suggest that TRF can disrupt the formation of high molecular weight Aβ42aggregates.
Next, we investigated the effect of TRF on the formation of fibrillar Aβ42 in vitro through ThT fluorescence assay after 0-, 6-, 24-, and 48-h incubation. The fluorescence intensity of Aβ42 incubated alone (5% ethanol in PBS) increased progressively with incubation time (Fig. 1B). Consistent with immunoblotting, formation of Aβ42 fibrils was significantly reduced in the presence of 0.1% v/v TRF after 24 h (p < 0.01) and 48 h (p < 0.05) compared with Aβ42 incubated alone and compared with PO after 24 h (p < 0.05). Further, fluorescence intensity decreased progressively at increasing TRF concentration (0.001%, 0.01%, and 0.1% v/v), with the reduction significant at 0.001% v/v after 48 h (p < 0.05), at 0.01% v/v after 24 and 48 h (p < 0.05 and p < 0.01, respectively), and at 0.1% after 24 and 48 h (p < 0.01) compared with Aβ42 incubated alone.
Amyloid-β oligomerization was inhibited by TRF under cell-free conditions
We examined the potential of TRF to inhibit Aβ oligomer formation under cell-free conditions using PICUP method. As shown in Fig. 2A, the cross-linked mixtures of 25 μM Aβ40 in PBS (control) resulted in oligomer formation from dimer to trimer, although uncross-linked samples were visualized as 1 band representing the monomeric form of Aβ. Cross-linked mixtures containing 25 μM Aβ40 with 0.001% v/v TRF showed a slight reduction of Aβ40 oligomer forms (dimer and trimer). Furthermore, 0.1% TRF markedly inhibited Aβ40 oligomer formation. Similarly, cross-linked mixtures containing 25 μM Aβ42 with TRF showed a dose-dependent reduction in Aβ42 oligomers. The Aβ40 and Aβ42 oligomer formations were not changed by PO treatment. In addition, we conducted PICUP experiment using glutathione-S-transferase (GST) to investigate the specificity of TRF on inhibiting PICUP oligomer formation. TRF did not inhibit GST oligomerization in PICUP assay (data not shown). Therefore, this finding confirmed the specificity of TRF to inhibit Aβ oligomerization.
TRF-treated Aβ aggregates did not promotes SH-SY5Y cells viability
We investigated the cell toxicity of Aβ42 aggregates that were prepared in the presence of TRF during the aggregation process. To evaluate cell toxicity, 10 μM Aβ42 were incubated for 24 h at 37°C to form aggregates, in the presence or absence of 0.1% v/v TRF or PO, and then the Aβ42 aggregates were treated in SH-SY5Y cells for 24 h at final concentrations of 0.3 μM Aβ42 and 0.003% v/v TRF or PO. A significant reduction of cell viability approximately up to 56% was detected in 0.3 μM Aβ42 aggregates, compared to vehicle (p < 0.001) (Fig. 3A). No significant difference was found in cell viability of 0.3 μM Aβ42 aggregates prepared in the presence or absence of TRF or PO compared to 0.3 μM Aβ42 aggregates alone. In addition, no significant difference was found in cell viability in the treatments with TRF (Fig. 3B) and PO (Fig. 3C) at final concentration of 0.00003%, 0.0003%, and 0.003% v/v, compared tovehicle.
TRF supplementation improved cognitive impairment in AβPP/PS1 mice
Novel object recognition test was conducted to study the effect of TRF supplementation on behavioral disorder of AβPP/PS1 mice. The test was performed according to the schematic diagram shown in Fig. 4. All mice spent equal time exploring the identical objects during the training trial, indicating no location preference (Fig. 5A). Mice were then tested in novel object recognition after 60 min by measure the time spent exploring a new object that replaced one of the two identical (familiar) objects. Wild-type mice spent a significantly longer time exploring the novel object compared with the familiar object (79% recognition index), indicating preserved memory for the familiar object. On the contrary, AβPP/PS1 mice supplemented with water (control) showed a significantly lower recognition index (approximately 60%), indicative of AD-related cognitive decline. In contrast, AβPP/PS1 mice administered 60 mg/kg TRF daily showed a recognition index comparable with WT mice (80%). Administration of PO slightly enhanced the recognition index compared with controls, but the increase did not reach significance. This finding demonstrates a therapeutic effect of TRF on age-related cognitive decline in AD model mice (Fig. 5B).
TRF reduced Aβ deposition in the brain of AβPP/PS1 mice
Immunohistochemical staining with anti-Aβ N-terminal antibody was measured in the brains of AβPP/PS1 mice to assess the effects of control, PO, and TRF supplementation on Aβ deposition (Fig. 6A). The immunoreactive area was uniformly higher in cortex than hippocampus (Fig. 6B). Hippocampal deposition of Aβ was slightly reduced in TRF-supplemented mice, although the difference was not significant (Fig. 6B, left), while a significant reduction was observed in the cortex of TRF-supplemented mice compared with control and PO groups (p < 0.05; Fig. 6B, right).
Thioflavin-s staining was also conducted to investigate fibrillar-type Aβ deposition in brain sections [35, 37]. Representative images of hippocampus and cortex stained with thioflavin-s are presented in Fig. 6C. Mice receiving daily TRF exhibited fewer thioflavin-s-positive Aβ deposits in both brain regions compared with control and PO groups. Semiquantitative analysis demonstrated a significant reduction in the number of amyloid plaques in the hippocampus and cortex of TRF group mice compared with control and PO groups (p < 0.05; Fig. 6D).
Microglial activity was not affected by TRF
Immunohistochemical staining for Iba1 was performed to evaluate microglial activation in the brain sections of WT mice and control, PO-treated, and TRF-treated AβPP/PS1 mice (Fig. 7A). Control AβPP/PS1 group mice exhibited significantly higher Iba1-immunoreactivity in the hippocampus and cortex compared with WT (p < 0.01), but there were no differences in staining among the control, PO, and TRF groups (Fig. 7B).
Aβ levels were not affected by TRF supplementation
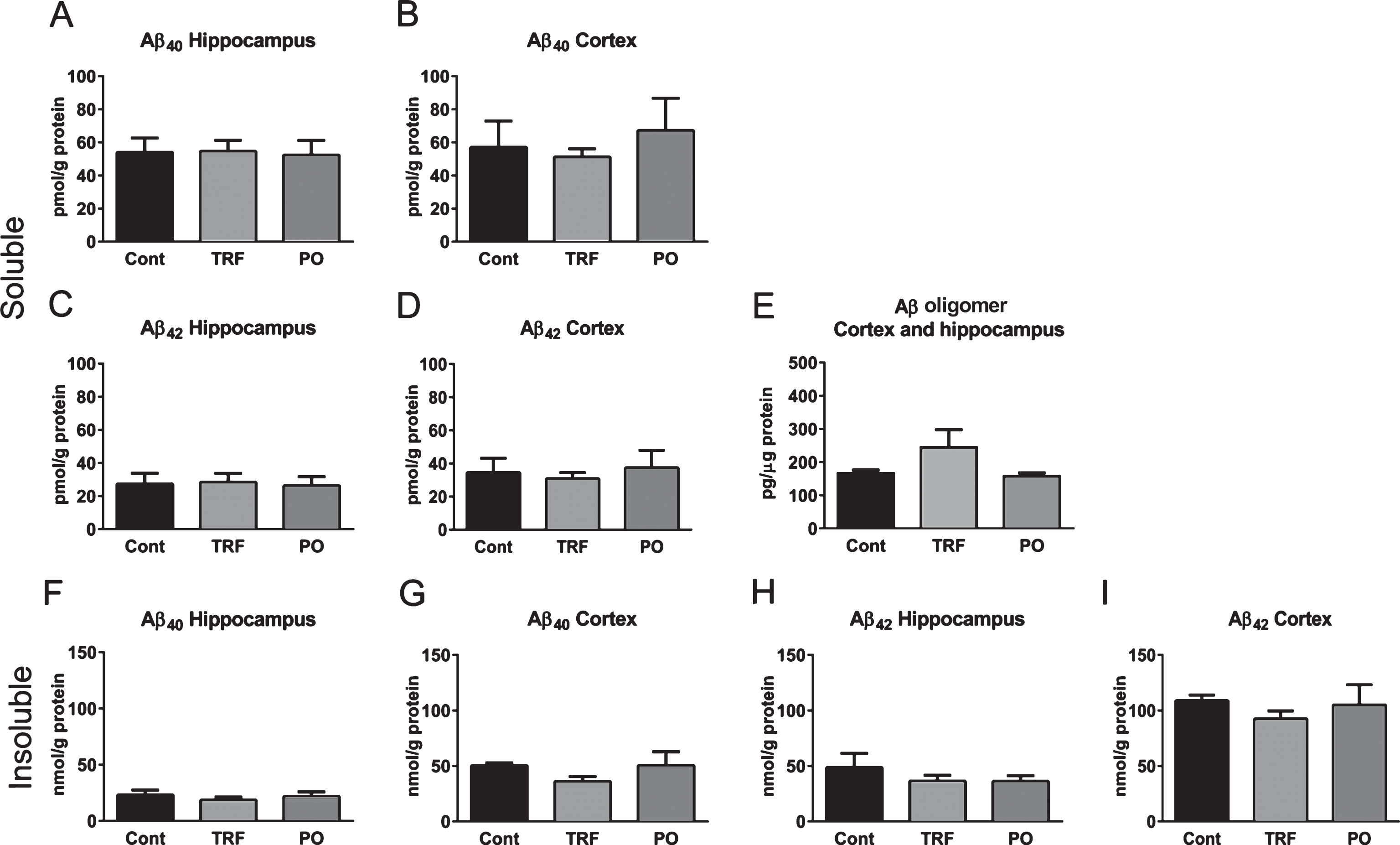
Finally, we examined the effects of TRF supplementation on Aβ levels in brain homogenates. The levels of soluble Aβ40, Aβ42, and Aβ oligomers in the TBS fraction from hippocampus and cortex did not differ between mouse groups (Fig. 8A–E). There were no statistically significant differences in the level of Aβ oligomers between groups; however, the level of Aβ oligomers appears to increase in the TRF group. Similarly, levels of insoluble Aβ40 and Aβ42 did not differ (Fig. 8F–I), although the TRF group displayed a trend for lower Aβ40 and Aβ42 in cortex compared with the control group (Fig. 8G, I).
DISCUSSION
The present study showed that TRF inhibited the formation of high molecular weight Aβ aggregates by reduction Aβ oligomers, and eventually attenuated Aβ fibrillization in vitro. These results were supported by a reduction of Aβ fibril and deposit in the brains of AβPP/PS1 mice supplemented with TRF. Collectively, these data suggested the modulation of Aβ by TRF, thus contributing to the recovery of cognitive impairment in AβPP/PS1 mice.
Amyloid fibril formation begins with polymerization of soluble Aβ oligomers to form protofibrils that coalesce into insoluble Aβ fibrils and plaques [38, 39]. The aggregation into Aβ fibrils results from interactions between aromatic residues of Aβ peptides, which stabilize the antiparallel β-sheet structure [40–42]. However, the aromatic interactions between Aβ peptides can be diminished by small aromatic molecules such as polyphenols that act as in vitro inhibitors of Aβ fibril formation [43, 44]. This notion was proved by our in vitro study that demonstrated TRF inhibited the formation of Aβ oligomers and increased lower molecular weight Aβ42 aggregates thus showing an inhibition of Aβ fibril formation in a dose-dependent manner. Although the mechanism of action was not studied, we speculate that the aromatic structure of tocopherol and tocotrienols specifically binds to Aβ peptides that prevent from polymerization into fibril form. Furthermore, these findings were in agreement with our in vivo results shown by a reduction of Aβ fibril in the hippocampus and cortex of AβPP/PS1 mice supplemented with TRF.
TRF was shown to inhibit Aβ42 fibrillogenesis in vitro and in vivo. As shown in our in vitro study, it is probable that the inhibition of Aβ42 fibrillogenesis induced the formation of lower molecular weight Aβ aggregates, including oligomers, known as the most toxic species of Aβ assemblies. Therefore, we investigated the cell toxicity of Aβ aggregates that were prepared in the presence of TRF during the aggregation process. However, no exacerbation of cell toxicity was detected between the Aβ aggregates prepared in the presence and absence of TRF. In addition, PICUP experiments showed that TRF inhibited both Aβ40 and Aβ42 oligomer formations. Based on these results, the inhibitory effect of TRF on Aβ aggregation would be favorable for improving pathological changes in Aβ in AD.
The possibility that TRF inversely make the already aggregated Aβ to form oligomeric species in vivo remains untested. Indeed, the TRF group showed an apparent increase in Aβ oligomers when compared with control; however, this effect was not statistically significant. Notably, one mouse in the TRF group showed a high level of Aβ oligomers that caused a larger standard deviation, as shown by the increased length of the error bar. However, the other three mice from the TRF group showed similar levels of Aβ oligomers when compared with the control and PO groups. Thus, TRF supplementation does not appear to increase oligomers via disaggregation of Aβ in vivo. This study is the first to investigate the effect of TRF on Aβ aggregation, and to date, no studies have examined the ability of TRF to disaggregate Aβ into oligomeric species in vivo or in vitro. Thus, further study is needed to clarify this potential effect.
According to Herrup [45], deposition of Aβ alone may not be the only pathogenic event required for the development and clinical manifestations of AD. Indeed, Aβ accumulation is also observed in the brains of cognitively healthy older people as reviewed by Jagust [46]. Therefore, in the present study, we also investigated the efficacy of TRF to preserve recognition memory in AβPP/PS1 mice using a spontaneous behavioral response (exploration of novel objects) that does not require stimulus presentation [47]. Our results demonstrated that the AβPP/PS1 mice supplemented daily with water showed a significant decline in novel object exploration time compared with aged-matched WT, consistent with other age-related memory impairment in AβPP/PS1 mice [48–50]. This decline in cognitive function was rescued to a greater extent by daily TRF supplementation than by PO, suggesting TRF is the active antifibrillogenic and neuroprotective component.
Using Aβ immunohistochemical and thioflavin-s staining, we demonstrated that TRF reduced Aβ deposition and fibrillization in vivo. However, neither Aβ40, Aβ42, and Aβ oligomers levels nor microglial activity indicative of brain inflammation was affected by TRF. A previous study reported that α-tocopherol administered as a chow diet supplement at a young age reduced Aβ levels and deposition in the brains of Tg2576 mice [28]. It is possible that TRF did not affect Aβ levels or microglial activity due to a few limitations such as insufficient dose, suboptimal method of administration, low bioavailability, late age of initiation (5 months), or inherent differences in AD animal models. These AβPP/PS1 mice exhibit a higher level of Aβ accumulation than Tg2576 mice and already show Aβ deposition at 5 months [51]. In contrast, supplementation of α-tocopherol was started as early as 6 months before Aβ deposition in Tg2576 mice [28]. Thus, earlier dosing may have more pervasive benefits. Furthermore, we did not measure TRF bioavailability under oral dosing. It was reported that the bioavailability of α-tocotrienol under chow diet supplementation was very low [52]. Alternatively, another study reported that oral α-tocotrienol reached the brains of pregnant rats and fetuses [53], consistent with our results showing that 60 mg/kg was sufficient to reduce both neuropathological and functional progression of AD in AβPP/PS1 mice. Further studies are necessary to determine the optimal dosing protocol for TRF and also to measure the concentration of TRF in specific brain regions that contributed to these findings.
Epidemiological studies suggest that intake levels of specific dietary micronutrients, including vitamin E, protect against AD and associated cognitive impairment [20]. Furthermore, food sources containing tocotrienols may be particularly effective for prevention of age-related neurodegenerative disorders due to antioxidant, anti-inflammatory, and cholesterol lowering capacities [19]. The RRR-α-tocopherol quinone component inhibited Aβ42 fibril formation while α-tocopherol failed to do so. [22]. In humans, higher plasma vitamin E was associated with lower AD incidence and combination of vitamin E forms related to neuroprotection [24]. It is possible that no medication with a single target or mode of action will ever prove sufficient for AD treatment. However, the multiple neurochemical and clinical effects of vitamin E mixtures, such as TRF found in palm oil, suggest great promise as an alternative treatment strategy.
As described by Petersen, et al. [54], regardless of the beneficial effects shown by TRF, previous randomized controlled trials with large sample sizes have indicated that α-tocopherol failed to rescue neither cognitive decline in AD patients nor the conversion from mild cognitive impairment to AD. This group demonstrated that supplementation of vitamin E to subjects with amnestic mild cognitive impairment showed no significant differences in the progression to AD when compared with placebo groups. In the TEAM-AD VA study [18], Dysken, et al. observed that α-tocopherol successfully slowed decline only in the Alzheimer’s Disease Cooperative Study/Activities of Daily Living (ADCS-ADL) Inventory scores, but not in the Mini-Mental State Examination (MMSE) scores nor the Alzheimer Disease Assessment Scale-Cognitive Subscale (ADAS-cog) scores. Therefore, this study revealed the beneficial effects of α-tocopherol on ADL but not cognitive function. Similar finding was also observed by Sano, et al. [55]. Overall, the randomized controlled trials with large sample size, all similarly demonstrated no beneficial effects of α-tocopherol on cognitive function in patients with AD.
The mechanisms in neuroprotection of TRF are likely due to tocotrienols because they are the major constituents of vitamin E. Unlike tocopherols, tocotrienols contain three trans double bond in the side chain. A previous study revealed that α-tocotrienol exhibits a significantly greater peroxyl radical scavenging potency than α-tocopherol in phosphatidylcholine liposomes [56]. In addition, at nanomolar concentration, α-tocotrienol, but not α-tocopherol, protects against glutamate-induced neuronal death by suppressing inducible pp60 c-Src kinase activation [57]. Another study showed that α-tocopherol had no effect on Aβ aggregation [22]; however, our results indicated that TRF inhibited the formation of Aβ aggregates, suggesting an inhibitory effect of tocotrienols on Aβ aggregation. Taken together, the TRF as well as tocotrienols have a higher potential for neuroprotection when compared with α-tocopherol.
In summary, the therapeutic effects of TRF on Aβ pathogenesis were evaluated in vitro and in vivo. The TRF successfully inhibited Aβ oligomers formation, and Aβ aggregation in vitro, and reduced Aβ accumulation in vivo. Although Aβ levels and microglial activity were not affected, cognitive impairment was rescued by TRF in AβPP/PS1 mice. Therefore, we propose that TRF could be a promising therapeutic agent for preventing AD.
ACKNOWLEDGMENTS
This study was supported by JSPS KAKENHI 26290022, I.T. and 15K16321, D.Y. from Japan Society for Promotion of Science, and a Long-Term Research Grant Scheme LRGS/BU/2012/UKM-UKM/K/04 from Ministry of Education Malaysia.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0685r2).
REFERENCES
[1] | Selkoe DJ ((1997) ) Alzheimer’s disease–genotypes, phenotypes, and treatments. Science 275: , 630–631. |
[2] | Auld DS , Kornecook TJ , Bastianetto S , Quirion R ((2002) ) Alzheimer’s disease and the basal forebrain cholinergic system: Relations to β-amyloid peptides, cognition, and treatment strategies. Prog Neurobiol 68: , 209–245. |
[3] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[4] | Blennow K , Leon MJd , Zetterberg H ((2006) ) Alzheimer’s Disease. Lancet 368: , 387–403. |
[5] | Duff K , Eckman C , Zehr C , Yu X , Prada CM , Perez-tur J , Hutton M , Buee L , Harigaya Y , Yager D , Morgan D , Gordon MN , Holcomb L , Refolo L , Zenk B , Hardy J , Younkin S ((1996) ) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383: , 710–713. |
[6] | Rovelet-Lecrux A , Hannequin D , Raux G , Le Meur N , Laquerriere A , Vital A , Dumanchin C , Feuillette S , Brice A , Vercelletto M , Dubas F , Frebourg T , Campion D ((2006) ) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38: , 24–26. |
[7] | Di Paolo G , Kim TW ((2011) ) Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat Rev Neurosci 12: , 284–296. |
[8] | Horvath G , Wessjohann L , Bigirimana J , Jansen M , Guisez Y , Caubergs R , Horemans N ((2006) ) Differential distribution of tocopherols and tocotrienols in photosynthetic and non-photosynthetic tissues. Phytochemistry 67: , 1185–1195. |
[9] | Sundram K , Sambanthamurthi R , Tan YA ((2003) ) Palm fruit chemistry and nutrition. Asia Pac J Clin Nutr 12: , 355–362. |
[10] | Sen CK , Rink C , Khanna S ((2010) ) Palm oil-derived natural vitamin E alpha-tocotrienol in brain health and disease. J Am Coll Nutr 29: (3 Suppl), 314S–323S. |
[11] | Chin SF , Ibahim J , Makpol S , Abdul Hamid NA , Abdul Latiff A , Zakaria Z , Mazlan M , Mohd Yusof YA , Abdul Karim A , Wan Ngah WZ ((2011) ) Tocotrienol rich fraction supplementation improved lipid profile and oxidative status in healthy older adults: A randomized controlled study. Nutr Metab (Lond) 8: , 42. |
[12] | Fukui K , Nakamura K , Shirai M , Hirano A , Takatsu H , Urano S ((2015) ) Long-term vitamin E-deficient mice exhibit cognitive dysfunction via elevation of brain oxidation. J Nutr Sci Vitaminol (Tokyo) 61: , 362–368. |
[13] | Khor SC , Mohd Yusof YA , Wan Ngah WZ , Makpol S ((2015) ) Tocotrienol-rich fraction prevents cellular aging by modulating cell proliferation signaling pathways. Clin Ter 166: , e81–e90. |
[14] | Zhang JS , Zhang SJ , Li Q , Liu YH , He N , Zhang J , Zhou PH , Li M , Guan T , Liu JR ((2015) ) Tocotrienol-rich fraction (TRF) suppresses the growth of human colon cancer xenografts in Balb/C nude mice by the Wnt pathway. PLoS One 10: , e0122175. |
[15] | Peh HY , Tan WS , Liao W , Wong WS ((2015) ) Vitamin E therapy beyond cancer: Tocopherol versus tocotrienol. Pharmacol Ther 162: , 152–169. |
[16] | Pearce BC , Parker RA , Deason ME , Qureshi AA , Wright JJ ((1992) ) Hypocholesterolemic activity of synthetic and natural tocotrienols. J Med Chem 35: , 3595–3606. |
[17] | Pearce BC , Parker RA , Deason ME , Dischino DD , Gillespie E , Qureshi AA , Volk K , Wright JJ ((1994) ) Inhibitors of cholesterol biosynthesis. 2. Hypocholesterolemic and antioxidant activities of benzopyran and tetrahydronaphthalene analogues of the tocotrienols. J Med Chem 37: , 526–541. |
[18] | Dysken MW , Sano M , Asthana S , Vertrees JE , Pallaki M , Llorente M , Love S , Schellenberg GD , McCarten JR , Malphurs J , Prieto S , Chen P , Loreck DJ , Trapp G , Bakshi RS , Mintzer JE , Heidebrink JL , Vidal-Cardona A , Arroyo LM , Cruz AR , Zachariah S , Kowall NW , Chopra MP , Craft S , Thielke S , Turvey CL , Woodman C , Monnell KA , Gordon K , Tomaska J , Segal Y , Peduzzi PN , Guarino PD ((2014) ) Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 311: , 33–44. |
[19] | Frank J , Chin XWD , Schrader C , Eckert GP , Rimbach G ((2012) ) Do tocotrienols have potential as neuroprotective dietary factors? Ageing Res Rev 11: , 163–180. |
[20] | Gillette-Guyonnet S , Secher M , Vellas B ((2013) ) Nutrition and neurodegeneration: Epidemiological evidence and challenges for future research. Br J Clin Pharmacol 75: , 738–755. |
[21] | La Fata G , Weber P , Mohajeri MH ((2014) ) Effects of vitamin E on cognitive performance during ageing and in Alzheimer’s disease. Nutrients 6: , 5453–5472. |
[22] | Yang SG , Wang WY , Ling TJ , Feng Y , Du XT , Zhang X , Sun XX , Zhao M , Xue D , Yang Y , Liu RT ((2010) ) Alpha-Tocopherol quinone inhibits beta-amyloid aggregation and cytotoxicity, disaggregates preformed fibrils and decreases the production of reactive oxygen species, NO and inflammatory cytokines. Neurochem Int 57: , 914–922. |
[23] | Mangialasche F , Xu W , Kivipelto M , Costanzi E , Ercolani S , Pigliautile M , Cecchetti R , Baglioni M , Simmons A , Soininen H , Tsolaki M , Kloszewska I , Vellas B , Lovestone S , Mecocci P ((2012) ) Tocopherols and tocotrienols plasma levels are associated with cognitive impairment. Neurobiol Aging 33: , 2282–2290. |
[24] | Mangialasche F , Kivipelto M , Mecocci P , Rizzuto D , Palmer K , Winblad B , Fratiglioni L ((2010) ) High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J Alzheimers Dis 20: , 1029–1037. |
[25] | Khanna S , Roy S , Parinandi NL , Maurer M , Sen CK ((2006) ) Characterization of the potent neuroprotective properties of the natural vitamin E alpha-tocotrienol. J Neurochem 98: , 1474–1486. |
[26] | Mazlan M , Johari RR , Hamid NAA ((2009) ) Induction of DNA damage and cell death by beta amyloid peptide and its modification by tocotrienol rich fraction (TRF). Med Health 4: , 8–15. |
[27] | Selvaraju TR , Khaza’ai H , Vidyadaran S , Abd Mutalib MS , Vasudevan R ((2014) ) The neuroprotective effects of tocotrienol rich fraction and alpha tocopherol against glutamate injury in astrocytes. Bosn J Basic Med Sci 14: , 195–204. |
[28] | Sung S , Yao Y , Uryu K , Yang H , Lee VM , Trojanowski JQ , Pratico D ((2004) ) Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J 18: , 323–325. |
[29] | Nesaretnam K , Khor HT , Ganeson J , Chong YH , Sundram K , Gapor A ((1992) ) The effect of vitamin E tocotrienols from palm oil on chemically induced mammary carcinogenesis in female rats. Nutr Res 12: , 879–892. |
[30] | Khee SG , Yusof YA , Makpol S ((2014) ) Expression of senescence-associated microRNAs and target genes in cellular aging and modulation by tocotrienol-rich fraction. Oxid Med Cell Longev 2014: , 725929. |
[31] | Yanagisawa D , Ibrahim NF , Taguchi H , Morikawa S , Hirao K , Shirai N , Sogabe T , Tooyama I ((2015) ) Curcumin derivative with the substitution at C-4 position, but not curcumin, is effective against amyloid pathology in APP/PS1 mice. Neurobiol Aging 36: , 201–210. |
[32] | Ono K , Condron MM , Teplow DB ((2009) ) Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc Natl Acad Sci U S A 106: , 14745–14750. |
[33] | Zhang R , Xue G , Wang S , Zhang L , Shi C , Xie X ((2012) ) Novel object recognition as a facile behavior test for evaluating drug effects in AbetaPP/PS1 Alzheimer’s disease mouse model. J Alzheimers Dis 31: , 801–812. |
[34] | Ly PT , Cai F , Song W ((2011) ) Detection of neuritic plaques in Alzheimer’s disease mouse model. J Vis Exp 15: , 108. |
[35] | Snellman A , Lopez-Picon FR , Rokka J , Salmona M , Forloni G , Scheinin M , Solin O , Rinne JO , Haaparanta-Solin M ((2013) ) Longitudinal amyloid imaging in mouse brain with 11C-PIB: Comparison of APP23, Tg2576, and APPswe-PS1dE9 mouse models of Alzheimer disease. J Nucl Med 54: , 1434–1441. |
[36] | Humpel C ((2015) ) Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front Aging Neurosci 7: , 47. |
[37] | Bussiere T , Bard F , Barbour R , Grajeda H , Guido T , Khan K , Schenk D , Games D , Seubert P , Buttini M ((2004) ) Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol 165: , 987–995. |
[38] | Serpell LC ((2000) ) Alzheimer’s amyloid fibrils: Structure and assembly. Biochim Biophys Acta 1502: , 16–30. |
[39] | Masters CL , Selkoe DJ ((2012) ) Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006262. |
[40] | Porat Y , Abramowitz A , Gazit E ((2006) ) Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des 67: , 27–37. |
[41] | Azriel R , Gazit E ((2001) ) Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. An experimental support for the key role of the phenylalanine residue in amyloid formation. J Biol Chem 276: , 34156–34161. |
[42] | Makin OS , Atkins E , Sikorski P , Johansson J , Serpell LC ((2005) ) Molecular basis for amyloid fibril formation and stability. Proc Natl Acad Sci U S A 102: , 315–320. |
[43] | Ono K , Yoshiike Y , Takashima A , Hasegawa K , Naiki H , Yamada M ((2003) ) Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J Neurochem 87: , 172–181. |
[44] | Ono K , Hasegawa K , Naiki H , Yamada M ((2004) ) Anti-amyloidogenic activity of tannic acid and its activity to destabilize Alzheimer’s beta-amyloid fibrils in vitro. Biochim Biophys Acta 1690: , 193–202. |
[45] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[46] | Jagust W ((2016) ) Is amyloid-beta harmful to the brain? Insights from human imaging studies. Brain 139: , 23–30. |
[47] | Moscardo E , Salvetti B , Becchi S , Bertini G , Fabene PF ((2012) ) The novel object recognition test in rodents: Which are the essential methodological aspects? Measuring Behavior 2012: , 476. |
[48] | Jin JL , Liou AK , Shi Y , Yin KL , Chen L , Li LL , Zhu XL , Qian L , Yang R , Chen J , Xu Y ((2015) ) CART treatment improves memory and synaptic structure in APP/PS1 mice. Sci Rep 5: , 10224. |
[49] | Jardanhazi-Kurutz D , Kummer MP , Terwel D , Vogel K , Dyrks T , Thiele A , Heneka MT ((2010) ) Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem Int 57: , 375–382. |
[50] | Guo HB , Cheng YF , Wu JG , Wang CM , Wang HT , Zhang C , Qiu ZK , Xu JP ((2015) ) Donepezil improves learning and memory deficits in APP/PS1 mice by inhibition of microglial activation. Neuroscience 290: , 530–542. |
[51] | Garcia-Alloza M , Robbins EM , Zhang-Nunes SX , Purcell SM , Betensky RA , Raju S , Prada C , Greenberg SM , Bacskai BJ , Frosch MP ((2006) ) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 24: , 516–524. |
[52] | Podda M , Weber C , Traber MG , Packer L ((1996) ) Simultaneous determination of tissue tocopherols, tocotrienols, ubiquinols, and ubiquinones. J Lipid Res 37: , 893–901. |
[53] | Roy S , Lado BH , Khanna S , Sen CK ((2002) ) Vitamin E sensitive genes in the developing rat fetal brain: A high-density oligonucleotide microarray analysis. FEBS Lett 530: , 17–23. |
[54] | Petersen RC , Thomas RG , Grundman M , Bennett D , Doody R , Ferris S , Galasko D , Jin S , Kaye J , Levey A , Pfeiffer E , Sano M , van Dyck CH , Thal LJ ((2005) ) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352: , 2379–2388. |
[55] | Sano M , Ernesto C , Thomas RG , Klauber MR , Schafer K , Grundman M , Woodbury P , Growdon J , Cotman CW , Pfeiffer E , Schneider LS , Thal LJ ((1997) ) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N Engl J Med 336: , 1216–1222. |
[56] | Suzuki YJ , Tsuchiya M , Wassall SR , Choo YM , Govil G , Kagan VE , Packer L ((1993) ) Structural and dynamic membrane properties of alpha-tocopherol and alpha-tocotrienol: Implication to the molecular mechanism of their antioxidant potency. Biochemistry 32: , 10692–10699. |
[57] | Sen CK , Khanna S , Roy S , Packer L ((2000) ) Molecular basis of vitamin E action. Tocotrienol potently inhibits glutamate-induced pp60(c-Src) kinase activation and death of HT4 neuronal cells. J Biol Chem 275: , 13049–13055. |
Figures and Tables
Fig.1
In vitro treatment with the tocotrienol-rich fraction (TRF) of palm oil prevents Aβ42 aggregation. A) Aβ42 samples treated with TRF, palm oil stripped of vitamin E (PO), or vehicle (5% ethanol in phosphate-buffered saline) for 24 h at 37°C were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. TRF-treated Aβ42 at final concentration of 0.1% v/v displayed a marked smear and intense bands compared to non-treated and PO-treated Aβ42. Amyloid-β42 incubated with 0.001%, 0.01%, or 0.1% v/v of TRF showed increasing in the density of smear and bands at concentration-dependent manner. Arrowhead indicates the top of the gel (A). B) Thioflavin T fluorescence intensity measured after 0-, 6-, 24-, and 48-h incubation of Aβ42 with vehicle (Aβ42 alone), PO, or TRF. Fluorescence intensity increased with time, indicating progressive Aβ42 fibril formation. This process was significantly slowed by TRF and PO. C) The inhibitory effect of TRF on fibril formation was concentration dependent. Data presented as mean±S.E.M. Significance (Bonferroni post hoc test after analysis of variance): *p < 0.05, **p < 0.01 versus Aβ alone; #p < 0.01 versus Aβ + 0.1% PO.
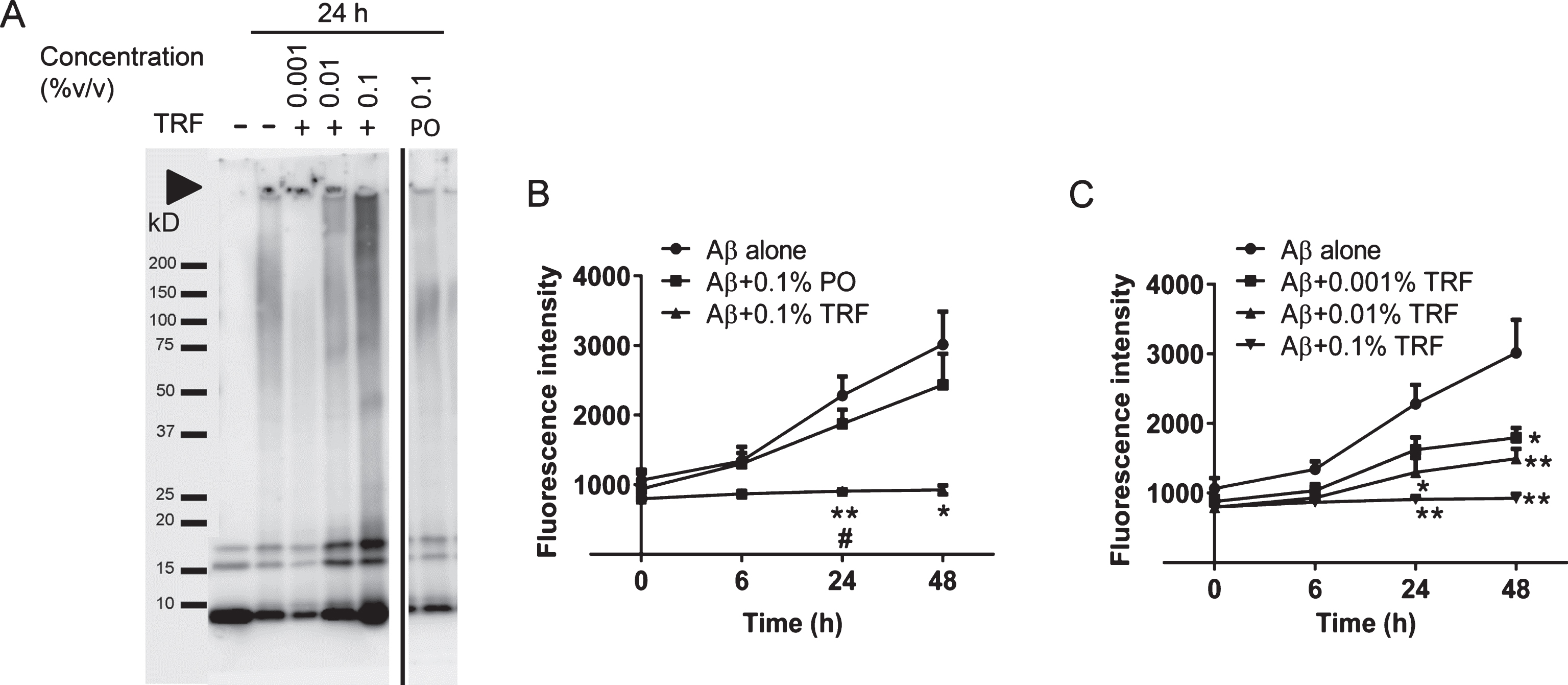
Fig.2
Tocotrienol-rich fraction (TRF) treatment inhibits Aβ40 and Aβ42 oligomerization under cell-free conditions. Aβ40 (A) and Aβ42 (B) peptides were mixed with phosphate-buffered saline (PBS) (control), TRF, palm oil stripped of vitamin E (PO), or 5% ethanol in PBS (vehicle) and cross-linked by PICUP. The samples were subjected to gel electrophoresis and visualized by silver staining. TRF inhibited oligomer formation in both Aβ40 and Aβ42 in a dose-dependent manner. No effects on Aβ oligomer formation were found with PO treatment. M = marker, UnXL = uncross-linked Aβ peptides prepared without light exposure.
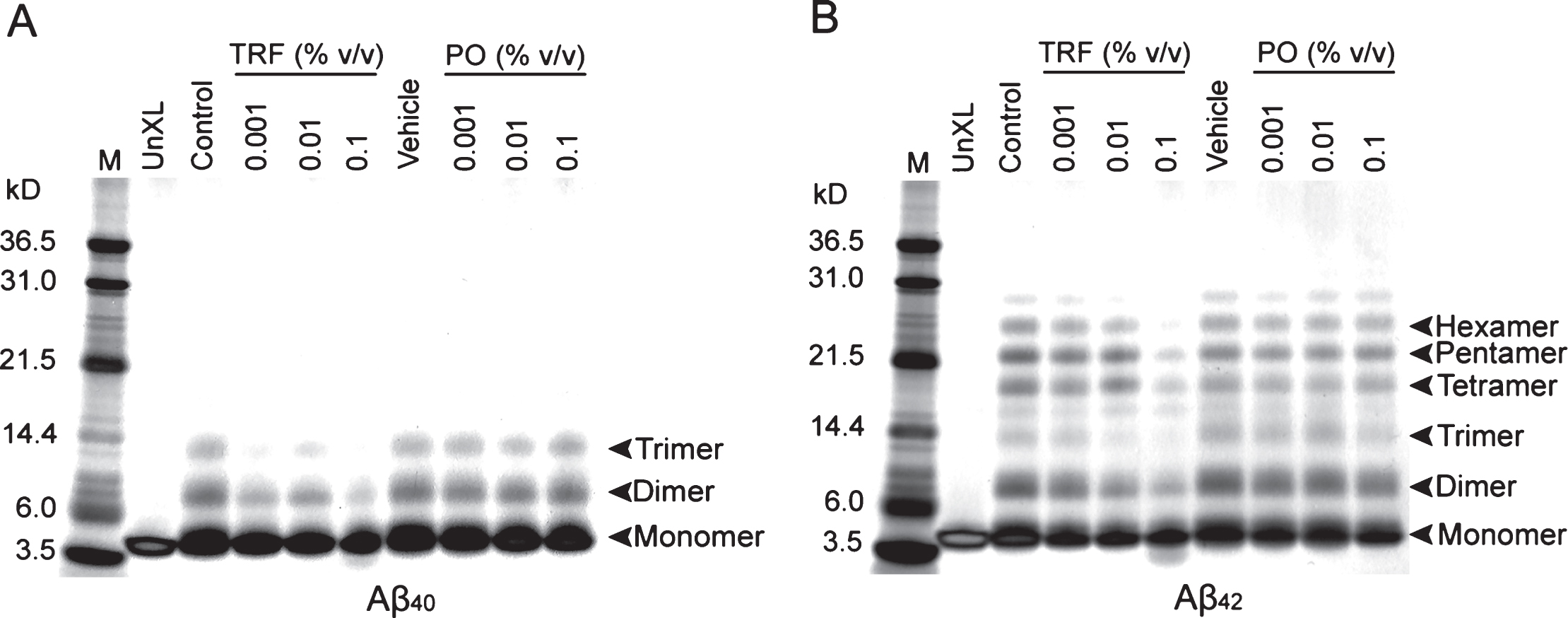
Fig.3
TRF-treated Aβ42 aggregates did not increased cell viability in SH-SY5Y cells. A) 10 μM Aβ42 were incubated for 24 h at 37°C to form aggregates, in the presence or absence of TRF or PO, and then the Aβ42 aggregates were treated to SH-SY5Y cells for 24 h at final concentrations of 0.3 μM Aβ42. A significant reduction of cell viability approximately up to 56% was detected in 0.3 μM Aβ42 aggregates, compared to vehicle. No significant difference was found in cell viability of 0.3 μM Aβ42 aggregates prepared in the presence and absence of TRF or PO. No inhibitory effect of TRF-treated Aβ42 aggregates at different dosage on cell viability. B,C) No significant difference in cell viability was found in TRF (B) and PO (C) treatments without Aβ42 aggregates. Data presented as mean±S.E.M. Significance (Student’s t-test): ***p < 0.001 0.3 μM Aβ42 versus vehicle. No significance by Bonferroni post hoc test after analysis of variance.
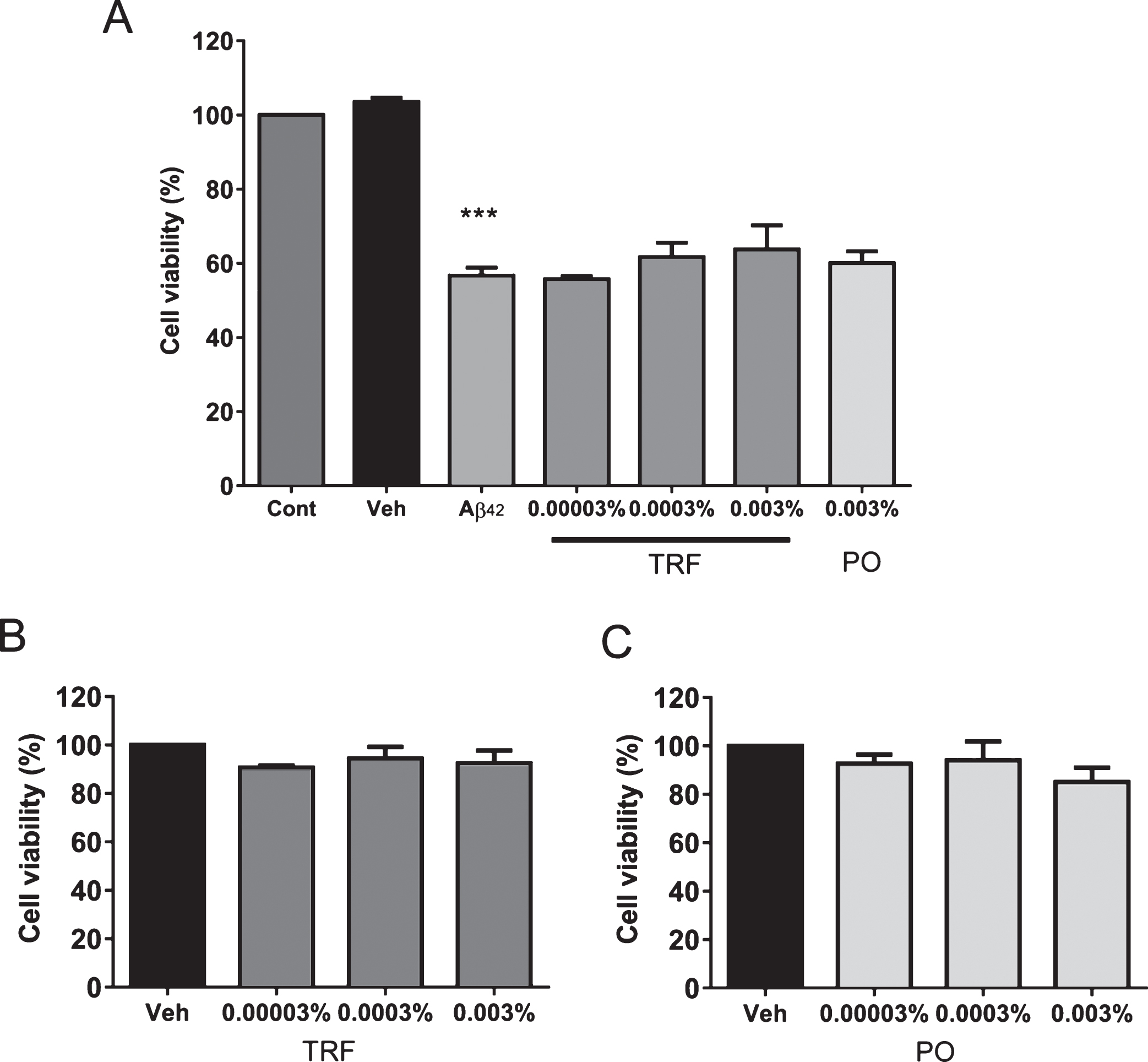
Fig.4
Schematic diagram of the novel object recognition procedure. Mice were divided into four groups: wild-type mice that received water (WT) and three groups of AβPP/PS1 mice that received water (Cont), tocotrienol-rich fraction (TRF), or palm oil stripped of vitamin E (PO). Daily supplementation started at 5 months and continued until 15 months of age. Four days before sacrifice and brain collection, the mice were subjected to the novel object recognition test as described in the text.
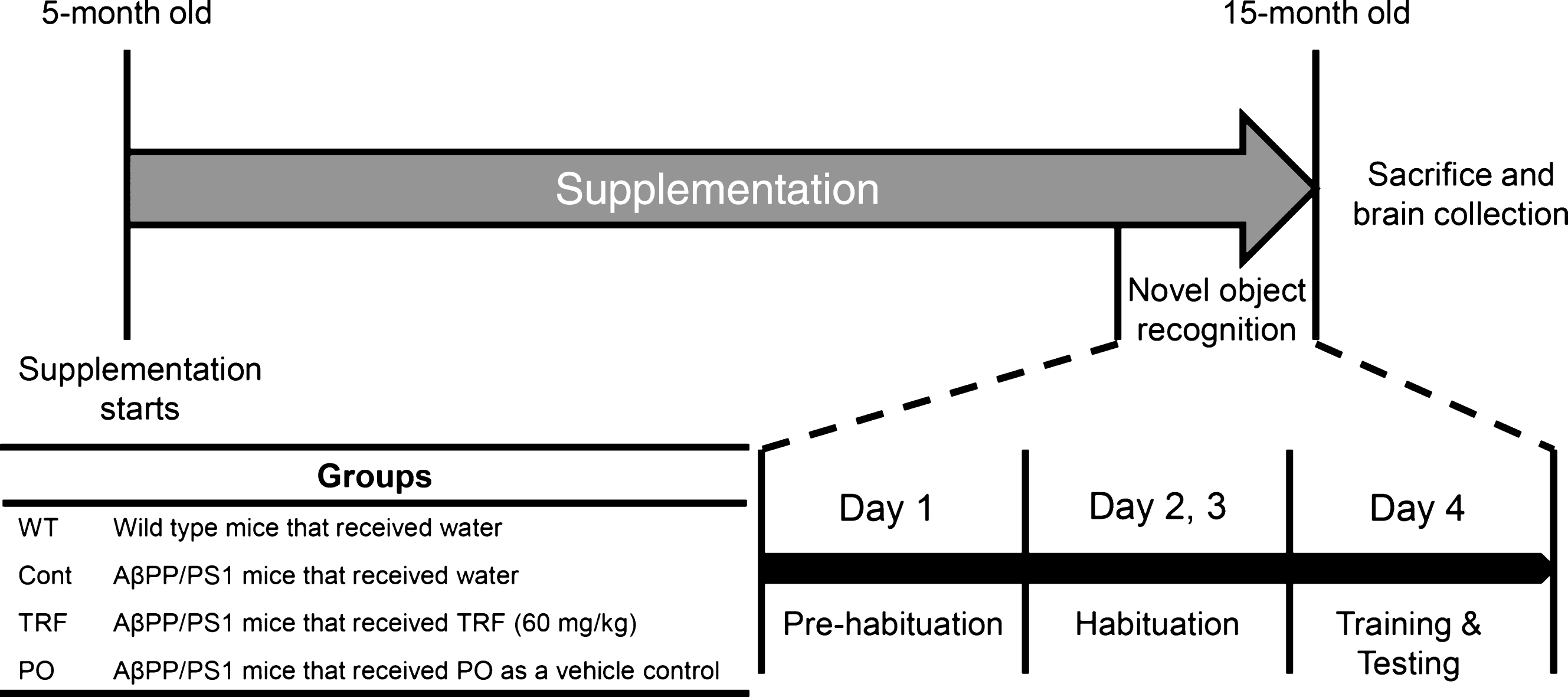
Fig.5
Supplementation with tocotrienol-rich fraction (TRF) prevents cognitive deficits in AβPP/PS1 mice as measured by the novel object recognition test. Four days before sacrifice, wild-type mice (WT; n = 13) and AβPP/PS1 mice supplemented daily with water (Cont; n = 9), TRF (n = 11), or palm oil stripped of vitamin E (PO; n = 10) for 10 months were subjected to the novel object recognition test. A) All groups spent approximately equal time exploring the two identical objects during the training trial, indicating no inherent place preference. B) Control mice exhibited a significantly lower recognition index compared with WT mice, while TRF rescued recognition index to the WT level. Data represent mean±S.E.M. Significance (Bonferroni post hoc test after analysis of variance): ‡p < 0.05 versus WT mice; *p < 0.05 versus TRF-supplemented mice.
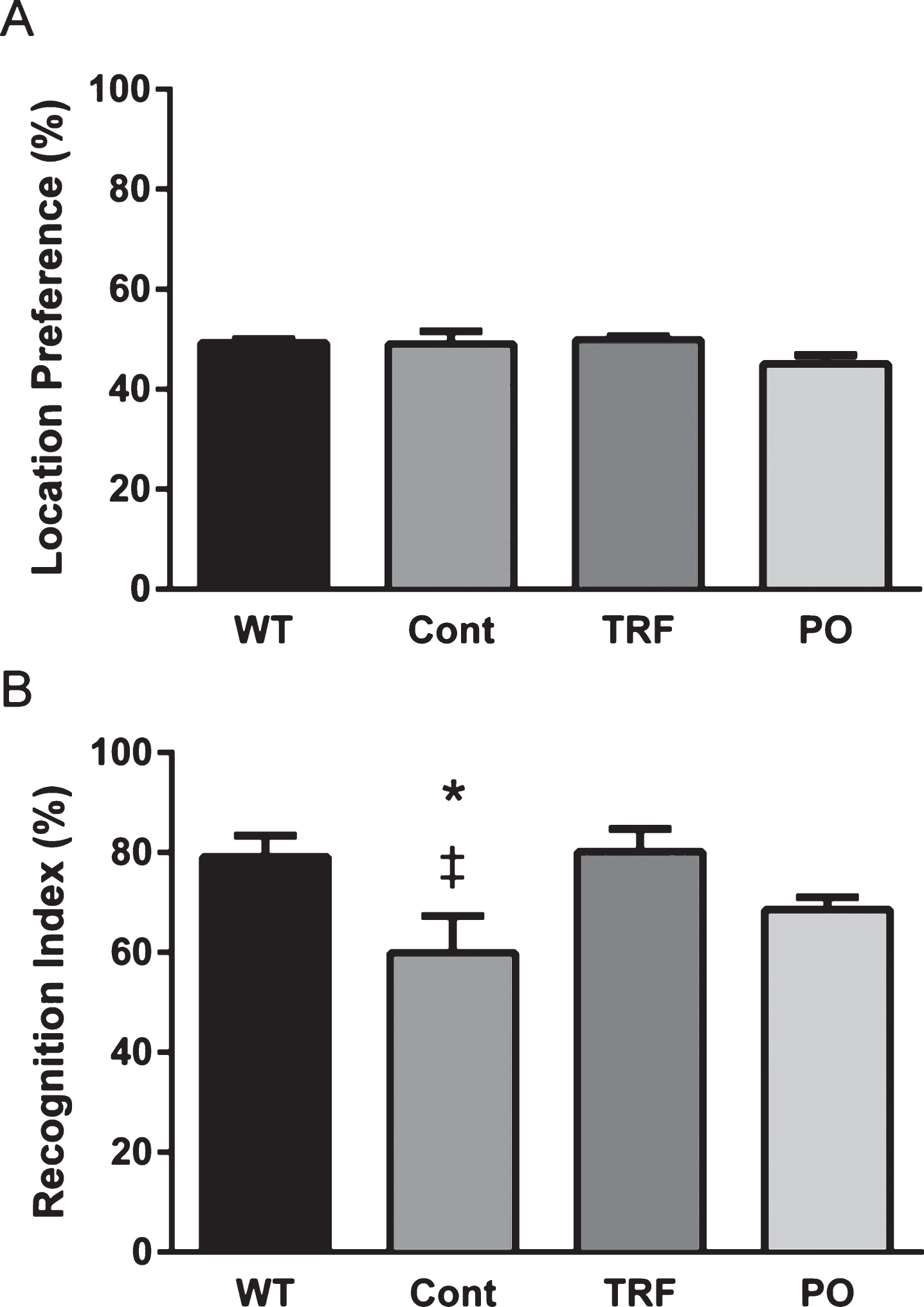
Fig.6
The tocotrienol-rich fraction (TRF) reduces Aβ deposits in the brain of AβPP/PS1 mice. A–D) Coronal brain sections from AβPP/PS1 mice supplemented with water (Cont; n = 4), TRF (n = 4), or palm oil stripped of vitamin E (PO; n = 4) were stained with anti-Aβ N-terminal antibody (A and B) or thioflavin S (C and D). A) Representative images of hippocampus and cortex from control, TRF, and PO groups stained with anti-Aβ N-terminal antibody. B) TRF-supplemented AβPP/PS1 mice show significantly decreased Aβ immunoreactive area compared to control and PO groups in the cortex. C) Representative images of hippocampus and cortex from control, TRF, and PO groups stained with thioflavin S. Brain sections stained without thioflavin S served as negative controls (Negative cont.) D) Brains from AβPP/PS1 mice supplemented with TRF showed a significant decrease in the number of plaques compared with control and PO groups in the hippocampus and cortex. Data represent mean±S.E.M. Significance (Bonferroni post hoc test after analysis of variance): *p < 0.05, **p < 0.01 versus control mice; #p < 0.05 versus PO-supplemented mice. Scale bar: 100 μm.
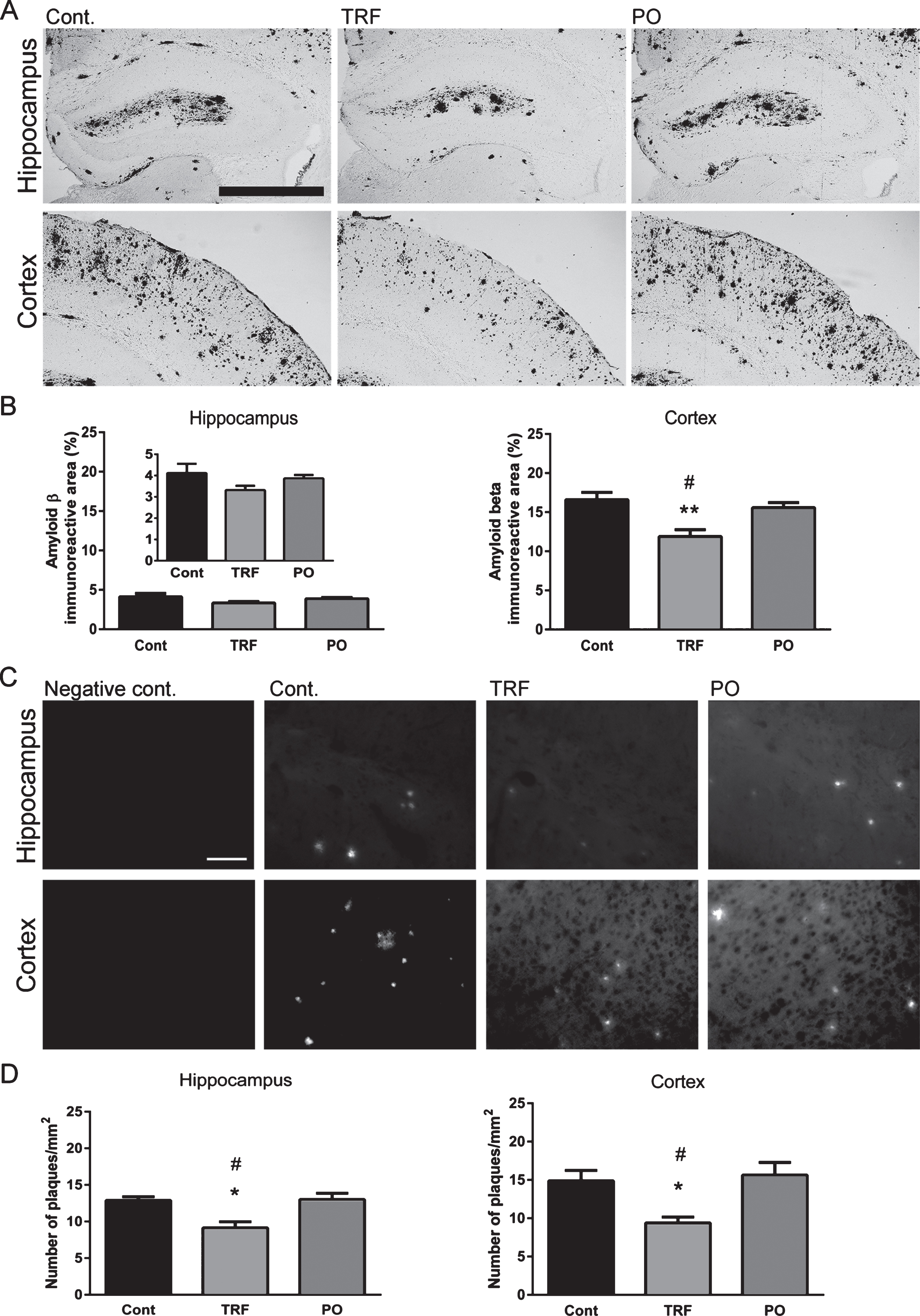
Fig.7
Treatment with tocotrienol-rich fraction (TRF) has no effect on microglial activity. Twenty-micrometer brain sections from wild-type mice (WT; n = 4) and AβPP/PS1 mice supplemented with water (Cont; n = 4), TRF (n = 4), or palm oil stripped of vitamin E (PO; n = 4) were stained with anti-Iba1 antibody for detection of activated microglia. A) Representative images of the hippocampus and cortex from WT, Cont, TRF, and PO groups. B) Control mice show significantly increased in the immunoreactivity in the hippocampus and cortex compared with WT, while immunoreactivity does not differ among Cont, TRF, and PO groups. Data represent mean±S.E.M. Significance (Bonferroni post hoc test after analysis of variance): ‡p < 0.05, ‡‡p < 0.01 versus WT mice. Scale bar: 100 μm.
Fig.8
Tocotrienol-rich fraction (TRF) supplementation does not affect brain levels of Aβ in AβPP/PS1 mice. Hippocampus and cortex from AβPP/PS1 mice supplemented with water (Cont; n = 4), TRF (n = 4), or palm oil stripped of vitamin E (PO; n = 4) were homogenized and soluble (Fig. 8A–E) and insoluble (Fig. 8F–I) Aβ fractions extracted. Levels of Aβ40, Aβ42, and Aβ oligomers were determined by enzyme-linked immunosorbent assay. No significant differences in soluble and insoluble Aβ levels were found among groups, although insoluble Aβ40 and Aβ42 in the cortex was lower in the TRF-supplemented group (Fig. 8G and I). Data represent mean±S.E.M. No significance by Bonferroni post hoc test after analysis of variance.