
Using Drugs as Molecular Probes: A Computational Chemical Biology Approach in Neurodegenerative Diseases
Abstract
Neurodegenerative diseases including Alzheimer’s disease are complex to tackle because of the complexity of the brain, both in structure and function. Such complexity is reflected by the involvement of various brain regions and multiple pathways in the etiology of neurodegenerative diseases that render single drug target approaches ineffective. Particularly in the area of neurodegeneration, attention has been drawn to repurposing existing drugs with proven efficacy and safety profiles. However, there is a lack of systematic analysis of the brain chemical space to predict the feasibility of repurposing strategies. Using a mechanism-based, drug-target interaction modeling approach, we have identified promising drug candidates for repositioning. Mechanistic cause-and-effect models consolidate relevant prior knowledge on drugs, targets, and pathways from the scientific literature and integrate insights derived from experimental data. We demonstrate the power of this approach by predicting two repositioning candidates for Alzheimer’s disease and one for amyotrophic lateral sclerosis.
INTRODUCTION
The human brain represents the most complex biological system, both structurally and functionally. Due to the inherent complexity, treating or even alleviating brain diseases, particularly neurodegenerative diseases, is not trivial. Development of drugs against neurodegenerative diseases has turned out to be among the greatest challenges in the pharmaceutical industry, as reflected by the high attrition rates and withdrawal of high profiled pharmaceutical companies from research on relevant indication areas [1]. A recent survey of success rates of drugs in clinical phases between 2003 and 2011 demonstrates that the likelihood of approval for drug candidates in the category of neurodegenerative diseases was only 9.8%, mainly limited by efficacy issues [2]. In fact, older empirical drug discovery methods are ignorant of mechanisms of actions and modern target-based drug discovery strategies follow a reductionist approach that excessively focuses on drug-receptor interactions and pharmacodynamics/pharmacokinetic properties of the candidate molecule. Both approaches do not consider the complex interplay of various biological entities across multiple biological scales and largely ignore the concept of polypharmacology [3].
Although new postgenomic technologies have produced a considerable amount of data at the molecular level, there has been little progress in inferring diseasemechanisms from these data. To overcome this hurdle, computational modeling methods, particularly model-driven systems analysis approaches, have opened up the opportunity to interpret biological datasets in a mechanistic context. Most of the relevant published studies, particularly in the data-rich field of cancer research, have used such methods to model the mechanism drug response around one or two signaling pathways using quantitative data and very limited amount of prior knowledge from literature [4, 5]. However, in a data-scarce field of research such as neurodegenerative diseases, aggregation of prior knowledge plays a key role in unraveling the puzzle of mechanisms underlying disease. This strategy leads to a second category of methods that aim at complementary integration of prior knowledge and experimental data to increase the interpretation power. Biological Expression Language (BEL) (http://www.openbel.org) is a comparably new and state-of-the-art mechanistic modeling syntax that offers a method to combine literature-derived ‘cause-and-effect’ relationships and data-driven results into a consolidated causal network model, which is amenable to further analysis for mechanistic biological interpretation. We have recently demonstrated the benefit of BEL modeling approaches in the area of neurodegenerative diseases by differential analysis of the mechanisms of Alzheimer’s disease (AD) [7].
For example, in the context of AD where most likely multiple mechanisms contribute to its pathology, systematic approaches such as a BEL-based mechanism-of-action discovery method instead of conventional drug-target-centric methods are more likely to deliver new, promising drug candidates. BEL-based modeling can help to repurpose other, already approved drugs with polypharmacological properties from other indications, as many of the drug targets are functionally pleiotropic and involved in multiple diseases. Drug repositioning, defined as the process of identifying and developing new indications for existing drugs, is also known as “drug redirecting”, “drug repurposing”, or “drug reprofiling” [8]. Off label use of Food and Drug Administration (FDA) approved drugs are very popular in many disease treatments; for instance, 50–75% of prescribed drug therapies for cancer are counted for off label uses [9]. One of the well-known examples for drug repositioning is sildenafil (Viagra), which is used in erectile dysfunction, but was initially developed to treat angina [10]. Another benefit of drug repositioning is that it offers very low risk, as repositioning candidates have already passed through several stages of clinical development. Therefore, repositioning can offer a better risk-versus-reward trade-off compared with other strategies in drug development.
Motivated by the capabilities that come with the cause-and-effect modeling approach, we have developed a causal model of drug-target interactions in the context of NDD. We demonstrate how scattered information existing in the scientific literature can be mechanistically linked to support the detection of putative drug action mechanisms in a defined disease context.
METHODS
Construction of drug-target interaction model around NDD
Using SCAIView (http://academia.scaiview.com/), our literature mining environment [11], we retrieved NDD related drugs based on PubMed abstracts with the query ([MeSH Disease: “Neurodegenerative Diseases”]) AND [Drug Names]. Next, we extracted related mechanistic information of all the drugs that reached clinical trials for NDDs using simple queries with the defined disease context (NDD) and specific drug names. For example, ([MeSH Disease: “Neurodegenerative Diseases”]) AND [Drug Names: “Donepezil”] has been performed to extract all PubMed articles containing information related to the mode-of-action of donepezil in the NDD context. We manually extracted causal information from these articles and coded into a BEL model. Then, we integrated this model with our in-house AD model [7], to achieve greater disease biology context for the analysis. In order to enrich this primary model by additional interactions for NDD related drugs, we extracted all interactions related to these drugs such as drug-drug interactions, target-target interactions, and drug-target interactions that occur within the brain from different drug interactions databases including Comparative Toxicogenomics Database (http://ctdbase.org/), Therapeutic Target Database (http://bidd.nus.edu.sg/group/cjttd/), DrugBank (http://www.drugbank.ca/), and STITCH database (http://stitch.embl.de/). The purpose of this enrichment was to integrate the biology context around drug targets, in particular those causal relationships that can be used to describe the physiological mode-of-action of a drug-target combination.
Model analysis and visualization
We used DAVID (http://david.abcc.ncifcrf.gov), a tool widely used for gene set enrichment analysis, to find the most significant pathways and biological processes associated to the drug targets in our model. The Cytoscape software was used for subnetwork extraction and visualization [12].
Target similarity approach
Using the query ([MeSH Disease: “Neurodegenerative Diseases”]) AND [Drug names] in SCAIView, PubMed abstracts were searched for all drugs mentioned in the context of neurodegenerative diseases. Targets of all NDD drugs were systematically compared against targets of five approved drugs for AD, and ranked based on the number of shared targets. Hence, NDD drugs having the highest number of shared targets with approved AD drugs were considered for further analysis in the context of AD. Only those targets having direct interactions are considered for this approach to avoid redundancy. Drugs from these lists were then spotted in the AD-specific BEL-based model for the prediction of similarity of their mechanism of action with five approved AD drugs.
RESULTS
Analysis of NDD-specific cause-and-effect model
The retrieved mechanistic information from the text mining tool SCAIView was manually inspected and filtered for relevant information. Both literature and data driven information were encoded into the NDD-specific BEL model and this model contains 9645 nodes and 26,660 edges including 7,215 genes/proteins, 442 biological processes, 101 disease concepts, and 1,081 chemical entities, coded into 34,403 BEL statements (Fig. 1). This model is comprised of several types of interactions such as drug-target interactions, drug-disease interactions, target-target interactions, target-pathway interactions, and drug-pathway interactions represented by mainly ‘increases’, or ‘decreases’ types of relationships in BEL.
GSEA for this model resulted in a list of significant pathways, in which Alzheimer’s disease pathway was on top of the list, followed by the Amyotrophic Lateral Sclerosis (ALS) pathway (Table 1). Two ‘target sets’ associated with these two pathways were selected from the model for further analyses with the intention of the identification of potential drug repurposing candidates from the model.
Model-based mechanistic analysis of drug repositioning candidates
We systematically analyzed our model in order to detect the mechanism of action of the drugs in the context of their causal relationships with the available targets, pathways, and biological processes in the NDD-specific mechanistic model. This sort of analysis helps to find possible interaction similarities between drugs of one indication to other indications within the disease context. In this study, we were able to predict three candidate drugs for drug repurposing by using our enriched NDD-specific BEL model:
Donepezil as potential repurposing candidate for ALS
Functional analysis of genes/proteins in our model revealed the “ALS disease pathway” as the putative shared pathway with AD, suggestive of evidence to explore the possibility of repositioning drugs between these two diseases. Further analysis of the ALS pathway sub-network based on our model led to the identification of the AD approved drug donepezil as a potential candidate for repositioning. Donepezil affects 26 proteins in the ALS mechanistic pathway sub-network in our model.
Mutant SOD1 protein is believed to be a key player in the pathology of ALS, which disturbs the normal physiological conditions and initiates a number of pathways that ultimately lead to the disease condition [13]. Our mechanistic analysis reveals that donepezil can prevent effects of mutant SOD1 by interfering the activities of many proteins that are altered by this mutation under ALS conditions. Mutant SOD1 protein in ALS mainly exerts its effect by three mechanisms that ultimately lead to neuronal cell death. Firstly, mutant SOD1 can exert its effect by stimulating pro-apoptotic proteins BAD and BAX and inhibiting the activity of anti-apoptotic proteins BCL2 and BCL2L1, which leads to an increase in cytochrome C (CYCS) release from the mitochondria [14, 15]. The activation of BAD and BAX can be also be achieved by recruiting TP53 proteins via mutant SOD1 [16]. The released CYCS interacts and forms a complex with APAF1 in the presence of ATP and activates the key player of the cell death CASP9, which subsequently activates CASP3 and initiates cell death [17]. Secondly, mutant SOD1 can initiate oxidative stress via P38 signaling pathway, which in turn inhibits EAAT2, a regulator of the glutamate concentration [18]. The inhibition of EAAT2 produces excess glutamate in synapses, which overstimulate glutamate receptors and initiate high calcium influx in the cytosol and produce reactive oxygen species (ROS), which will ultimately activate CASP9 [19]. In the third pathway, mutant SOD1 inhibits the activity of XIAP, which regulates CASP9 in normal condition, ultimately leading to activation of CASP9 and CASP3 and initiating neuronal death [20].
According to our model, donepezil can interact with several significant targets in each of these three routes of the ALS pathway. Donepezil can increase the activity of BCL2, BCL2L1, and XIAP, which are inhibited by mutant SOD1 in ALS. It is also able to reduce the level of Ca2 + and ROS production in oxidative stress and to inhibit the activation of CASP9 and CASP3 too (Fig. 2).
Epidemiological evidences suggest that early treatment of donepezil in mild cognitive impairment plays a neuroprotective role by preventing neuronal cell death in the hippocampus, hence, reduces the likelihood of disease progression to AD [21, 22]. Interestingly, cognitive dysfunction and inflammation in ALS are broadly associated with morphological changes in the hippocampal region due to excessive neuronal cell death [23–25]. Therefore, based on mechanistic analysis of our model along with the evidences presented; we can hypothesize that donepezil may be a promising repurposing candidate for treating ALS and absolutely worthy of further investigations.
Riluzole as potential repurposing candidate for AD
Being motivated by the donepezil reposition prospect for ALS, we investigated the likelihood of repurposing any ALS drugs for AD with the help of our model. Using our literature-mining environment SCAIView, we found riluzole as the most prominent and effective drug for ALS treatment until now, which helps to prolong the survival of ALS patients. Therefore, we inspected all interactions related to riluzole in our model to investigate its potential influences on the AD pathology. Interestingly, our model was able to bring together some mechanisms of actions of riluzole, which interfere with some crucial mechanisms of AD etiology as described below (Fig. 3).
Riluzole is usually known to prolong life longevity of ALS patients by inhibiting Ca2 + and Na + channel activities and increasing K + channel activity, which result in decreased glutamate release in the cell [26]. Decreased glutamate concentration contributes to TNF inactivation, which leads to inhibition of inflammatory processes in the brain. The model predicts that riluzole can be beneficial in AD by interfering with a number of mechanistic routes in AD pathology, as follows: in addition to glutamate release inhibition, riluzole can stop neuronal excitotoxicity by inhibiting NMDA and Kainate binding to the NMDA receptor. Riluzole can impede oxidative stress by inhibiting lipid peroxidation via blocking PLA2 activity. Riluzole can also suppress VEGFA and PRKC activities, which are found to be upregulated in AD [27, 28] and believed to be involved in tau phosphorylation [29, 30]. Moreover, riluzole can inhibit pro-apoptotic CASP3 and stimulate anti-apoptotic BCL2 protein to prevent apoptosis [31]. Excessive N-methyl-D-aspartate (NMDA) receptor activation is believed to mediate calcium-dependent glutamate excitotoxicity in different neurodegenerative disorders like AD [32]. Our mechanistic model predicts that glutamate release inhibitor riluzole can provide further therapeutic benefits in AD when used in combination with memantine, the first-in-class approved drug for AD, by modifying excess transmission of synaptic glutamate. Additionally, an ongoing clinical trial NCT01703117, where riluzole is being tested for treating mild stage AD patients, provides further supporting evidence for the mechanism hypothesis we present here. Therefore, we feel encouraged to speculate that riluzole might have therapeutic benefitsfor AD.
Identification of potential drugs for AD by a target similarity approach
The mechanistic prediction capability of our model inspired us to pursue this slightly different approach for exploring the repositioning potential of drugs present in our model. Analysis for finding common targets between NDD drugs in our model and 5 approved AD drugs identified resveratrol and simvastatin, as drugs that share the highest number of targets with approved AD drugs. Interestingly, these two drugs are already being proposed or investigated for their therapeutics effects in AD (Supplementary Table 1), which points to the legitimacy of our approach to find drugs that can be used for similar therapeutic approaches based on their target similarity. To this end, we have selected another top-ranked drug “cyclosporine”, from our target similarity list, for further mechanistic analysis.
Cyclosporine mode-of-action analysis in the context of AD
Cyclosporine is a drug against rheumatoid arthritis and has common targets with all five AD approved drugs (Fig. 4), but has not been previously considered for its potential effects in AD.
Instead of being an immunosuppressive agent, the NDD-specific BEL model predicts that cyclosporine can exert neuroprotective effects in various alternative ways. Cyclosporine mainly inhibits immunocompetent lymphocytes (T1 helper cells) in a specific and reversible manner so that T1H cells decrease IL2 and IFNG production and release, which leads to suppression of the immune system response partially (Fig. 5).
According to the model prediction in Fig. 5, cyclosporine can also repress the apoptosis of neuronal cells by inhibiting Cyclophilin D, a member of mitochondrial permeability transition complex (MPTP) [33]. Cyclophilin D inhibition results in regulation of the MPTP complex and decreased Cytochrome C (CYCS) release from mitochondria [34, 35]. This inhibition of CYCS prevents CASP9 and CASP3 mediated apoptotic cell death [20]. The apoptosis inhibition through prevention of CYCS release is also facilitated by the inhibitory effects of cyclosporine on the anti-apoptotic protein BCL2, pro-apoptotic BAD, BAX, and also AKT [36, 37]. Cyclosporine activity inhibits BAX and BAD via stimulation of AKT activity [38], which in turn inhibits GSK3β that phosphorylates and activates BAX [39]. It also inhibits calcineurin, which results in repression of inflammation [40] and down-regulation of ACHE and BCHE, potentially via increasing AKT activity [41, 42], while AKT degeneration leads to increased ACHE and BCHE levels in AD [32]. There is also evidence that cyclosporine decreases ABCB1 and ABCC2 activity [38, 43], which has been reported to increase amyloid-β (Aβ) accumulation in the brain of AD patients [44].
Further support for this hypothesis was provided by a number of patents that explain the putative mechanisms we reconstructed for the potential role of cyclosporine in AD. The claims sections of these patents state clearly, that apart from immunosuppressive activity, cyclosporines could also be effective to improve disease condition by interfering cyclophilins activity and Aβ accumulation. According to US Patents US6583265 and US7538084, cyclosporines can have therapeutic effect in AD by inhibiting the catalytic activity of cyclophilins. A European Patent, EP1893226, recommends the use of cyclosporine to treat AD by preventing Aβ accumulation in the brain in addition to their cyclophilin inhibition activity. Therefore, cyclosporine can be proposed as a multipotent therapeutic agent for AD treatment andthis hypothesis bears potential for further clinical investigation.
DISCUSSION
Structural and functional complexity of the human brain has posed serious challenges to the development of novel therapeutics against neurodegenerative diseases. Capturing this complexity across different molecular entity types and various biological scales can be assisted by computational systems modeling approaches that aim at linking molecular mechanisms to clinical phenotypes. Particularly, in complex diseases like AD, integrating all the entities and bioprocesses involved in the disease into consolidated, cause-and-effect models bears some potential to shed light on interdependent processes and pathways that remain unnoticed in the shadow of disease complexity otherwise. In fact, representing a priori relevant knowledge in the form of causal relationship models confers enhanced interpretation power that is well suited to back up experimental data and generate new testable hypotheses. Once such mechanistic, context-sensitive models are available, the molecular space can be enriched for chemical entities to facilitate prediction of mode-of-action for drugs and biomarkers.
As demonstrated in this work, disease-specific mechanistic models that are enriched with chemical entities can be used not only to explain the physiological action mode of approved drugs or candidate drugs, but also to explore the multi-targeting nature of potent compounds and predict the suitability of existing drugs for repurposing in another indication area as well. Based on their role in our cause-and-effect drug-target network, two FDA approved drugs, riluzole and cyclosporine, may be repurposing candidates for AD. Another FDA approved drug, donepezil, could be a potential repurposing candidate for ALS. Although our inferences are based upon the aggregated a priori knowledge consolidated in BEL models, further functional or translational validation can be provided by integration of experimental data such as gene expression values. Cross-validation of our models with the signature-based results of Siavelis et al. [14] indicates that rilozule and cyclosporine belong to PKC and GSK3 inhibitor classes of repurposing candidates for AD.
Our approach of using drugs as molecular probes supports the notion that integration of literature-driven information into a formalized model can be instrumental for prediction, analysis, and interpretation of possible biological mechanisms underlying a disease process. Using this approach, we could demonstrate that potential new roles of existing approved drugs can be predicted based on a meaningful functional context. Nevertheless, BEL based mechanistic models, of course, cannot be considered as a replacement for any structure-activity relationship (SAR) model based drug discovery approach. On the contrary, the BEL model presented here merely provides a common platform to put drug-target information into a functional, mechanistic context that focuses on causes and effects and allows for prediction of the repurposing potential of drugs.
It should be noted here, however, that computational models like the ones presented in this study assist hypothesis generation and candidate prioritization. Indeed, these models are merely precursors to clinical and laboratory research findings so that predicted candidates enriched with supporting evidence should be ultimately confirmed by experimental and clinical studies. But such prioritized candidates at this stage can guide future validation efforts in experimental research settings with lower decision-making and investment risk.
CONCLUSION
Failure of conventional drug discovery and development approaches to deliver new drugs for complex disease like, AD or ALS, has proven that the “one size fits all” paradigm can no longer hold true for chronic and complex idiopathic diseases, particularly in the area of neurodegenerative diseases. This is because of the inherent multitargeting nature of therapeutic agents that modify often unknown pathways with unwanted effects. However, this property can be used positively for repositioning of already approved drugs if the mechanism of action for these drugs can be shown in the context of other diseases. Thus, consolidating the mechanistic information within causal computational models lends support to scientists and decision makers to substantiate their hypotheses based on collective information from both knowledge- and data-driven approaches. It is foreseen that, with the consistent growth of published knowledge and advent of big data, such mechanistic models will play an increasingly important role in the future generation of drug discovery and repurposing pipelines.
ACKNOWLEDGMENTS
The research leading to these results has received support from the EU/EFPIA Innovative Medicines Initiative Joint Undertaking under AETIONOMY grant agreement n°115568, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013) and in kind contributions from EFPIA companies.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0222r2).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-160222.
REFERENCES
[1] | Wegener G , Rujescu D ((2013) ) The current development of CNS drug research. Int J Neuropsychopharmacol 16: , 1687–1693. |
[2] | Hay M , Thomas DW , Craighead JL , Economides C , Rosenthal J ((2014) ) Clinical development success rates for investigational drugs. Nat Biotechnol 32: , 40–51. |
[3] | Enna SJ , Williams M ((2009) ) Challenges in the search for drugs to treat central nervous system disorders. J Pharmacol Exp Ther 329: , 404–411. |
[4] | Wierling C , Kühn A , Hache H , Daskalaki A , Maschke-Dutz E , Peycheva S , Li J , Herwig R , Lehrach H ((2012) ) Prediction in the face of uncertainty: A Monte Carlo-based approach for systems biology of cancer treatment. Mutat Res 746: , 163–170. |
[5] | Chen WW , Schoeberl B , Jasper PJ , Niepel M , Nielsen UB , Lauffenburger DA , Sorger PK ((2009) ) Input-output behavior of ErbB signaling pathways as revealed by a mass action model trained against dynamic data. Mol Syst Biol 5: , 239. |
[6] | Subramanian A , Subramanian A , Tamayo P , Tamayo P , Mootha VK , Mootha VK , Mukherjee S , Mukherjee S , Ebert BL , Ebert BL , Gillette MA , Gillette MA , Paulovich A , Paulovich A , Pomeroy SL , Pomeroy SL , Golub TR , Golub TR , Lander ES , Lander ES , Mesirov JP , Mesirov JP ((2005) ) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: , 15545–15550. |
[7] | Kodamullil AT , Younesi E , Naz M , Bagewadi S , Hofmann-Apitius M ((2015) ) Computable cause-and-effect models of healthy and Alzheimer’s disease states and their mechanistic differential analysis. Alzheimer’s Dement 11: , 1329–1339. |
[8] | Ashburn TT , Thor KB ((2004) ) Drug repositioning: Identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3: , 673–683. |
[9] | Pfister DG ((2012) ) Off-label use of oncology drugs: The need for more data and then some. J Clin Oncol 30: , 584. |
[10] | Novac N ((2013) ) Challenges and opportunities of drug repositioning. Trends Pharmacol Sci 34: , 267–272. |
[11] | Friedrich CM , Dach H , Gattermayer T , Engelbrecht G , Benkner S , Hofmann-Apitius M ((2008) ) @neuLink: A service-oriented application for biomedical knowledge discovery. Stud Health Technol Inform 138: , 165–172. |
[12] | Smoot ME , Ono K , Ruscheinski J , Wang PL , Ideker T ((2011) ) Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 27: , 431–432. |
[13] | Andersen PM ((2006) ) Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep 6: , 37–46. |
[14] | Vukosavic S , Dubois-Dauphin M , Romero N , Przedborski S ((1999) ) Bax and Bcl-2 interaction in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 73: , 2460–2468. |
[15] | Ido A , Fukuyama H , Urushitani M ((2011) ) Protein misdirection inside and outside motor neurons in amyotrophic lateral sclerosis (ALS): A possible clue for therapeutic strategies. Int J Mol Sci 12: , 6980–7003. |
[16] | González de Aguilar JL , Gordon JW , René F , de Tapia M , Lutz-Bucher B , Gaiddon C , Loeffler JP ((2000) ) Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: Evidence for the implication of the p53 signaling pathway. Neurobiol Dis 7: , 406–415. |
[17] | Guégan C , Vila M , Rosoklija G , Hays AP , Przedborski S ((2001) ) Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 21: , 6569–6576. |
[18] | Lee JK , Hwang SG , Shin JH , Shim J , Choi E-J ((2014) ) CIIA prevents SOD1(G93A)-induced cytotoxicity by blocking ASK1-mediated signaling. Front Cell Neurosci 8: , 179. |
[19] | Boillée S , Vande Velde C , Cleveland DW ((2006) ) ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 52: , 39–59. |
[20] | Inoue H , Tsukita K , Iwasato T , Suzuki Y , Tomioka M , Tateno M , Nagao M , Kawata A , Saido TC , Miura M , Misawa H , Itohara S , Takahashi R ((2003) ) The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J 22: , 6665–6674. |
[21] | Petersen RC , Thomas RG , Grundman M , Bennett D , Doody R , Ferris S , Galasko D , Jin S , Kaye J , Levey A , Pfeiffer E , Sano M , van Dyck CH , Thal LJ ((2005) ) Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med 352: , 2379–2388. |
[22] | Min D , Mao X , Wu K , Cao Y , Guo F , Zhu S , Xie N , Wang L , Chen T , Shaw C , Cai J ((2012) ) Donepezil attenuates hippocampal neuronal damage and cognitive deficits after global cerebral ischemia in gerbils. Neurosci Lett 510: , 29–33. |
[23] | Westeneng H-J , Verstraete E , Walhout R , Schmidt R , Hendrikse J , Veldink JH , van den Heuvel MP , van den Berg LH ((2015) ) Subcortical structures in amyotrophic lateral sclerosis. Neurobiol Aging 36: , 1075–1082. |
[24] | Raaphorst J , van Tol MJ , de Visser M , van der Kooi AJ , Majoie CB , van den Berg LH , Schmand B , Veltman DJ ((2015) ) Prose memory impairment in amyotrophic lateral sclerosis patients is related to hippocampus volume. Eur J Neurol 22: , 547–554. |
[25] | Brownell A-L , Kuruppu D , Kil K-E , Jokivarsi K , Poutiainen P , Zhu A , Maxwell M ((2015) ) PET imaging studies show enhanced expression of mGluR5 and inflammatory response during progressive degeneration in ALS mouse model expressing SOD1-G93A gene. J Neuroinflammation 12: , 217. |
[26] | Albo F , Pieri M , Zona C ((2004) ) Modulation of AMPA receptors in spinal motor neurons by the neuroprotective agent riluzole. J Neurosci Res 78: , 200–207. |
[27] | Tang H , Mao X , Xie L , Greenberg DA , Jin K ((2013) ) Expression level of vascular endothelial growth factor in hippocampus is associated with cognitive impairment in patients with Alzheimer’s disease. Neurobiol Aging 34: , 1412–1415. |
[28] | Yoo MH , Hyun HJ , Koh JY , Yoon YH ((2005) ) Riluzole inhibits VEGF-induced endothelial cell proliferationin vitro and hyperoxia-induced abnormal vessel formation in vivo. Investig. Ophthalmol Vis Sci 46: , 4780–4787. |
[29] | Hoshi M , Nishida E , Miyata Y , Sakai H , Miyoshi T , Ogawara H , Akiyama T ((1987) ) Protein kinase C phosphorylates tau and induces its functional alterations. FEBS Lett 217: , 237–241. |
[30] | Correas I , Díaz-Nido J , Avila J ((1992) ) Microtubule-associated protein tau is phosphorylated by protein kinase C on its tubulin binding domain. J Biol Chem 267: , 15721–15728. |
[31] | Hassanzadeh K , Roshangar L , Habibi-asl B , Farajnia S , Izadpanah E , Nemati M , Arasteh M , Mohammadi S ((2011) ) Riluzole prevents morphine-induced apoptosis in rat cerebral cortex. Pharmacol Reports 63: , 697–707. |
[32] | Parihar MS , Brewer GJ ((2010) ) Amyloid-β as a modulator of synaptic plasticity. J Alzheimers Dis 22: , 741–763. |
[33] | Kajitani K , Fujihashi M , Kobayashi Y , Shimizu S , Tsujimoto Y , Miki K ((2008) ) Crystal structure and putative function of small Toprim domain-containing protein from Bacillus stearothermophilus. Proteins 70: , 311–319. |
[34] | Xie J-R , Yu L-N ((2007) ) Cardioprotective effects of cyclosporine A in an in vivo model of myocardial ischemia and reperfusion. Acta Anaesthesiol Scand 51: , 909–913. |
[35] | Duan X , Ji B , Yu K , Hei F , Liu J , Long C ((2011) ) Acidic buffer or plus cyclosporine A post-conditioning protects isolated rat hearts against ischemia-reperfusion injury. Perfusion 26: , 245–252. |
[36] | Hage-Sleiman R , Esmerian MO , Kobeissy H , Dbaibo G ((2013) ) p53 and ceramide as collaborators in the stress response. Int J Mol Sci 14: , 4982–5012. |
[37] | Weon JB , Yun BR , Lee J , Eom MR , Ko HJ , Lee HY , Park DS , Chung HC , Chung JY , Ma CJ ((2014) ) Neuroprotective effect of steamed and fermented Codonopsis lanceolata. Biomol Ther (Seoul) 2014: , 246–253. |
[38] | Yamazaki M , Li B , Louie SW , Pudvah NT , Stocco R , Wong W , Abramovitz M , Demartis A , Laufer R , Hochman JH , Prueksaritanont T , Lin JH ((2005) ) Effects of fibrates on human organic anion-transporting polypeptide 1B1-, multidrug resistance protein 2- and P-glycoprotein-mediated transport. Xenobiotica 35: , 737–753. |
[39] | Linseman DA , Butts BD , Precht TA , Phelps RA , Le SS , Laessig TA , Bouchard RJ , Florez-McClure ML , Heidenreich KA ((2004) ) Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24: , 9993–10002. |
[40] | Norris CM , Kadish I , Blalock EM , Chen K-C , Thibault V , Porter NM , Landfield PW , Kraner SD ((2005) ) Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci 25: , 4649–4658. |
[41] | Herink J , Krejčová G , Bajgar J , Svoboda Z , Květina J , Živnú P , Palička V ((2003) ) Cyclosporine A inhibits acetylcholinesterase activity in selected parts of the rat brain. Neurosci Lett 339: , 251–253. |
[42] | Jennen DGJ , Magkoufopoulou C , Ketelslegers HB , van Herwijnen MHM , Kleinjans JCS , van Delft JHM ((2010) ) Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol Sci 115: , 66–79. |
[43] | Luo L , Sun YJ , Yang L , Huang S , Wu YJ ((2013) ) Avermectin induces P-glycoprotein expression in S2 cells via the calcium/calmodulin/NF-??B pathway. Chem Biol Interact 203: , 430–439. |
[44] | Song JS , Chae J-W , Lee K-R , Lee BH , Choi EJ , Ahn SH , Kwon K-I , Bae MA ((2011) ) Pharmacokinetic characterization of decursinol derived from Angelica gigas Nakai in rats. Xenobiotica 41: , 895–902. |
Figures and Tables
Fig.1
AD-specific BEL model enriched with drug-target interactions. The extract represents various interaction types for Riluzole such as drug-protein, drug-bioprocess, and drug-pathology interactions encoded into the BEL model.
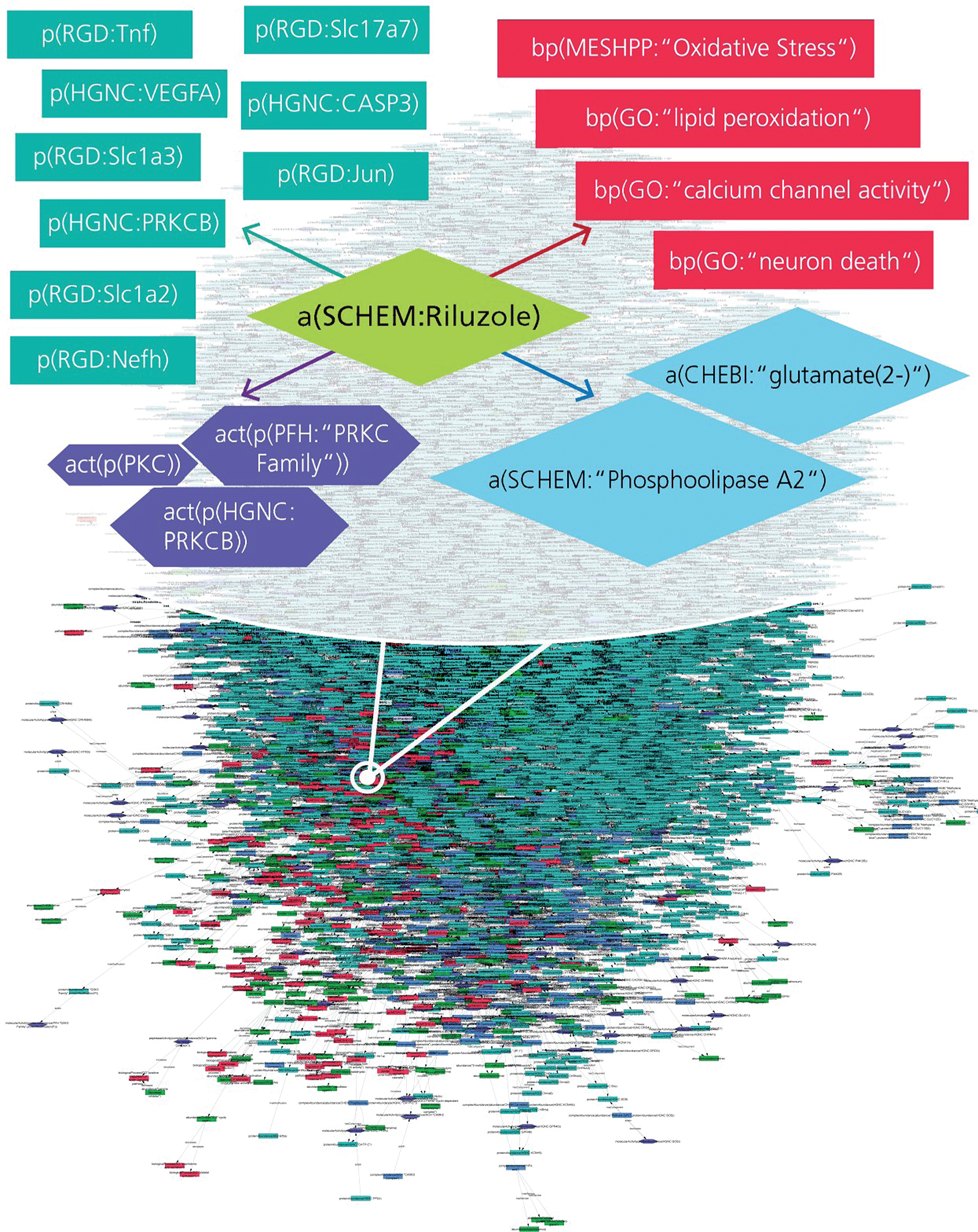
Fig.2
Model-based prediction of donepezil’s mode-of-action in the context of the ALS Pathway. The figure illustrates how donepezil modifies the ALS shared pathway. Red lines represent perturbations in disease condition and green lines indicate normal physiological processes, while blue lines indicate drug effects on targets. Arrows represent increased activities of entities while T lines stand for decreased activities of entities and dotted lines represent intermediate interactions.
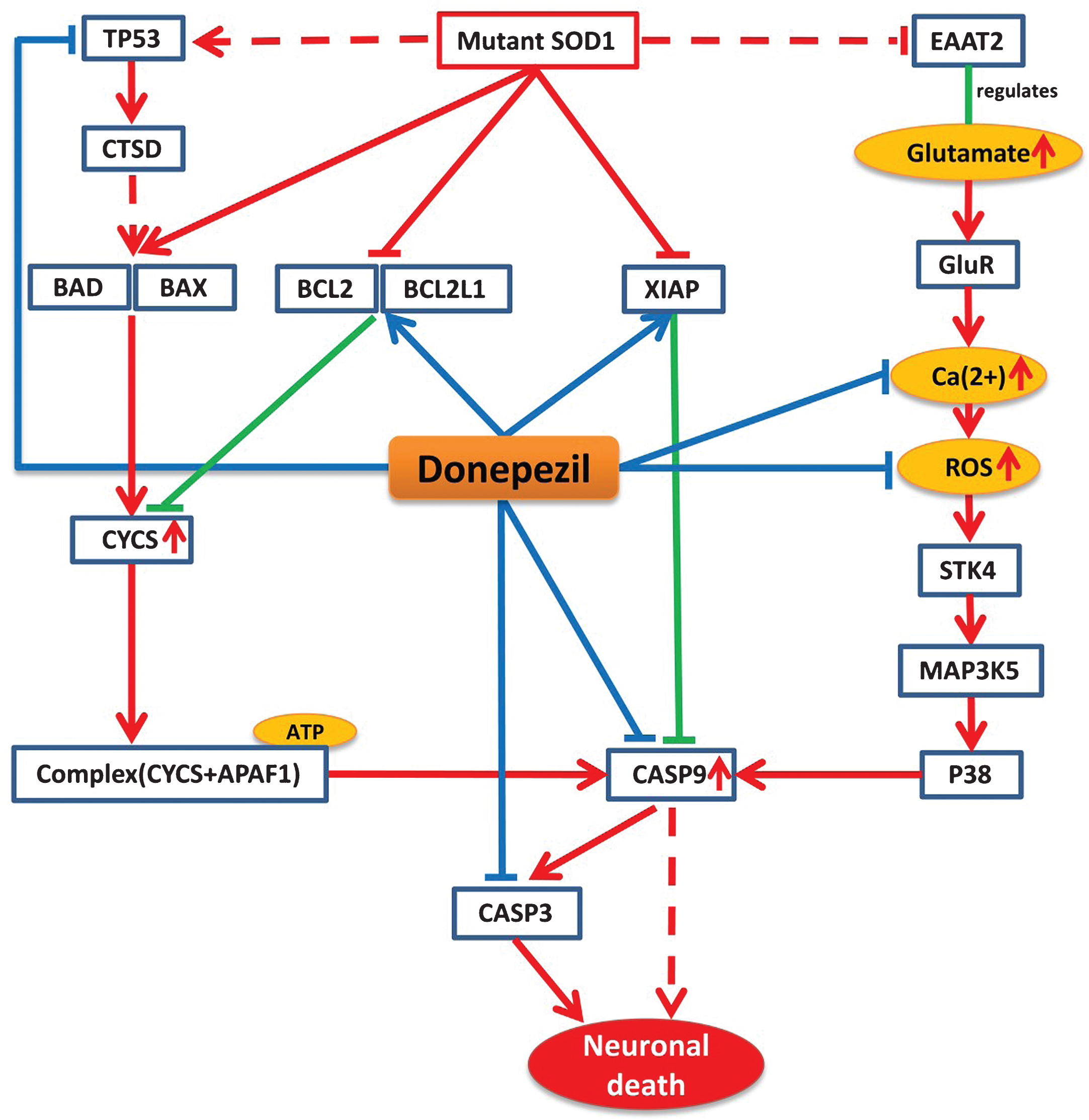
Fig.3
Schematic representation of Riluzole mechanism of action and its neuroprotective Effect in the context of AD. Blue lines here represent only the alternative effect of riluzole on these pathways. Purple boxes represent the direct protein target and green nodes represent channels and receptors, which can be targeted by riluzole. Yellow nodes represent targeted ions/chemicals and red nodes represent biological processes.
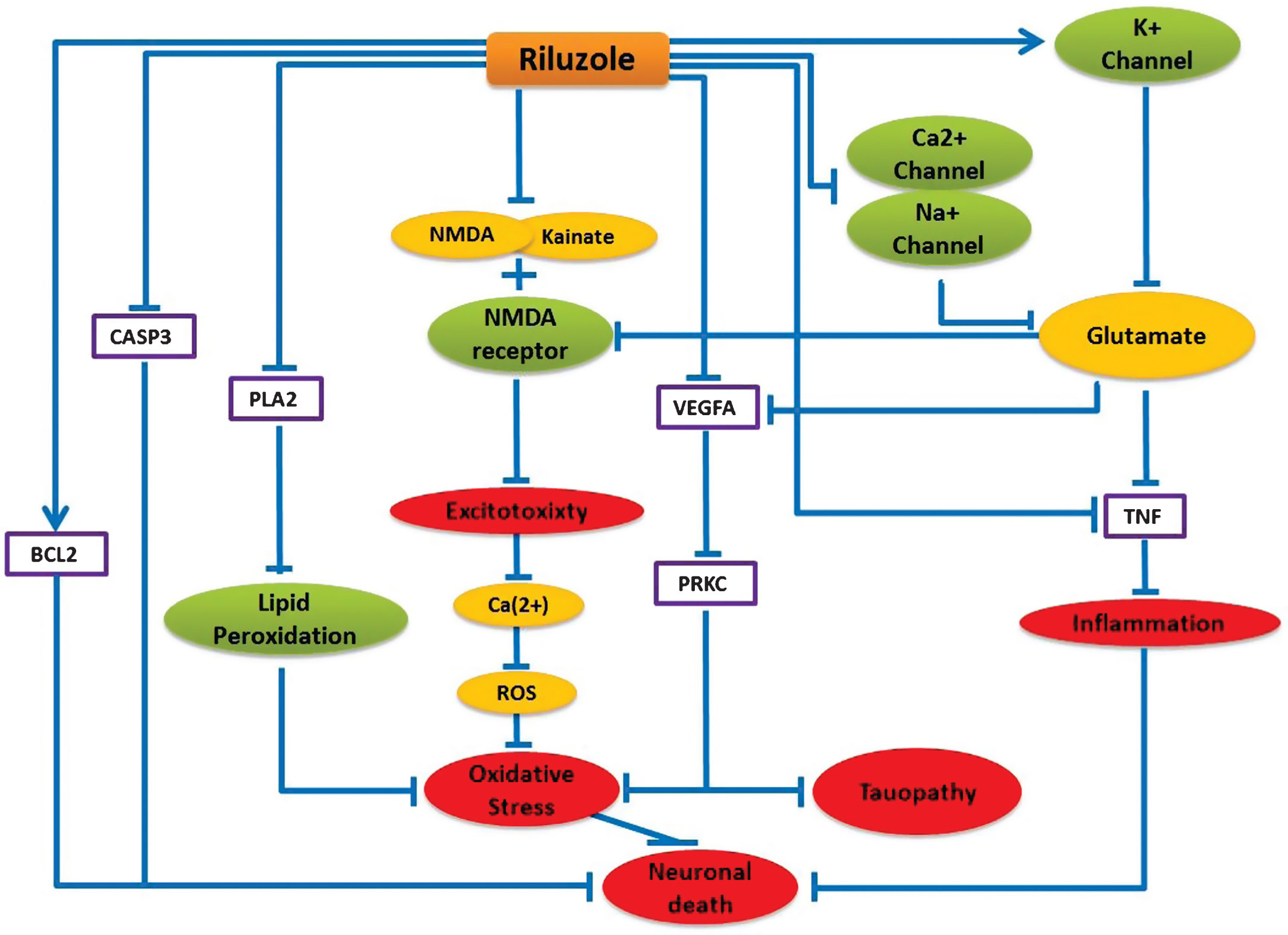
Fig.4
Distribution of common targets between cyclosporine and five AD approved drugs. The pie chart shows the number of targets that cyclosporine shares with 5 different approved AD drugs.
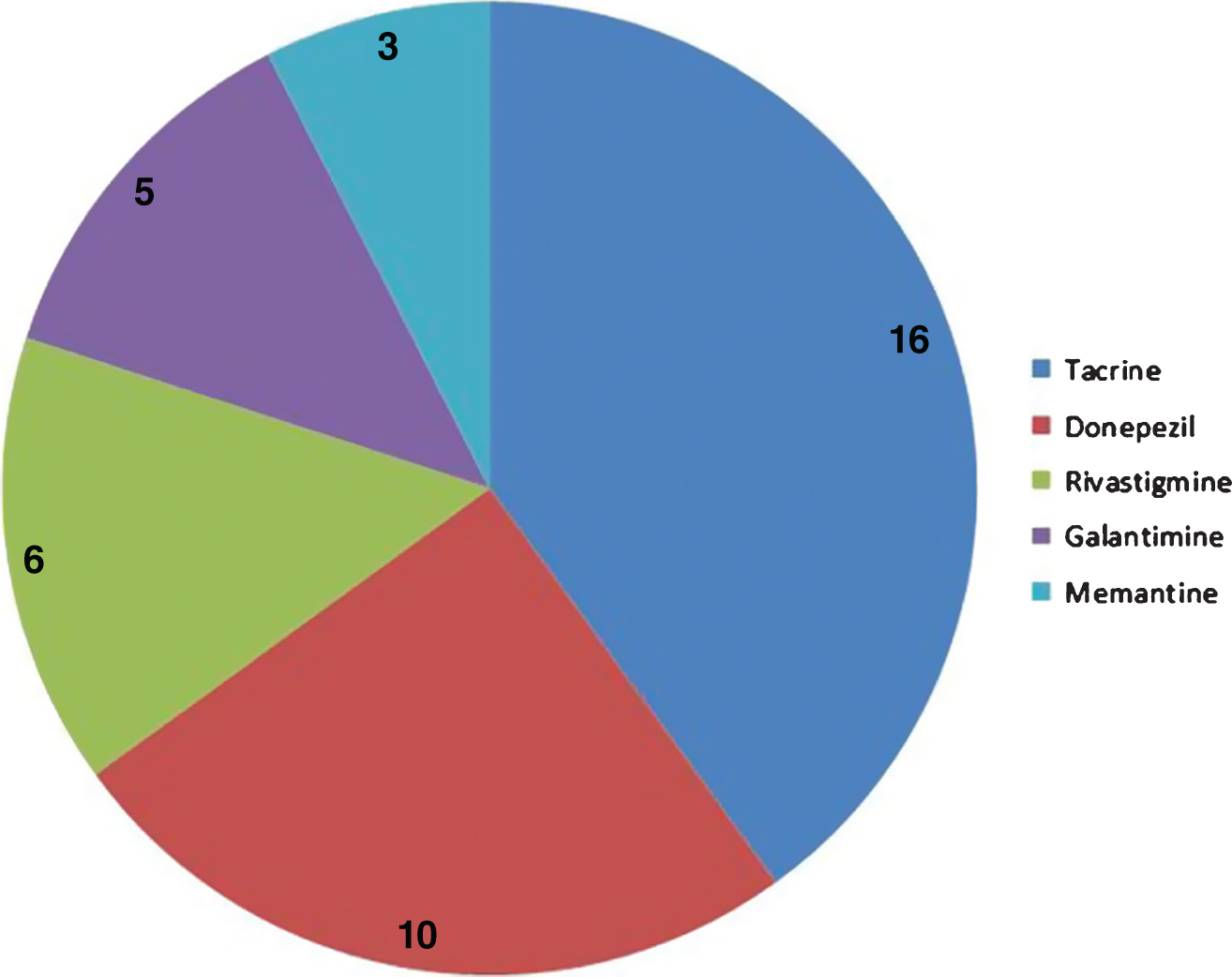
Fig.5
Neuroprotective effects of cyclosporine in the context of AD. This cartoon demonstrates the mode of action of cyclosporine explicitly. The blue lines here represent the alternative effect of cyclosporine on these different pathways. Cyclosporine mainly inhibits T helper cells to suppress the immune system. Cyclosporine found to affect neuronal cell death by inhibiting cyclophilin D that prevents the cytochrome C release and CASP9, CASP3 activation. Cyclosporine can also down regulate ACHE and BCHE which can provide improved cholinergic function. Moreover, cyclosporine might be useful to prevent amyloid beta accumulation by preventing ABCC2 and ABCB1 proteins.
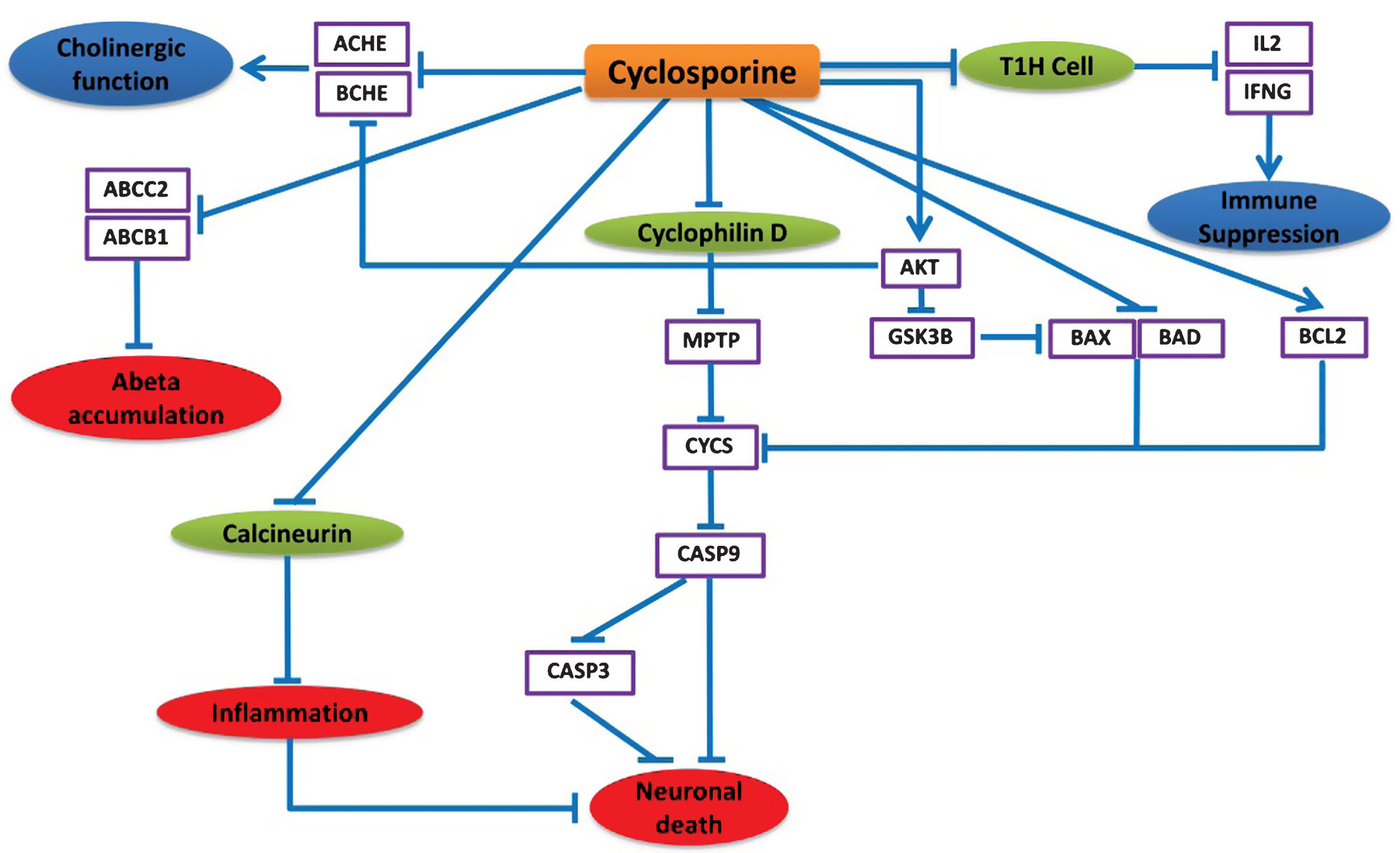
Table 1
Top Pathways from the gene set enrichment analysis (GESA)
| Pathway Names | Count | p-value | Bonferroni | FDR |
| Alzheimer’s disease | 30 | 2.02E–29 | 2.43E–27 | 2.32E–26 |
| Amyotrophic lateral sclerosis (ALS) | 24 | 7.85E–18 | 9.41E–16 | 9.01E–15 |
| Pathways in cancer | 50 | 1.98E–14 | 2.37E–12 | 2.27E–11 |
| Prostate cancer | 26 | 4.82E–14 | 5.78E–12 | 5.53E–11 |
| MAPK signaling pathway | 39 | 1.20E–10 | 1.43E–08 | 1.37E–07 |
| Neurotrophin signaling pathway | 26 | 1.46E–10 | 1.75E–08 | 1.67E–07 |
| Bladder cancer | 16 | 1.52E–10 | 1.83E–08 | 1.75E–07 |
| Calcium signaling pathway | 31 | 1.55E–10 | 1.86E–08 | 1.78E–07 |
| Pancreatic cancer | 20 | 2.04E–10 | 2.44E–08 | 2.34E–07 |
| Toll-like receptor signaling pathway | 23 | 4.13E–10 | 4.96E–08 | 4.75E–07 |


