
AVN-101: A Multi-Target Drug Candidate for the Treatment of CNS Disorders
Abstract
Lack of efficacy of many new highly selective and specific drug candidates in treating diseases with poorly understood or complex etiology, as are many of central nervous system (CNS) diseases, encouraged an idea of developing multi-modal (multi-targeted) drugs. In this manuscript, we describe molecular pharmacology, in vitro ADME, pharmacokinetics in animals and humans (part of the Phase I clinical studies), bio-distribution, bioavailability, in vivo efficacy, and safety profile of the multimodal drug candidate, AVN-101. We have carried out development of a next generation drug candidate with a multi-targeted mechanism of action, to treat CNS disorders. AVN-101 is a very potent 5-HT7 receptor antagonist (Ki = 153 pM), with slightly lesser potency toward 5-HT6, 5-HT2A, and 5HT-2C receptors (Ki = 1.2–2.0 nM). AVN-101 also exhibits a rather high affinity toward histamine H1 (Ki = 0.58 nM) and adrenergic α2A, α2B, and α2C (Ki = 0.41–3.6 nM) receptors. AVN-101 shows a good oral bioavailability and facilitated brain-blood barrier permeability, low toxicity, and reasonable efficacy in animal models of CNS diseases. The Phase I clinical study indicates the AVN-101 to be well tolerated when taken orally at doses of up to 20 mg daily. It does not dramatically influence plasma and urine biochemistry, nor does it prolong QT ECG interval, thus indicating low safety concerns. The primary therapeutic area for AVN-101 to be tested in clinical trials would be Alzheimer’s disease. However, due to its anxiolytic and anti-depressive activities, there is a strong rational for it to also be studied in such diseases as general anxiety disorders, depression, schizophrenia, and multiple sclerosis.
INTRODUCTION
Multi-target drug discovery is an emerging area of increasing interest to the drug discovery community [1]. It has been shown that mono-target drugs are usually do not work well for curing complex diseases due to the fact that many diseases are complex and multi-factorial [2]. AVN-101 is a novel drug candidate designed as a multi-targeted therapy for the treatment of cognitive, neurodegenerative, and anxiety disorders, which affect millions of people worldwide. Alzheimer’s disease, epilepsy, schizophrenia, depression, obsessive-compulsive disorder, migraine, circadian rhythm disorders, and sleep disorders are the most common types of these diseases. An estimated 5.3 million Americans of all ages will have Alzheimer’s disease in 2015 [3]; the prevalence rate for schizophrenia is approximately 1.1% of the population over the age of 18 (source: NIMH) or, in other words, at any one time as many as 51 million people worldwide suffer from schizophrenia, including 2.2 million people in US [4]; and anxiety disorders are the most common mental illness in the US, affecting 40 million adults at the age of 18 and older, or 18% of the population [5].
Though no cure exists currently for Alzheimer’s disease, serotonin receptors attract considerable interest as potential targets to alleviate living conditions of patients. Also, the serotonin receptors are being intensely investigated as targets for other diseases of central nervous system (CNS) [6].
Quite recently, one of the newest receptors, 5-HT6 receptor, has been extensively studied as potentially valuable therapeutic target [6–8]. 5-HT6 receptor has been cloned and pharmacologically characterized in 1993 as a receptor with moderate to low affinity to serotonin, positively coupled with adenylyl cyclase [9, 10]. Its highest expression was detected in such brain areas as striatum, olfactory tubercle, nucleus accumbens, and hippocampus [9].
A plethora of highly selective and structurally diverse 5-HT6 receptor antagonists has been developed since the receptor discovery [11–17]. Much hope was attributed to the receptor blockade as a means to cognitive enhancement, in general, and in Alzheimer’s disease patients, in particular [8, 17].
However, early clinical trials proved to be a decisive negative filter for most of them to transit to more advanced phases. Only two 5-HT6 receptor antagonists, SB-742457 [17] and Lu AE58054 [18], have progressed to the Phase III clinical trials in patients with mild to moderate Alzheimer’s disease [18–23].
Lu AE58054 was shown to statistically improve cognitive function in patients with moderate Alzheimer’s disease being treated with donepezil [21]. These data in combination with earlier Phase I studies, encouraged Lundbeck to begin multiple Phase III clinical trials of LU-AE-58054 with donepezil or with an acetyl cholinesterase inhibitor in patients with mild to moderate Alzheimer’s disease [22, 24].
GSK has been developing 5-HT6 receptor antagonists for quite a long time and its highly potent (PKi = 9.63) and selective drug candidate, SB-742457, was under development for the treatment of Alzheimer’s disease and demonstrated some preliminary efficacy in Phase II clinical trials [18, 19]. Based on the preclinical and Phase I and Phase II clinical studies performed by GSK, Axovant Sciences Ltd., which purchased the rights for the SB-742457 [25] entered it in 2015 into Phase III 12-month open-label study under the name RVT-101 for the treatment of mild-to-moderate Alzheimer’s disease [23, 26].
Another serotonin receptor, 5-HT7, with moderate to high affinity for serotonin binding and positive coupling to adenylyl cyclase has also been cloned and characterized in 1993 [9, 27]. Several splice variants of the 5-HT7 receptor have been identified with no major differences in their pharmacology and ability to activate adenylyl cyclase [28]. Its major anatomical localization in rat brain areas such as cerebral cortex, hippocampal formation, tenia tecta, thalamus, and hypothalamus, has been established employing multiple strategies, such as immunocytochemistry, in situ hybridization, and agonist stimulated cFos expression [29]. Involvement of the 5-HT7 receptor in the modulation of learning and memory processes has been discussed to provide a basis to obtain new therapeutic agents and strategies for the treatment of learning and memory disorders [30]. It should be noted that literature on the potential role of the 5-HT6 and 5-HT7 agonists and antagonists is quite controversial [31, 32]. In mice models lacking 5-HT7 receptor or where the receptor was blocked with a selective antagonists, the hippocampus-dependent place learning was subtly impaired [33]. On the other hand, positive effects of the 5-HT7 antagonists were reported for depression [34], working memory deficit [35], and schizophrenia-like deficit [36]. One must keep in mind though that the conclusions have been reached based on results obtained in animal models, which do not always correctly reflect real CNS diseases in humans. Taking into consideration that the anatomical brain distribution areas of both the 5-HT6 and 5-HT7 receptors partially overlap with each other in brain zones responsible for cognition, memory, and anxiety and both receptors are coupled to the same signal transduction mechanism (adenylyl cyclase stimulation), it is feasible to suggest the role of 5-HT7 receptors in age-dependent cognitive impairments and in other CNS disorders such as depression [37], mild to moderate Alzheimer’s disease, schizophrenia [32], and others.
In this work, we summarize the data on the orally bio-available CNS drug candidate, AVN-101, designed and developed based on the multi-target approach, with an emphasis on dual 5-HT6 and 5-HT7 receptors antagonism. We also compared its characteristics to those available for Lu AE58054 and SB-742457.
One of potential disorders to be treated with the multi-target drugs is a host of CNS disorders collectively called anxiety. Among them are: panic disorder, generalized anxiety disorder, social anxiety disorder, agoraphobia, specific phobia, separation anxiety disorder, selective mutism, substance/medication-induced anxiety disorder, anxiety disorder due to another medical condition. Anxiety disorders are widespread psychiatric conditions with significant social and professional disability, poor quality of life, an increased risk of suicide, and frequent attendance of medical services. Despite the high prevalence rates of these anxiety disorders, they often are under recognized and undertreated clinical problems. The etiology of anxiety disorders is complex and they share genetic and environmental risk factors [38]. They often occur in conjunction with other mental disorders, particularly major depressive disorder, bipolar disorder, certain personality disorders, and eating disorders. The anxiety disorders cover mainly four aspects of experiences that an individual may have: mental apprehension, physical tension, physical symptoms, and dissociative anxiety [39]. The emotions manifested by anxiety disorders range from simple nervousness to bouts of terror [40].
There are various forms of anxiety disorders, including generalized anxiety disorder, phobic disorder, and panic disorder. While each has its own characteristics and symptoms, they all include symptoms of anxiety. As of 2010, approximately 273 million (total prevalence –3.96% of the population) had an anxiety disorders and the number of sufferers increased 36.4% compared to 1990 [41]. The prevalence of anxiety disorders is significantly higher in females (5.18%) than in males (2.76%).
Besides the psychological treatment of patients, “the goal of pharmacological treatment of anxiety disorders is to relieve the symptoms, prevent relapses, as well as the lasting effects, all with the best possible toleration of the medication” [42]. According to the guidelines, antidepressants for long-term and tranqilizers for short-term treatment have proved efficacious for anxiety disorders. Current medications used in both treatment stratagems have significant limitations in particular due to their ineffectiveness in a substantial number of patients [43] and in some cases intolerable side effects [44]. Therefore, discovery of new medication for anxiety disorders with additional modes of action is warranted.
In this paper, we present the results of preclinical and early Phase I clinical studies of a new drug candidate, AVN-101, which has been developed on the basis of SAR research of numerous hydrogenated gamma-carbazoles [12] and their heterocyclic analogues [45]. Based on drug design, synthesis, and extensive medicinal chemistry efforts [13, 47–69], we came up with several small molecule candidates for further pre-clinical evaluation. Multiple iterations of γ-carboline substitutions brought to bear a promising multi-modal molecule, AVN-101, with a superior 5-HT7 receptor affinity, good bioavailability, low toxicity, and reasonable efficacy in animal models of CNS-related activity. The drug candidate has a potential to treat different CNS diseases in general and Alzheimer’s disease in particular. For the treatment of Alzheimer’s disease with AVN-101, we concentrate on the role of the serotonin receptors in influencing (improving) symptoms associated with the progression of the disease (such as anxiety, depression, sleep disorder, and decline in memory and cognition functions) and not on potential association with AβPP metabolism or tau phosphorylation [46].
RESULTS AND DISCUSSION
Molecular pharmacology of AVN-101
Physicochemical descriptors
AVN-101 (2,8-dimethyl-5-penethyl-2,3,4,5-tetra-hydro-1h-pyrido[4,3-B]indole hydrochloride) (see figure of the AVN-101 structure) is a multi-modal receptor antagonist with promising pre-clinical characteristics, which is under development as an orally available drug candidate to treat anxiety, memory disorders, and, potentially, Alzheimer’s disease. Physicochemical characteristics of the AVN-101 are in a good compliance with the Lipinski rule of 5 [70].
Major physicochemical descriptors have been assessed using calculator built-in into Accelrys’ DS ViewerPro 6 and are presented in Table 1.
Target specificity/selectivity. Ability of AVN-101, as well as that of its M1 and M2 metabolites, to interact with multiple targets was tested in a competitive radio-labeled ligand displacement assays against multiple therapeutic targets including GPCRs, neuromediator transporters, ion channels, and enzymes (measured as inhibition of their enzymatic activity) [54, 67, 71]. The affinity values, binding pKi, were measured for those targets that demonstrably responded to the AVN-101 in a primary specificity profile at its concentration of 1μM, are presented in Fig. 1.
Serotonin receptors. AVN-101 is selective toward 5-HT7 receptor with the affinity in a low picomolar range, Ki = 153 pM. It also exhibits rather strong affinity toward 5-HT2 C (Ki = 1.17 nM), 5-HT2A (Ki = 1.56 nM), and 5-HT6, (Ki = 2.04 nM), receptors, followed by 100-fold to 200-fold lower affinity to 5-HT2B (Ki = 10.6 nM), 5-HT5A (Ki = 20.8) nM and 5-HT1A (Ki >30 nM) receptors.
It should be mentioned that while both the SB-742457 and Lu AE58054 show higher affinity to 5-HT6 receptor (Ki = 234 pM [72] and 830 pM [73], respectively) than that of AVN-101 (Ki = 2.04 nM), neither exhibits comparable affinity toward the 5-HT7 receptor.
The SB-742457 is claimed to be highly selective antagonist with at least a 100-fold selectivity against other receptors. However, we failed to locate an original paper on SB-742457 to be able to clarify if the 5-HT7 receptor was among those “other receptors” claimed to be tested. Therefore, we have tested its affinity in parallel with AVN-101. In our hands (Fig. 29 in [13]), the receptor binding profile of SB -742457 at 1μM, showed very low affinity to the 5-HT7 receptor (∼25% binding inhibition). Assessment of the AVN-101 and SB -742457 affinities to 5-HT7 receptor was performed in parallel (Fig. 2A) and produced IC50 values of 0.28 nM and 1.27μM, correspondingly. We should mention though that based on the data of target panel (Fig. 29 in [13]) SB-742457 was not inordinately specific. Indeed, at 1μM, SB-742457 displayed competition for 5-HT2A (91% radioligand displacement), 5-HT1B (86% displacement), 5-HT2B (88% displacement), and 5-HT2C (85% displacement). We have also compared affinities of the AVN-101 and SB -742457 to the 5-HT2A receptor (Fig. 2B).
The data shows that while the AVN-101 has ∼4-orders of magnitude higher affinity to the 5-HT7 receptor, the affinities of both compounds to the 5-HT2A receptor are practically equal to each other. Alterations of 5-HT2A receptor activity may be a contributing factor to the development of cognitive dysfunction in mental disorders such as schizophrenia and depression, and thus, may provide an important target for drug therapy of memory dysfunctions [74]. Antagonists of 5-HT2A receptor are considered to be useful in treatment of psychosis, and possibly alcohol and cocaine dependence [75] and the receptor importance in mediating CNS functions is consistent with its considerable expression in various regions of nervous system [76].
Similar to SB-742457, the Lu AE58054 has very low, if any, binding affinity to the 5-HT7 receptor [73]. However, unlike the SB-742457, it does not bind well with 5-HT2A receptor (Table 2). This data shows that while Lu AE58054 is highly selective toward the 5-HT6 receptor, AVN-101 is highly selective toward the 5-HT7 receptor (Ki = 153 pM).
Thus the AVN-101 specificity pattern is distinctly different from that of both the Lu AE58054 and SB-742457. In particular, the AVN-101 has highest selectivity toward the 5-HT7 receptor with an order of magnitude lower affinity toward the 5-HT6, 5-HT2A, and 5-HT2C receptors. It has been speculated (see in [32]) that blockade of 5-HT7 receptor could reverse memory deficits. Also, it was shown that additive combination of 5-HT6 and 5-HT2A specific antagonists but not the individual antagonists by themselves, was able to prevent MK-801-induced prepulse inhibition (PPI) deficit in rats [77]. It seems very appealing to have a candidate with ability to block all three potential targets (5-HT7, 5-HT6, and 5-HT2A) for treatment of CNS disorders.
It is still to be seen what role the 5-HT2C ligand could play in a combination with inhibition of other serotonin receptors but it was shown that 5-HT2C receptor antagonists stimulate dopaminergic and adrenergic pathways and exert antidepressant and anxiolytic actions [78].
Affinity of AVN-101 to another serotonin receptor, 5-HT2B, is also rather high (Ki = 10.6 nM, Table 2). Though major expression of this receptor is found in smooth muscles of stomach, esophagus, duodenum, small and large intestine, myocardium, and lung, some expression has also been detected in brain cerebellum [79]. The role of 5-HT2B receptor in CNS diseases is not quite established yet, and potential negative effect of its stimulation was associated with valvular heart disease [80]. Therefore, the AVN-101 was tested in both the cell-based functional assay (Fig. 3A, intracellular Ca2 + mobilization in HEK 297 cells with extragenously expressed human recombinant 5-HT2B receptor) and in a rat stomach fundus contraction assay (Fig. 3).
Addition of AVN-101 alone produced neither increase in the intracellular free Ca2 + ions (Fig. 3A) nor contraction of the rat stomach fundus (Fig. 3B), meaning that it lacked intrinsic agonistic activity on the 5-HT2B receptor while effectively blocking both the α methyl-serotonin-induced spike in the intracellular Ca2 + concentration and α methyl-serotonin-induced fundus contraction. This finding makes AVN-101 safe from the potentially developing cardio vulvopathy in patients.
Histamine receptors. AVN-101 shows some ability to compete with radio-labeled ligands for both the histamine H1 (Ki = 0.58 nM) and H2 receptors (Ki = 89 nM) while being totally inactive on H3 receptor. H1 receptor is widely distributed in different brain regions and its activity is associated with Gq11 [81] and, hence, modulation of intracellular Ca2 + mobilization. H2 receptor is positively coupled to adenylate cyclase via Gs. and hence, its stimulation produces increase in cAMP production.
To test AVN-101 for its activity, either agonistic or antagonistic, we used cell-based functional assays with human neuroblastoma cell line, SK-N-SH, which endogenously expressed H1 receptor and CHO-K1 (AequoZen cells, Eurofin), which exogenously expressed H2 receptor. SK-N-SH cells were stimulated with 10μM histamine and intracellular spike in free Ca2 + concentration was registered. CHO-K1 cells were stimulated with 50 nM amthamine and intracellular accumulation of cAMP was registered.
We found that the AVN-101 activated neither H1 nor H2 receptors but effectively blocked the corresponding agonist-induced cell responses (Fig. 4A) and competed with both H1 and H2 receptors for their radio-labeled ligands (Fig. 4B).
Histamine functions in CNS as a neuromediator, and its actions are associated with wakefulness, alertness, and reaction time. Antagonists (reverse agonists) of H1 receptor are suggested to lead to drowsiness [82]. On the other hand, it was shown [83] that histamine and its receptors have been implicated in the multiple sclerosis, a chronic inflammatory demyelinating disease of the CNS. In mice knockout experiments, the authors showed that only a dual H1/H2 knockout was effective in preventing or reducing severity of experimental allergic encephalomyelitis, the autoimmune model of multiple sclerosis. Thus, the dual H1/H2 antagonistic activity is a very helpful feature of the AVN-101 that could be useful in preventing CNS inflammation. Both the SB-742457 and Lu AE58054 lack such a feature.
Imidazoline I2 receptor. AVN-101 interacts with inidazoline I2 receptor (Fig. 1). The ability of some β-carboline derivatives to interact with imidazoline receptors has previously been published [84], and it was shown that the imidazoline receptors could be involved in a multitude of neurophysiologic and neuropathologic functions [85]. Therefore, the ability of the AVN-101 to interact with these receptors could also be an advantageous feature of the AVN-101 treatment; however, its affinity to the receptor, Ki = 179 nM, is a thousand times as high as the one for 5-HT-7 receptor, Ki = 0.153 nM.
Adrenergic receptors. From the data of Fig. 1, one can see that AVN-101 also binds to α2 receptors with higher affinity than to α1 receptors (Table 3).
The AVN-101 demonstrates selectivity toward α2B receptor followed by α2A and α2C (in the order of affinity) and then by α1B, α1A, and α1D. In CNS, adrenergic α2A and α2C receptors are predominantly localized in pre-synaptic areas while α2B receptors localized post-synaptically [86]. Due to a lack of selective antagonists, the role of adrenergic α2B receptors in behavioral disorders is not clear yet. Adrenergic α2A and α2C receptors control the CNS physiologic functions based on modulation of negative feedback circuitry controlling inhibition of the noradrenalin release. It was postulated that α2A- and α2C-receptors complement each other to integrate CNS function and behavior [86] and hence, drugs acting via the α2A+α2C receptors may have therapeutic value in disorders associated with enhanced startle responses and sensorimotor gating deficits, such as schizophrenia, attention deficit disorder, posttraumatic stress disorder, and drug withdrawal, depression, and anxiety.
Though the expression of adrenergic α1 receptors are rather abundant throughout the different tissues of human body [87] the most established role of the receptor blockers was reported for treatment of hypertension and symptomatic relief of prostatic hypertrophy [88]. The elderly have a much higher hypertension prevalence rate than younger people [89] and Alzheimer’s disease develops later in the life, which makes it beneficial for the AVN-101 to also control blood pressure through its anti-adrenergic activity.
Dopaminergic receptors. As seen from Fig. 1, AVN-101 has very low affinity toward these receptors compared to the 5-HT7 receptor.
Other targets. AVN-101 does not bind to the following tested targets (see Fig. 1): hERG channel, sodium channel (Site 2), calcium channels of L and N type, dopamine and norpinephrine transporters, muscarinic M2 receptor, sigma 1 receptor, and histamine H3 receptor.
Pre-clinical and early clinical studies of AVN-101
All animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committee and institutional guidelines for the proper, humane use of animals in research were properly followed.
Metabolism
Stability in aqueous solutions. AVN-101 is rather stable upon prolong (72 h) incubation at room temperature in aqueous solutions. After the 72 h incubation, the fraction of original AVN-101 concentration was equal to: in distilled water: 97.03%; in salt solution at pH 4.0 : 98.05%; in salt solution at pH 7.0 : 99.95%; and in physiological solution at pH 7.4 : 99.38%.
Stability in mouse, simian, and human blood plasma. AVN-101 is poorly stable in murine plasma: by the end of 30-min incubation at room temperature, the residual amount of the original compound is reduced to 16%. At the same time, AVN-101 shows better stability in plasma samples taken from monkey and human: by the end of the 30-min incubation at room temperature, the residual amount of the original compound is reduced to only 45% and 68%, respectively.
Human plasma protein binding. Assessment of an ability of AVN-101 to bind to human plasma proteins was performed with whole plasma at AVN-101 concentrations of 0.5 mg/mL and 5 mg/mL using equilibrium dialysis approach (Fig. 5).
It appeared that ∼41% of the AVN-101 was bound to the plasma proteins and this percentage did not depend on the AVN-101 concentration in the range between 0.5 and 5μg/mL.
Permeability in Caco-2 model. Caco-2 Permeability assay uses an established method for predicting the in vivo absorption of drugs across the gut wall by measuring the rate of transport of a compound across a monolayer Caco-2 cell line [90]. In characterizing ability of potential drugs to interact with and be transported by the ATP-binding cassette transporters (ABC), known also as multidrug resistance protein 1, MDR1, or P-glycoprotein, Pgp, it is suggested to use three approaches: i) measure activation of ATPase activity as an indication of being a Pgp substrate, ii) measure efflux rate of the test drug as an indication of being actually transported out by the Pgp, and iii) measure effect of the drug on efflux of known compound with asymmetric influx/efflux characteristics as an indication of potential inhibition of the Pgp transport [91].
The Caco-2 cells are derived from a human colon carcinoma, which have characteristics similar to intestinal epithelial cells— formation of a polarized cell monolayer, well-defined brush border on the apical surface, and intercellular junctions. Assessment of a drug transport in both directions [apical-to-basolateral (A-B) and basolateral-to-apical (B-A)] across the CACO-2 cell monolayer enables determination of the efflux asymmetry and hence, indication of either the drug is being actively transported by the Pgp pump. The data in Table 4 shows that AVN-101 belongs to a category of drugs with rather high CACO-2 permeability in both directions with no apparent permeability asymmetry of the transport in both directions, A⟶B and B⟶A, asymmetry ratio = 1.1. Hence, it is not considered to be expelled from the cells by the Pgp-pump and as a result, would have a good bioavailability. The lack of the asymmetry in permeability is quite common for highly permeable drugs and is attributed to assay limitations [91] rather than the inability of the highly permeable drugs to interact with the Pgp. Therefore, we have investigated effects of the AVN-101 on Pgp ATPase activity as well on permeability of Rhodamine123 known to undergo active efflux through the Pgp pump.
For studies of ATPase activity, we used Pgp-Glotrademark Assay System with P-glycoprotein (Promega Corporation, USA). Effect of the AVN-101 on orthovanadate-sensitive ATPase activity was measured in accordance with manufacturer’s protocol and data is presented in Fig. 6. The AVN-101 causes strong activation of the Pgp ATP-ase activity, which is an indication of the AVN-101 being a non-transportable substrate for the Pgp [91].
Next, we have tested ability of the AVN-101 to inhibit the Pgp pump by assessing its effect on an asymmetric permeability of Rhodamine123 through the Caco-2 monolayer (Table 4). Indeed, the AVN-101 has considerable effect on the asymmetry of the Rhodamine123 permeability for the monolayer comparable with that of known inhibitor of the Pgp pump, verapamil. This presents an additional advantage of using AVN-101 as a co-drug that would enhance overall permeability of the accompanied drug, which could be Pgp-dependent. Of course, one must keep in mind that the apparent potency of the AVN-101 to stimulate Pgp-related ATPase activity (EC50 ∼50μM) is many orders of magnitude lower than that needed to block both the 5-HT6 and 5-HT7 receptors, and further research will need to be done to show if such concentrations of the AVN-101 even achievable in the gut with therapeutically relevant dose formulations.
In vitro metabolism in rat and human microsomes. When tested at a concentration of 1μM, AVN-101 rapidly metabolized by both the human (HLM) and rat (RLM) microsomes (Fig. 7 and Table 5).
Ten probable metabolites have been structurally identified, M1-M10 (Fig. 8). M1, M2, M4-M6, M9, and M10 were found in quantifiable amounts only in reactions with human microsomes. M7 was identified only with rat microsomes, and M3, M8, and a few other unidentifiable metabolites were found in both the rat and human microsomes.
By the end of a 30-min incubation time, the remaining amount of AVN-101 comprised only 2% and <1% for human and rat microsomes, respectively (Table 6). However, at 2μM the decomposition AVN-101 in human microsomes was substantially diminished; only 57% of the original AVN-101 was consumed by the end of 30-min incubation (Table 6). It seemed that at the higher concentration, AVN-101 might have inhibited some cytochromes(s) participating in the AVN-101metabolism.
Indeed, the quantity of a major metabolite, M3, in HLM reaction (83%) when measured at 1μM AVN-101, substantially dropped (6%) when 2μM AVN-101 was used (Table 6).
To determine which cytochromes could participate in the AVN-101 metabolism, we have tested six most common human recombinant cytochromes, and relative quantities of AVN-101 remaining after 30-min incubation with each cytochrome at 37°C are presented in Table 7. One can see that AVN-101 is most probably metabolized by CYP2C19, CYP2D6, CYP3A4, and, partially, by CYP1A2. Neither CYP2C8 nor CYP2C9 participate in the AVN-101 metabolism.
Three metabolites, M1 (2-(2,8-dimethyl-1,2,3,4-tetrahydro-pyrido[4,3-b]indol-5-yl)-1-phenyl-ethan-ol), M2 (2-methyl-5-phenethyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indol-8-yl)-methanol)), and M3 (2-methyl-5-phenethyl-2,3,4,5-tetrahydro-1H-pyri-do[4,3-b]indol-8-yl)-carboxilic acid)), have been synthesized and their structures established. As our PK and blood-brain barrier permeability studies showed, metabolite M3, containing carboxylic group, did not accumulate in the brain in reasonable concentrations, hence, we found it unnecessary to characterize its affinity to the receptors at this point.
Compared to AVN-101, M1 has practically same affinity to the adrenergic α1A receptor, 3-fold lower affinity to α2A receptor, 83-fold lower affinity (Ki =∼48 nM) to histamine H1 receptor, 9-fold lower affinity to H2 receptor, 2-fold lower affinity to 5-HT2A receptor, 7-fold lower affinity to 5-HT2C receptor, 4-fold lower affinity to 5-HT6 receptor, and 50-fold lower affinity (Ki = 7.72 nM) to 5-HT7 receptor.
M2 metabolite exhibits 22-fold reduction in affinity to the 5-HT6 receptor and 9-fold reduction in affinity to histamine H2 receptor (Fig. 1).
Neither AVN-101 nor its metabolites, M1 and M2, seem to interact with 5-HT3 and 5-HT4 receptors. In Phase I PK studies, we found that in humans, the metabolite M2 was not detectable in blood and, hence, we did not profile it further.
Metabolism in human liver S9 fraction. Next, we assessed if the AVN-101 could be metabolized not only with phase 1 metabolism enzymes but can also be susceptible for phase 2 metabolism, in particular, through glucuronidation. For that, we used human and rat isolated S9 fractions that contain enzymes of both the phase 1 and phase 2 metabolism. The data of Fig. 9 shows that the AVN-101 easily metabolized in the presence of NADPH, a phase 1 metabolism co-factor, being more stable with the human S9 fraction (see also half life of metabolism in Table 8). In the presence of uridine-diphosphate-glucuronic acid (UDPGA), a co-factor of phase 2 metabolism, there was no metabolism of AVN-101 observed. This indicates that AVN-101 is not subject to direct glucuronidation. In the presence of both the NADPH and UDPGA (1st and 2nd metabolism phases), no apparent difference in the metabolism rate was observed compared to only NADPH.
Noteworthy,increased concentration of AVN-101 led to a slower rate of metabolism with the S9 fraction (Table 8), similar to what we have observed with its metabolism by purified microsomes.
Pharmacokinetics of AVN-101 and its metabolites, M1 and M2
Pharmacokinetics of AVN-101 in mice. We have assessed pharmacokinetics of AVN101 in mice upon intraperitoneal (IP) administration (Fig. 10).
Several doses were used to see if there is a pattern of dose-related overload of the drug (Table 9, Fig. 11).
The data presented in Fig. 11 shows that there is a linear relation between the drug dosage used (Fig. 11A) and either maximal concentration (CMAX) or exposure (AUC). Interesting that the half-life of the drug elimination was also dose-dependent and it reached a plateau at T1/2 = 130 min at a dose of about 2 mg/kg (Fig. 11B).
Pharmacokinetics of AVN-101 in rats. Bioavailability of the AVN-101 (5 mg/kg) was assessed in Wistar rats upon its administration through oral (PO) and IP routes as compared to the intravenous (IV) route (Fig. 12, Table 10). AVN-101 was detected in the blood plasma using LC-MS/MS.
Summary of the PK parameters in rats is shown in Table 10. The AVN-101 time-dependent concentrations in the blood were registered using LC-MS/MS method.
Based on this data, bioavailability of AVN-101 in rats (as calculated based on values of AUC0 ⟶240) is 26.3% upon IP administration and 8.5% upon PO administration. It should be noted though that the drug elimination determined in the in vivo experiment, Kel = 0.0121 min–1 (IV route, Table 10), is substantially slower than that observed with the rat microsomes (Kel = 0.335 min–1, see Table 5).
Biodistribution of AVN-101 in rat organs. Distribution of AVN-101 and its metabolites in main organs of the rats was assessed with [3H]AVN-101 as a marker. Male Wistar rats (180–200 g; n = 36) were administered, either orally or intravenously, with the AVN-101 containing [3H]AVN-101 (22.2×106 dpm/mg) at a dose of, respectively, 10 mg/kg and 5 mg/kg. 4 h after the drug administration, the animals were killed by placing them into CO2 chamber, tissues were collected immediately after the death (3–5 min), washed with ice-chilled saline, homogenized, and their radioactivity was analyzed using a MicroBeta LSC counter (PerkinElmer, USA).
The data, presented in Table 11 and Fig. 13A, showed that 4 h after IV or PO administration, AVN-101 redistributed into all the organs studied, irrespective of the administration route. The lowest AVN-101 concentration was observed in the blood. The highest accumulation of the drug was observed in the brain, 13-fold (PO) and 26-fold (IV) higher than in the blood. Concentration of the AVN-101 in all other organs was also higher than that in the blood thus indicating a high volume of distribution. When comparing the AVN-101 tissue accumulation as a function of the drug administration route (Fig. 13B and Table 11), one can conclude that brain, kidney, liver, and spleen show similar to each other accumulation pattern with the relative oral bioavailability of 31.5% –36.2%, while accumulation of the AVN-101 in heart and lung tissues was less effective upon PO administration (oral availability of 17.7% –17.9%). The higher efficiency of the AVN-101 distribution into heart and lungs upon IV administration compared to the PO administration could be responsible for the lower IV/PO ration of the AVN-101 concentrations in the blood, 1.57, which provides a good explanation of the relatively high oral bioavailability in the blood, 63.9% (Table 11).
Brain/plasma distribution of the AVN-101 and its metabolites, M1 and M3, in rats. As distribution data above showed, the major AVN-101 accumulation compartment is the brain, which is the organ of our interest of developing the drug for CNS diseases. Therefore, the brain/plasma distribution was studied in more details using LC-MS/MS method for detection of both the non-metabolized AVN-101 and its two major metabolites that are produced by microsomes. The Sprague Dawley rats were administered with the AVN-101 (2 mg/kg through the IV route and after specified time intervals, the animals were euthanized by placing them into CO2-filled chamber for 3–5 min. The blood, cerebrospinal fluid (CSF), and brain samples were taken at different time points after IV administration, from 3 rats in each time group. The blood was drawn through cardio puncture into vacutainer tubes filled with heparin, CSF was aspirated from cisterna magna [92], and brains were removed from the crania and placed on ice. Blood samples were centrifuged to remove blood cells and brains were thoroughly washed with an ice-chilled saline solution and homogenized in four volumes of purified water. All plasma and CSF samples as well as brain homogenates were frozen and kept under liquid nitrogen until sample preparation for the analyses was initiated.
Kinetics of the AVN-101 distribution as well as that of its metabolites, M1 and M3, is shown for the rat blood plasma (Fig. 14A), brain homogenates (Fig. 14B), and CSF (Fig. 14C).
In compliance with the previous data (Table 11 and Fig. 13), the AVN-101 exposure in the brain was substantially higher than in the blood starting with very first time point, 5 min (Fig. 14A, B), which indicated that the AVN-101 very rapidly distributed into the brain. Generation of M1 product roughly followed the course of the AVN-101 elimination, though the concentrations of both the M1 and M3 products were substantially lower. Interestingly, the AVN-101 exposure in CSF (Fig. 14C) was very low even comparing with that of blood.
Exposure of the AVN-101 in brain was 20-fold higher than that in blood (compare AUC values in Table 12). In the CSF, the AVN-101 exposure was dramatically lower than in blood and brain.
Elimination rate of the AVN-101 in the Sprague Dawley rats was similar to that of Wistar rats (Fig. 12) and substantially slower (kel = 0.0048 min–1, Table 12) than its in vitro transformation rate by both the microsomes (Fig. 7, Table 5) and S9 fraction (Fig. 9). In both the brain and CSF, the AVN-101 is metabolized very slowly (kel = 0.0092 min–1, and 0.0071 min–1, respectively, Table 12) with formation of M1 metabolite while M3 metabolite can be detected only at very low concentrations (<1 ng/mL) at all time points.
Potential mechanism underlying the slow AVN-101 elimination will be discussed below in the light of discovered fast transport of AVN-101 from the blood stream into the brain and thus shielding a balk of it from systemic circulation.
The analysis of the AVN-101 kinetics in blood plasma (Fig. 14A) and in brain tissue (Fig. 14B) unexpectedly revealed that the AVN-101 permeated the blood-brain barrier extremely quickly and at a substantially higher concentration relative to one in blood plasma. Indeed, the maximal AVN-101 concentration, Cmax, in brain was achieved at the very first time point, 5 min, after the IV administration. Both the Cmax and exposure (AUC) values in the brain were much higher, ca. 20-fold, compared to that in plasma. To explain the relatively low plasma concentration of the AVN-101, we have tested a hypothesis that it could be a result of a strong retention of the compound with the blood cell elements. We measured the AVN-101 concentration in the whole blood and in plasma and blood cells after centrifugal separation of the blood into plasma and cell elements (Fig. 15). From the data in Fig. 15, one can clearly see that the compound detected in the blood is mainly distributed in plasma, and the blood cells have low binding capacity for the AVN-101. At the same time, the AVN-101 concentration in brain was more than an order of magnitude higher than in the blood.
Thus the low plasma concentration of the AVN-101 is caused by fast redistribution of the drug into brain and other organs. The very high concentration of the AVN-101 in the brain even at a very short time (5 min) after IV injection indicates very easy permeation of the compound through the brain-blood barrier, possibly through some transport mechanisms present in the cells forming the blood-brain barrier. Noteworthy and also unexpectedly, the AVN-101 concentration in CSF was substantially lower than that in either blood or brain tissues, which indicates very poor permeation through the blood/CSF and brain/CSF barriers. Though the movement of substances from the blood into the CSF is, in many ways, analogous to that from the blood into the brain, with many of the same transporter systems present in both tissues [93], it appears that the AVN-101 might be effectively prevented from the entry into CSF by some efflux transport mechanisms. The very high concentration of the drug in brain compared to that in both the CSF and blood achieved just 5 min after the drug injection could be a result of high and tight absorption of the compound by brain matter and/or due to energy-dependent pump mechanisms quickly transporting the AVN-101 against its concentration gradient. To test the hypothesis of high absorption of the compound by a brain matter as a potential mechanism of this counter gradient movement of the AVN-101 into brain, we performed a simulation experiment, whereby the rat brain tissue homogenized in four volumes ofphosphate buffered saline (PBS, pH 7.2) was dialyzed against plasma 5-fold diluted with PBS to maintain concentrations of possible AVN-101 binding substances of brain and plasma tissues at approximately same relative concentrations. The AVN-101 was spiked (1μM final) into the diluted plasma. Dialysis was performed in triplicates using the 48-well Teflon® Rapid Equilibrium Dialysis (RED) plate and disposable inserts with regenerated cellulose dialysis membrane, a cutoff size of pores of ca. 8,000 MW (Thermo Fisher Scientific, USA). After diluted plasma containing the 1μM AVN-101 and brain homogenates were placed into corresponding chambers of the dialysis device, the samples of both plasma and brain homogenate were taken out for analysis at different incubation time points to simulate the same time interval as in the animal experiments (Fig. 16A). The samples after removal were matrix-equilibrated; in other words, brain homogenate was added to the plasma samples and diluted plasma was added to the brain homogenate samples. As controls for the membrane permeability, we have also measured the equilibration pattern of the AVN-101 when both chambers of the dialysis apparatus contained either 20% plasma (Fig. 16B) or PBS buffer(Fig. 16C).
The data obtained and presented in Fig. 16 clearly showed that unlike the in vivo experiments, transfer of the AVN-101 through the semi-permeable membrane was, as expected, rather slow and its concentration in brain-containing chamber never exceeds that of the plasma chamber. Based on this behavior of the AVN-101, the fast transfer from blood to brain against its concentration gradient in the in vivo experiments while having normal distribution between the plasma and brain in the ex vivo experiments, we speculate that an active AVN-101 transport mechanism exists in the cells forming the blood-brain barrier, which the AVN-101 exploits.
It is also surprising that the AVN-101 has very low permeability through both blood/CSF and brain/CSF barrier as evident from its very low concentration in CSF (Figs. 14 and 15). This potentially could be due to the effective efflux mechanisms present in the cells of the blood/CSF and brain/CSF borders [94].
Thus, one can conclude that in rats, the AVN-101 accumulates very efficiently in the brain through a yet unidentified energy-dependent pump.
Pharmacokinetics of AVN-101 in dogs. A dose of the AVN-101 or its synthetic metabolite M1, as shown in Table 13, is given to dogs (Beagle) either through IV or PO route of administration as a bolus and blood samples were collected through 48 h of observation.
It is interesting to note that the exposure, AUC, of the metabolite M1 in the blood upon the AVN-101 PO gavage was significantly higher than when the drug was administered through the IV route (8,995 (min×ng/mL)/10 (mg/kg) versus 264.6 (min×ng/mL)/2 (mg/kg)). Based on the PK and tissue distribution data obtained for rats, one could speculate that the systemic administration of AVN-101 allows for more compound to distribute into organs thus shielding it from metabolism in the liver.
Bioavailability of the synthetic metabolite M1 was somewhat better, 8.4% versus 4.6%, than that of its precursor, AVN-101. Metabolite M1 has also exhibited higher levels of exposure, AUC, upon both routs of administration than those of AVN-101. Noteworthy, the M1 was able to “regenerate” back into AVN-101. Judging by higher exposure of “regenerated” AVN-101 (1,896 (min×ng/mL)/5 (mg/kg)) formed upon PO administration of the M1 compared to that upon IV administration (137.5 (min×ng/mL) /1 (mg/kg)), the “regeneration”, or major part of it, is taking place in the liver.
Metabolite M2 was practically undetectable irrespective of either AVN-101 or M1 was administered.
Pharmacokinetics of AVN-101 in monkeys. Rhesus monkeys, two for each IV and PO routes of administration, were used for the pharmacokinetics experiments. The animals had not been used in other studies for at least 2.5 months prior to the pharmacokinetics experiments. Animals were kept in cages and feeding was performed with usual balanced fodders, eggs, vegetables, and fruits. Clinical status of animals and their behavior were normal. Main pharmacokinetic parameters are shown in Table 14.
Preclinical Efficacy Studies of the AVN-101
Anxiolytic effect. There are several different test systems developed to assess anxiety status in rodents and to screen compounds for their effects as pro- or anti-anxiety drugs. To characterize AVN-101, we used the following test systems: i) Elevated Plus-Maze, ii) Elevated Platform, and iii) Open FieldPlatform.
The Elevated Plus-Maze is one of most broadly used test systems to assess level of anxiety of animals and anxiolytic effects of experimental drugs. Behavior of the male BALB/c mice was recorded with a video-tracking system allowing for subsequent automated analyses of the animal movements using the Any-Maze software package by Stoelting Co., USA.
The AVN-101 or its synthetic metabolites, M1 or M2, were administered IP 5 min before the test and a positive control, Buspirone, was administered 30 min before start of the test. Each test group was comprised of at least 8 animals. The drugs were dissolved into saline and the saline was used as a vehicle in the control group. During the procedure, an animal was removed from its cage and placed onto the junction between open and closed arms of the Elevated Plus-Maze always facing the same open arm. The video-tracking system was turned on immediately after placing the animal and recording duration was set for 5 min. At the end of the test period, number of defecations was also recorded. The data is presented in Figs. 17 and 18.
Buspirone (positive control), AVN-101, and its metabolite, M1, all produced clear anxiolytic effects. Buspirone (5 mg/kg) increased both the time spent on open arm as compared to closed arm (Fig. 17A) and number of visits to the open bridge (Fig. 17B). Buspirone substantially decreased number of defecations (Fig. 17C) but had no effect on total number of arm visits (Fig. 17D) indicating that at this dose, it did not produce any effect on the mice locomotion. Both the AVN-101 and its metabolite, M1, were as effective as Buspirone. However, M1 at a dose of 5 or 20 mg/kg induced some reduction in mice locomotor activity (Fig. 17D).
Another AVN-101 metabolite, M2, was also tested in the Elevated Plus Maze test system. M2 produced an anxiolytic effect at all tested doses, 0.05, 0.5, 1.0, and 5.0 mg/kg (Fig. 18A–C), comparable to that of Buspirone. However, at the doses of 1 and 5 mg/kg, significant increase in the mice motor activity was observed (Fig. 18D).
The Elevated Platform test system is another approach to study anxiety [96] and is considered to be suitable for assessment of anxiolytic and/or antidepressant drugs [97]. In the elevated platform test a mouse was placed onto round ceramic platform (14 cm in diameter, 20 cm above surface) for 5 min and allowed to exploring the environment. Episodes of freezing behavior were recorded by video tracking system with the automated analysis of the animal movements with the Any-maze program. Normally, the most active exploration combined with episodes of freezing behavior is seen during first 1-2 min of test. Saline solutions of Lorazepam (positive control) and AVN-101 were administered intraperitoneally with Lorazepam (0.05 mg/kg) being injected 30 min and AVN-101 (0.2 mg/kg) 5 min prior to the beginning of the test.
Cumulative “freezing time” during the test duration is shown in Fig. 19.
The data in Fig. 19 confirm the anxiolytic effect of AVN-101 observed in the Elevated Plus-Maze test. Both Lorazepam and AVN-101 significantly reduced duration of the freezing behavior. However, in the Elevated Platform test, the anxiolytic effect with both drugs was observed only during the initial 2 min of the total of 5 min of exploration.
Open Field Platform: Anxiolytic effect of AVN-101 has also been confirmed with BALB/c mice tested in the Open Field Test system [98, 99]. We used round platform made of clear plastic (62 cm in diameter and 31 cm high wall) and brightly illuminated from above the platform. In the experiment, a mouse was placed onto the center of the platform and allowed to explore the environment for 10 min. The animal movements were recorded by a video-system and analyzed by Any-maze software. The overall animal activity and its activity in a central zone (31 cm in diameter) were analyzed separately. The drugs, Lorazepam (positive control) and AVN-101, were dissolved in saline and administered intraperitoneally. Lorazepam (0.05 mg/kg) was administered 30 min before the test, and AVN-101 (0.2 mg/kg) was administered 5 min before the test. Control animals were injected with vehicle (saline). Each group included at least 8 animals.
Normally, mice avoid visiting the central zone for a long time as a consequence of the environment-induced anxiety. Anxiolytic drugs would increase the time the animals spent in the central zone. The effect of the Lorazepam (positive control) and AVN-101 on the duration of the time spent by mice in the centralzone of the open field platform during a 10 min exploration is documented in Fig. 20. In this test, both the positive control, Lorazepam, and experimental drug candidate, AVN-101, show significant anxiolytic effect by increasing a time spent in the central area of the open-field platform.
Geller-Seifter Conflict Test: Anxiolytic effect of AVN-101 has also been tested with male Long Evans rats as an object, using Geller-Seifter conflict paradigm test system [100]. The rats were trained for a few weeks to be reworded with a food when pressing a lever when a white light was on –unpunished rewards session. In a conflict reward session, when a red light was on, the rats were getting electric shock of increasing power while pressing the lever to get the food reward. The animals were food-deprived for 24 h before the test. Each group consisted of at least 5 animals. The test consisted of two sessions of 30 min each, unpunished rewards and conflict rewards, during which a number of lever presses was registered (Fig. 21).
AVN-101 was administered IV through a tail vein 5 min before the start of the test. In a control (vehicle) group of the animals, the rat activity to press the lever was substantially decreased during the conflict session as compared to the unpunished rewards session. Relative changes in the rat unpunished and conflict responses sessions induced by different concentrations of AVN-101, compared to the control (vehicle) group, are presented in Fig. 21. There was no effect observed of the AVN-101 at the tested concentrations on the activity of the rats during the unpunished rewards session. Contrary, during the punished conflict rewards session, activity of the rats received the drug was significantly increased compared to the vehicle group. In accordance with this model [100], such behavior is representation of anxiolytic effect of the AVN-101 in the rats.
Antidepressant Activity: Potential antidepressant activity of the AVN-101 was tested on Balb/C mice in both the Porsolt Forced Swim and the Tail Suspension tests [101].
In the Porsolt forced swim test, a reservoir (height 300 mm, diameter 480 mm) was filled with water at ∼25°C to 70% of its capacity. An animal was placed in the center of the reservoir for 15 min. Data of last 5 min of the swimming activity was used for theanalysis.
In the tail suspension test, the animal was suspended for 3 min in the air with its tail fixed to the support holder 400 mm above the table surface.
In both tests, the animals were considered immobile if there was no any motion for at least 1.5 s. Monitoring of the animal movements was performed with a video tracking system operated under control of Any-maze program.
Positive controls, Fluoxetine (15 mg/kg) and Desipramine (15 mg/kg), and AVN-101 (0.05 mg/kg) were dissolved in saline and administered intraperitoneally once daily for 4 days. The last injection of positive control drugs was made 30 min before the test start, while AVN-101 was last administered 5 min before the test. The control animals were injected with the vehicle (saline). Each experimental group included at least 8 animals.
The antidepressant activity of AVN-101 seen as a decrease in the immobility time in both the Porsolt forced swim (Fig. 22A) and tail suspension (Fig. 22B) tests was comparable to that observed with either Fluoxetine or Desipramine.
Memory/cognition enhancing effect. Test of the AVN-101 on its ability to improve cognition, learning, and memory was performed with a Passive Avoidance paradigm [102], Morris Water Maze [103], Novel Object Recognition Test [104], and Mouse Contextual Fear Conditioning Test [105].
Passive avoidance test: Both the male BALB/c mice and Sprague Dawley rats were used with amnesiac models based on either scopolamine-induced [106] or MK-801-induced [107] memory impairment and its reversal with the AVN-101.
The tests were performed using Ugo Basile passive avoidance step-through apparatus consisting of a cage (48×21×23 cm) with two, white and dark, chambers separated by an automatically operated vertical door. The bars of the dark compartment floor were wired to the constant-current shock generator delivering 10 ms pulses of 0.6 mA amplitude for mice and 1.2 mA for rats with a frequency of 5 Hz for 3 s. During experiment, the white chamber was brightly illuminated.
Test was performed in two stages. During the first learning stage, an animal was placed into the white illuminated compartment and latency time of entry into the dark chamber was registered. Then, while the animal was still in the black chamber, the door was closed and the animal was given an electric shock. Twenty-four hours later, the animal was placed again in the white illuminated chamber and latency time to enter the black chamber, overall duration of staying in the white compartment, and the number of entries into the dark compartment was monitored for 5 min of the test time with no punishing shock in the dark chamber.
Scopolamine, MK-801, and drugs of comparison were diluted in saline and administered to the animals 30 min before the training. The AVN-101 was given 5 min before the training. Control animals were injected with saline as a vehicle. All the drugs were administered intraperitoneally. Each group included at least 8 animals.
Effect of AVN-101 in scopolamine-induced amnesia model
Mice. Three parameters, namely, Latency to enter into black chamber (Fig. 23A), Time spent in the brightly lit chamber (Fig. 23B), and Number of entries into the black chamber (Fig. 23C) were measured. Naïve mice had both the low latent time to enter into the dark chamber and a short time spent in the brightly lit chamber while number of entries into the dark chamber was high. After the learning phase, when the electric shock has been provided in the dark chamber, the latent time to enter the dark chamber and time spent in the light chamber significantly increased and the number of entries into the dark chamber decreased (placebo 1). Pre-treatment of the mice with scopolamine (0.3 mg/kg), non-selective muscarinic acetylcholine receptor antagonist, substantially reduced effect of the learning by inducing so-called anterograde amnesia (Fig. 23, placebo 2). The preventative effect on all three scopolamine-induced amnesic parameters was observed at the AVN-101 doses as low as 0.02 mg/kg, substantially increasing at the dose of 0.05 mg/kg and 0.2 mg/kg (Fig. 23), and then slightly decreasing at higher doses up to 5 mg/kg with a bell shape-like dose behavior. Even at the highest dose of 5 mg/kg, the receding part of the preventative dose curve, AVN-101 was equal to or more effective than either memantine or tacrine in reversing the scopolamine-induced amnesia, especially when considering number of entries into the dark chamber (Fig. 23C).
Interestingly, the highly potent and highly selective 5-HT6 receptor antagonist, SB-742457, [72] in our hands produced a very mild preventative effect in this model at highest dose tested, 1 mg/kg. The authors demonstrated an effectiveness of the acute SB-742457 pre-dosing in a scopolamine-induced amnesia passive avoidance model in a dose range of 0.5–4.5 mg/kg. The much higher efficiency and potency of the AVN-101 in this model indicates a likelihood that it is 5-HT7 receptor blocking ability of the AVN-101 that is responsible for its anti-amnesic effect at such low doses.
Rats. The AVN-101 was also effective in the scopolamine-induced amnesia in rats, as can be seen in Fig. 24. The AVN-101 at a dose of 0.05 mg/kg, was able to significantly reverse pro-amnesic action of Scopolamine.
Effect of AVN-101 in MK-801-induced amnesia model
Similar to scopolamine, treatment of the Balb/c mice with MK-801, NMDA glutamate receptor antagonist, also produced anterograde amnesia (Fig. 25). Judging by the three measured parameters, latency to enter the dark compartment, time spent in the white compartment, and number of entries into the dark compartment, the AVN-101 dose-dependently prevented the MK-801-induced amnesic effect. It should be noted, that the MK-801-induced memory impairment model was less sensitive to AVN-101 than the scopolamine model, the anti-amnesic dose of AVN-101 being approximately 10-fold as high in the MK-801 model (0.5 mg/kg) as in the scopolamine model (0.05 mg/kg) of the amnesia. Memantine was not active in this model and the effect of the AVN-101was similar to that of SB-742457. It is reasonable to assume, that similar to the SB-742457, in the MK-801-induced amnesia model, it is 5-HT6 receptor blocking ability of the AVN-101, which is responsible for its efficacy.
Morris water maze test. The AVN-101 was tested in amnesiac model of scopolamine-induced memory impairment and its reversal [106] using Morris Water Maze with BALB/c mice.
An 85 cm in diameter and 55 cm high pool filled with water (20–22°C) to the depths of 30 cm was used in the experiments. A round ceramic escape platform, 14 cm in diameter, was located in one of four quadrants 0.5 cm below the water surface (hidden). A video tracking system for automated analysis of the animal movements was operated by Any-maze program (Stoelting Co., USA). The environment included asymmetric external cues that were kept constant throughout the experiment.
The mice with learning capabilities were pre-selected using the following selection process. An animal was placed on the platform located 1 cm above the water surface for 20 s. Then the mouse was removed from the platform and placed in the water at the opposite side in the pool and allowed to find the platform within 60 s. If the mouse failed to find the escape platform within two consecutive trials, it was excluded from the further experiments.
The training session comprised of two consecutive days of training, with four trials per day. The platform was placed 0.5 cm below the water surface and its location was kept constant during the training. The time intervals between consecutive trials were kept at 20 s, during which time, the mice were allowed to stay on the platform. Before the first trial, the animal was placed onto the platform for 20 s after which time it was immersed into the water. The place where the animal was immersed into water was alternated between the trials at two different points located in each half-side of the pool opposite to the platform. After that, the animal was allowed to stay on the submersed platform for an additional 20 s and the procedure was repeated. The latency time to reach the platform was registered during each of the four trials.
On the third day, the test session, the platform was removed and the mice were allowed to swim for 60 s to determine their search bias. The data from the video-tracking system was analyzed to determine the time spent in the quadrant of the pool that previously contained the platform. This parameter served as a measure of training efficacy.
The drugs under the study were given on each day of training session in accordance with following schedule. Scopolamine (1.5 mg/kg) was administered 30 min before start of training. The drugs of comparison, tacrine (3 mg/kg) and donepezil (3 mg/kg), were administered 60 min before training, i.e., 30 min before scopolamine. AVN-101 was administered 5 min before the start of the training session. In other words, the drugs of comparison were tested in scopolamine prevention mode (prophylactic mode) whereas AVN-101 was tested in a scopolamine reversal mode (therapeutic mode).
All the drugs were dissolved in saline and administered intraperitoneally. The control animals were injected with the saline vehicle. Each group contained at least 8 animals.
The data of Morris water maze test (Fig. 26) shows that mice without scopolamine treatment, Placebo 1 group, were able to learn and memorize spatial position of the “safety platform”, which translated into longer time spent in the quadrant that contained the hidden platform during the training sessions. Treatment with scopolamine (Placebo 2 group) led to amnesia, as manifested by reduced time spent in the “platform quadrant”. AVN-101 at a dose of 0.2 mg/kg given 25 min after the scopolamine, effectively reversed the scopolamine-induced amnesia manifested by increased time spent in the “platform”. Tacrine (3 mg/kg), the positive control drug, showed significant prevention of the scopolamine-induced amnesia in this model. On the other hand, donepezil (1 mg/kg) was not as effective.
Novel object recognition (NOR) test. Effectiveness of the AVN-101 was tested on both mice (BALB/c) and rats (Wistar) in the NOR tests [108] with scopolamine-induced [109] and MK-801-induced [110] model of memory impairment and its reversal with the drugs.
An apparatus for the NOR was a modification of Y maze [111] that is suited for both the object recognition and temporal memory testing. We used Cross-Maze, which consisted of 4 closed chambers made of black Plexiglas with clear top covers instead of Y-shaped maze. The chambers, 14×14×14 cm, were connected to a central compartment through 7×7 cm holes. An animal was placed into the central compartment of the maze and allowed for free exploration of all the chambers. Time and sequence of visits into each arm were recorded by an observer until total of 12 visits had been made. The criterion for a visit was entry into a compartment with all four paws inside a given chamber.
During a training session, the animals were allowed exploring the cross-maze arms with four identical plastic cups (3 cm in diameter × 7 cm height) in each. During a test session performed 1 h after the training session, the cups in two opposite arms were replaced by glass conical flasks (4 cm bottom diameter and 7 cm height) and the animals were allowed to explore the environment again. The relative time spent by animals in arms with the novel objects, conical flasks, compared to total time of experiment (12 visits) was used as the novel object recognition index.
Either scopolamine or MK-801 was administered 30 min before start of training session. Drugs of comparison were also administered 30 min before training, and AVN-101 was administered 5 min before training. All the drugs were dissolved in saline and administered intraperitoneally. The vehicle, saline, was administered to control animals. Each group included at least 8 animals. The data is presented in Fig. 27.
In this test, control mice (Placebo 1) were able to distinguish the novel objects as evidenced by approximately 70% time spent in the arms with the novel objects. The 1.0 mg/kg scopolamine as well as 0.2 mg/kg MK-801 (Placebo 2) reduced the animal ability to recognize the novel objects (the time spent in the arms with novel objects was approximately same as that with the old objects, recognition index close to 50%). The AVN-101 was able to partially reverse the impairment effects of both the scopolamine and MK-801. Among the comparative drugs, memantine (10 mg/kg) or SB-742457 (0.2 or 1.0 mg/kg) were not statistically effective in preventing the drug-induced deficit in novel object recognition.
Antipsychotic activity. The antipsychotic efficacy of the AVN-101 was tested in a Pre-Pulse Inhibition (PPI) model. PPI is the normal diminution of a startle response that occurs when the startling stimulus is preceded by a weak lead stimulus (“pre-pulse”). Schizophrenic patients exhibit abnormally low levels of PPI; therefore, animal models of deficient PPI may provide information regarding neural dysfunctions underlying schizophrenia [112]. In the PPI model, systemic administration of the direct dopamine agonist, apomorphine, reliably disrupts PPI, and such disruption is antagonized by antipsychotic drugs [113]. Apomorphine-induced PPI disruption has therefore been commonly employed as a screening test for potential antipsychotic compounds[114].
The apparatus consisted of acoustic startle chamber (Columbus Instruments, USA) made of clear Plexiglas plastic attached to a platform, which was in turn resting on a solid base inside a sound-attenuated isolation cubicle. A high-frequency loudspeaker located at a distance of 2 cm from startle chamber produced various acoustic stimuli. Vibrations of the Plexiglas enclosure caused by the whole-body startle response of the animal were converted into digitized analog signals by a piezoelectric unit attached to the platform. Background noise level was 65 dB. There were four repetitions of the startle stimulus in pulse-alone and pre-pulse-plus-pulse trials. First presentation of each type of acoustic stimuli was used as habituation period and the corresponding data were not included into analysis. The maximal response amplitude during 200 ms after a pulse was used to determine the stimulus reactivity. Both pre-pulse and pulse stimuli were 20 ms in duration separated by 50 ms interval. The pre-pulse stimulus intensity was 85 dB, while the pulse stimulus amplitude was 105 dB.
An interval between presentations of different types of acoustic stimuli was 10 s. Startle pre-pulse inhibition was calculated as percentage of decrease in mean startle amplitude in response to pre-pulse plus pulse stimuli to mean startle amplitude in response to pulse stimulus alone.
All experiments were conducted on male SHK mice during the light phase of the dark-light cycle. Apomorphine HCl was dissolved in 0.1% ascorbic acid in sterilized water. Haloperidol was dissolved in Tween 80. AVN-101 was dissolved in sterilized water. The appropriate vehicle solution included 0.1% ascorbic acid and Tween 80. Haloperidol was administered 60 min prior to testing. Apomorphine was administered 15 min before testing. AVN-101 was administered 5 min before testing. The volume of injection was 10 ml/kg. All drugs were administered via IP. Apomorphine (20 mg/kg) administration served as a model of disrupted PPI and the effectiveness of the AVN-101 and test compounds were assessed for their ability to normalize the apomorphine-induced abnormality(Fig. 28).
In the group of non-treated mice (Placebo 1), PPI was around 40%. Pro-psychotic agent, apomorphine (Placebo 2), reduced PPI to approximately 20% thus confirming its ability to impair filtration of sensory signals in SHK mice. Haloperidol (1 mg/kg) was able to prevent such effect of the Apomorphine. The AVN-101 was effective in reversing of the apomorphine-induced disruption of PPI with bell-shaped dose dependence. The optimal dose of the AVN-101 in restoration of PPI was 0.1–0.5 mg/kg.
Safety Pharmacology of AVN-101
Assessment of the median lethal dose after single administration of AVN-101. Median lethal dose of AVN-101 was measured to establish a safety window for its therapeutic use. The toxicity after acute administration of AVN-101 was determined in experiments with male CD1 mice and the LD50 values were calculated in accordance with [115]. The AVN-101 was dissolved in a saline and administered intraperitoneally (80–200 mg/kg) or orally (200–700 mg/kg). Each dose was applied to an independent group of mice (n = 6). The animals were observed for 2 weeks after single administration of the test compound and incidents of death were tabulated. The LD50 value and standard error of LD50 were calculated by probit-analysis using Biostat 2006 software and were determined to be 105 ± 5 mg/kg for IP administration and 331 ± 101 mg/kg for PO administration route.
Assessment of maximum tolerated dose after multiple administrations of AVN-101. Newell et al. showed that preclinical toxicology studies in rodents are predictive to identify a safety clinical starting dose and potential human toxicity [116].
Male CD1 mice were used to determine the maximum tolerated dose (MTD), for the PO administration of the AVN-101. The test compound was dissolved in sterile water and administered orally at sub-toxic doses of 10, 30, and 60 mg/kg. Each dose was administered orally once daily for 5 days to independent groups of mice (n = 8 per group). The body weights and number of dead mice were registered daily during 5-day treatment period and the monitoring was continued for subsequent 2 weeks of an observation phase. The highest dose that produced neither mortality nor body weight loss was considered as MTD.
AVN-101 caused 50% mortality and produced statistically significant weight loss starting from the 4th day of injection at a dose of 60 mg/kg. At a dose of 30 mg/kg, AVN-101 caused 12.5% mortality (1 animal out of eight in the group). Our results suggest that the MTD of AVN-101 is somewhere between 10 mg/kg and 30 mg/kg.
Estimation of a chronic toxicity of AVN-101.
Chronic toxicity in rodents: In these studies, we used male adult SHK mice (14 days toxicity), Wistar rats (30 days and 180 days toxicity), and laboratory rabbits (30 days toxicity). AVN-101 was administered orally every day during the specified time frame.
In the 14 days toxicity study, the mice were randomly selected into two dose groups: the vehicle and 5 mg/kg. No signs of toxicity were observed.
In the 30 days toxicity study, Wistar rats and healthy laboratory rabbits were used.
Rats were randomly selected into four dose groups: vehicle, 0.5 mg/kg, 5 mg/kg, and 25 mg/kg; with 10 male and 10 female animals per group. No signs of toxicity were observed at any of the tested doses.
Rabbits were randomly selected into two dose groups: vehicle and 4 mg/kg (3 males and 3 females per group). No changes in the body weight, parameters of plasma biochemistry markers, macro- and microscopic observations were observed. However, one of 3 males in AVN-101 group had developed small infarctions in cortical substance of the kidneys and one of 3 females in the same group had developed ovary hypo function.
The 180 days toxicity study of AVN-101 at the doses of 0.25 mg/kg, 0.5 mg/kg, and 5 mg/kg in 8-week-old rats suggests severe hepatotoxic and moderate cardiotoxic effects of the AVN-101 administered in the dose of 5 mg/kg. The toxic effects were substantially less noticeable when lower doses, 0.25 and 0.5 mg/kg, were used. The morphology studies have been performed within an hour after decapitation. Samples of thymus, lungs, heart, liver, kidneys, spleen, testicles, brain, and cerebellum were collected for histological analysis. Clinical observation revealed no signs of toxicity: no hair loss, glossy and shiny hair; skin is elastic, sub-epidermal cellular tissue moderately expressed. All internal organs were located regularly.
Chronic toxicity in monkeys: In these studies, we used 5-year-old rhesus macaques (Macaca Mulatta, 3 males and 3 females). Food and water consumption were checked regularly during the 30 days study. AVN-101 was administered at the dose of 5 mg/kg orally every day during the 30 days time frame. Blood samples for hematological and biochemical analysis were taken just before first gavage of the AVN-101 and on the 31st day of the experiment. Samples taken before the experiment were used as reference point. Body weight measurements were performed daily.
AVN-101 did not show any signs of toxicity during the 30 days study in all 6 animals.
Cardiotoxicity.
Interaction AVN-101 and M1 with cardiac ion channels: Drugs belonging to diverse classes have been shown to be cause heart QT prolongation leading, in some cases, to serious ventricular arrhythmias and even death. The most common mechanism for these adverse events is an inhibition of one or more cardiac potassium channels, in particular hERG channel. Potassium currents controlled by this channel are important for re-polarization of cardiac myocytes and unintended blockage of these channels by some drugs causes prolonged myocardial QT interval with potentially lethal consequences.
AVN-101 and M1 were tested for their ability to block hERG channel using HEK cells stably expressing the hERG potassium channel and measuring potassium currents in the single cell patch clamp experiments. It was shown that both AVN-101 and M1 can block the hERG channel with IC50 of 0.58μM and 1.63μM, respectively. However, upon the IP route of administration in mice, the blood concentration of c.a. 0.65μM can be achieved at a dose of 5 mg/kg, which is substantially higher than the therapeutic dose of AVN-101.
We have also tested ability of the AVN-101 to block a few other cardiac channels (Table 15).
Q-T interval prolongation assessment AVN-101: Due to the fact that the AVN-101 has an ability to interact with the hERG channel in in vitro patch-clump experiments, albeit at concentrations substantially higher than those provided by therapeutic doses, it was necessary to directly asses its effect on the heart ECG in in vivo experiments. The testing has been performed with guinea pigs and rhesus macaque (Macaca mulatta).
Guinea pigs. AVN-101 has been administered intraperitoneally at a dose of 10 mg/kg. The positive control drug, amiodaron [117], was administered through the same route and at the same dose. Q-T interval prolongation (Fig. 29) is expressed as a percentage increase over the normal (vehicle) QT interval. As one can see, AVN-101 even a dose of 10 mg/kg does not induce higher than 10% increase in the QT interval, while the amiodaron caused expected QT prolongation compared to the vehicle. Taking into consideration that the therapeutic dose of AVN-101 in rodents is close to 0.05 mg/kg, we concluded that most likely AVN-101 will not present a danger of cardiac toxicity, at least in rodents.
Monkeys (Macaca mulatta). AVN-101 and the positive control, amiodaron, were administered intravenously at the doses of 1 mg/kg (3 animals) and 15 mg/kg (2 animals), respectively.
The ECG signals and pulse were recorded 15 min before (background) and at several time points after the injection (Fig. 30). AVN-101 at the dose of 1 mg/kg affected neither the QT interval (0.22 ± 0.04 s) nor heart rate (ca. 175 beats/min, not shown). IV administration of amidaron caused a 34% prolongation of the QT interval by 60 min, which then returned to normal by 6-h post injection. Amidaron caused no effect on the heart rate.
Dogs (beagles). We have performed more detailed of AVN-101 safety studies in dogs with monitoring femoral and aortic arterial blood pressure, heart rate, left ventricular pressure, 6-lead ECG, femoral and coronary blood flow. Four male dogs were anaesthetized (propofol/alfentanil) and surgically prepared for the measurements. Dogs received sequentially a vehicle and then 4 ascending doses of the AVN-101 (0. 05, 0.5, 2.0, and 10 mg/kg) through IV infusions. The infusions were continuing for 15 min (the infusion volumes were 2.0 ml/kg for doses 0.05–2.0 mg/kg and 5.0 ml/kg for the dose of 10 mg/kg). After each infusion session, the dogs were monitored for 30 min, after which time the infusion of the next dose was started. Monitoring of the PD parameters after the last dose was performed for at least 60 min. After each monitoring cycle, the small portion of the blood was taken for subsequent analysis of the AVN-101 blood content.
The peak effects of the AVN-101 infused on the cardiovascular parameters usually occurred during the first several min of the observations for all dose levels of AVN-101 (Table 16), unless otherwise stated in the text.
At the 0.05 mg/kg (blood plasma concentration = 0.14μM), the AVN-101 caused no significant changes in all monitored parameters: femoral, left ventricular (LV), and aortic blood pressure, femoral blood flow, heart rate (HR), and ECG recordings.
At the 0.5 mg/kg (blood plasma concentration = 0.99μM), the AVN-101 caused a 28 bpm (39%, not significant) increase in HR with corresponding reduction in femoral systolic blood pressure (SBP) of 17 mm Hg (13%, p < 0.05) with no change in femoral blood flow. At this dose, the AVN-101 caused no significant effects on either aortic blood pressure or LV or ECG parameters.
At the 2.0 mg/kg (blood plasma concentration = 3.28μM), the AVN-101 increased HR by 39 bpm (56%, p < 0.05), shortening of QT and QTcV (both at 30 min) of 40 and 24 ms, respectively (15% and 8%, p < 0.01), and reductions in femoral SBP and mean blood pressure (MABP) of 21 and 13 mm Hg, respectively (16 and 14%, p < 0.05). Aortic SBP was also significantly (p < 0.05) decreased by 8 mmHg (8%). No significant effects of the AVN-101 on LV parameters were observed.
At the 10.0 mg/kg (blood plasma concentration = 11.1μM), the AVN-101 caused an increase in HR (at 30 min) by 118 bpm (164%, p < 0.001) and significant reductions in QT and QTcV of 80 and 35 ms (30 and 13%, p < 0.001), respectively. Femoral SBP, DBP, and MABP were reduced by 15 mm Hg (11%, NS), 12 mm Hg (17%, p < 0.05), and 13 mm Hg (16%, p < 0.05), respectively. Aortic DBP also fell by 9 mm Hg (13%, p < 0.05). LVSBP (at 45 min) increased by 16 mm Hg (16%, p < 0.01) and LVDBP was decreased by 2 mmHg (40%, p < 0.05). Femoral flow increased by 35 ml/min (27%, p < 0.05) accompanied by a decrease in femoral resistance of 0.3 mm Hg/ml (38%, p < 0.05).
Mean peak plasma concentrations in the blood samples obtained at the end of the infusions of the AVN-101 (0.14 ± 0.02, 0.99 ± 0.04, 3.28 ± 0.30, and 11.1 ± 2.15μM) were linearly correlated with the infused dose.
In summary, infusion of the dogs with the AVN-101 at different doses caused dose-dependent changes in multiple cardio-vascular parameters. Unexpectedly, the AVN-101 caused dose-dependent QT shortening instead of its prolongation, which was not entirely due to the increase in a heart rate, as QTcV also decreased with increasing dose.
Overall, the data obtained clearly shows that the AVN-101 causes no QT prolongation in any of the studied species, guinea pigs, beagles, and Rhesus Macaca mulatta, even when the compound was given intravenously at a dose as high as 10 mg/kg. In dogs, we have observed the shortening of the QT at high doses, increase in heart rate, and a systemic vasodilation.
Mutagenicity
Mutagenicity of the AVN-101 was assessed in both the Ames test [118] in MPFtrademark format [119] and chromosomal aberration bone marrow cells test [120] with C57BL/6 mice.
Ames test. The test was performed in accordance with the Xenometrix manual, Ames MPFtrademark 98/100/1535/1537 Mutagenicity Assay, using three histidine-dependent Salmonella typhimurium strains: TA98, TA1535, and TA1537 [121, 122]. As positive controls, 15μg/mL 4-nitro-o-phenylenediamine (4-NOPD) was used for strain TA98, 2μg/mL sodium azide for strain TA1535, and 50μg/mL 9-aminoacridine (9-AA) for TA1537 (Fig. 31).
Data presented in Fig. 31 shows that while positive control compounds produce significant mutagenicity in corresponding strains of Salmonella, AVN-101 produced no mutagenesis in either strain even at the highest concentration, 50μM, tested.
Chromosomal aberrations. Potential pro-mutage-nesis effect of the AVN-101 was tested on 8–12-week-old male and female mice, CD-1. The animals were housed in standard cages. 10 animals per cage were housed before experiments and 5 animals per cage, for each experimental group, were housed when the experiments have started. The housing was maintained on 12 h light/dark regimen with free access to both the food and drink. The test compound, AVN-101, and a positive control compound, cyclophosphamide, were given orally. In the single dose experiments, the animals were decapitated 24 h after the drug administration. In the 5 days dosing experiments, the animals were decapitated 6 h after the last drug administration. In both cases, 2.5 h before the decapitation, the animals were orally administered with 4 mg/kg colhicine to arrest cells in metaphase-blocked mitoses state [123]. Immediately after decapitation, femur bone marrow cells were isolated into warm (37°C) hypotonic potassium chloride (0.55%) solution. After an additional incubation at 37°C for 15 min, the cell suspension was spanned down for 5 min at 1,000 rev/min. The cell precipitate was re-suspended into ice-called fixative comprised of 3 : 1 ethanol/glacial acetic acid, incubated for 20 min at 4°C and spanned down. After several cycles of fixation and precipitation, the cells were applied onto microscope slides and dyed with azure-eosin for cytogenetic analysis of the chromosome aberrations (Table 17). Analysis of the chromosome aberrations was performed using 100 cells from each animal preparation (total of 500 cells per group) and number of the aberrant cells was expressed as percentage of cells with altered metaphases.
In the placebo group, there were 1.8 ± 0.6% cells with chromosomal aberrations. In the animals treated with positive control (cyclophosphamide, 20 mg/kg), there were 20.8 ± 1.8% chromosomal aberrations. Single PO treatment of male mice with AVN-101 at doses of 1 mg/kg and 20 revealed no increase in the chromosomal aberrations, 1.2 ± 0.5 and 1.6 ± 0.6% chromosomal aberrations respectively, relative to the placebo group. Semi-chronic administration of AVN-101 (5 days once a day) in the dose of 1 mg/kg did not affect the amount of chromosomal aberrations either in male (1.4 ± 0.5%) or female (0.8 ± 0.4%) mice.
Conclusion: AVN-101 does not pose mutagenicity as evident from both the Ames MPFtrademark (Fig, 31) and chromosomal aberration (Table 17) tests.
Reproductive toxicology of AVN-101 in rats
AVN-101 was tested for its possible reproductive toxicity [124] by assessing embryo toxicity, influence on the development of posterity and the reproductive (generative) function. The testing was performed on outbreed male and female rats, 220–250 g weight. AVN-101 in 0.9% physiological solution was administered intragastrically at the doses of 1.0 mg/kg or 25.0 mg/kg.
When the AVN-101 was administered daily for 48 days to male rats, no reproductive function alteration was observed at both doses, 1.0 mg/kg and 25.0 mg/kg. The drug neither influenced a capability of animals for pairing and fertilization, nor did it change the level of spontaneous embryonic mortality. When the AVN-101 was administered to female rats daily for 15 days prior to pairing, reduction in the body mass increase of the pregnant rats and their posterity was observed, with no affect on the generative function. Mechanism of such an effect of the AVN-101 on the female rats is not clear yet and requires additional studies.
The study also showed that the AVN-101 administered to either male or female animals at the doses up to 25.0 mg/kg, caused neither embryotoxic nor teratogenic effect and did not produce any significant influence on the postnatal development of the rat posterity.
Phase I clinical trial
The Phase I clinical trial of the AVN-101 was carried out at the clinic of the Mental Health Research Center of Russian Academy of Medical Sciences. Study protocol was developed to comply with GCP guidelines of the International Conference on Harmonization and in accordance with the Helsinki Declaration of 1975, and approved by the Review Board of the Ministry of Health and Social Development of Russian Federation on 04/24/2009.
Tolerability to AVN-101. Tolerability to the AVN-101 was tested on physically and mentally healthy volunteers, recruited for this Phase I study with their signed consent. All volunteers were white Caucasian 20–50-year-old males with body mass index of 20–25. Each dose group (2, 4, 10, and 20 mg, single dose) consisted of 8 volunteers. The study was performed in succession starting with the lowest dose group, 2 mg, followed by next group after evaluating previous group for signs of adverse effects. The PO route was selected for the drug administration. The drug was taken in the morning after night fasting as gel capsules containing indicated quantity of the active ingredient. Venous blood samples were taken through an IV catheter just before and at different time points after the drug administration. Urine samples have also been collected before and after (4 h and 24 h) the drug administration.
In the group with dose 10 mg, one person developed a slightly elevated temperature (37.3°C) 30 h after the drug administration. The systolic and diastolic pressure as well as heart rate was normal for all the volunteers. In the dose 20 mg group, one person also developed a slightly elevated temperature (37.1°C) 30 h after drug administration and another person displayed transient increase in systolic pressure from 141/88 before drug administration to 157/87 (2 h–4 h) and 155/77 (6 h–7.5 h) after the drug administration, respectively.
Biochemical analysis on blood samples consisted of the following: total bilirubin, creatinine, AST, ALT, urea, albumin, alkaline phosphatase, glucose. Clinical cellular analysis of blood samples consisted of determination of hemoglobin, erythrocytes, leucocytes, platelets, eosinophils, basophils, lymphocytes, and monocytes. Urine analysis comprised of measurements of urine specific gravity, pH, protein, glucose, ketone bodies, bile acids, epithelial cells, leucocytes, and erythrocytes.
All biochemical and clinical parameters of blood and urine samples of volunteers in all dose groups were in a normal range.
AVN-101 does not significantly influence plasma and urine biochemical parameters. No QT interval prolongation was observed in ECG of individuals from each dose group. No substantial deviation in the functional state of the volunteers was observed upon objective and subjective observations during and 6 days after the drug administration. At the AVN-101 dose of 20 mg, some adverse effects were detected in five volunteers during the first half of the day, sleepiness (3 volunteers), excitation (one volunteer), and paresthesia (one volunteer).
Pharmacokinetics of AVN-101 and its metabolites, M1 and M2. Blood samples were collected from the subjects following single PO dose of AVN 101 and frozen in liquid nitrogen. Analysis was performed by MicroConstants, Inc., San Diego, CA.
Kinetics of the AVN-101 and its metabolites after single PO administration in human is presented in Fig. 32. Both the original compound, AVN-101, and its metabolite, M1, reach maximum concentration in the blood of patients at around 1 h after PO drug administration, with the M1 concentration and, hence, its exposure being much higher than those of the original compound. Concentration of M2 was near or at the low detection limit at all AVN-101 doses.
Summary of major PK parameters for AVN-101, M1, and M2 is presented in Table 18. Increase in the AVN-101 dose leads to almost linear increase in both the maximal concentration (Cmax) and level of exposure (AUC) of the original compound, AVN-101, as well as of its metabolite, M1 (Fig. 33).
The fact that M1 appears in the blood stream simultaneously (Tmax ∼1 h) with and at a significantly higher concentration (Cmax) than the AVN-101, is in agreement with both the PK in dogs (Table 13) and fast metabolism of AVN-101 in microsome suspensions (Fig. 7). However, when AVN-101 entered the systemic circulation, its half-life was rather long (8–14 h; T1/2 in Table 18). At the same time, elimination of M1 was somewhat faster, the T1/2 was between 2.5 h and 3.8 h (Table 18). Noteworthy, the blood concentration of AVN-101 in human subjects is rather low and could result from fast redistribution into brain analogously to what we have observed with the rat blood/brain distribution.
Conclusions
A novel orally bio available drug candidate, AVN-101, with facilitated and predominant distribution into brain is a multi-target drug candidate that has an advantageous target fingerprint of activities with prevalent affinity to serotonin receptors, mainly 5-HT7, 5-HT6, 5-HT2A, and 5-HT2C, as well as to adrenergic α2B, α2A, and α2C and histamine H1 and H2 receptors.
The AVN-101 has demonstrated positive effects in the animal models of both impaired and innate cognition. It also exhibited significant anxiolytic and anti-depressant capabilities. The primary therapeutic area for AVN-101 to be tested in clinical trials would be Alzheimer’s disease. However, due to its anxiolytic and anti-depressive activities, there is a strong rational for it to also be studied in such diseases as general anxiety disorders, depression, schizophrenia, and multiple sclerosis.
ACKNOWLEDGMENTS
We would like to extend our sincere gratitude to Drs. A. Koryakova and R. Karapetyan (Chemical Diversity Research Institute, Moscow, Russia) and R. M. Salimov (Incuron LLC, Russia), who were responsible for preclinical evaluation of the AVN-101, for their excellent work in setting up and supervising all animal work including disease models and pharmacokinetics analyses. We also wish to thank Dr. V.I. Kazey (Exactelabs, Moscow) for PK/PD studies, Dr. O.D. Mitkin (Chemical Diversity Research Institute, Moscow, Russia) for developing synthetic protocols and synthesis of AVN-101 for preclinical and clinical trials as well as Dr. M. Morozova (National Center of Mental Health of Russian Academy of Medical Sciences) for supervising Phase I clinical trials.
The work was fully funded by Avineuro Pharmaceuticals Inc.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1146r2).
REFERENCES
[1] | Lipinski CA ((2012) ) Phenotypic and in vivo screening: Lead discovery and drug repurposing. In, Designing Multi-Target Drugs, Morphy JR, Harris CJ, eds. Royal Society of Chemistry, pp, 86–93. |
[2] | Hornberg JJ ((2012) ) Simple drugs do not cure complex diseases: The need for multi-targeted drugs. In Designing Multi-Target Drugs, Morphy JR, Harris CJ, eds. Royal Society of Chemistry, pp, 1–13. |
[3] | ((2015) ) 2015 Alzheimer’s disease facts and figures. Alzheimers Dement 11: , 332–384. |
[4] | Schizophrenia Facts and Statistics, http://www.schizophrenia.com/szfacts.htm. |
[5] | Anxiety and Depression Association of America. Facts & Statistics, http://www.adaa.org/about-adaa/press-room/facts-statistics, Last updated September, (2014) . |
[6] | Glennon RA ((2003) ) Higher-end serotonin receptors: 5-HT 5, 5-HT 6, and 5-HT 7. J Med Chem 46: , 2795–2812. |
[7] | Johnson CN , Ahmed M , Miller ND ((2008) ) 5-HT6 receptor antagonists: Prospects for the treatment of cognitive disorders including dementia. Curr Opin Drug Discov Devel 11: , 642–654. |
[8] | Ramírez MJ ((2013) ) 5-HT6 receptors and Alzheimer’s disease. Alzheimers Res Ther 5: , 15–22. |
[9] | Ruat M , Traiffort E , Arrang JM , Tardivel-Lacombe J , Diaz J , Leurs R , Schwartz JC ((1993) ) A novel rat serotonin (5-HT6) receptor: Molecular cloning, localization and stimulation of cAMP accumulation. Biochem Biophys Res Commun 193: , 268–276. |
[10] | Monsma FJ , Shen Y , Ward RP , Hamblin MW , Sibley DR ((1993) ) Cloning and expression of a novel serotonin receptor with high affinity for tricyclic psychotropic drugs. Mol Pharmacol 43: , 320–327. |
[11] | Codony X , Vela JM , Ramírez MJ ((2011) ) 5-HT(6) receptor and cognition. Curr Opin Pharmacol 11: , 94–100. |
[12] | Ivachtchenko AV ((2014) ) Sulfonyl-containing modulators of serotonin 5-HT 6 receptors and their pharmacophore models. Russ Chem Rev 83: , 439–473. |
[13] | Ivachtchenko AV , Ivanenkov YA ((2013) ) Small molecule 5-HT6R ligands: A comprehensive insight into their selectivity and activity. Curr Bioact Compd 9: , 64–100. |
[14] | Ivachtchenko AV , Ivanenkov YA ((2012) ) 5HT(6) receptor antagonists: A patent update. Part 1. Sulfonyl derivatives. Expert Opin Ther Pat 22: , 917–964. |
[15] | Ivachtchenko AV , Ivanenkov YA , Skorenko AV ((2012) ) 5-HT(6) receptor modulators: A patent update. Part 2. Diversity in heterocyclic scaffolds. Expert Opin Ther Pat 22: , 1123–1168. |
[16] | Ivachtchenko AV , Ivanenkov YA , Tkachenko SE ((2010) ) 5-hydroxytryptamine subtype 6 receptor modulators: A patent survey. Expert Opin Ther Pat 20: , 1171–1196. |
[17] | Upton N , Chuang TT , Hunter AJ , Virley DJ ((2008) ) 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics 5: , 458–469. |
[18] | Rossé G , Schaffhauser H ((2010) ) 5-HT6 receptor antagonists as potential therapeutics for cognitive impairment. Curr Top Med Chem 10: , 207–221. |
[19] | Maher-Edwards G , Zvartau-Hind M , Hunter AJ , Gold M , Hopton G , Jacobs G , Davy M , Williams P ((2010) ) Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr Alzheimer Res 7: , 374–385. |
[20] | Maher-Edwards G , Watson C , Ascher J , Barnett C , Boswell D , Davies J , Fernandez M , Kurz A , Zanetti O , Safirstein B , Schronen JP , Zvartau-Hind M , Gold M ((2015) ) Two randomized controlled trials of SB742457 in mild-to-moderate Alzheimer’s disease. Alzheimers Dement (N Y) 1: , 23–36. |
[21] | Wilkinson D , Windfeld K , Colding-Jørgensen E ((2014) ) Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 13: , 1092–1099. |
[22] | Clinicaltrials.gov, Study of Lu AE58054 in Patients With Mild - Moderate Alzheimer’s Disease Treated With Donepezil (STARSHINE), https://clinicaltrials.gov/ct2/show/NCT01955161. |
[23] | Clinicaltrials.gov, 12 Month Open-label Study of RVT-101 in Subjects With Mild to Moderate Alzheimer’s Disease: MINDSET Extension, https://clinicaltrials.gov/ct2/show/NCT02586909, Last updated 2015, Accessed on, (2015) . |
[24] | Clinicaltrails.gov, Lu AE58054, https://clinicaltrials.gov/ct2/results?term=Lu+AE58054. |
[25] | Roivant Sciences ((2014) ) Roivant Acquires SB742457 from GlaxoSmithKline, http://roivant.com/roivant-acquires-sb742457-glaxosmithkline. |
[26] | RVT-101 - Axovant Sciences, http://axovant.com/rvt-101/clinical-trial/mindset. |
[27] | Ruat M , Traiffort E , Leurs R , Tardivel-Lacombe J , Diaz J , Arrang JM , Schwartz JC ((1993) ) Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation. Proc Natl Acad Sci U S A 90: , 8547–8551. |
[28] | Gellynck E , Laenen K , Andressen KW , Lintermans B , De Martelaere K , Matthys A , Levy FO , Haegeman G , Vanhoenacker P , Van Craenenbroeck K ((2008) ) Cloning, genomic organization and functionality of 5-HT(7) receptor splice variants from mouse brain. Gene 426: , 23–31. |
[29] | Neumaier JF , Sexton TJ , Yracheta J , Diaz AM , Brownfield M ((2001) ) Localization of 5-HT(7) receptors in rat brain by immunocytochemistry, hybridization, and agonist stimulated cFos expression. J Chem Neuroanat 21: , 63–73. |
[30] | Cifariello A , Pompili A , Gasbarri A ((2008) ) 5-HT(7) receptors in the modulation of cognitive processes. Behav Brain Res 195: , 171–179. |
[31] | Meneses A ((2014) ) 5-HT7 receptor stimulation and blockade: A therapeutic paradox about memory formation and amnesia. Front Behav Neurosci 8: , 207. |
[32] | Meneses A ((2014) ) Memory formation and memory alterations: 5-HT6 and 5-HT7 receptors, novel alternative. Rev Neurosci 25: , 325–356. |
[33] | Roberts AJ , Hedlund PB ((2012) ) The 5-HT(7) receptor in learning and memory. Hippocampus 22: , 762–771. |
[34] | Mnie-Filali O , Faure C , Lambás-Señas L , El Mansari M , Belblidia H , Gondard E , Etiévant A , Scarna H , Didier A , Berod A , Blier P , Haddjeri N ((2011) ) Pharmacological blockade of 5-HT7 receptors as a putative fast acting antidepressant strategy. Neuropsychopharmacology 36: , 1275–1288. |
[35] | Bonaventure P , Aluisio L , Shoblock J , Boggs JD , Fraser IC , Lord B , Lovenberg TW , Galici R ((2011) ) Pharmacological blockade of serotonin 5-HT7 receptor reverses working memory deficits in rats by normalizing cortical glutamate neurotransmission. PLoS One 6: , e20210. |
[36] | Nikiforuk A , Kos T , Fijał K , Hołuj M , Rafa D , Popik P ((2013) ) Effects of the selective 5-HT7 receptor antagonist SB-269970 and amisulpride on ketamine-induced schizophrenia-like deficits in rats. PLoS One 8: , e66695. |
[37] | Mnie-Filali O , Lambás-Señas L , Zimmer L , Haddjeri N ((2007) ) 5-HT7 receptor antagonists as a new class of antidepressants. Drug News Perspect 20: , 613–618. |
[38] | Tambs K , Czajkowsky N , Roysamb E , Neale MC , Reichborn-Kjennerud T , Aggen SH , Harris JR , Orstavik RE , Kendler KS ((2009) ) Structure of genetic and environmental risk factors for dimensional representations of DSM-IV anxiety disorders. Br J Psychiatry 195: , 301–307. |
[39] | Healy D ((2008) ) Psychiatric Drugs Explained. Elsevier Health Sciences, pp. 136–170. |
[40] | Barker P ((2008) ) Psychiatric and Mental Health Nursing: The craft of caring. Second Edition, CRC Press. |
[41] | Vos T , Barber RM , Bell B , Bertozzi-Villa A , Biryukov S , Bolliger I , Charlson F , Davis A , Degenhardt L , Dicker D , Duan L , Erskine H , Feigin VL , Ferrari AJ , Fitzmaurice C , Fleming T , Graetz N , Guinovart C , Haagsma J , Hansen GM , Hanson SW , Heuton KR , Higashi H , Kassebaum N , Kyu H , Laurie E , Liang X , Lofgren K , Lozano R , MacIntyre MF , Moradi-Lakeh M , Naghavi M , Nguyen G , Odell S , Ortblad K , Roberts DA , Roth GA , Sandar L , Serina PT , Stanaway JD , Steiner C , Thomas B , Vollset SE , Whiteford H , Wolock TM , Ye P , Zhou M , Ãvila MA , Aasvang GM , Abbafati C , Ozgoren AA , Abd-Allah F , Aziz MIA , Abera SF , Aboyans V , Abraham JP , Abraham B , Abubakar I , Abu-Raddad LJ , Abu-Rmeileh NM , Aburto TC , Achoki T , Ackerman IN , Adelekan A , Ademi Z , Adou AK , Adsuar JC , Arnlov J , Agardh EE , Al Khabouri MJ , Alam SS , Alasfoor D , Albittar MI , Alegretti MA , Aleman A V , Alemu ZA , Alfonso-Cristancho R , Alhabib S , Ali R , Alla F , Allebeck P , Allen PJ , AlMazroa MA , Alsharif U , Alvarez E , Alvis-Guzman N , Ameli O , Amini H , Ammar W , Anderson BO , Anderson HR , Antonio CAT , Anwari P , Apfel H , Arsenijevic VSA , Artaman A , Asghar RJ , Assadi R , Atkins LS , Atkinson C , Badawi A , Bahit MC , Bakfalouni T , Balakrishnan K , Balalla S , Banerjee A , Barker-Collo SL , Barquera S , Barregard L , Barrero LH , Basu S , Basu A , Baxter A , Beardsley J , Bedi N , Beghi E , Bekele T , Bell ML , Benjet C , Bennett DA , Bensenor IM , Benzian H , Bernabe E , Beyene TJ , Bhala N , Bhalla A , Bhutta Z , Bienhoff K , Bikbov B , Abdulhak A Bin , Blore JD , Blyth FM , Bohensky MA , Basara BB , Borges G , Bornstein NM , Bose D , Boufous S , Bourne RR , Boyers LN , Brainin M , Brauer M , Brayne CE , Brazinova A , Breitborde NJ , Brenner H , Briggs AD , Brooks PM , Brown J , Brugha TS , Buchbinder R , Buckle GC , Bukhman G , Bulloch AG , Burch M , Burnett R , Cardenas R , Cabral NL , Nonato IRC , Campuzano JC , Carapetis JR , Carpenter DO , Caso V , Castaneda-Orjuela CA , Catala-Lopez F , Chadha VK , Chang J-C , Chen H , Chen W , Chiang PP , Chimed-Ochir O , Chowdhury R , Christensen H , Christophi CA , Chugh SS , Cirillo M , Coggeshall M , Cohen A , Colistro V , Colquhoun SM , Contreras AG , Cooper LT , Cooper C , Cooperrider K , Coresh J , Cortinovis M , Criqui MH , Crump JA , Cuevas-Nasu L , Dandona R , Dandona L , Dansereau E , Dantes HG , Dargan PI , Davey G , Davitoiu D V , Dayama A , De la Cruz-Gongora V , de la Vega SF , De Leo D , del Pozo-Cruz B , Dellavalle RP , Deribe K , Derrett S , Des Jarlais DC , Dessalegn M , DeVeber GA , Dharmaratne SD , Diaz-Torne C , Ding EL , Dokova K , Dorsey ER , Driscoll TR , Duber H , Durrani AM , Edmond KM , Ellenbogen RG , Endres M , Ermakov SP , Eshrati B , Esteghamati A , Estep K , Fahimi S , Farzadfar F , Fay DF , Felson DT , Fereshtehnejad S-M , Fernandes JG , Ferri CP , Flaxman A , Foigt N , Foreman KJ , Fowkes FGR , Franklin RC , Furst T , Futran ND , Gabbe BJ , Gankpe FG , Garcia-Guerra FA , Geleijnse JM , Gessner BD , Gibney KB , Gillum RF , Ginawi IA , Giroud M , Giussani G , Goenka S , Goginashvili K , Gona P , de Cosio TG , Gosselin RA , Gotay CC , Goto A , Gouda HN , Guerrant R l , Gugnani HC , Gunnell D , Gupta R , Gupta R , Gutierrez RA , Hafezi-Nejad N , Hagan H , Halasa Y , Hamadeh RR , Hamavid H , Hammami M , Hankey GJ , Hao Y , Harb HL , Haro JM , Havmoeller R , Hay RJ , Hay S , Hedayati MT , Pi IBH , Heydarpour P , Hijar M , Hoek HW , Hoffman HJ , Hornberger JC , Hosgood HD , Hossain M , Hotez PJ , Hoy DG , Hsairi M , Hu H , Hu G , Huang JJ , Huang C , Huiart L , Husseini A , Iannarone M , Iburg KM , Innos K , Inoue M , Jacobsen KH , Jassal SK , Jeemon P , Jensen PN , Jha V , Jiang G , Jiang Y , Jonas JB , Joseph J , Juel K , Kan H , Karch A , Karimkhani C , Karthikeyan G , Katz R , Kaul A , Kawakami N , Kazi DS , Kemp AH , Kengne AP , Khader YS , Khalifa SEA , Khan EA , Khan G , Khang Y-H , Khonelidze I , Kieling C , Kim D , Kim S , Kimokoti RW , Kinfu Y , Kinge JM , Kissela BM , Kivipelto M , Knibbs L , Knudsen AK , Kokubo Y , Kosen S , Kramer A , Kravchenko M , Krishnamurthi R V , Krishnaswami S , Defo BK , Bicer BK , Kuipers EJ , Kulkarni VS , Kumar K , Kumar GA , Kwan GF , Lai T , Lalloo R , Lam H , Lan Q , Lansingh VC , Larson H , Larsson A , Lawrynowicz AE , Leasher JL , Lee J-T , Leigh J , Leung R , Levi M , Li B , Li Y , Li Y , Liang J , Lim S , Lin H-H , Lind M , Lindsay MP , Lipshultz SE , Liu S , Lloyd BK , Ohno SL , Logroscino G , Looker KJ , Lopez AD , Lopez-Olmedo N , Lortet-Tieulent J , Lotufo PA , Low N , Lucas RM , Lunevicius R , Lyons RA , Ma J , Ma S , Mackay MT , Majdan M , Malekzadeh R , Mapoma CC , Marcenes W , March LM , Margono C , Marks GB , Marzan MB , Masci JR , Mason-Jones AJ , Matzopoulos RG , Mayosi BM , Mazorodze TT , McGill NW , McGrath JJ , McKee M , McLain A , McMahon BJ , Meaney PA , Mehndiratta MM , Mejia-Rodriguez F , Mekonnen W , Melaku YA , Meltzer M , Memish ZA , Mensah G , Meretoja A , Mhimbira FA , Micha R , Miller TR , Mills EJ , Mitchell PB , Mock CN , Moffitt TE , Ibrahim NM , Mohammad KA , Mokdad AH , Mola GL , Monasta L , Montico M , Montine TJ , Moore AR , Moran AE , Morawska L , Mori R , Moschandreas J , Moturi WN , Moyer M , Mozaffarian D , Mueller UO , Mukaigawara M , Murdoch ME , Murray J , Murthy KS , Naghavi P , Nahas Z , Naheed A , Naidoo KS , Naldi L , Nand D , Nangia V , Narayan KMV , Nash D , Nejjari C , Neupane SP , Newman LM , Newton CR , Ng M , Ngalesoni FN , Nhung NT , Nisar MI , Nolte S , Norheim OF , Norman RE , Norrving B , Nyakarahuka L , Oh IH , Ohkubo T , Omer SB , Opio JN , Ortiz A , Pandian JD , Panelo CIA , Papachristou C , Park E-K , Parry CD , Caicedo AJP , Patten SB , Paul VK , Pavlin BI , Pearce N , Pedraza LS , Pellegrini CA , Pereira DM , Perez-Ruiz FP , Perico N , Pervaiz A , Pesudovs K , Peterson CB , Petzold M , Phillips MR , Phillips D , Phillips B , Piel FB , Plass D , Poenaru D , Polanczyk G V , Polinder S , Pope CA , Popova S , Poulton RG , Pourmalek F , Prabhakaran D , Prasad NM , Qato D , Quistberg DA , Rafay A , Rahimi K , Rahimi-Movaghar V , Rahman S ur , Raju M , Rakovac I , Rana SM , Razavi H , Refaat A , Rehm J , Remuzzi G , Resnikoff S , Ribeiro AL , Riccio PM , Richardson L , Richardus JH , Riederer AM , Robinson M , Roca A , Rodriguez A , Rojas-Rueda D , Ronfani L , Rothenbacher D , Roy N , Ruhago GM , Sabin N , Sacco RL , Ksoreide K , Saha S , Sahathevan R , Sahraian MA , Sampson U , Sanabria JR , Sanchez-Riera L , Santos IS , Satpathy M , Saunders JE , Sawhney M , Saylan MI , Scarborough P , Schoettker B , Schneider IJ , Schwebel DC , Scott JG , Seedat S , Sepanlou SG , Serdar B , Servan-Mori EE , Shackelford K , Shaheen A , Shahraz S , Levy TS , Shangguan S , She J , Sheikhbahaei S , Shepard DS , Shi P , Shibuya K , Shinohara Y , Shiri R , Shishani K , Shiue I , Shrime MG , Sigfusdottir ID , Silberberg DH , Simard EP , Sindi S , Singh JA , Singh L , Skirbekk V , Sliwa K , Soljak M , Soneji S , Soshnikov SS , Speyer P , Sposato LA , Sreeramareddy CT , Stoeckl H , Stathopoulou VK , Steckling N , Stein MB , Stein DJ , Steiner TJ , Stewart A , Stork E , Stovner LJ , Stroumpoulis K , Sturua L , Sunguya BF , Swaroop M , Sykes BL , Tabb KM , Takahashi K , Tan F , Tandon N , Tanne D , Tanner M , Tavakkoli M , Taylor HR , Te Ao BJ , Temesgen AM , Have M Ten , Tenkorang EY , Terkawi AS , Theadom AM , Thomas E , Thorne-Lyman AL , Thrift AG , Tleyjeh IM , Tonelli M , Topouzis F , Towbin JA , Toyoshima H , Traebert J , Tran BX , Trasande L , Trillini M , Truelsen T , Trujillo U , Tsilimbaris M , Tuzcu EM , Ukwaja KN , Undurraga EA , Uzun SB , van Brakel WH , van de Vijver S , Van Dingenen R , van Gool CH , Varakin YY , Vasankari TJ , Vavilala MS , Veerman LJ , Velasquez-Melendez G , Venketasubramanian N , Vijayakumar L , Villalpando S , Violante FS , Vlassov V V , Waller S , Wallin MT , Wan X , Wang L , Wang J , Wang Y , Warouw TS , Weichenthal S , Weiderpass E , Weintraub RG , Werdecker A , Wessells KRR , Westerman R , Wilkinson JD , Williams HC , Williams TN , Woldeyohannes SM , Wolfe CDA , Wong JQ , Wong H , Woolf AD , Wright JL , Wurtz B , Xu G , Yang G , Yano Y , Yenesew MA , Yentur GK , Yip P , Yonemoto N , Yoon S-J , Younis M , Yu C , Kim KY , Zaki MES , Zhang Y , Zhao Z , Zhao Y , Zhu J , Zonies D , Zunt JR , Salomon JA , Murray CJ ((2015) ) Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 386: , 743–800. |
[42] | ((2008) ) Guideline Working Group for the Treatment of Patients with Anxiety Disorders in Primary Care. National Plan for the NHS of the MSC. Health technology Assessment Unit. Laín Entralgo Agency. Community of Madrid; Clinical Practice Guidelines in the NHS. UETS N° 2006/10, http://portal.guiasalud.es/GPC/GPC_430_Anxiety_Lain_Entr_compl_en.pdf. |
[43] | de Menezes GB , Fontenelle LF , Mululo S , Versiani M ((2007) ) Resistência ao tratamento nos transtornos de ansiedade: Fobia social, transtorno de ansiedade generalizada e transtorno do pânico. Rev Bras Psiquiatr 29: , S55–S60. |
[44] | Zahreddine N , Richa S ((2013) ) Non-antidepressant treatment of generalized anxiety disorder. Curr Clin Pharmacol 10: , 86–96. |
[45] | Ivachtchenko AV , Frolov EB , Mitkin OD , Tkachenko SE , Khvat AV ((2010) ) Synthesis of hydrogenated 2,7-dimethylpyrrolo[3,4-b]indoles –analogs of dimebon. Chem Heterocycl Compd 46: , 170–178. |
[46] | Butzlaff M , Ponimaskin E ((2016) ) The role of serotonin receptors in Alzheimer’s disease. Opera Med Physiol 1: , 91–100. |
[47] | Frolov EB , Khvat AV , Malarchuk S , Mitkin OD , Okun I , Kyselev AS , Savchuk NP , Ivashchenko AA , Ivachtchenko AV ((2012) ) Substituted azepino[4,3-b]indoles, pharmacological composition and a method for the production and use thereof.. World Patent No. WO2008026965. |
[48] | Frolov EB , Ivachtchenko AV , Ivashchenko AA , Kravchenko DV , Mitkin OD , Okun I , Savchuk NP , Tkachenko SE ((2008) ) Substituted 2,3,4,5-tetrahydro-1h-pyrido[4,3-b]indoles.. World Patent No. WO2008123800. |
[49] | Frolov EB , Khvat AV , Malarchuk S , Mitkin OD , Okun I , Kyselev AS , Savchuk NP , Ivashchenko AA , Ivachtchenko AV ((2012) ) Substituted azepino[4,3-b]indoles, pharmacological composition and a method for the production and use thereof.. US Patent No. US 20120040965. |
[50] | Ivachtchenko AV , Ivashchenko AA , Kravchenko DV , Mitkin OD , Okun I , Savchuk NP , Tkachenko SE ((2008) ) Substituted 2,3,4,5-tetrahydro-1h-pyrido[4,3-b]indoles, methods for the production and the use thereof.. World Patent No. WO2008115098. |
[51] | Ivachtchenko AV , Ivashchenko AA , Savchuk NP ((2009) ) Substituted 3-sulphonyl-[1,2,3]triazolo [1,5-a]pyrimidines-antagonists of serotonin 5-ht6 receptors and methods for the production thereof.. World Patent No. WO2009093934. |
[52] | Ivachtchenko AV , Mitkin OD ((2013) ) Substituted hydrogenated thieno-pyrrolo[3,2-c]pyridine, ligands, a pharmaceutical composition and a method for using the above.. US Patent No. US 20130267551. |
[53] | Ivachtchenko AV , Mitkin OD , Kadieva MG ((2014) ) Substituted methyl amines, serotonin 5-HT6 receptor antagonists, methods for production and use thereof... US US8829002. |
[54] | Ivachtchenko AV , Frolov EB , Mitkin OD , Tkachenko SE , Okun IM , Khvat A V ((2010) ) Synthesis and biological activity of 5-styryl and 5-phenethyl-substituted 2,3,4,5-tetrahydro-1H-pyrido[4,3-b]indoles. Bioorg Med Chem Lett 20: , 78–82. |
[55] | Ivachtchenko AV , Frolov EB , Mitkin OD , Kysil VM , Khvat A V , Tkachenko SE ((2009) ) Synthesis and biological evaluation of novel 5,8-disubstituted-2-methyl-2,3,4,5-tetrahydro-1H-pyrido[4,3-b] indoles as 5-HT(6) and H(1) receptors antagonists. Arch Pharm (Weinheim) 342: , 740–747. |
[56] | Ivachtchenko AV , Mitkin OD , Tkachenko SE , Okun IM , Kysil VM ((2010) ) 8-Sulfonyl-substituted tetrahydro-1H-pyrido[4,3-b]indoles as 5-HT6 receptor antagonists. Eur J Med Chem 45: , 782–789. |
[57] | Ivashchenko AA , Ivachtchenko AV , Lavrovsky YV , Mitkin OD , Savchuk NP , Tkachenko SE , Okun I ((2009) ) Ligands of α-adrenoceptors and of dopamine, histamine, imidozaline and serotonin receptors and the use thereof.. World Patent No. WO2009082268. |
[58] | Ivashchenko AA , Ivachtchenko AV , Savchuk NP ((2009) ) Substituted 2-amino-3-sulfonyl-pyrazolo[1,5-a] pyrimidines/antagonists of serotonin 5-ht6 receptors, methods for the production and the use thereof.. World Patent No. WO2009093208. |
[59] | Ivashchenko AA , Ivachtchenko AV , Savchuk NP ((2009) ) 3-sulfonyl-pyrazolo[1,5-a] pyrimidines/antagonists of serotonin 5-ht6 receptors, methods for the production and the use thereof.. World Patent No. WO2009093206. |
[60] | Ivashchenko AA , Ivachtchenko AV , Savchuk NP ((2014) ) Substituted cycloalcano[e and d] pyrazolo [1,5-a]pyrimidines/antagonists of serotonin 5-HT6 receptors and methods for production and the use thereof.. US Patent No. US 8629154. |
[61] | Ivashchenko AA , Kysil VM , Savchuk NP , Ivachtchenko AV ((2009) ) Tetrahydro-pyrazolo[1,5-a]pyrido-pyrimidines as antagonists of serotonin 5-ht6 receptors, methods for the production and use thereof.. World Patent No. WO2009136814. |
[62] | Ivashchenko AA , Kysil VM , Savchuk NP , Ivachtchenko AV ((2013) ) Tetrahydro-pyrazolo[1,5-a]pyrido-pyrimidines as antagonists of serotonin 5-HT6 receptors, methods for the production and use thereof.. US Patent No. US 8569318. |
[63] | Ivashchenko AA , Savchuk NP , Ivachtchenko AV ((2011) ) Ligand with a broad spectrum of pharmacological activity, a pharmaceutical composition, a medicinal agent and a method of treatment.. US Patent No. US 20110136853. |
[64] | Ivashchenko AA , Savchuk NP , Ivachtchenko AV , Lavrovsky Y , Mitkin OD , Kadieva MG ((2013) ) Substituted 3-arylsulfonyl-pyrazolo[1,5-A]pyrimidines, serotonin 5-HT6 receptor antagonists and methods for the production and use thereof.. US Patent No. US 8618114. |
[65] | Ivashchenko AA , Savchuk NP , Ivachtchenko AV , Mitkin OD , Kysil VM ((2012) ) Substituted 8-sulfonyl-2,3,4,5-tetrahydro-1h-gamma-carbolines, ligands and pharmaceutical composition; method for the production and use of same.. US Patent No. US 20120071493. |
[66] | Ivashchenko AA , Ivashchenko AV , Tkachenko SY , Okun IM , Savchuk , Nikolay Filippovich ((2008) ) Ligands of 5-HT6 receptors, a pharmaceutical composition, method for the production and use thereof.. US Patent No. US 20110046368. |
[67] | Lavrovsky Y , Ivachtchenko AV , Morozova M , Salimov RM , Kasey V ((2010) ) Preclinical and early clinical studies of AVN-101, a novel balanced molecule for the treatment of Alzheimer’s disease. Alzheimers Dement 6: , S583. |
[68] | Tkachenko S ((2008) ) Discovery and in vivo evaluation of potent 5-HT6 receptor antagonists for cognition enhancement in treating Alzheimer’s disease. Alzheimers Dement 4: , T514. |
[69] | Tkachenko S , Ivachtchenko A , Okun I , Lavrovsky Y , Mitkin O , Salimov R ((2009) ) Comparison of highly selective and non-selective 5-HT6 receptor antagonists in in vitro and in vivo neurodegenerative models. Alzheimers Dement 5: , P492. |
[70] | Lipinski CA ((2004) ) Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov Today Technol 1: , 337–341. |
[71] | Okun I , Tkachenko SE , Khvat A , Mitkin O , Kazey V , Ivachtchenko AV ((2010) ) From anti-allergic to anti-Alzheimer’s: Molecular pharmacology of Dimebon. Curr Alzheimer Res 7: , 97–112. |
[72] | Chuang ATT , Foley A , Pugh PL , Sunter D , Tong X , Regan C , Dawson LA , Medhurst AD , Upton N ((2006) ) P4-387. Alzheimers Dement 2: , S631–S632. |
[73] | Arnt J , Bang-Andersen B , Grayson B , Bymaster FP , Cohen MP , DeLapp NW , Giethlen B , Kreilgaard M , McKinzie DL , Neill JC , Nelson DL , Nielsen SM , Poulsen MN , Schaus JM , Witten LM ((2010) ) Lu AE58054, a 5-HT6 antagonist, reverses cognitive impairment induced by subchronic phencyclidine in a novel object recognition test in rats. Int J Neuropsychopharmacol 13: , 1021–1033. |
[74] | Williams GV , Rao SG , Goldman-Rakic PS ((2002) ) The physiological role of 5-HT2A receptors in working memory. J Neurosci 22: , 2843–2854. |
[75] | de Angelis L ((2002) ) 5-HT2A antagonists in psychiatric disorders. Curr Opin Investig Drugs 3: , 106–112. |
[76] | Raote I , Bhattacharya A , Panicker MM ((2007) ) Serotonin 2A (5-HT2A) receptor function: Ligand-dependent mechanisms and pathways. In Serotonin Receptors in Neurobiology, Chattopadhyay A, ed. CRC Press, Boca Raton, FL, pp. 1–17. |
[77] | Fijał K , Popik P , Nikiforuk A ((2014) ) Co-administration of 5-HT6 receptor antagonists with clozapine, risperidone, and a 5-HT2A receptor antagonist: Effects on prepulse inhibition in rats. Psychopharmacology (Berl) 231: , 269–281. |
[78] | Millan MJ Serotonin 5-HT2C receptors as a target for the treatment of depressive and anxious states: Focus on novel therapeutic strategies. Therapie 60: , 441–460. |
[79] | Choi D-S , Maroteaux L ((1996) ) Immunohistochemical localisation of the serotonin 5-HT2B receptor in mouse gut, cardiovascular system, and brain. FEBS Lett 391: , 45–51. |
[80] | Rothman RB , Baumann MH , Savage JE , Rauser L , McBride A , Hufeisen SJ , Roth BL ((2000) ) Evidence for possible involvement of 5-HT2B receptors in the cardiac valvulopathy associated with fenfluramine and other serotonergic medications. Circulation 102: , 2836–2841. |
[81] | Hill SJ , Ganellin CR , Timmerman H , Schwartz JC , Shankley NP , Young JM , Schunack W , Levi R , Haas HL ((1997) ) International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev 49: , 253–278. |
[82] | Lieberman P ((2009) ) Histamine, antihistamines, and the central nervous system. Allergy Asthma Proc 30: , 482–486. |
[83] | Saligrama N , Noubade R , Case LK , del Rio R , Teuscher C ((2012) ) Combinatorial roles for histamine H1-H2 and H3-H4 receptors in autoimmune inflammatory disease of the central nervous system. Eur J Immunol 42: , 1536–1546. |
[84] | Husbands SM , Glennon RA , Gorgerat S , Gough R , Tyacke R , Crosby J , Nutt DJ , Lewis JW , Hudson AL ((2001) ) β-carboline binding to imidazoline receptors. Drug Alcohol Depend 64: , 203–208. |
[85] | Halaris A , Piletz JE ((2003) ) Relevance of imidazoline receptors and agmatine to psychiatry: A decade of progress. Ann N Y Acad Sci 1009: , 1–20. |
[86] | Philipp M , Brede M , Hein L ((2002) ) Physiological significance of alpha(2)-adrenergic receptor subtype diversity: One receptor is not enough. Am J Physiol Regul Integr Comp Physiol 283: , R287–295. |
[87] | Price DT , Lefkowitz RJ , Caron MG , Berkowitz D , Schwinn DA ((1994) ) Localization of mRNA for three distinct alpha 1-adrenergic receptor subtypes in human tissues: Implications for human alpha-adrenergic physiology. Mol Pharmacol 45: , 171–175. |
[88] | Alpha1AdrenergicReceptorAntagonists, http://livertox.nih.gov/Alpha1AdrenergicReceptorAntagonists.htm. |
[89] | Ong KL , Cheung BMY , Man YB , Lau CP , Lam KSL ((2007) ) Prevalence, awareness, treatment, and control of hypertension among United States adults 1999-2004. Hypertension 49: , 69–75. |
[90] | Press B ((2011) ) Optimization of the Caco-2 permeability assay to screen drug compounds for intestinal absorption and efflux. Methods Mol Biol 763: , 139–154. |
[91] | Polli JW , Wring SA , Humphreys JE , Huang L , Morgan JB , Webster LO , Serabjit-Singh CS ((2001) ) Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther 299: , 620–628. |
[92] | Nirogi R , Kandikere V , Mudigonda K , Bhyrapuneni G , Muddana N , Saralaya R , Benade V ((2009) ) A simple and rapid method to collect the cerebrospinal fluid of rats and its application for the assessment of drug penetration into the central nervous system. J Neurosci Methods 178: , 116–169. |
[93] | Laterra J , Keep R , Betz LA , Goldstein GW ((1999) ) Blood— cerebrospinal fluid barrier. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, Siegel GJ, Agranoff BW, Albers RW, ed., Lippincott–Raven, Philadelphia. |
[94] | Bickel U ((2005) ) How to measure drug transport across the blood-brain barrier. NeuroRx 2: , 15–26. |
[95] | Walf AA , Frye CA ((2007) ) The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2: , 322–328. |
[96] | Ennaceur A ((2012) ) Open space anxiety test in rodents: The elevated platform with steep slopes. Methods Mol Biol 829: , 177–191. |
[97] | Miyata S , Shimoi T , Hirano S , Yamada N , Hata Y , Yoshikawa N , Ohsawa M , Kamei J ((2007) ) Effects of serotonergic anxiolytics on the freezing behor in the elevated open-platform test in mice. J Pharmacol Sci 105: , 272–278 avi. |
[98] | Prut L , Belzung C ((2003) ) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur J Pharmacol 463: , 3–33. |
[99] | Holmes A , Yang RJ , Lesch K-P , Crawley JN , Murphy DL ((2003) ) Mice lacking the serotonin transporter exhibit 5-HT(1A) receptor-mediated abnormalities in tests for anxiety-like behavior. Neuropsychopharmacology 28: , 2077–2088. |
[100] | Pollard GT , Howard JL ((1979) ) The Geller-Seifter conflict paradigm with incremental shock. Psychopharmacology (Berl) 62: , 117–121. |
[101] | Castagné V , Moser P , Porsolt RD ((2009) ) Behavioral assessment of antidepressant activity in rodents. In Methods of Behavior Analysis in Neuroscience, Buccafusco JJ, ed., CRC Press, Boca Raton, FL. |
[102] | Graham JH , Jerry J , Buccafusco ((2001) ) Inhibitory avoidance behavior and memory assessment. In Methods of Behavior Analysis in Neuroscience, Buccafusco JJ, ed., Boca Raton, FL, CRC Press, pp. 153–163. |
[103] | Vorhees CV , Williams MT ((2006) ) Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1: , 848–858. |
[104] | Ennaceur A , Delacour J ((1988) ) A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res 31: , 47–59. |
[105] | Wehner JM , Radcliffe RA ((2004) ) Cued and contextual fear conditioning in mice. Curr Protoc Neurosci Chapter 8, Unit 8.5C. |
[106] | Klinkenberg I , Blokland A ((2010) ) The validity of scopolamine as a pharmacological model for cognitive impairment: A review of animal behavioral studies. Neurosci Biobehav Rev 34: , 1307–1350. |
[107] | Brown JW , Rueter LE , Zhang M ((2014) ) Predictive validity of a MK-801-induced cognitive impairment model in mice: Implications on the potential limitations and challenges of modeling cognitive impairment associated with schizophrenia preclinically. Prog Neuropsychopharmacol Biol Psychiatry 49: , 53–62. |
[108] | Antunes M , Biala G ((2012) ) The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn Process 13: , 93–110. |
[109] | Dodart JC , Mathis C , Ungerer A ((1997) ) Scopolamine-induced deficits in a two-trial object recognition task in mice. Neuroreport 8: , 1173–1178. |
[110] | Bubeníková-Valesová V , Horácek J , Vrajová M , Höschl C ((2008) ) Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev 32: , 1014–1023. |
[111] | Briones TL , Hala Darwish ((2012) ) Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J Neuroinflammation 9: , 244–256. |
[112] | Caine SB , Geyer MA , Swerdlow NR ((1992) ) Hippocampal modulation of acoustic startle and prepulse inhibition in the rat. Pharmacol Biochem Behav 43: , 1201–1208. |
[113] | Swerdlow NR ((1994) ) Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry 51: , 139–154. |
[114] | Swerdlow NR , Geyer MA ((1998) ) Using an animal model of deficient sensorimotor gating to study the pathophysiology and new treatments of schizophrenia. Schizophr Bull 24: , 285–301. |
[115] | Kalish LA ((1990) ) Efficient design for estimation of median lethal dose and quantal dose-response curves. Biometrics 46: , 737–748. |
[116] | Newell DR , Burtles SS , Fox BW , Jodrell DI , Connors TA ((1999) ) Evaluation of rodent-only toxicology for early clinical trials with novel cancer therapeutics. Br J Cancer 81: , 760–768. |
[117] | Torres V , Tepper D , Flowers D , Wynn J , Lam S , Keefe D , Miura DS , Somberg JC ((1986) ) QT prolongation and the antiarrhythmic efficacy of amiodarone. J Am Coll Cardiol 7: , 142–147. |
[118] | Mortelmans K , Zeiger E ((2000) ) The Ames Salmonella/microsome mutagenicity assay.. Mutat Res 455: , 29–60. |
[119] | Flückiger-Isler S , Kamber M ((2012) ) Direct comparison of the Ames microplate format (MPF) test in liquid medium with the standard Ames pre-incubation assay on agar plates by use of equivocal to weakly positive test compounds. Mutat Res 747: , 36–45. |
[120] | Julian Preston R , Dean BJ , Galloway S , Holden H , McFee AF , Shelby M ((1987) ) Mammalian in vivo cytogenetic assays Analysis of chromosome aberrations in bone marrow cells. Mutat Res Toxicol 189: , 157–165. |
[121] | The Salmonella/E. coli Mutagenicity Test or Ames Test –NTP, http://ntp.niehs.nih.gov/testing/types/genetic/invitro/sa/index.html |
[122] | Ames BN , Lee FD , Durston WE ((1973) ) An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Natl Acad Sci U S A 70: , 782–786. |
[123] | Taylor EW ((1965) ) The mechanism of colchicine inhibition of mitosis. I. Kinetics of inhibition and the binding of H3-colchicine. (Suppl). J Cell Biol 25: , 145–160. |
[124] | Lamb JC ((1985) ) Reproductive toxicity testing: Evaluating and developing new testing systems. Int J Toxicol 4: , 163–171. |
Figures and Tables
Fig.1
Affinities, pKi, of AVN-101 and its major metabolites, M1 and M2, to different targets, as measured in radio-ligand binding competition assay. Assessment of the binding affinity was performed by the Contract Research Organization, Ricerka, in accordance with their protocols briefly described in [71].
![Affinities, pKi, of AVN-101 and its major metabolites, M1 and M2, to different targets, as measured in radio-ligand binding competition assay. Assessment of the binding affinity was performed by the Contract Research Organization, Ricerka, in accordance with their protocols briefly described in [71].](https://content.iospress.com:443/media/jad/2016/53-2/jad-53-2-jad151146/jad-53-jad151146-g001.jpg)
Fig.2
Affinities of AVN-101 and SB742457 to 5-HT7 (A) and 5-HT2A (B) receptors in competitive radioligand binding assay. Radio-labeled 5.5 nM [3H] lysergic acid diethylamide was used for 5-HT7 receptor and 0.5 nM [3H] ketanserin was used for 5-HT2A receptor.
![Affinities of AVN-101 and SB742457 to 5-HT7 (A) and 5-HT2A (B) receptors in competitive radioligand binding assay. Radio-labeled 5.5 nM [3H] lysergic acid diethylamide was used for 5-HT7 receptor and 0.5 nM [3H] ketanserin was used for 5-HT2A receptor.](https://content.iospress.com:443/media/jad/2016/53-2/jad-53-2-jad151146/jad-53-jad151146-g002.jpg)
Fig.3
A) Kinetics of Ca2 + mobilization induced by αMet5-HT (5μM) without (dashed line) and in the presence of 10μM AVN-101 (solid line) in HEK 297 cells extragenously expressing the human recombinant 5-HT2B receptor. Arrows indicate additions of the corresponding compounds. B) Activation (squares) and inhibition of the αMet5-HT-induced rat stomach fundus contraction (circles) with AVN-101.
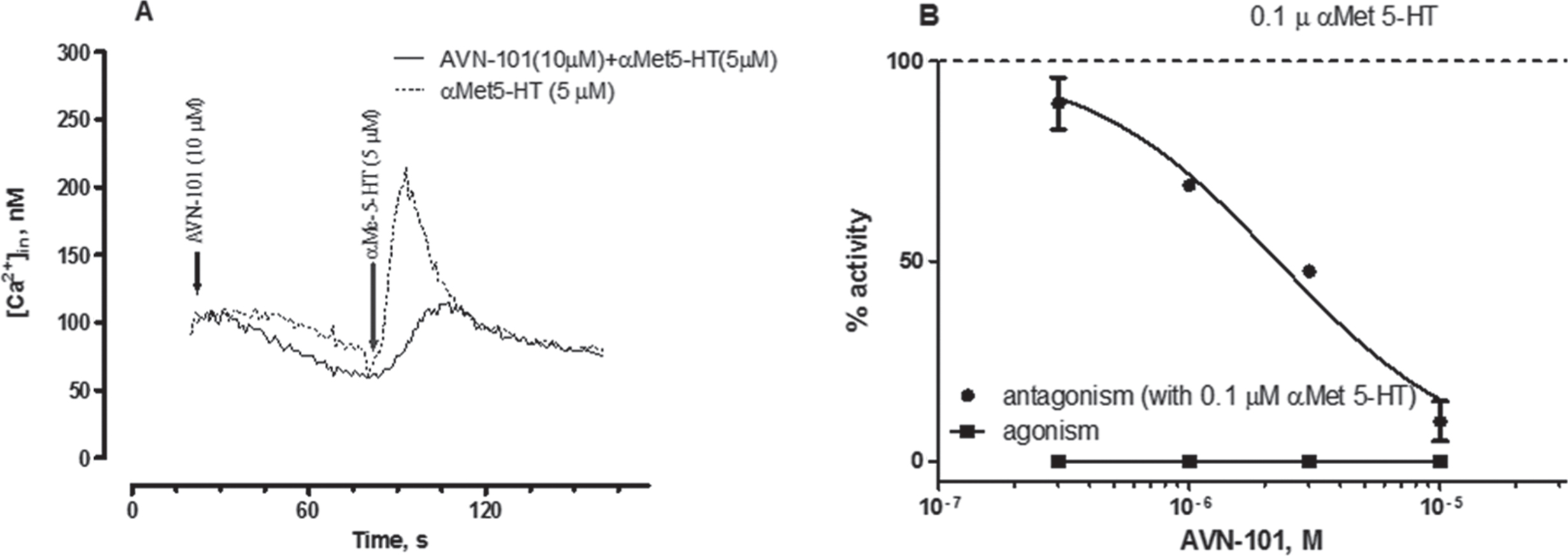
Fig.4
A) AVN-101-induced blockade of cell responses (SK-N-SH endogenously expressing H1 receptors and CHO-K1 exogenously expressing human recombinant H2 receptors) to corresponding agonists, 10μM histamine and 50 nM amthamine. Histamine induced Ca2 + mobilization in SK-N-SH cells and amthamine induced cAMP accumulation in CHO-1K cells. B) AVN-101 effectively competes for the H1 and H2 receptors, expressed in CHO-K1 cells, with corresponding radio-labeled ligands, 1.2 nM [3H]Pyrilamine and 0.1 nM [125I]Aminopotentidine.
![A) AVN-101-induced blockade of cell responses (SK-N-SH endogenously expressing H1 receptors and CHO-K1 exogenously expressing human recombinant H2 receptors) to corresponding agonists, 10μM histamine and 50 nM amthamine. Histamine induced Ca2 + mobilization in SK-N-SH cells and amthamine induced cAMP accumulation in CHO-1K cells. B) AVN-101 effectively competes for the H1 and H2 receptors, expressed in CHO-K1 cells, with corresponding radio-labeled ligands, 1.2 nM [3H]Pyrilamine and 0.1 nM [125I]Aminopotentidine.](https://content.iospress.com:443/media/jad/2016/53-2/jad-53-2-jad151146/jad-53-jad151146-g004.jpg)
Fig.5
4 h equilibrium concentrations of AVN-101 in dialysis apparatus, whole human blood plasma in one chamber and PBS (pH 7.2) in another. Fb –AVN-101 fraction bound to plasma proteins.
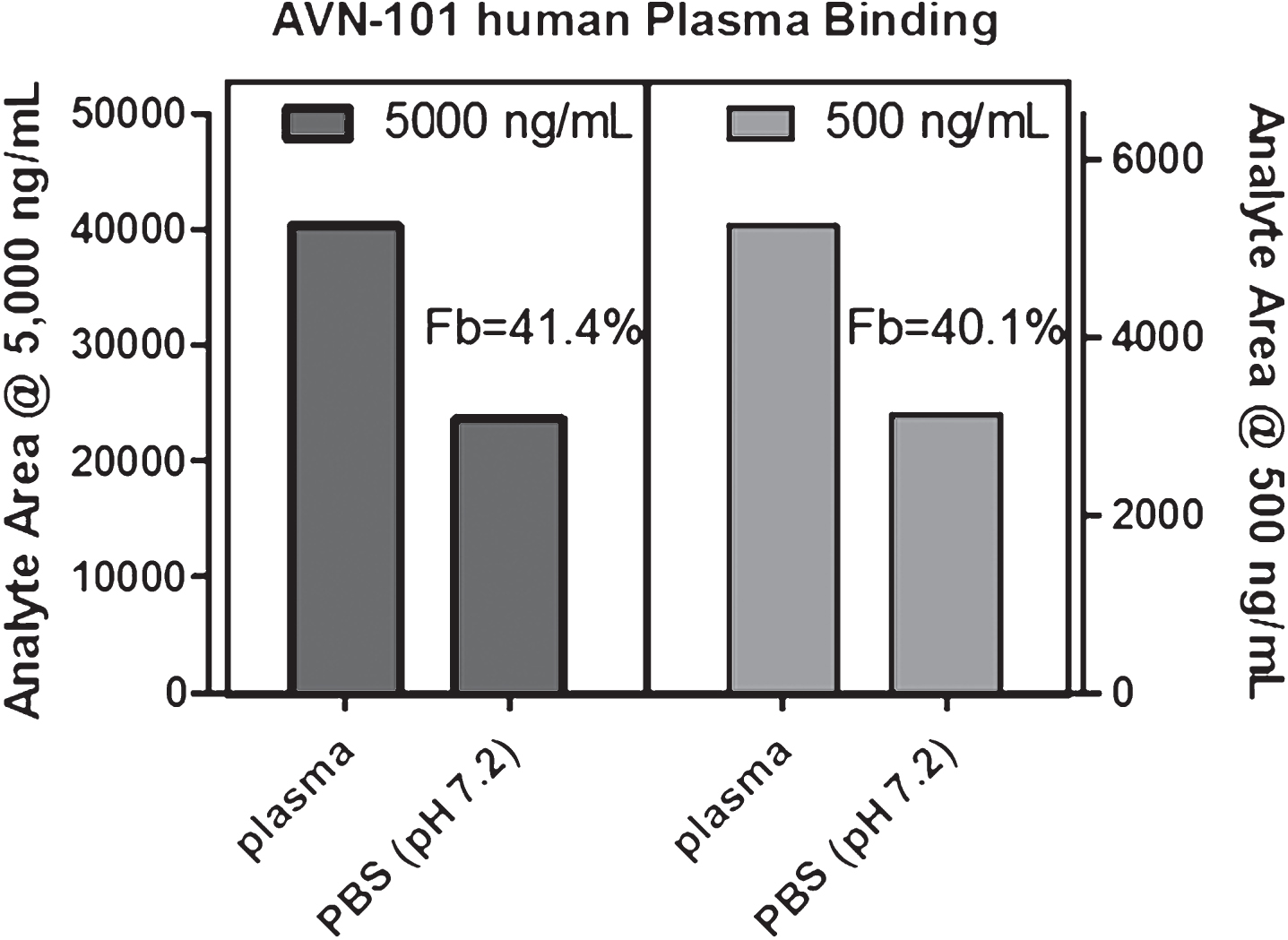
Fig.6
AVN-101-induced activation of Pgp ATPase activity.
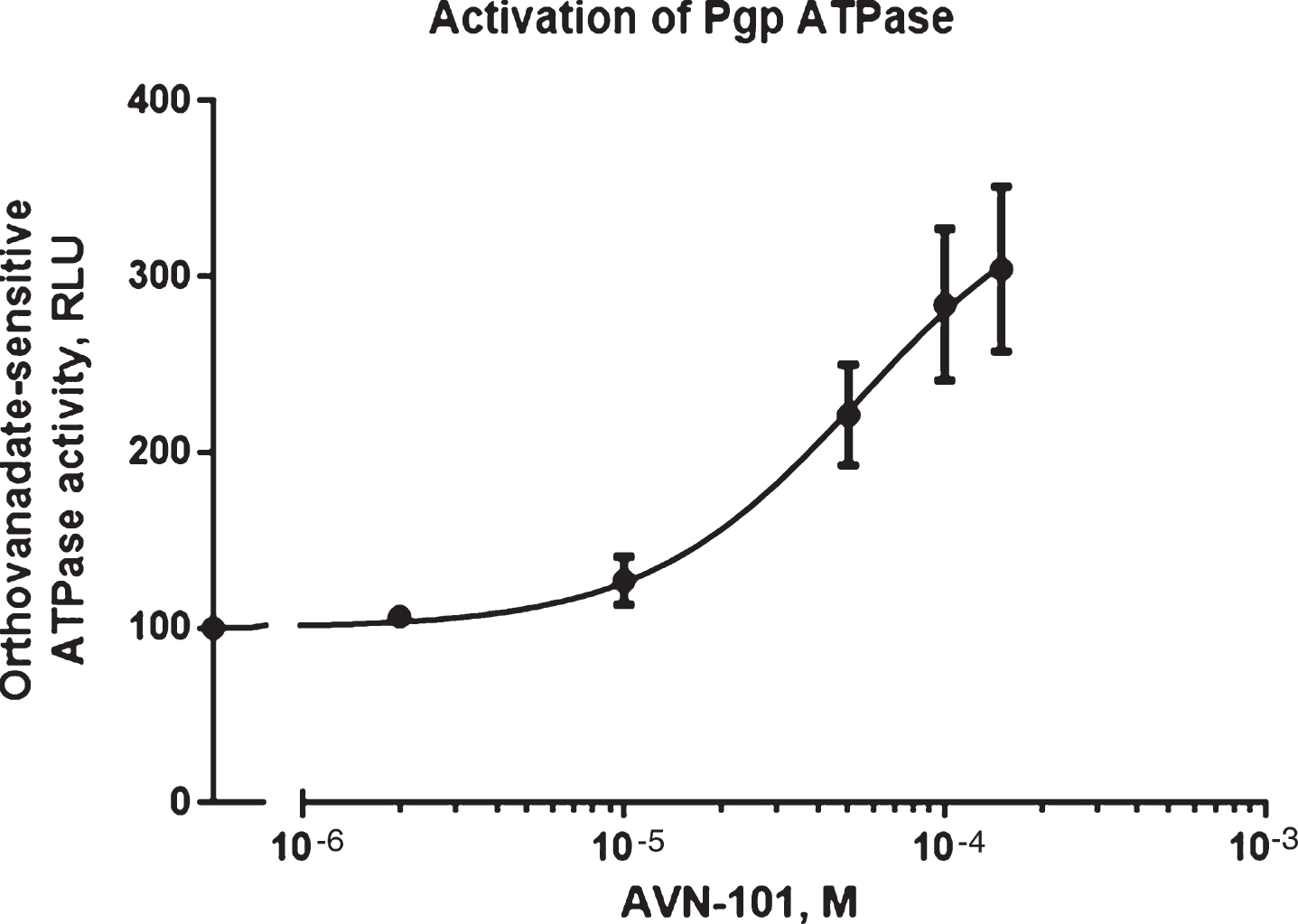
Fig.7
Kinetics of AVN-101 (1μM) decomposition by rat and human microsomes.
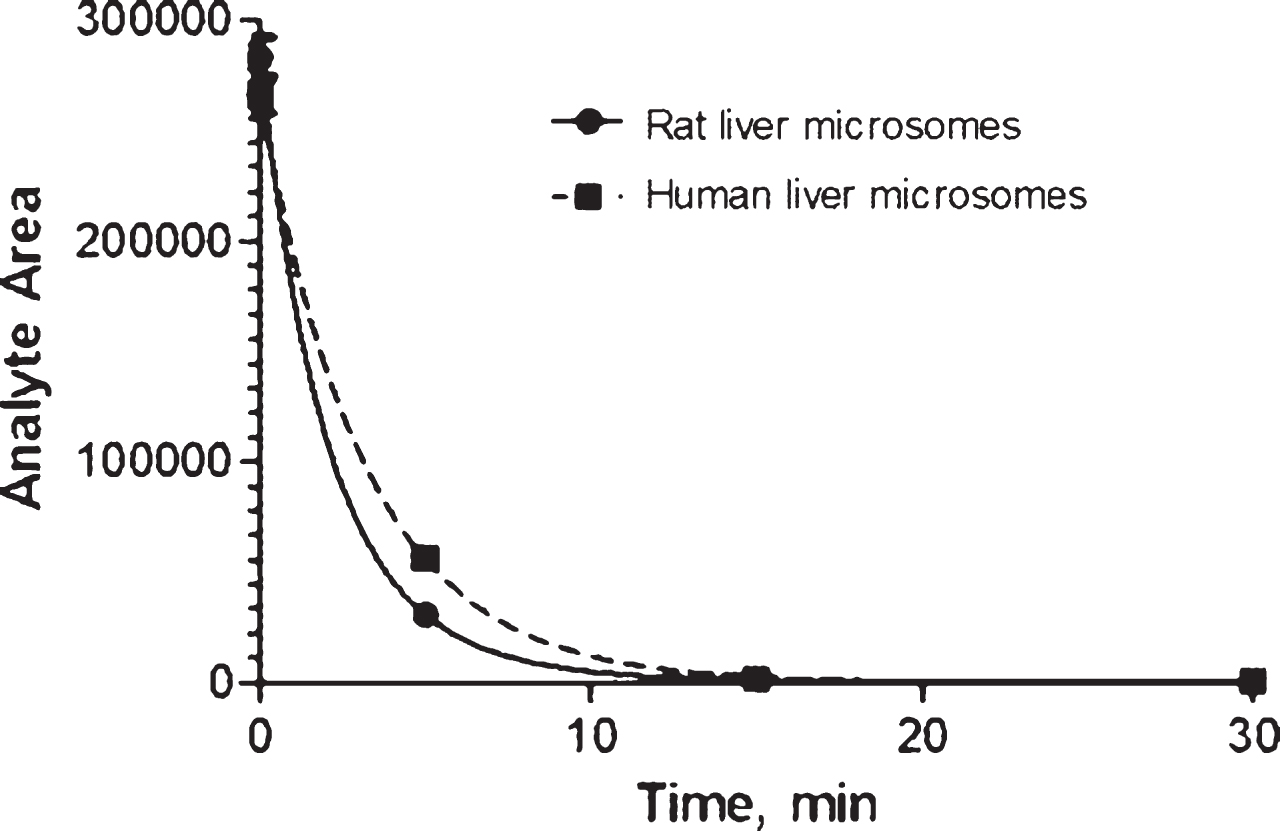
Fig.8
Metabolites of AVN-101digestion identified in human (HLM) and rat (RLM) microsomes. Solid arrows indicate metabolites determined in the HLM “soup” and dashed ones indicate metabolites identified in the RLM. Grayed out areas on the structures indicate possible oxidation points.
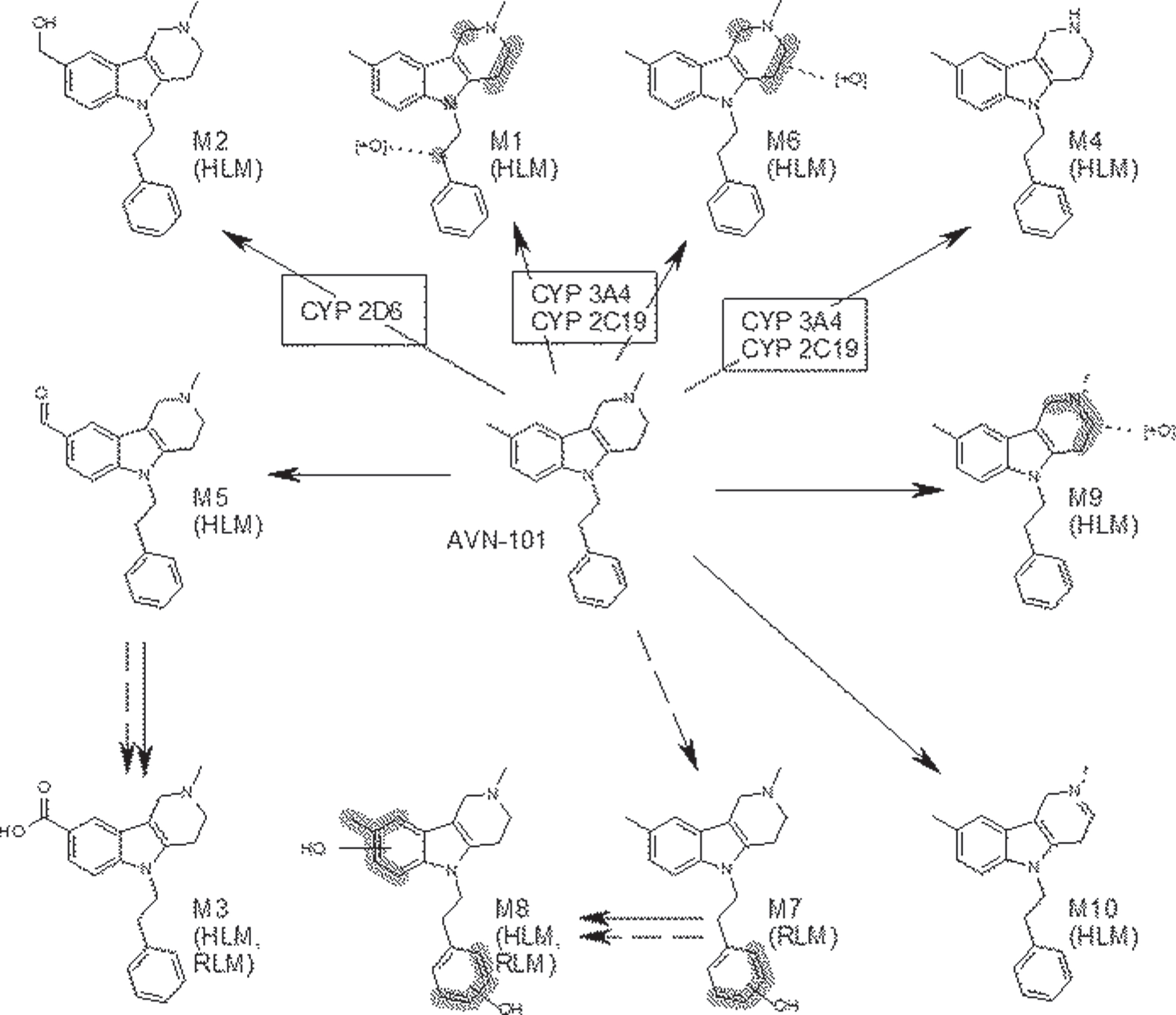
Fig.9
Metabolism of AVN-101 in a suspension of S9 fraction isolated from rat (A and B) and human (C and D) liver in the presence of i) only phase 1 metabolism co-factor, NADPH (closed circles), ii) only phase 2 metabolism co-factor, UDPGA (closed squares), and iii) when both phase 1 and phase 2 co-factors were present (open circles). AVN-101 was tested at a concentration of 1μM (A and C) and 2μM (B and D).
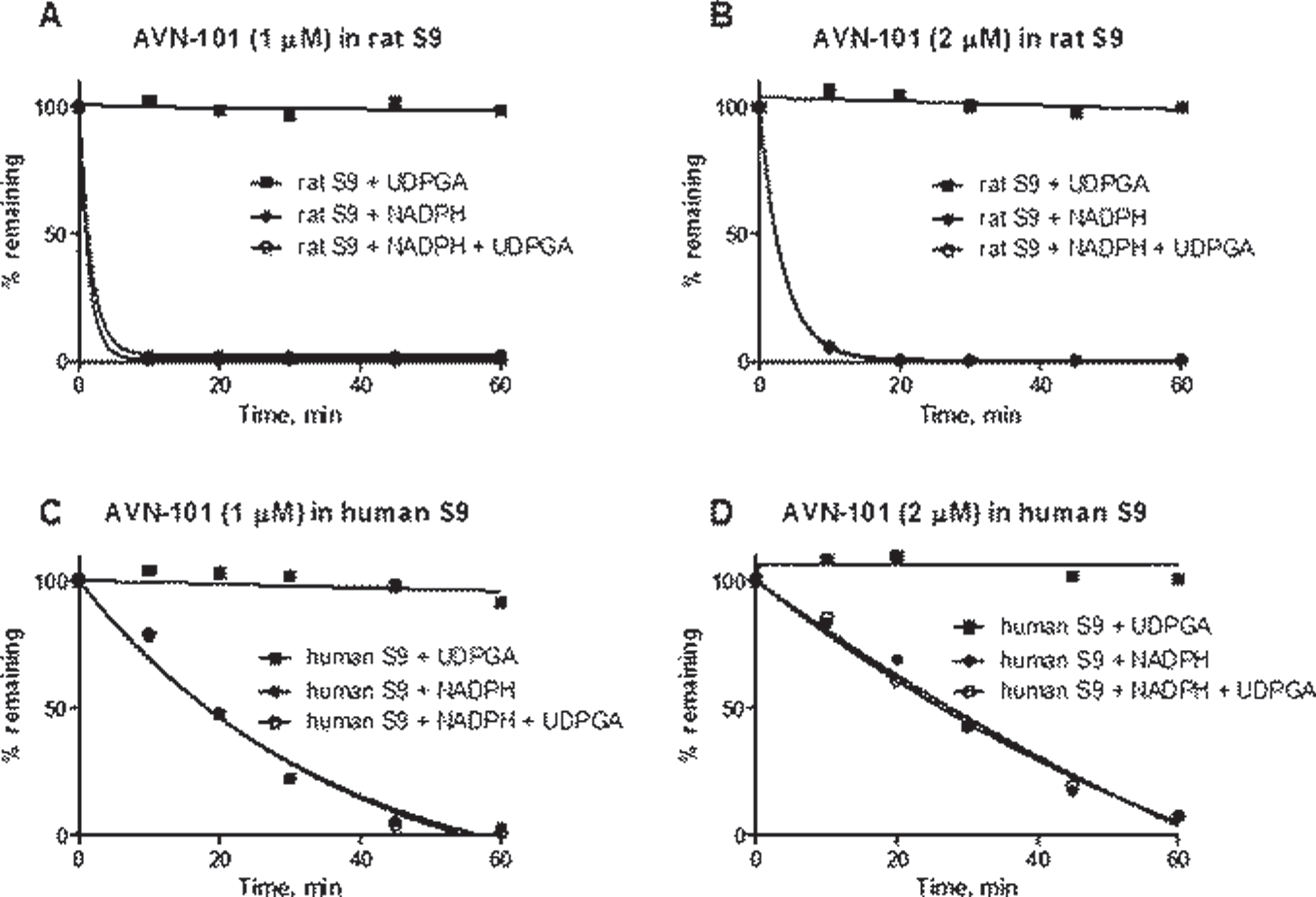
Fig.10
Pharmacokinetics of AVN-101 in mice upon IP administration at a dose of 5 mg/kg.
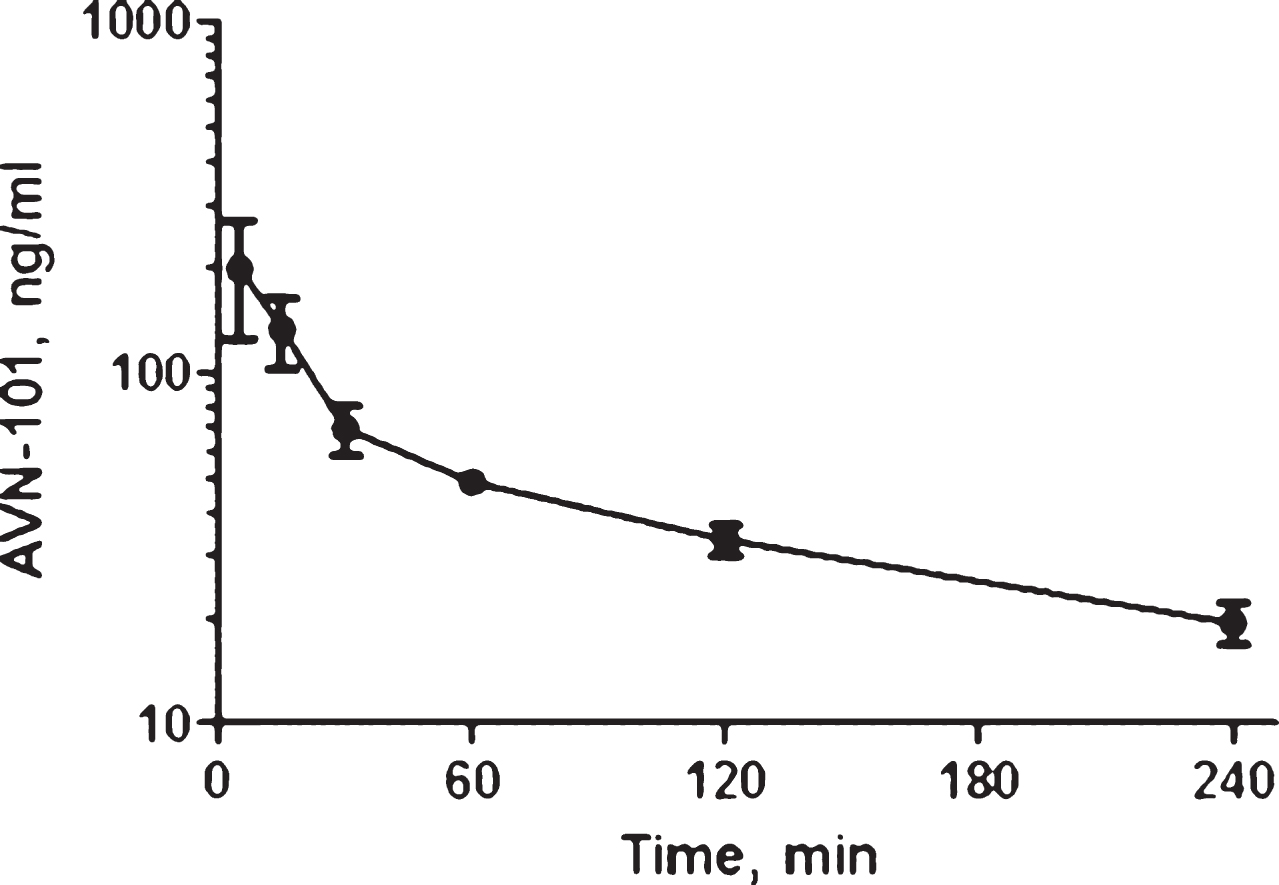
Fig.11
AVN-101 PK in mice blood upon IP administration. A) Dose dependence of maximal concentration (CMAX) and exposure (AUC0 - >∞) of the AVN-101. B) Half-life of elimination (T1/2) of the AVN-101.
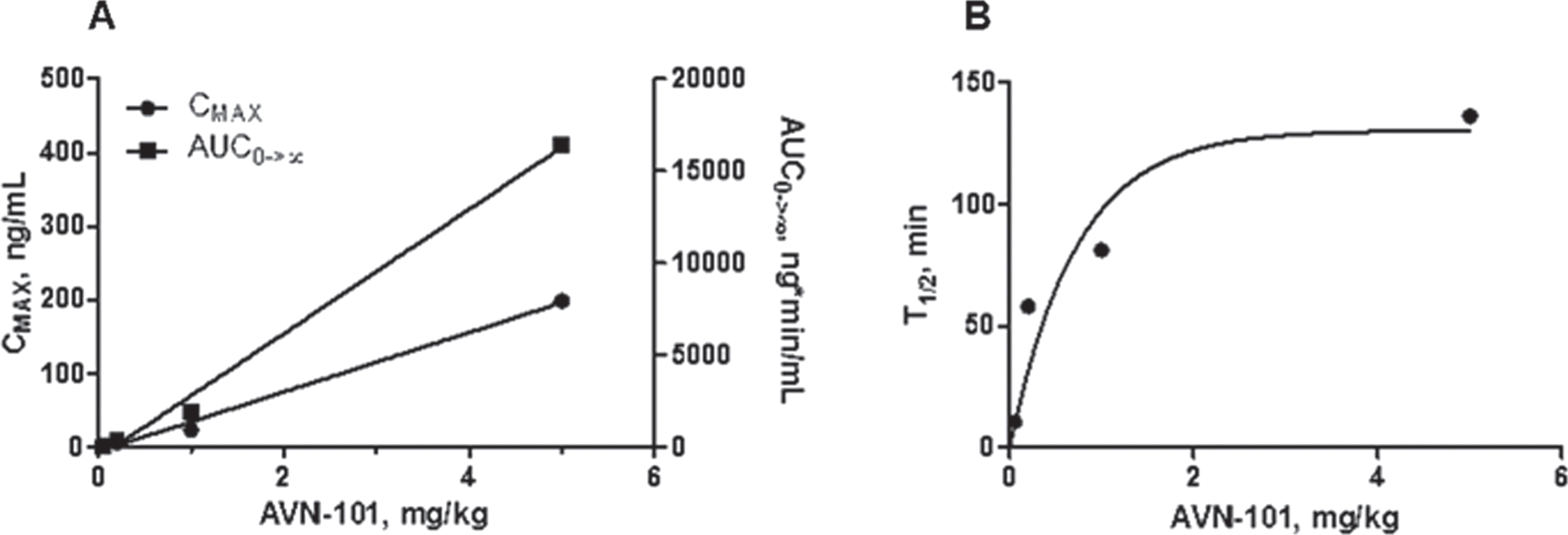
Fig.12
Pharmacokinetics of AVN-101 (dose 5 mg/kg) in Wistar rats, administered through IV, IP, and PO routes.
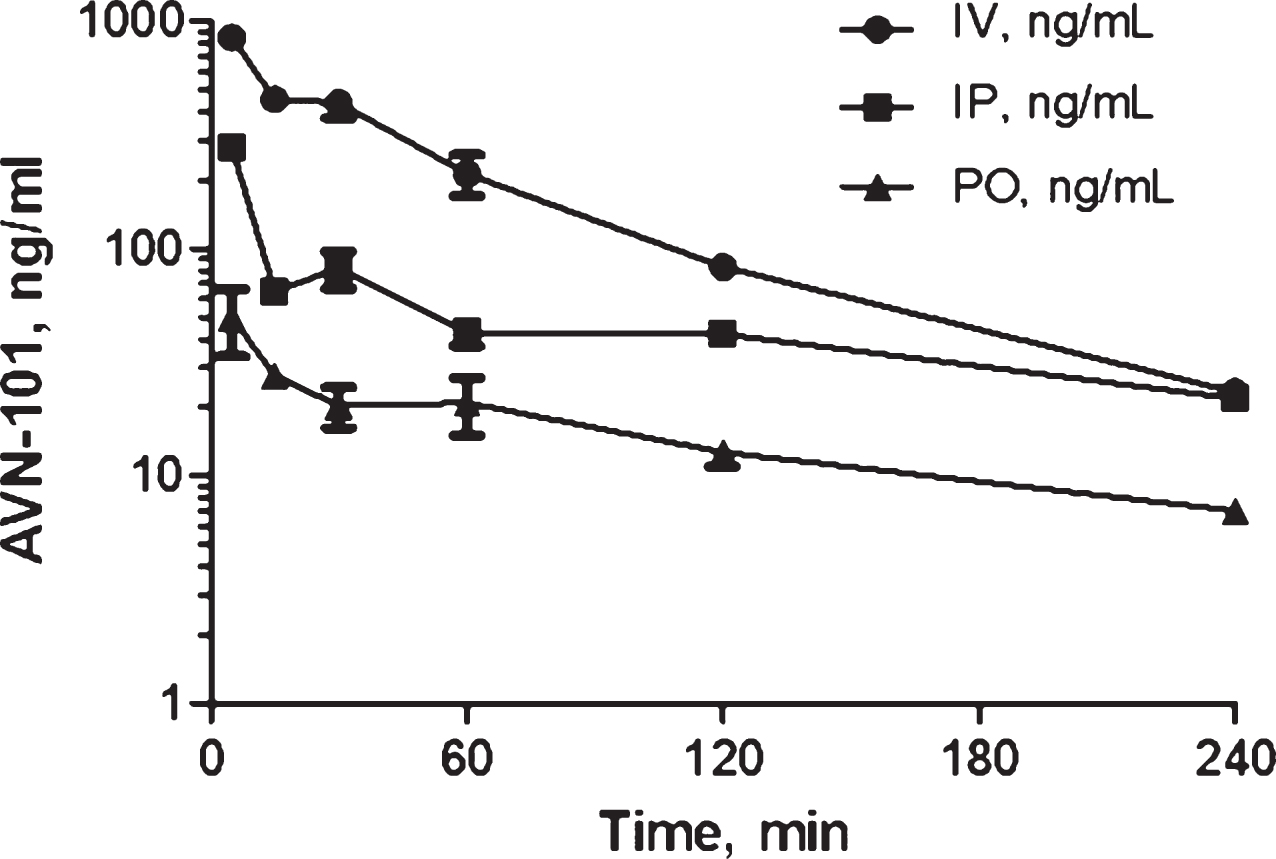
Fig.13
A) Content of AVN-101 (μg/mL) in Wistar rat tissues 4 h after either IV (5 mg/kg) or PO (10 mg/kg) administration. B) Ratio of tissue AVN-101 concentrations, IV administration over PO administration, normalized by the corresponding dose.
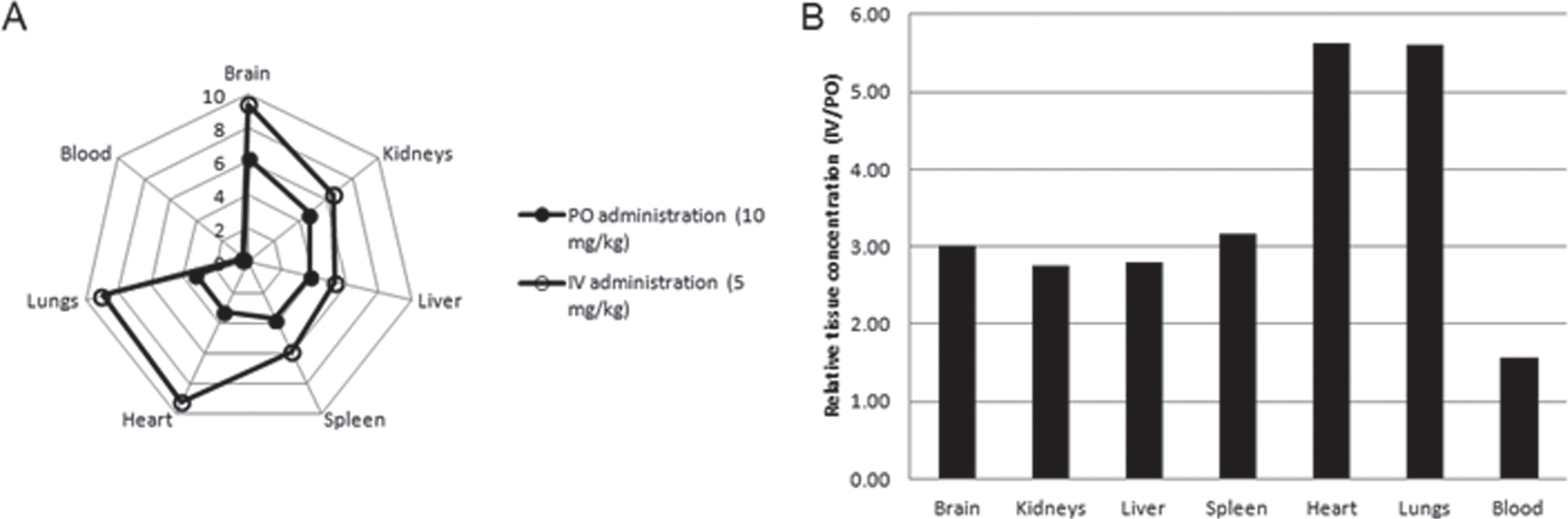
Fig.14
Pharmacokinetics of AVN-101 and its metabolites, M1 and M3, in plasma (A), brain (B), and CSF (C) of Sprague Dawley rats. AVN-101 was administered as a bolus in physiologic saline solution at a dose of 2 mg/kg through the tail vein.

Fig.15
The AVN-101 distribution in blood, plasma, and cells as well as in CSF and brain of Sprague Dawley male rats administered IV with 2 mg/kg dose of the drug. The tissue samples were collected 5 min after the drug injection.
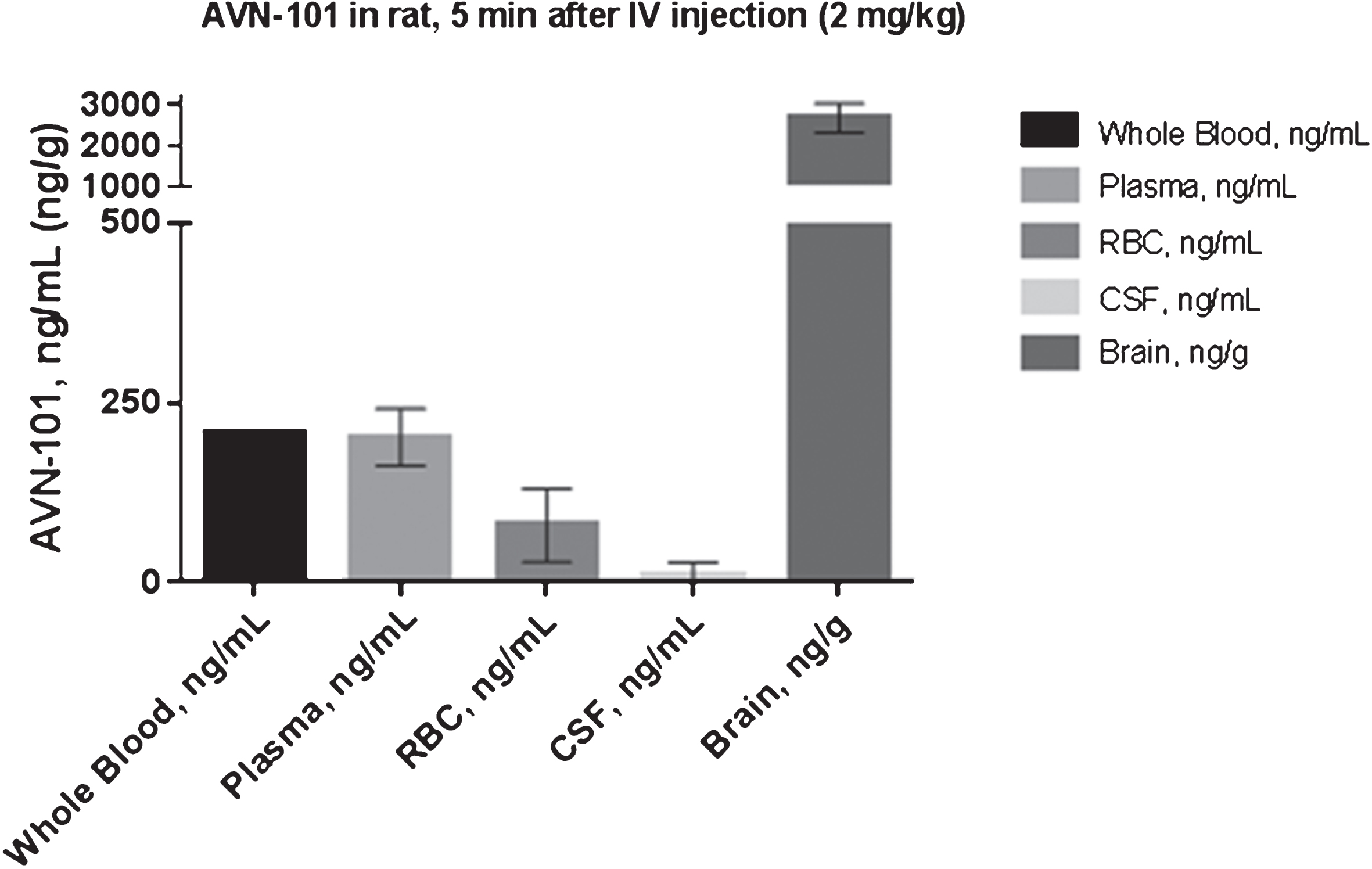
Fig.16
Kinetics of AVN-101 diffusion through semi-permeable membrane of a 48-well dialysis plate. A) Donor chamber contained diluted with PBS 20% plasma with 1μM AVN-101 and acceptor chamber contained rat brain homogenate (A), both chambers contained 20% rat plasma with 1μM AVN-101 in the donor chamber (B), and both chambers contained PBS with 1μM AVN-101 in the donor chamber (C).
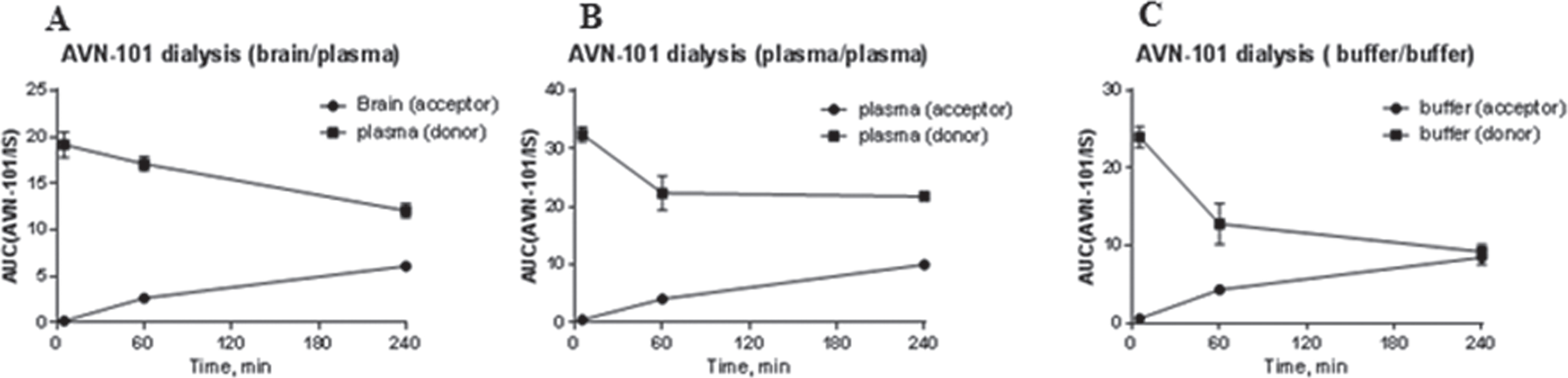
Fig.17
Ratio of time spent in open arm over the closed arm (A), ratio of number of visits into open arm over closed arm (B), number of defecations (C), and total number of visits (D) in the elevated plus-maze test. Significance (Student t-test) of difference between each of the experimental groups and placebo (control) group: *p < 0.05; **p < 0.01; ***p < 0.005; and ****p < 0.001.
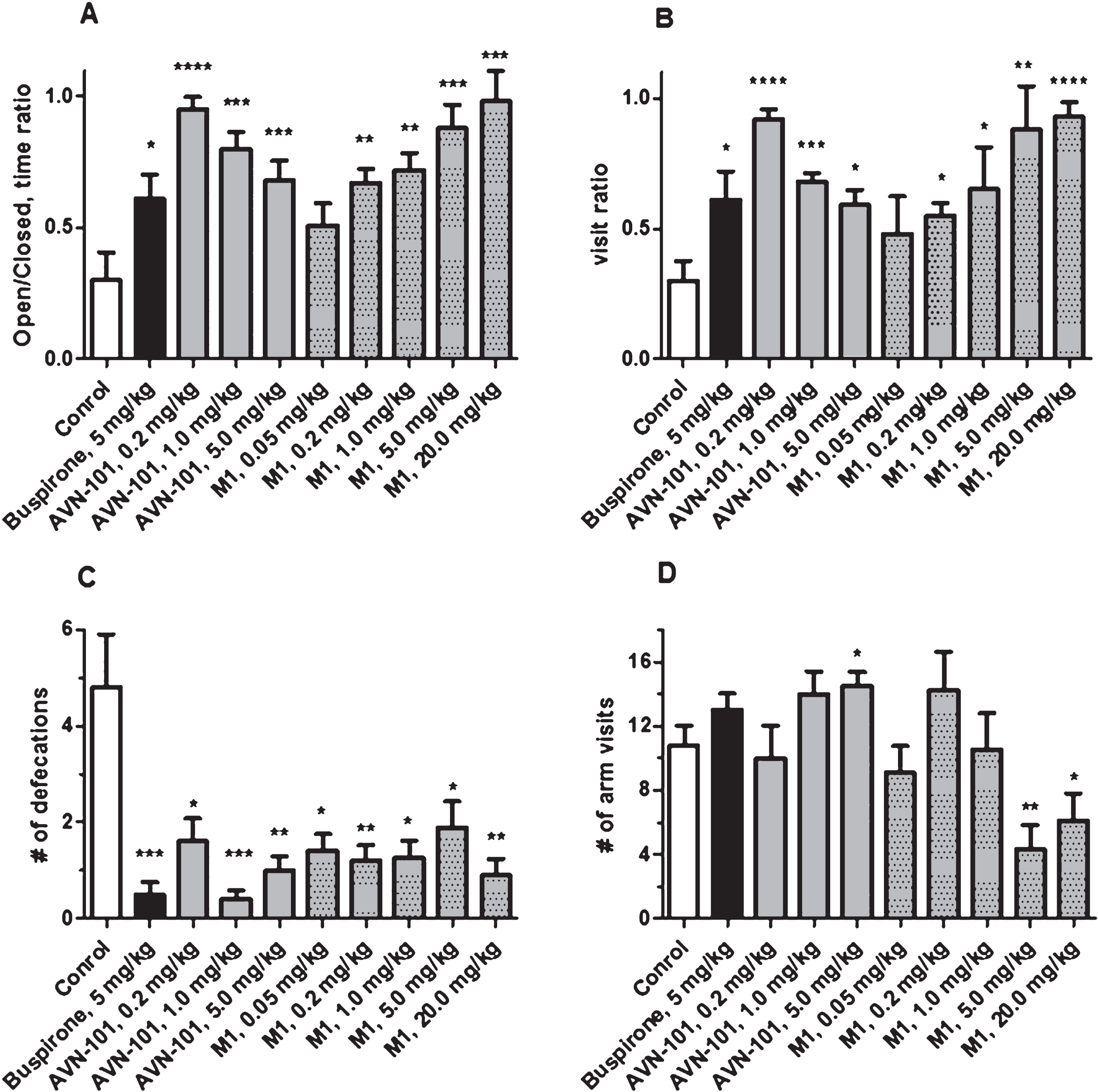
Fig.18
Ratio of time spent in open arm over the closed arm (A), ratio of number of visits into open arm over closed arm (B), number of defecations (C), and total number of visits (D) in the elevated plus-maze test. Significance (Student t-test) of differences between each of the experimental groups and placebo (control) group: *p < 0.05; **p < 0.01; ***p < 0.005.
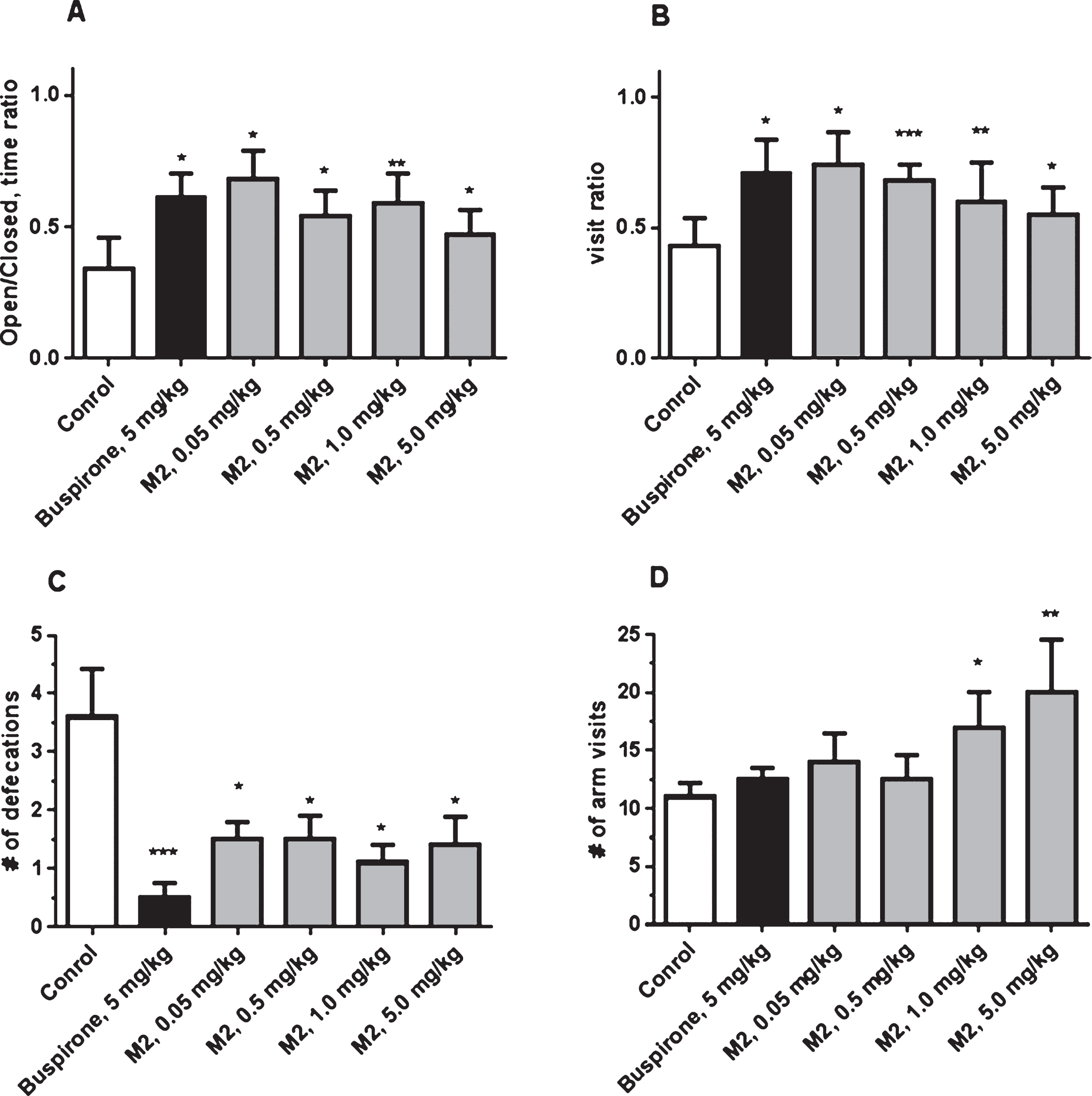
Fig.19
Cumulative time in frozen state of male BALB/c mice in the elevated platform test. Group 1 –control (saline vehicle), Group 2 - Lorazepam (0.05 mg/kg) and Group 3 - AVN-101 (0.2 mg/kg). Each group included at least 8 animals. Mean values ± SE. Difference from placebo group: *p < 0.05 (ANOVA’s Fisher LS test).
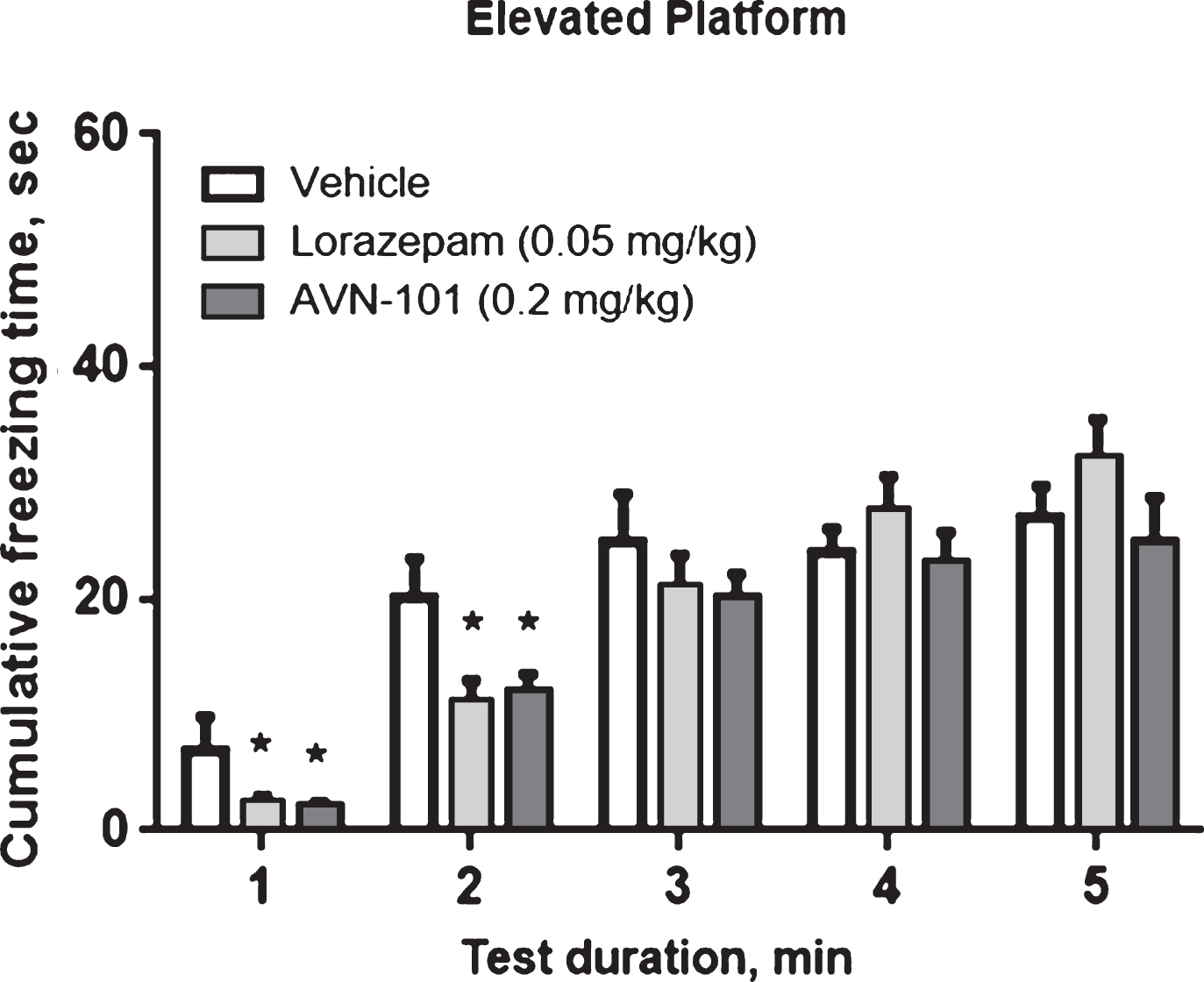
Fig.20
Time duration spent by male BALB/c mice in the central zone of open-field platform. Significance of the difference of test groups, Lorazepam and AVN-101 from control (placebo) group: *p < 0.05; **p < 0.01 (ANOVAs Fisher LS test).
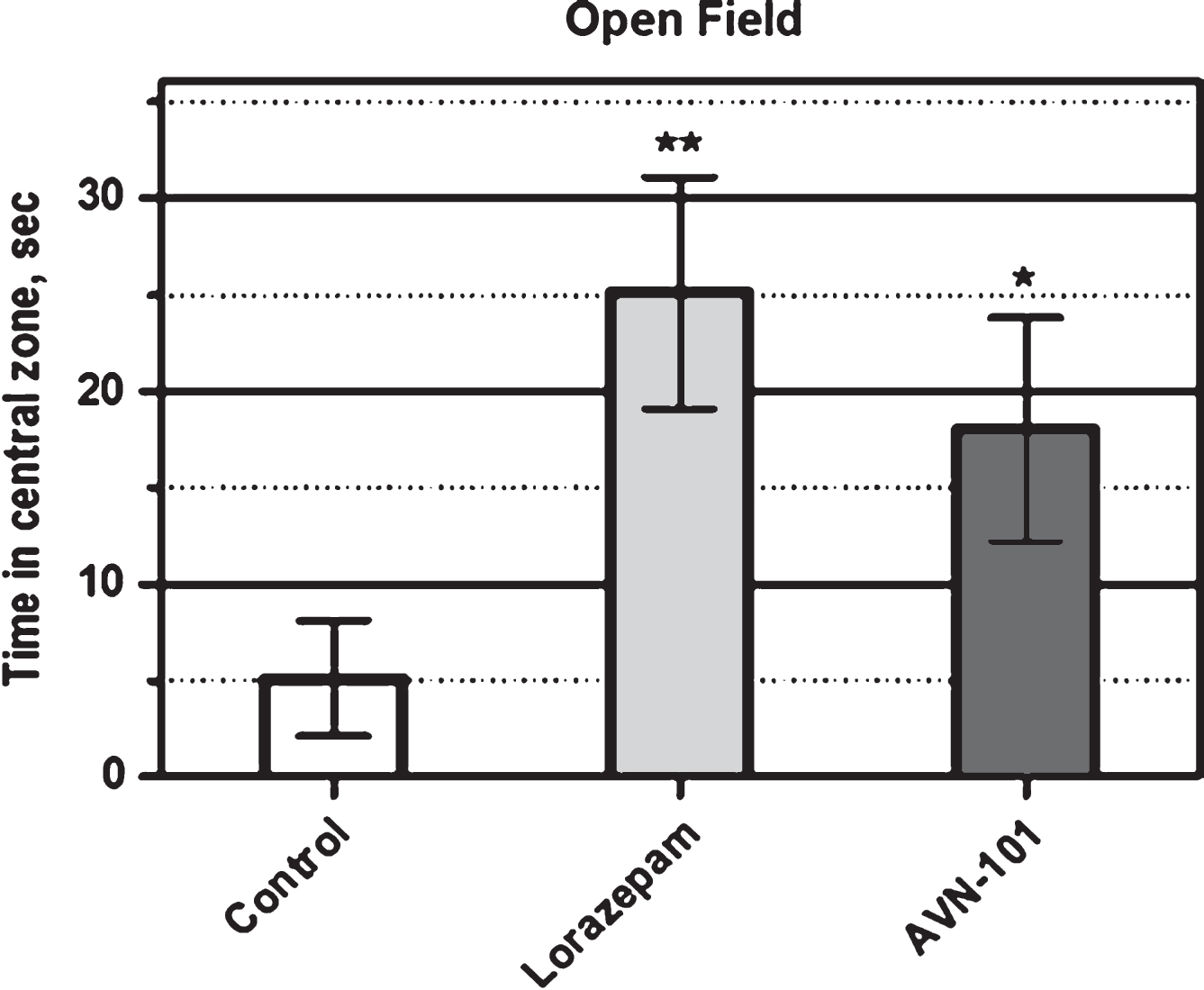
Fig.21
Relative changes of rat responses during unpunished (open bars) and punished (closed bars) sessions in the Geller-Seifter conflict test.
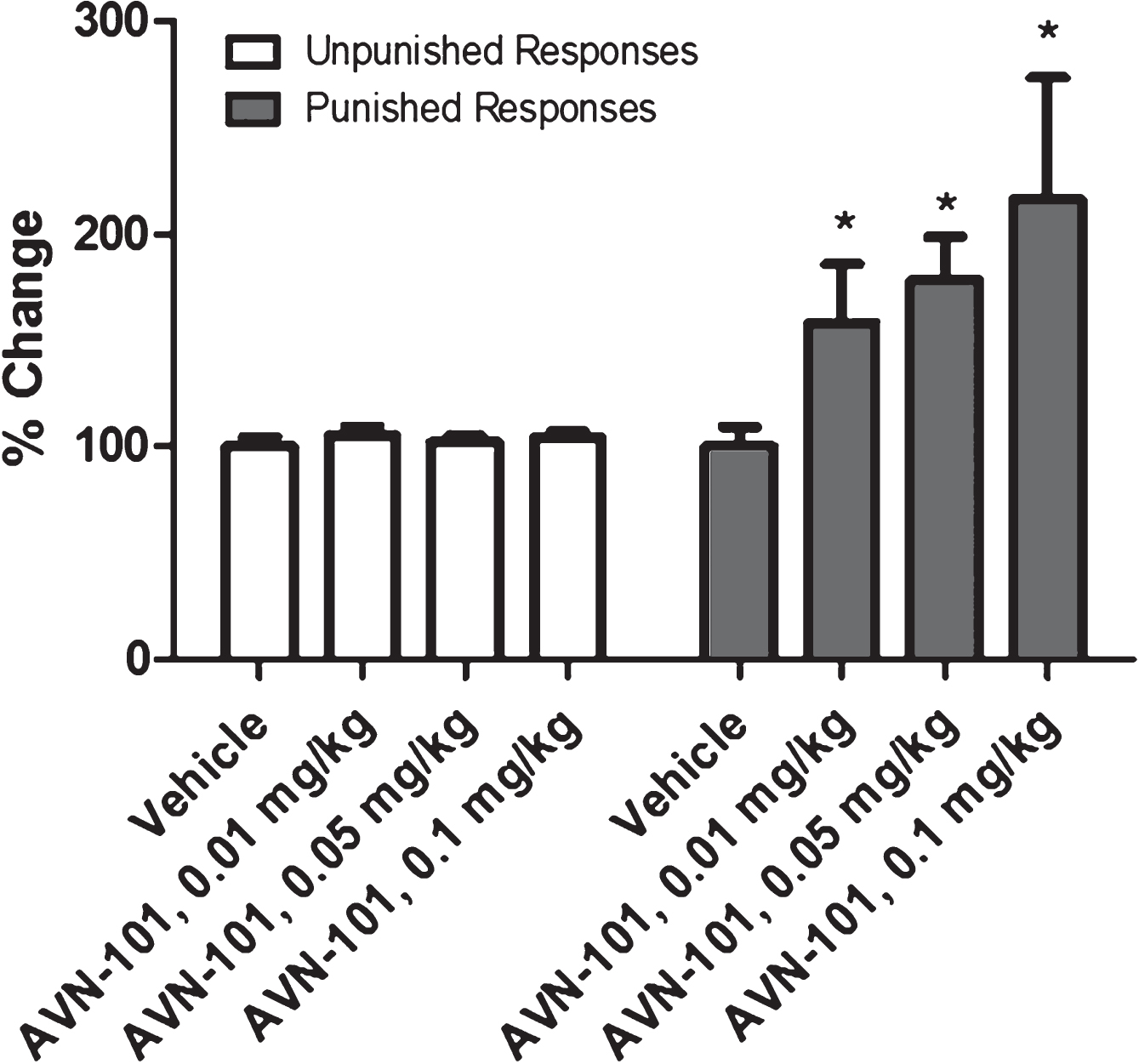
Fig.22
Effect of Fluoxetine and Desipramine (both at 15 mg/kg) and AVN-101 (0.05 mg/kg) on mice immobility in (A) the Porsolt forced swim test and (B) tail suspension test. *p < 0.05, **p < 0.01, ***p < 0.001 (Student t-test).
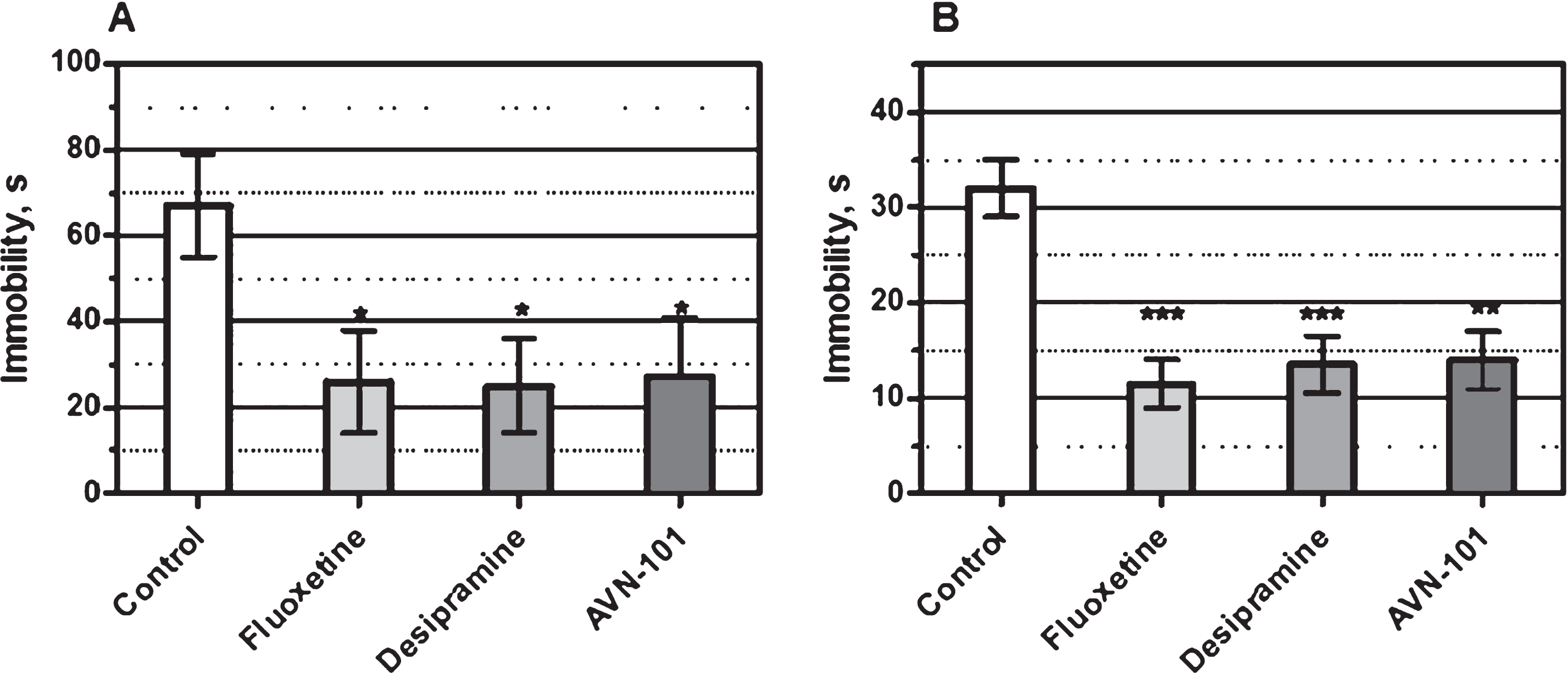
Fig.23
Latency time before the first entry of BALB/c mice into the dark box (A), duration of staying in the white compartment (B) and number of entries into the dark compartment (C) measured 24 h after the electric shock was given (mean ± SEM). Difference from scopolamine-treated placebo group: *p < 0.05; **p < 0.01; ***p < 0.001 (Student t-test).
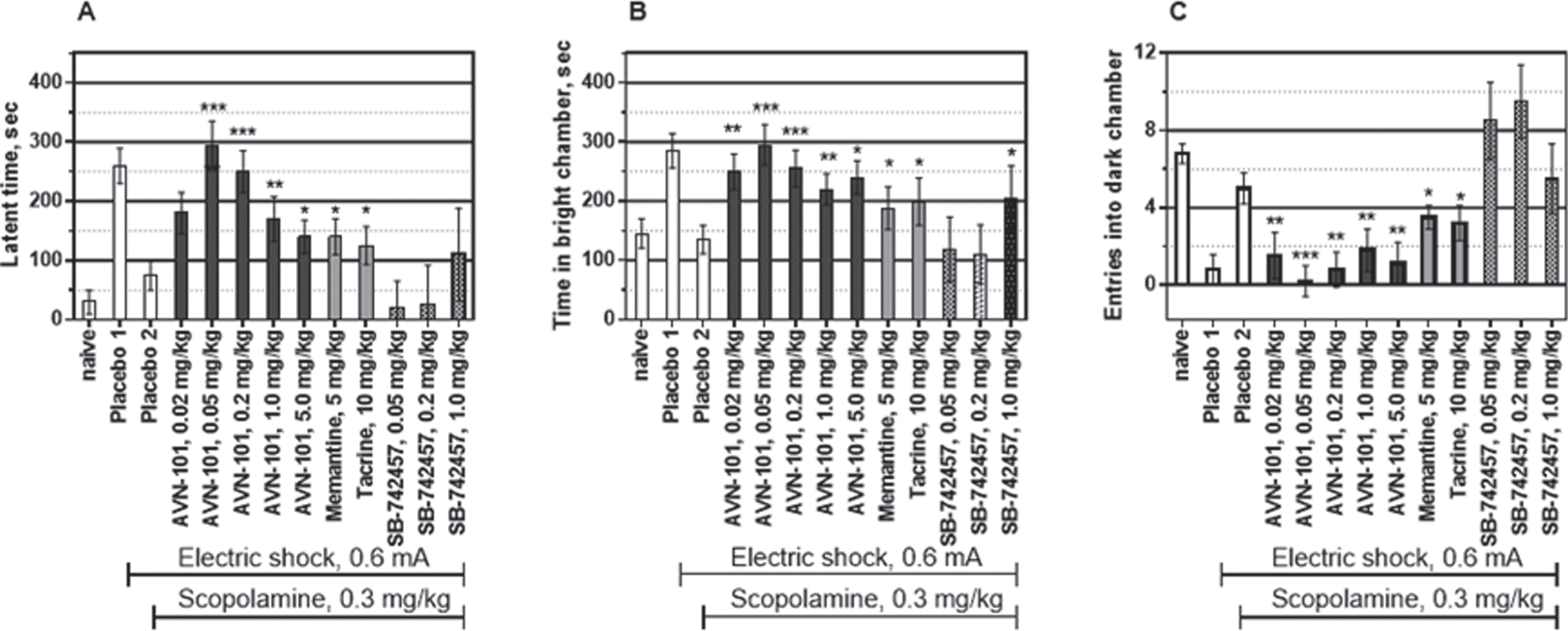
Fig.24
Latency time before the first entry of Spraque Dawley rats into the dark box (A), duration of staying in the white compartment (B), and number of entries into the dark compartment (C) measured 24 h after the electric shock was given (mean ± SEM). Difference from scopolamine-treated placebo group: *p < 0.05; **p < 0.01; (Student t-test).
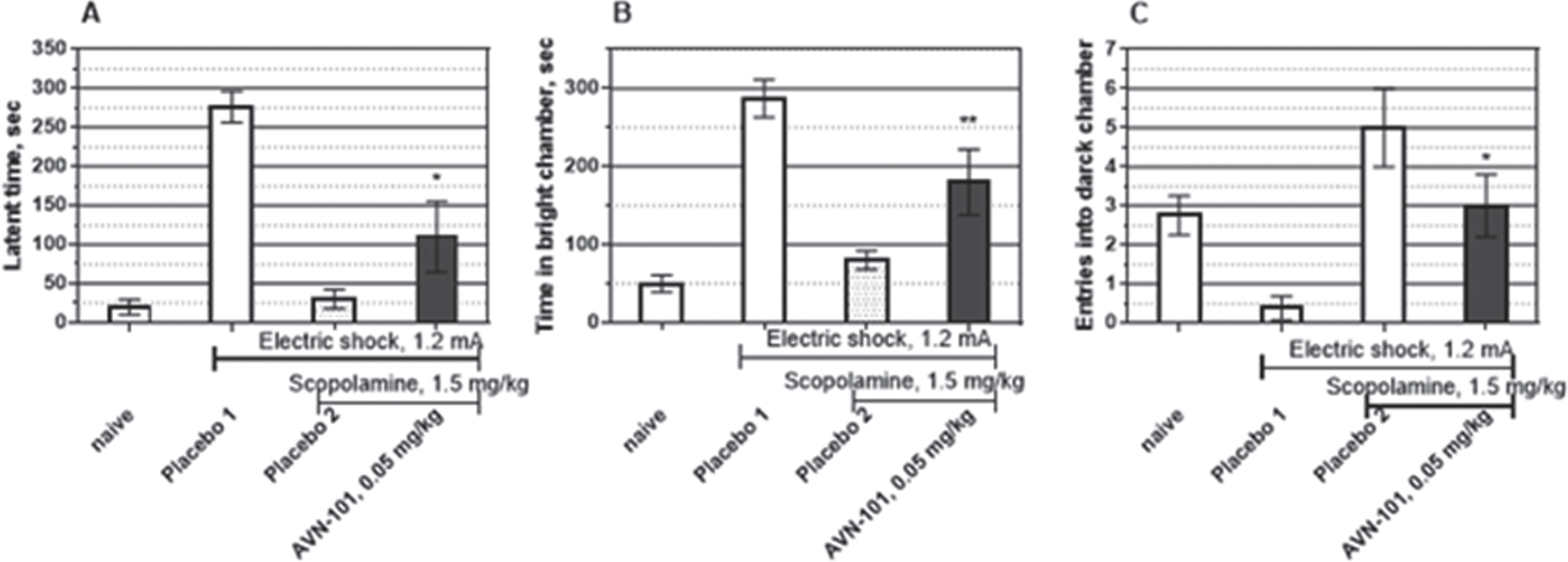
Fig.25
Latency time for the first entry into the dark box (A), duration of staying in the white compartment (B) and number of entries into the dark compartment (C) measured 24 h after the electric shock was given (mean ± SEM). Difference from MK-801-treated placebo group (Placebo 2): *p < 0.05 (Student t-test).
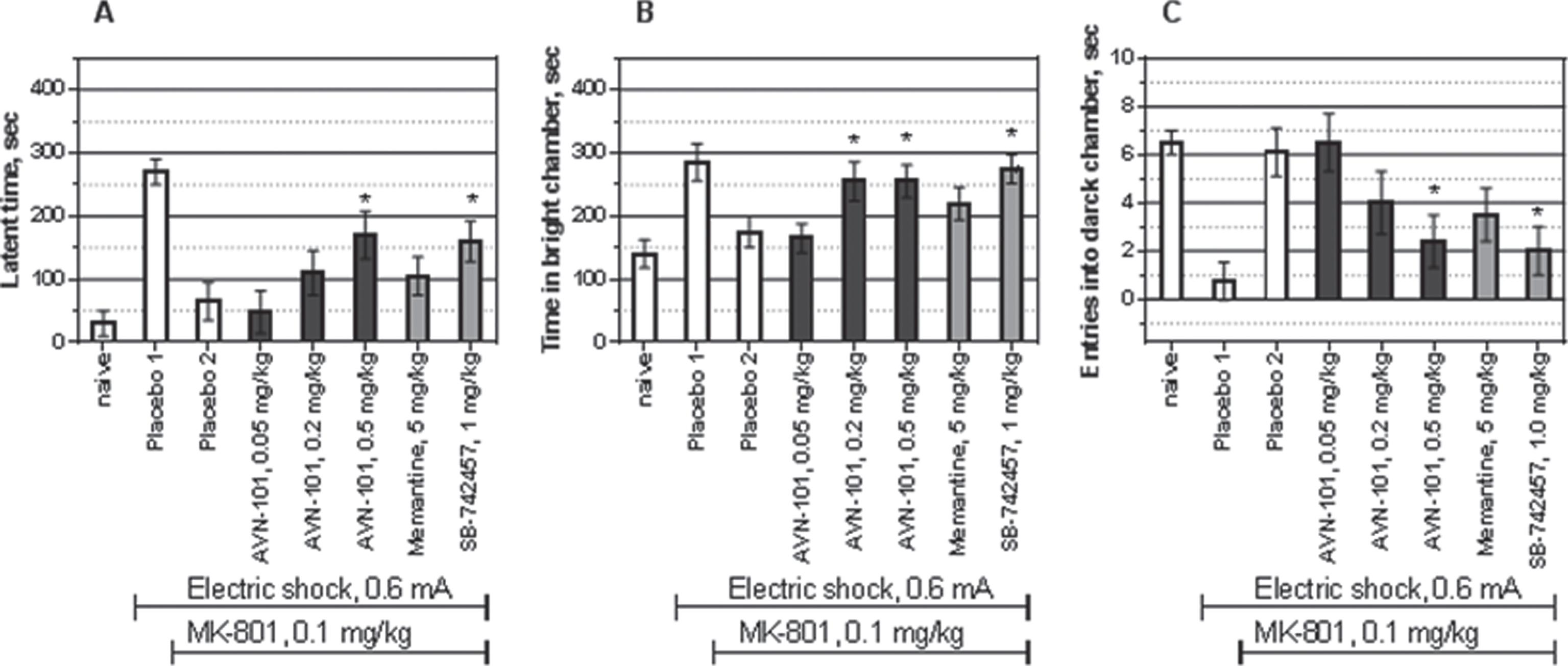
Fig.26
The Morris water maze test for reversal of scopolamine-induced amnesia in male BALB/c mice. Ordinate axis: the time (mean ± SE) spent in the quadrant where the “safe” platform was located during two previous training days. Difference from placebo 2 group: *p < 0.05; **p < 0.01 (Student t-test).
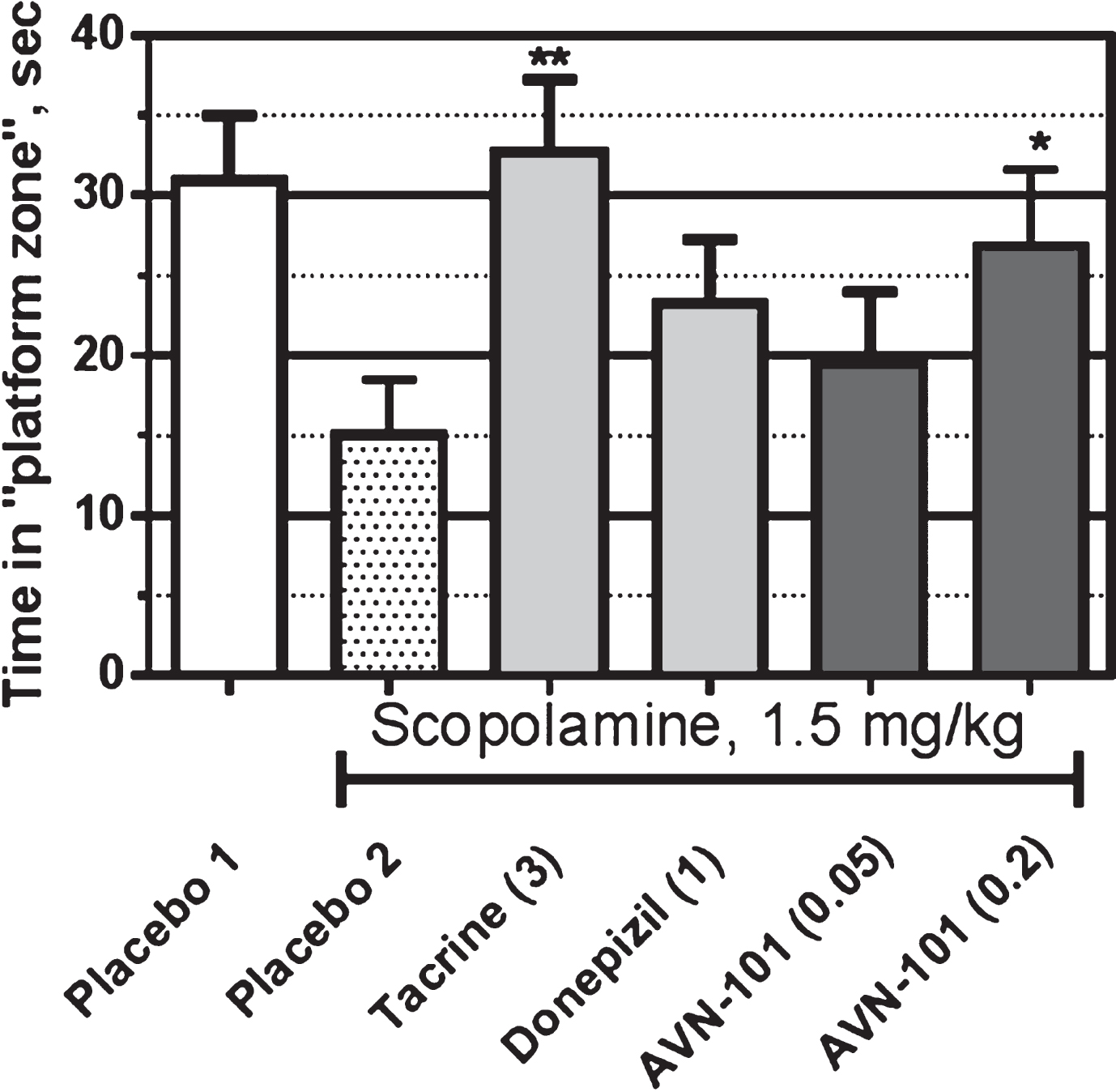
Fig.27
Effect of the AVN-101 and positive control drugs, memantine and SB-742457, on novel object recognition index in male BALB/c mice (mean ± SE) upon scopolamine (A) or NK-801-induced amnesia (B). Difference from group administered with scopolamine (Placebo 2): *p < 0.05; **p < 0.01 (Student test).
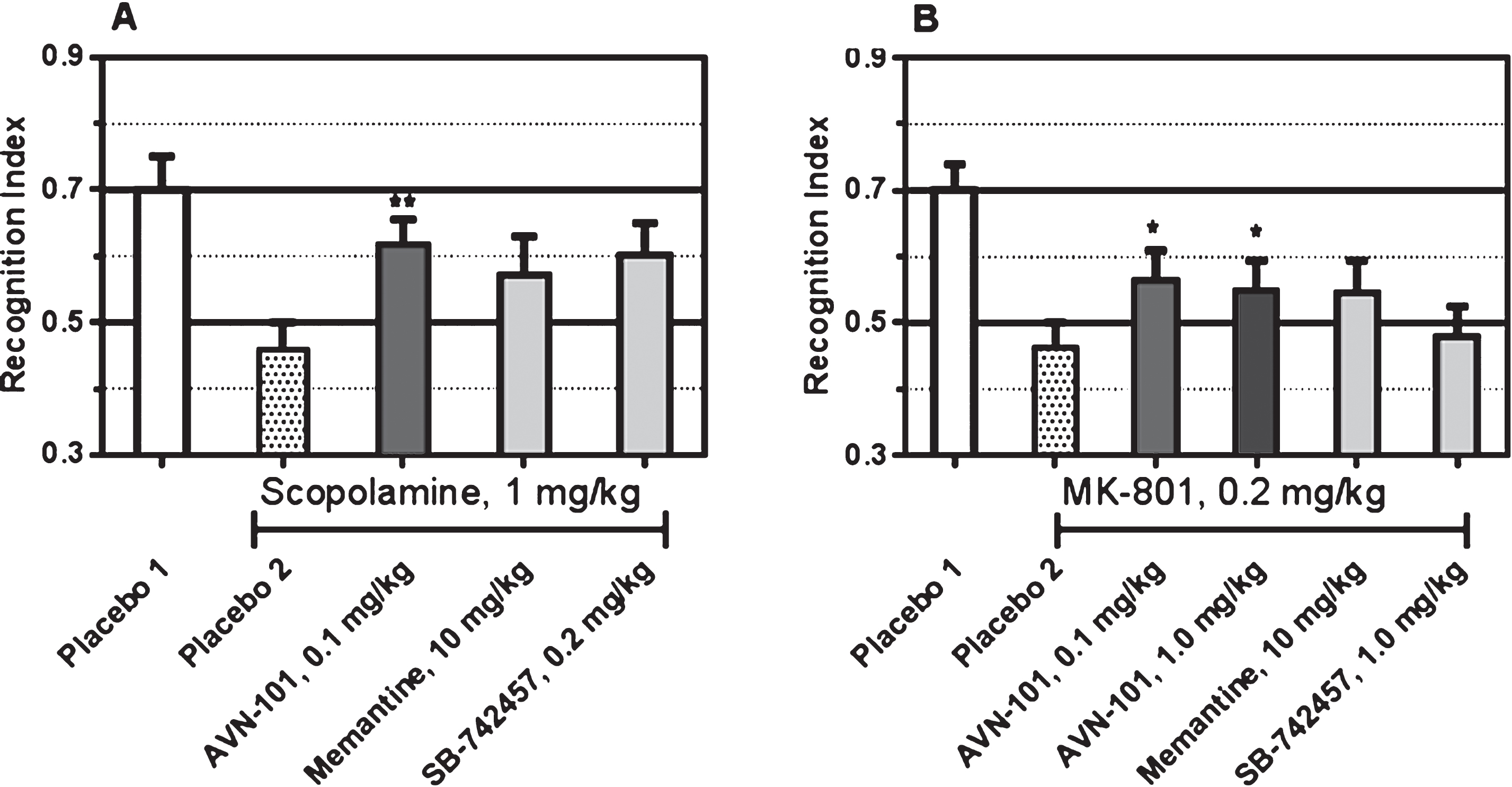
Fig.28
Effect of AVN-101 and Haloperidol on apomorphine-induced disruption of pre-pulse inhibition in SHK male mice. Difference from group administered with Apomorphine (Placebo 2): *p < 0.05; **p < 0.01 (Student test).
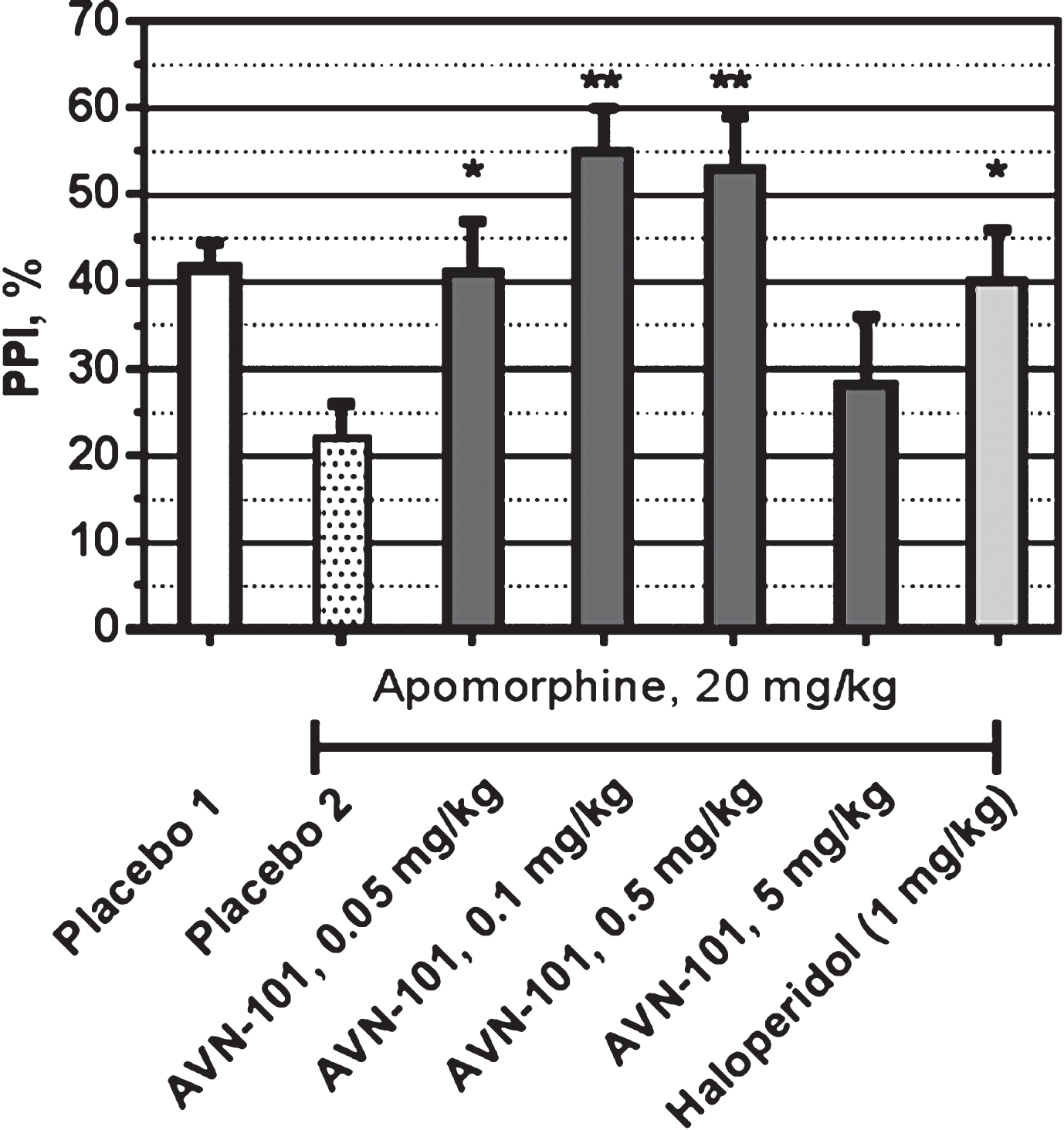
Fig.29
Q-T prolongation in guinea pigs by AVN-101 and positive control amiodaron.
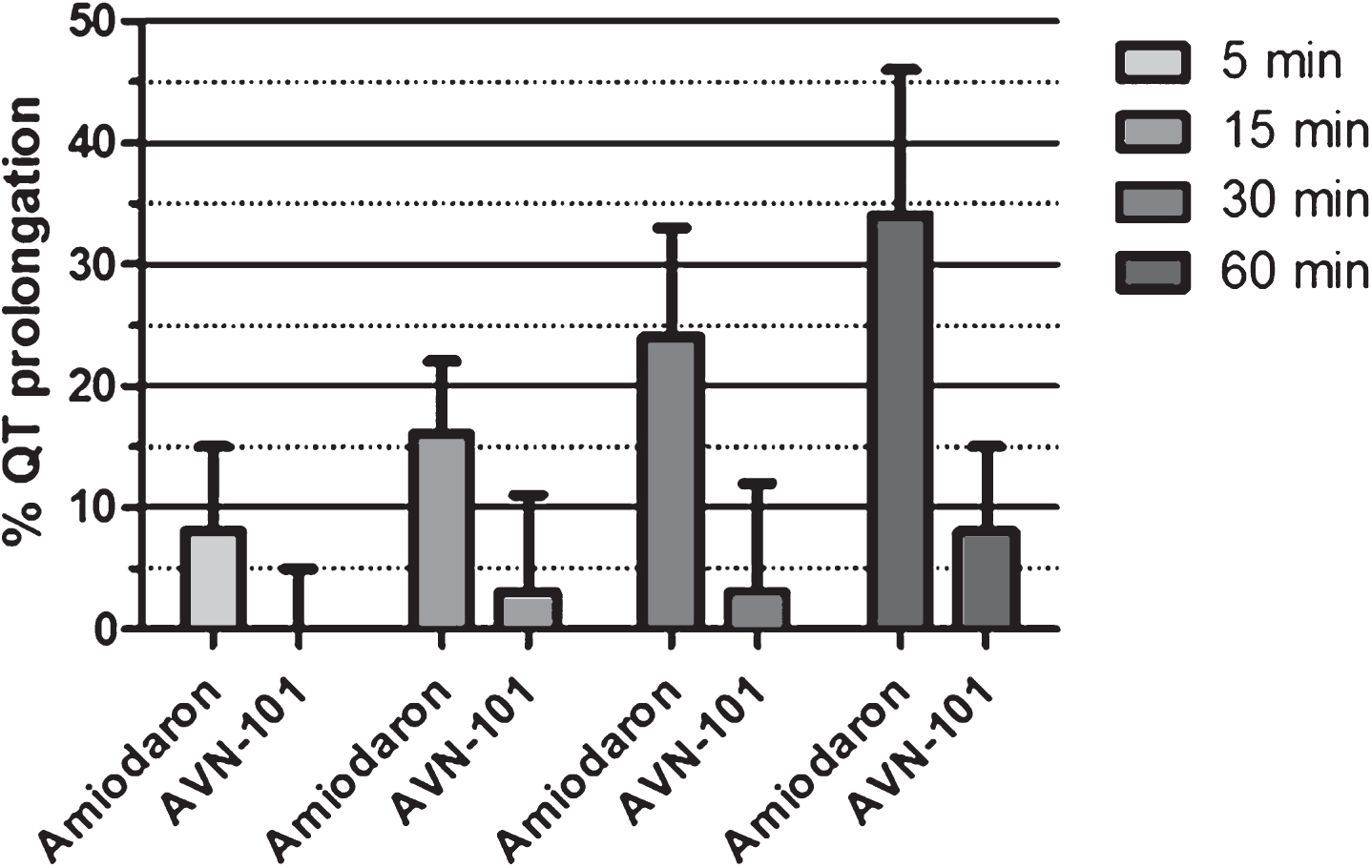
Fig.30
QT interval in Rhesus Macaca mulatta upon IV administration of positive control drug amiodaron (15 mg/kg) or AVN-101 (1 mg/kg).
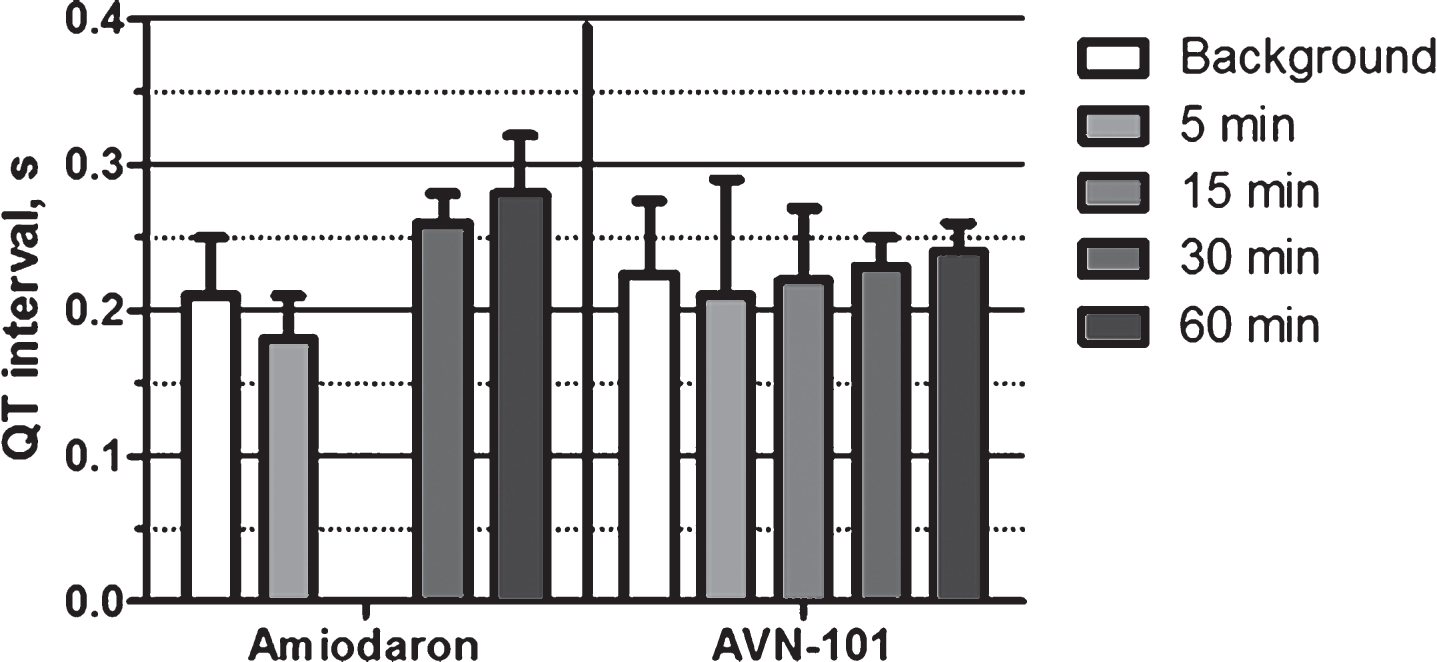
Fig.31
Effect of AVN-101 and control substances on reverse mutation rate (Ames test) of Salmonella typhimurium strains TA98 (light gray), TA1535 (dark gray), and TA1537 (black). Results for the highest AVN-101 concentration tested, 50μM, are shown. Positive controls for TA98 (4-NOPD), TA1535 (Na-azide), and TA1537 (9-AA), were tested at respective concentrations of 15μg/mL, 2μg/mL, and 50μg/mL. Effects of the compounds are presented as a fold increase over negative control, non-treated bacteria cultures (mean ± SE). Dotted line shows negative control level and dashed line represents the 3-fold threshold that is considered as a significant level for mutagenic effect.
Fig.32
Kinetics of AVN-101 and its metabolites, M1 and M2, in plasma of human healthy volunteers after single AVN-101 PO administration at different doses.
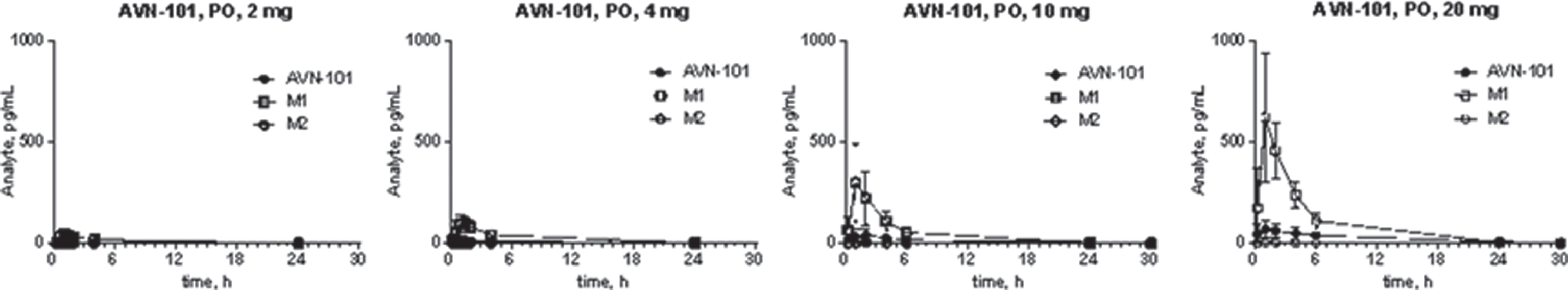
Fig.33
Relation between an AVN-101 dose upon PO administration and blood concentration of AVN-101 and its two metabolites, M1 and M2, in humans. A) Peak concentration, Cmax (mean ± SD). B) Exposure, AUC (mean ± SD).
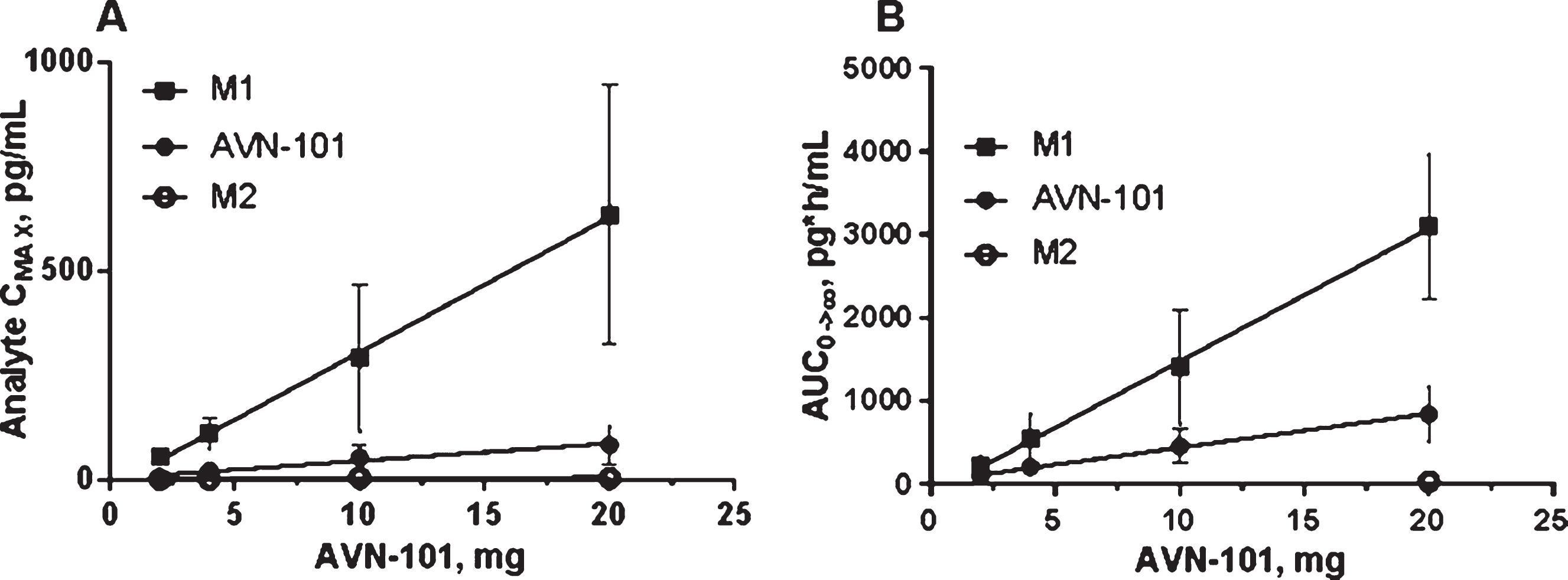
Table 1
Physicochemical characteristics of AVN-101
| Parameter | Value |
| MW | 304.439 |
| Log P (pH 4) | 2.47 |
| Log P (pH 9.5) | 5.25 |
| Log Sw | 5.94 |
| N+O | 2 |
| H Acceptors | 2 |
| H Donors | 0 |
| B rotN | 3 |
| PSA | 8.17 |
| Solubility (H2O), pH7.4 | 194 mg/mL |
| Solubility (H2O), pH 4.0 | >200 mg/mL |
Table 2
Comparative selectivity profiles for AVN-101 and Lu AE8054 against serotonin receptors
| Receptor | AVN-101 Ki, nM [13] | Lu AE58054 Ki, nM [73] |
| 5-HT1A | 61 | 2300 |
| 5-HT1B | 720 | >10,000 |
| 5-HT2A | 1.56 | 83 |
| 5-HT2B | 10.6 | >4100 |
| 5-HT2C | 1.17 | 250 |
| 5-HT6 | 2.04 | 0.83 |
| 5-HT7 | 0.153 | >10000 |
Table 3
Affinity of AVN-101 to bind to adrenergic receptors measured in radio ligand competition assay
| Target | Ki, nM | Relative Selectivitya |
| Adrenergic α2B | 0.41 | 1 |
| Adrenergic α2A | 1.77 | 4.3 |
| Adrenergic α2C | 3.55 | 8.7 |
| Adrenergic α1B | 9.4 | 22.9 |
| Adrenergic α1A | 18.9 | 46.1 |
| Adrenergic α1D | 30.2 | 73.7 |
aKi for Adrenergic α2B is equated to 1.
Table 4
Permeability of AVN-101 (200μM) through CACO-2 cell monolayer and Rhodamine123 (30μM) alone and of Rhodamine123 in the presence of either pgP inhibitor Verapamil (100μM) or AVN-101 (100μM)
| Compound | Papp×106 cm/s | B-A/A-B ratio | |
| A⟶B | B⟶A | ||
| AVN-101 (200μM) | 19 ± 1.8 | 21 ± 2.1 | 1.1 |
| Rhodamine123 (30μM)a | 0.6 ± 0.2 | 6.8 ± 0.4 | 11.0 |
| Rhodamine123+Verapamilb | 0.6 ± 0.2 | 1.0 ± 0.2 | 1.7 |
| Rhodamine123+AVN-101 | 0.6 ± 0.1 | 1.2 ± 0.3 | 2.0 |
aRhodamine123 was used as Pgp pump substrate. bVerapamil (100μM) was used as Pgp pump inhibitor.
Table 5
Main kinetics parameters of the AVN-101 degradation by rat and human microsomes
| Species | t1/2 (min) | kel | Clint (μl/min/mg protein) |
| rat | 1.97 | 0.352 | 703 |
| human | 2.07 | 0.335 | 335 |
Table 6
Relative amounts of the original compound, AVN-101, and its metabolites in suspensions of human and rat microsomes, determined by the indicated incubation time
| Analyte | AVN-101, μM | microsomes | Incubation | Remaining |
| time | quantity, % | |||
| AVN-101 | 1 | HLM | 7.9 min | 3.0 |
| AVN-101 | 1 | HLM | 30 min | 2.0 |
| AVN-101 | 2 | HLM | 7.9 min | 40.0 |
| AVN-101 | 2 | HLM | 30 min | 43.0 |
| AVN-101 | 1 | RLM | 7.9 min | <1 |
| AVN-101 | 1 | RLM | 30 min | <1 |
| M1 | 1 | HLM | 6.2 min | <1 |
| M1 | 2 | HLM | 6.2 min | 30 |
| M2 | 1 | HLM | 7.0 min | <1 |
| M2 | 2 | HLM | 7.0 min | 10 |
| M3 | 1 | HLM | 83 | |
| M3 | 2 | HLM | 6 | |
| M3 | 1 | RLM | 6 | |
| M4 | 1 | HLM | 7.8 min | 9 |
| M4 | 2 | HLM | 7.8 min | 5 |
| M5 | 1 | HLM | 3 | |
| M5 | 2 | HLM | 1 | |
| M6 | 1 | HLM | 7.8 min | 2 |
| M6 | 2 | HLM | 7.8 min | 2 |
| M7 | 1 | RLM | 28 | |
| M8 | 1 | HLM | 5.2 min | <1 |
| M8 | 2 | HLM | 5.2 min | <1 |
| M8 | 1 | RLM | 5.2 min | 30.0 |
| M9 | 1 | HLM | 6.9 min | <1 |
| M9 | 2 | HLM | 6.9 min | 2.0 |
| M10 | 1 | HLM | 6.8 min | <1 |
| M10 | 2 | HLM | 6.8 min | 3 |
| Others | 2 | HLM | 1.5 | |
| Others | 1 | RLM | 33 |
Table 7
Metabolism of AVN-101 with human recombinant cytochromes
| Enzyme | AVN-101 (2μM), |
| % remaining | |
| HLM | 43 |
| CYP1A2 | 46 |
| CYP2C8 | 118 |
| CYP2C9 | 110 |
| CYP2C19 | 1 |
| CYP2D6 | 0 |
| CYP3A4 | 2 |
Table 8
Half-life of AVN-101 metabolism with rat or human S9 fractions in the presence of phase 1 metabolism co-factor, NADPH, and phase 2 metabolism co-factor, UDPGA
| S9 fraction | +NADPH | +NADPH+UDPGA | ||
| 1μM | 2μM | 1μM | 2μM | |
| rat S9, T1/2 min | 1.22 | 2.36 | 0.96 | 2.34 |
| human S9, T1/2 min | 24.8 | 87.1 | 26.2 | 58.9 |
Table 9
AVN-101 main pharmacokinetic parameters in mice upon IP bolus administration
| AVN-101, mg/kg | Cmax, ng/ml | Tmax, min | AUC0 - >∞ ng*min/ml | AUC0 - >t, ng*min/ml | T1/2, min |
| 5.0 | 198 | 5 | 16438 | 11049 | 136 |
| 1.0 | 23 | 15 | 1921 | 1552 | 81 |
| 0.2 | 5 | 15 | 365 | 339 | 58 |
| 0.05 | 1.9 | 5 | 29.9 | 29.2 | 10.5 |
Table 10
AVN-101 PK parameters in Wistar rats with LC-MS/MS detection of the original substance
| Route | Dose, mg/kg | Tmax, min | Cmax, ng/mL | AUC0 ⟶240, ng*min/mL | Fabs, % | AUC0 ⟶∞, ng*min/mL | T1/2, min | Kel, min–1 |
| IV | 5 | 5 | 846.7 | 43269 | 452049 | 57.5 | 0.0121 | |
| IP | 5 | 5 | 278.3 | 113829 | 26.3 | 169899 | 176.6 | 0.0039 |
| PO | 5 | 5 | 50 | 36839 | 8.5 | 48629 | 116.7 | 0.0059 |
Table 11
AVN-101 concentrations in rat liver, heart, lungs, spleen, kidneys, brain, and blood 4 h after either PO (10 mg/kg) or IV (5 mg/kg) administration
| Organs | AVN-101, μg/ml | IV/PO, normalized* | Organ bioavailability, % | |
| PO administration | IV administration | |||
| Brain | 6.24 ± 0.24 | 9.44 ± 0.36 | 3.03 | 33.0 |
| Kidneys | 4.70 ± 0.22 | 6.50 ± 0.20 | 2.77 | 36.2 |
| Liver | 3.78 ± 0.13 | 5.30 ± 0.16 | 2.80 | 35.6 |
| Spleen | 3.74 ± 0.15 | 5.94 ± 0.15 | 3.18 | 31.5 |
| Heart | 3.28 ± 0.15 | 9.24 ± 0.23 | 5.63 | 17.7 |
| Lungs | 3.20 ± 0.23 | 8.96 ± 0.11 | 5.60 | 17.9 |
| Blood | 0.46 ± 0.01 | 0.36 ± 0.01 | 1.57 | 63.9 |
*AVN-101 organ concentrations were normalized by corresponding dose.
Table 12
AVN-101 PK parameters in blood plasma, brain tissue, and cerebrospinal fluid (CSF) of Sprague Dawley rats upon IV administration at a dose of 2 mg/mL
| PK Parameter | Units | Plasma | Brain | CSF |
| k el | Min–1 | 0.0048 | 0.0092 | 0.0071 |
| T1/2 | min | 143.42 | 75.66 | 97.490 |
| Tmax | h | 0.083 | 0.08 | |
| Cmax | ng/ml | 274 | 4665 | 11.0 |
| C0 | ng/ml | 308 | ||
| AUClast | min*ng/ml | 22589 | 480129 | 1520.4 |
| AUCINF | min*ng/ml | 31296 | 537361 | 1904.9 |
| Vz | mL/kg | 13222.7 | ||
| Cl | ml/min/kg | 63.905 | ||
| Vss_obs | mL/kg | 11613.6 |
Table 13
PK parameters of AVN-101 and its metabolites, M1 and M2, in Dogs (LC-MS/MS method)
| Administration | Substance analyzed | T1/2, min | Tmax, min | Cmax, ng/mL | AUC0 - >∞, min×ng/mL. | Fabs, % | ||
| Compound | Route | Dose, mg/kg | ||||||
| AVN-101 | IV | 2 | AVN-101 | 29.8 | 1 | 3,740 | 29,869 | 4.6 |
| AVN-101 | PO | 10 | AVN-101 | 366 | 60 | 23 | 6,866 | |
| AVN-101 | IV | 2 | M1 | 51.4 | 5 | 5.7 | 264.6 | |
| AVN-101 | PO | 10 | M1 | 154.9 | 60 | 30.1 | 8,995 | |
| AVN-101 | IV | 2 | M2 | Below detection limit | ||||
| AVN-101 | PO | 10 | M2 | 168.7 | 60 | 0.23 | 39.1 | |
| M1 | IV | 1 | M1 | 57.5 | 1 | 4,550 | 25,894 | 8.4 |
| M1 | PO | 5 | M1 | 138.5 | 120 | 43 | 10,828 | |
| M1 | IV | 1 | AVN-101 | 20.9 | 1 | 13.7 | 137.5 | |
| M1 | PO | 5 | AVN-101 | – | 240 | 9.1 | 1,896 | |
| M1 | IV | 1 | M2 | Below detection limit | ||||
| M1 | PO | 10 | M2 | Below detection limit | ||||
Table 14
AVN-101 PK parameters in Rhesus monkeys
| Monkey # | Route of | Dose, | Tmax, | Cmax, | T1/2, | Kel, | AUC0 ⟶t, | AUC0 ⟶∞, | Fabs, % |
| Admin. | mg/kg | min | ng/ml | min | min–1 | ng×min/ml | ng×min/ml | ||
| 34887 | IV | 2 | 1 | 304 | 33 | 0.0208 | 21,375.5 | 23,298 | |
| 35188 | IV | 2 | 1 | 345 | 162 | 0.0043 | 29,733.2 | 71,708 | |
| 34920 | PO | 10 | 120 | 53 | 438 | 0.0016 | 19,275 | 38,227 | |
| 35189 | PO | 10 | 120 | 79 | 255 | 0.0027 | 23,865 | 34,518 | |
| Mean: | IV | 2 | 1 | 324 | 98 | 0.0125 | 25,554.3 | 47,503 | 15.3 |
| Mean: | PO | 10 | 120 | 66 | 346 | 0.0022 | 21,570 | 36,373 |
Table 15
Inhibition of cardiac channels by AVN-101
| Channel | % Inhibition at 31.6μM | IC50 (μM)hNav1.5 | 97 | 2.4 |
| hKv4.3-hKCh1P2.2 (Ito) | 66 | 14.8 | ||
| hKvLQT1-hminK (IKs) | 55 | 22.5 |
Table 16
Effect of AVN-101 on cardio-vascular parameters of dogs (beagle)
| AVN-101 dose, mg/kg | 0.05 | 0.5 | 2 | 10 |
| Blood plasma concentration, μM | 0.14 ± 0.02 | 0.99 ± 0.04 | 3.28 ± 0.30 | 11.1 ± 2.15 |
| femoral SBP | – | down 17 mm Hg, 13% (p < 0.05) | down 21 mm Hg, 16% (p < 0.05) | down 15 mm Hg,11% (p > 0.05) |
| femoral DBP | – | – | – | down 12 mm Hg,17% (p < 0.05) |
| femoral BF | – | – | – | up 35 ml/min (27%, p < 0.05) |
| aortic SBP | – | – | down 8 mmHg, 8% (p < 0.05) | – |
| aortic DBP | – | – | – | down 9 mmHg, 13% (p < 0.05) |
| left ventricular SBP | – | – | – | up 16 mmHg (16%, p < 0.01) |
| left ventricular DBP | – | – | – | down 2 mmHg (40%, p < 0.05) |
| HR | – | up 28 bpm (39%, p > 0.05) | up 39 bpm, 56% (p < 0.05) | up 118 bpm, 164% (p < 0.001) |
| QT interval | – | – | down 40 ms (p < 0.01) at 30 min | down 80 ms (p < 0.001) at 30 min |
| QTcV interval | – | – | down 24 ms (p < 0.01) at 30 min | down 35 ms (p < 0.001) at 30 min |
SBP, systolic blood pressure; DBP, diastolic blood pressure; BF, blood flow; HR, heart rate.
Table 17
Chromosomal aberrations in femur bone marrow cells in male and female mice, F1 CBA×C57BL/6
| Compound | Dose, mg/kg | cells with aberration, % * | Total | p | ||||
| gaps | Single | Double | exchanges | Cells with | altered | |||
| fragment | fragment | multiple | metaphases | |||||
| aberrations | (%) | |||||||
| Males | ||||||||
| Placebo | 0.4 | 1.4 | 0 | 0 | 0 | 1.8 ± 0.6 | ||
| cyclophosphomide (positive control) | 20a | 0.2 | 19.8 | 0.4 | 1.0 | 6.2 | 20.8 ± 1.8 | <0.001 |
| AVN-101 | 1a | 0 | 1.2 | 0 | 0 | 0 | 1.2 ± 0.5 | >0.05 |
| AVN-101 | 20a | 1.0 | 0.6 | 0 | 0 | 0 | 1.6 ± 0.6 | >0.05 |
| AVN-101 | 1b | 0.2 | 1.2 | 0 | 0 | 0 | 1.4 ± 0.5 | >0.05 |
| Females | ||||||||
| Placebo | 0.2 | 1.4 | 0 | 0 | 0 | 1.6 ± 0.6 | ||
| AVN-101 | 1b | 0 | 0.8 | 0 | 0 | 0 | 0.8 ± 0.4 | >0.05 |
asingle dose, bqd for 5 days. *500 cells per experimental group were analyzed.
Table 18
Summary of PK parameters following single oral dose of AVN-101 in humans
| Administration | Substance analyzed | T1/2a, h | Cmaxa, pg/mL | AUC0 - >∞a, h*pg/mL | ||
| Compound | Route | Dose, mg | ||||
| AVN-101 | PO | 2 | AVN-101 | 14.1 ± 10.2 | 5.79 ± 2.90 | 94.6 ± 26.8 |
| AVN-101 | PO | 2 | M1 | 2.49 ± 1.2 | 56.1 ± 18.2 | 220 ± 116 |
| AVN-101 | PO | 2 | M2 | nd | 2.10 ± 3.16 | nd |
| AVN-101 | PO | 4 | AVN-101 | 8.15 ± 2.23 | 22.9 ± 16.4 | 211 ± 86.2 |
| AVN-101 | PO | 4 | M1 | 2.48 ± 1.27 | 112 ± 38.1 | 555 ± 297 |
| AVN-101 | PO | 4 | M2 | nd | 1.7 ± 2.17 | nd |
| AVN-101 | PO | 10 | AVN-101 | 10.2 ± 2.55 | 53.7 ± 31.8 | 462 ± 207 |
| AVN-101 | PO | 10 | M1 | 2.8 ± 1.71 | 292 ± 176 | 1410 ± 688 |
| AVN-101 | PO | 10 | M2 | nd | 5.08 ± 6.96 | nd |
| AVN-101 | PO | 20 | AVN-101 | 8.18 ± 2.09 | 84.1 ± 46.2 | 843 ± 333 |
| AVN-101 | PO | 20 | M1 | 3.85 ± 0.56 | 635 ± 311 | 3100 ± 870 |
| AVN-101 | PO | 20 | M2 | 4.74 ± 1.36 | 4.67 ± 1.9 | 35.2 ± 0.44 |
aMeans ± SD. nd, could not be calculated due to low content in the blood.


