Possible Involvement of Adiponectin, the Anti-Diabetes Molecule, in the Pathogenesis of Alzheimer’s Disease
Abstract
Adiponectin (APN) is protective in animal models of neurodegenerative diseases, but the role of APN in human brain has not been established. Using an enzyme-linked immunosorbent assay, we found that APN was significantly decreased in cerebrospinal fluid (CSF) of patients with Alzheimer’s disease (AD), compared to those in patients with mild cognitive impairment (MCI) and in normal controls (NC), despite elevation of APN in serum of patients with MCI and AD compared to that in NC. The discrepancy of CSF APN from serum APN in AD may suggest some critical actions of APN in the pathogenesis of AD. Indeed, it was histologically observed that APN was co-localized with tau in neurofibrillary tangles and immunoblot analysis showed that the functional trimers of APN were significantly decreased in AD compared to those in NC. Collectively, a loss of function of APN may be involved in the pathogenesis of AD.
INTRODUCTION
Adiponectin (APN) is an adipokine with various biological functions, including insulin sensitization and anti-inflammatory activities [1]. APN is homologous to collagen VIII and X and complement C1q, and APN signaling has been compared to an “exercise mimetic” that may be protective against a wide range of metabolic disorders, including hypertension, type II diabetes, dyslipidemia, atherosclerosis, and obesity [1]. These findings indicate that APN may be a promising therapy for various metabolic diseases.
The concentration of APN in cerebrospinal fluid (CSF) is extremely low compared with that in peripheral tissue (∼0.1%), and most APN in the central nervous system is present in a trimeric form that may be important for neuronal function [2, 3]. Emerging evidence suggests that APN plays an important role in Alzheimer’s disease (AD) [4]. Consistent with this view, we recently showed that APN modulates a metabolic pathway to suppress neurodegeneration in cellular and mouse models of α-synucleinopathies, including Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) [5]. Moreover, Osmotin, the plant homologue of mammalian APN, attenuates amyloid-β (Aβ)42-induced memory impairment, in addition to tau hyperphosphorylation, inflammation and neurodegeneration in mouse hippocampus [6]. Thus, these results suggest that APN may have therapeutic potential for neurodegenerative diseases.
In contrast to the protective effect of APN on neurodegeneration in model systems, the role of APN in human diseases is poorly understood. Previous clinical studies have shown that lifestyle-related diseases such as hypertension, type II diabetes, dyslipidemia, and obesity may enhance the risks for AD and other neurodegenerative diseases [7–10]. However, only one study has examined APN in this context, with Une et al. finding increased serum APN in patients with mild cognitive impairment (MCI) and AD compared to age-matched normal controls (NC) and increased CSF APN in MCI compared to NC [8].
Given this background, the main objective of this study was to better understand the role of APN in the pathology of AD. For this purpose, serum, CSF, and postmortem brains were analyzed using an enzyme-linked immunosorbent assay (ELISA), immunohistochemistry, and immunoblot analyses. The results showed that CSF APN in AD was significantly decreased compared to those in MCI and NC, whereas serum APN in MCI and AD were increased compared to that in NC. Histological studies showed APN co-localized with tau in neurofibrillary tangles (NFTs), and biochemical studies suggested alterations of APN, such as misfolding, in the AD brain. These results suggest that a loss of function of APN may play a role in the pathogenesis of AD.
MATERIALS AND METHODS
Evaluation of APN in serum and CSF
A cross-sectional study was performed in 189 subjects recruited at the memory clinic center at Higashi Matsudo Municipal Hospital, including patients with amnestic MCI (n = 64), patients with AD (n = 63), and age matched NC (n = 62). The study was approved by the Ethics Committees of Higashi Matsudo Municipal Hospital. Informed consent was obtained from all subjects. Diagnoses of MCI and AD were based on clinical criteria [11–13]. Patients with systemic diseases, including type II diabetes, that may affect circulating APN levels were strictly excluded. The demographic information of the MCI, AD, and NC subjects is summarized in Supplementary Table 1.
CSF samples were obtained from all subjects by lumbar puncture. The concentrations of APN in serum, and Aβ42 and phosphorylated tau (p-tau) in CSF were measured by ELISA [14]. Severity of brain atrophy of the grey matter in the medial temporal lobe, including the hippocampus, was determined by Z-score on VSRAD, a voxel-based morphometry-MRI method [15, 16].
Statistical analysis was performed using GraphPad Prism® (GraphPad Software Inc., San Diego, CA). Values are shown as the mean ± standard deviation and median ± interquartile deviation. Groups were compared using χ2 or Kruskal-Wallis test, followed by Spearman rank correlation analysis. A linear model was used to obtain correlation coefficients (r) and p values. Statistical significance was defined as p < 0.05.
Histological and biochemical analyses of APN in AD brain
Immunohistochemistry, confocal immunofluorescence, and immunoblot analyses of autopsied brains (neurologically healthy: NC and AD) were performed [5, 17, 18]. Brain tissue blocks were collected from the frontal cortex (n = 3), parietal cortex (n = 3), and temporal cortex (n = 3), and processed and embedded in paraffin at 10- μm thickness for histological analyses, in which about 200–300 cells per region were used for quantification. For biochemical studies, the brain blocks were subjected to homogenization, followed by fractionation by ultracentrifugation.
Anti-tau antibodies (AT8, PHF-1), anti-amyloid 1-42 antibody (Sigma), and polyclonal anti-C terminal APN antibody (Novus Biologicals, Littleton, CO) and anti-N-terminal APN antibody (Abcam, Cambridge, UK) were used as primary antibodies for histological and biochemical studies. The secondary antibodies were Alexa Fluor 488-conjugated anti-rabbit and Alexa Fluor 594-conjugated anti-mouse antibodies (Invitrogen, Carlsbad, CA). Brain sections were stained with secondary antibody alone to show the specificity of the anti-APN antibody, and were also stained with AT8 and PHF-1. To confirm the specificity of primary antibodies, control experiments were performed in which sections were incubated overnight in the absence of primary antibody or with pre-immune serum and primary antibody alone.
In biochemical studies, SDS-PAGE was performed using the standard Laemmli method. The sample buffer for reducing conditions was 3% SDS, 50 mM Tris-HCl pH 6.8, 5% and 10% glycerol without 2-mercaptoethanol. Denaturing was performed at 70°C for 10 min. Proteins separated by SDS-PAGE were transferred to nitrocellulose membranes for immunoblotting. Images were analyzed using ImageJ software (ver. 1.451; National Institutes of Health, http://imagej.nih.gov/ij/).
RESULTS
Serum and CSF APN levels in MCI and AD
Serum and CSF APN levels in the pathogenesis of AD were examined by ELISA (Fig. 1). Serum APN was significantly higher in patients with MCI and AD than in NC (MCI: p < 0.05, AD: p < 0.001; Fig. 1a), whereas CSF APN was significantly lower in patients with AD compared to those with MCI and NC (Fig. 1b).
Correlation of serum and CSF APN levels with AD biomarkers
To determine whether changes in serum and CSF APN correlated with neurodegenerative features in AD, ELISAs were performed for Aβ42 and p-tau and patients were evaluated by MRI and using the Mini-Mental State Examination (MMSE) (Fig. 1c–f). CSF APN levels were positively correlated with MMSE score (r = 0.4247, p < 0.001) (Fig. 1c) and negatively correlated with severity of medial temporal atrophy (MTA), reflecting hippocampal atrophy, on MRI (r = –0.2910, p < 0.0001) (Fig. 1d). CSF APN was significantly lower in AD compared to NC (Fig. 1b), and CSF APN levels were significantly correlated with declined cognitive function (MMSE score) (Fig. 1c) and severity of hippocampal atrophy (Fig. 1d). CSF APN levels were also correlated with CSF Aβ42 levels (r = 0.1917, p < 0.01) (Fig. 1e) and negatively correlated with CSF p-tau levels (r = –0.2979, p < 0.0001) (Fig. 1f).
In contrast to CSF APN, serum APN levels were not significantly correlated with AD biomarkers such as MMSE score, MTA, CSF Aβ42 level and CSF p-tau level (data not shown). Among metabolic factors, plasma insulin and TG were lower and HDL and LDL were higher in patients with AD compared to NC (Supplementary Figure 1), but CSF APN did not show a significant correlation with these factors.
Expression of APN in autopsied AD brain
To test the possibility that changes in CSF APN may mirror those of APN in AD brains, APN expression was investigated in postmortem brains. In an immunohistochemical study, APN was strongly stained in NFTs in AD brains (Fig. 2a, middle panel, arrows), but not in NC brains (Fig. 2a, upper panel). Use of the secondary antibody without anti-APN antibody gave no staining, showing the specificity of the anti-APN antibody (Fig. 2a, lower panel).
The number of APN-positive NFTs was significantly higher in AD brains compared to NC in the frontal, parietal, and temporal cortex (Fig. 2b). Consistent with this result, confocal laser scanning microscopy using double staining with APN and anti-tau antibodies (AT8 or PHF-1) showed that the immunoreactivity of anti-APN antibody merged with those of AT8 and PHF-1 (Fig. 2c). Co-localization of APN and NFTs in AD brains was significantly more frequent (an average of 46%) compared to that in NC brains (p < 0.01) (Fig. 2d upper and lower panel).
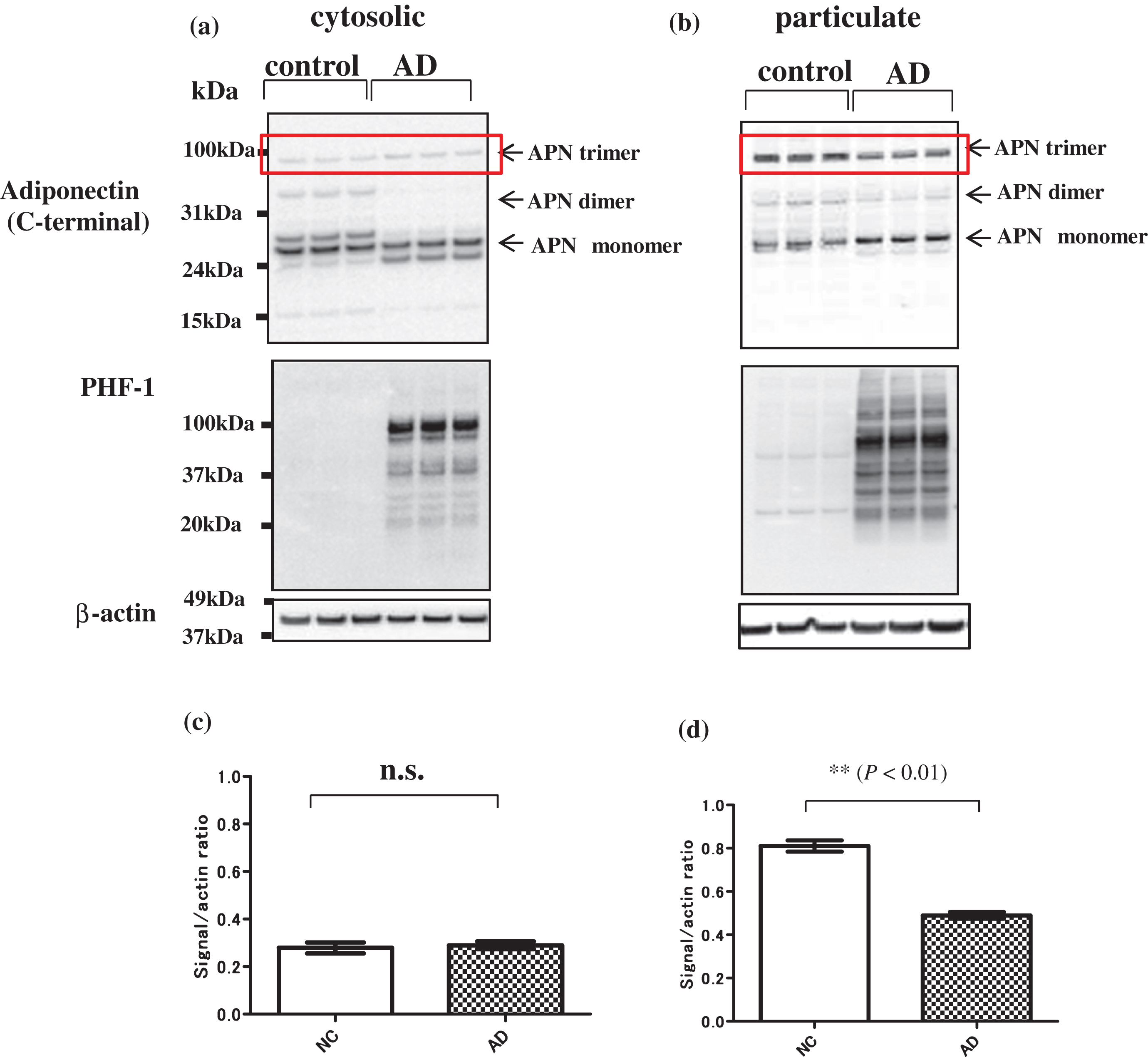
Immunoblot analysis of brain extracts using anti-C terminal APN antibody gave three major bands at about 100, 60, and 30 kDa, which correspond to the trimer, dimer, and monomer of APN, respectively (Fig. 3a cytosolic fraction, Fig. 3b particulate fraction). The monomer appeared as a double band and the mobility was higher in AD compared to that in NC in cytosolic fractions (Fig. 3a), which suggests that APN may be structurally altered in AD, possibly due to misfolding. A similar pattern was observed using anti-N-terminal APN antibodies (data not shown), indicating that APN is not N-terminally cleaved in AD brain. Notably, the 100 kDa band (trimeric APN) in AD was significantly weaker than that in NC in particulate fractions (Fig. 3d), but not in cytosolic fractions (Fig. 3c). A similar result was obtained by immunoblot with anti-N terminal APN antibody (data not shown). Given that the trimer is the functional unit of APN [19], this suggests that APN signaling may be downregulated in AD compared to NC.
DISCUSSION
In this study, changes in APN levels were found in serum, CSF, and postmortem brains of patients with AD. Consistent with previous studies [7–10], serum APN levels in MCI and AD were significantly higher than in controls. A large-scale study in 2012 showed that increased plasma APN was an independent risk factor for development of all-cause dementia and AD in women [7]. Gender may be a critical factor in the role of APN in the pathogenesis of AD, but we did not find a gender difference in changes of APN in AD (data not shown). The potential significance of gender may require a study in a larger number of samples.
The increase in serum APN in AD may reflect systemic and compensatory mechanisms against neurodegeneration. In contrast, CSF APN was significantly lower in AD compared to MCI and controls. This result differs from the findings in Une et al., which showed increased CSF APN in MCI, but not in AD, compared to controls [8]. The reason for this discrepancy is unclear, but the number of subjects in our study (n = 189) was more than double that in Une et al. (n = 73) [8]. Furthermore, we found correlations of CSF APN levels with AD specific CSF biomarkers, including CSF Aβ42, CSF p-tau, and hippocampal atrophy. Therefore, our results may better reflect the role of APN in the pathogenesis of MCI and AD.
Although the CSF APN level in AD group was statistically decreased, the CSF APN level of each AD patient was slightly scattering (Fig. 1b). It was estimated that the level of CSF APN might be changeable in various pathological stage of AD.
The findings raise the question of how APN is increased in serum and decreased in CSF of AD. This difference between serum and CSF APN in AD is reminiscent of that for Aβ42 in CSF [20, 21]. Serum APN produced from adipocytes may be increased for protection against neurodegeneration. Thus, APN levels in serum and CSF may be elevated in the initial stage of the disease. However, such compensatory mechanisms may be limited. Given that formation of inclusion bodies may be protective, it may be speculated that sequestration of APN by pathological tau into NFTs may result in suppression of the neurotoxicity of pathological tau. In support of this view, co-localization of APN with NFTs in AD brains was shown histologically. Furthermore, imunoblot analyses showed that expression of functional trimeric APN was decreased, while monomer APN may be misfolded in AD. The exact mechanisms are elusive, but it is possible that altered APN-APN receptor signaling may be pathologically changed and that APN signaling may eventually be downregulated in AD brain. A similar loss of function of APN may also occur in α-synucleinopathies, since we previously showed that APN was localized in Lewy bodies in the brains of patients with PD and DLB [5]. Our histological analyses (Fig. 2) showed apparent stronger immunoreactivities of APN in AD compared to those in NC, although imunoblot analyses showed decrease of the APN band corresponding trimer (100 kD) in the particulate fractions of AD brain compared to NC (Fig. 3). The results of histological analyses (Fig. 2) showed apparent stronger immunoreactivities of APN in AD compared to those in NC, although imunoblot analyses showed decrease of the APN band corresponding to trimer (100 kD) in the particulate fractions of AD brain compared to NC (Fig. 3). Given that histology may reflect the total immunoreactivity of the monomer and multimers of APN, one possible explanation is that immunoreactivity of the monomer of APN might be stronger than that of multimer of APN. Indeed, it is probable that adiponectin monomer (30 kD) may be denatured to expose the antigen of anti-APN antibody. However, further studies are required to demonstrate this possibility.
In summary, differential regulation of APN in serum and CSF according to the stage of AD suggests that APN may be a unique biomarker for AD. Furthermore, if loss of function of APN is critical in the pathology of AD, APN may be a target for disease-modifying therapy for AD. Further studies are warranted to investigate these interesting possibilities.
ACKNOWLEDGMENTS
This work was supported in part by a grant-in-aid for Science Research and Basic Science Research C 15K09820 (to M.W.) and Basic Science Research B 25290019 (to M.H.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan,; and by NIH Grants AG18440, AG022074, AG10435, and ES016731 (to E.M.).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1116r2).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-151116.
REFERENCES
[1] | Kadowaki T , Yamauchi T , Kubota N , Hara K , Ueki K , Tobe K ((2006) ) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116: , 1784–1792. |
[2] | Psilopanagioti A , Papadaki H , Kranioti EF , Alexandrides TK , Varakis JN ((2009) ) Expression of adiponectin and adiponectin receptors in human pituitary gland and brain. Neuroendocrinology 89: , 38–47. |
[3] | Kusminski CM , McTernan PG , Schraw T , Kos K , O’Hare JP , Ahima R , Kumar S , Scherer PE ((2007) ) Adiponectin complexes in human cerebrospinal fluid: Distinct complex distribution from serum. Diabetologia 50: , 634–642. |
[4] | Giordano V , Peluso G , Iannuccelli M , Benatti P , Nicolai R , Calvani M ((2007) ) Systemic and brain metabolic dysfunction as a new paradigm for approaching Alzheimer’s dementia. Neurochem Res 32: , 555–567. |
[5] | Sekiyama K , Waragai M , Akatsu H , Sugama S , Takenouchi T , Takamatsu Y , Fujita M , Sekigawa A , Rockenstein E , Inoue S , La Spada AR , Masliah E , Hashimoto M ((2014) ) Disease-modifying effect of adiponectin in model of α-synucleinopathies. Ann Clin Transl Neurol 1: , 479–489. |
[6] | Ali T , Yoon GH , Shah SA , Lee HY , Kim MO ((2015) ) Osmotin attenuates amyloid beta-induced memory impairment, tau phosphorylation and neurodegeneration in the mouse hippocampus. Sci Rep 5: , 11708. |
[7] | van Himbergen TM , Beiser AS , Ai M , Seshadri S , Otokozawa S , Au R , Thongtang N , Wolf PA , Schaefer EJ ((2012) ) Biomarkers for insulin resistance and inflammation and the risk for all-cause dementia and Alzheimer disease: Results from the Framingham Heart Study. Arch Neurol 69: , 594–600. |
[8] | Une K , Takei YA , Tomita N , Asamura T , Ohrui T , Furukawa K , Arai H ((2011) ) Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur J Neurol 18: , 1006–1009. |
[9] | Teixeira AL , Diniz BS , Campos AC , Miranda AS , Rocha NP , Talib LL , Gattaz WF , Forlenza OV ((2013) ) Decreased levels of circulating adiponectin in mild cognitive impairment and Alzheimer’s disease. Neuromolecular Med 15: , 115–121. |
[10] | Khemka VK , Bagchi D , Bandyopadhyay K , Bir A , Chattopadhyay M , Biswas A , Basu D , Chakrabarti S ((2014) ) Altered serum levels of adipokines and insulin in probable Alzheimer’s disease. J Alzheimers Dis 41: , 525–533. |
[11] | Petersen RC , Smith GE , Waring SC , Ivnik RJ , Tangalos EG , Kokmen E ((1999) ) Mild cognitive impairment: Clinical characterization and outcome. Arch Neurol 56: , 303–308. |
[12] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[13] | Yesavage JA , Brink TL , Rose TL , Lum O , Huang V , Adey M , Leirer VO ((1982) ) Development and validation of a geriatric depression screening scale: A preliminary report. J Psychiatr Res 17: , 37–49. |
[14] | Ebinuma H , Miida T , Yamauchi T , Hada Y , Hara K , Kubota N , Kadowaki T ((2007) ) Improved ELISA for selective measurement of APN multimers and identification of APN in human cerebrospinal fluid. Clin Chem 53: , 1541–1544. |
[15] | Matsuda H , Mizumura S , Nemoto K , Yamashita F , Imabayashi E , Sato N , Asada T ((2012) ) Automatic voxel-based morphometry of structural MRI by SPM8 plus diffeomorphic anatomic registration through exponentiated lie algebra improves the diagnosis of probable Alzheimer Disease. AJNR Am J Neuroradiol 33: , 1109–1114. |
[16] | Waragai M , Hata S , Suzuki T , Ishii R , Fujii C , Tokuda T , Arai H , Ohrui T , Higuchi S , Yoshida M , Igarashi K , Moriya M , Iwai N , Uemura K ((2014) ) Utility of SPM8 plus DARTEL (VSRAD) combined with magnetic resonance spectroscopy as adjunct techniques for screening and predicting dementia due to Alzheimer’s disease in clinical practice. J Alzheimers Dis 41: , 1207–1222. |
[17] | Hashimoto M , Takeda A , Hsu LJ , Takenouchi T , Masliah E ((1999) ) Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J Biol Chem 274: , 28849–28852. |
[18] | Takeda A , Mallory M , Sundsmo M , Honer W , Hansen L , Masliah E ((1998) ) Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am J Pathol 152: , 367–372. |
[19] | Waki H , Yamauchi T , Kamon J , Ito Y , Uchida S , Kita S , Hara K , Hada Y , Vasseur F , Froguel P , Kimura S , Nagai R , Kadowaki T ((2003) ) Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem 278: , 40352–40363. |
[20] | DeMattos RB , Bales KR , Parsadanian M , O’Dell MA , Foss EM , Paul SM , Holtzman DM ((2002) ) Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem 81: , 229–236. |
[21] | Fagan AM , Mintun MA , Mach RH , Lee SY , Dence CS , Shah AR , LaRossa GN , Spinner ML , Klunk WE , Mathis CA , DeKosky ST , Morris JC , Holtzman DM ((2006) ) Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 59: , 512–519. |
Figures and Tables
Fig.1
ELISAs of serum and CSF adiponectin (APN) in patients with MCI and AD and normal controls (NC). a) Serum APN was significantly higher in MCI and AD compared to NC (*p < 0.05, **p < 0.001). b) In contrast, CSF APN was statistically lower in AD compared to MCI and NC (***p < 0.0001). c–f) CSF APN levels were positively correlated with MMSE scores (r = 0.4247, p < 0.001) (c) and CSF Aβ42 levels (r = 0.1917, p < 0.01) (e), and negatively correlated with severity of medial temporal lobe atrophy (MTA) (r = –0.2910, p < 0.0001) (d) and CSF phosphorylated tau (p-tau) levels (r = –0.2979, p < 0.0001) f). These results suggest that a decreased CSF APN level may be associated with changes in CSF biomarkers for AD: Decreased CSF Aβ42 and increased CSF p-tau, and brain atrophy. There were no correlations between serum APN and AD biomarkers (data not shown). n.s., not significant.
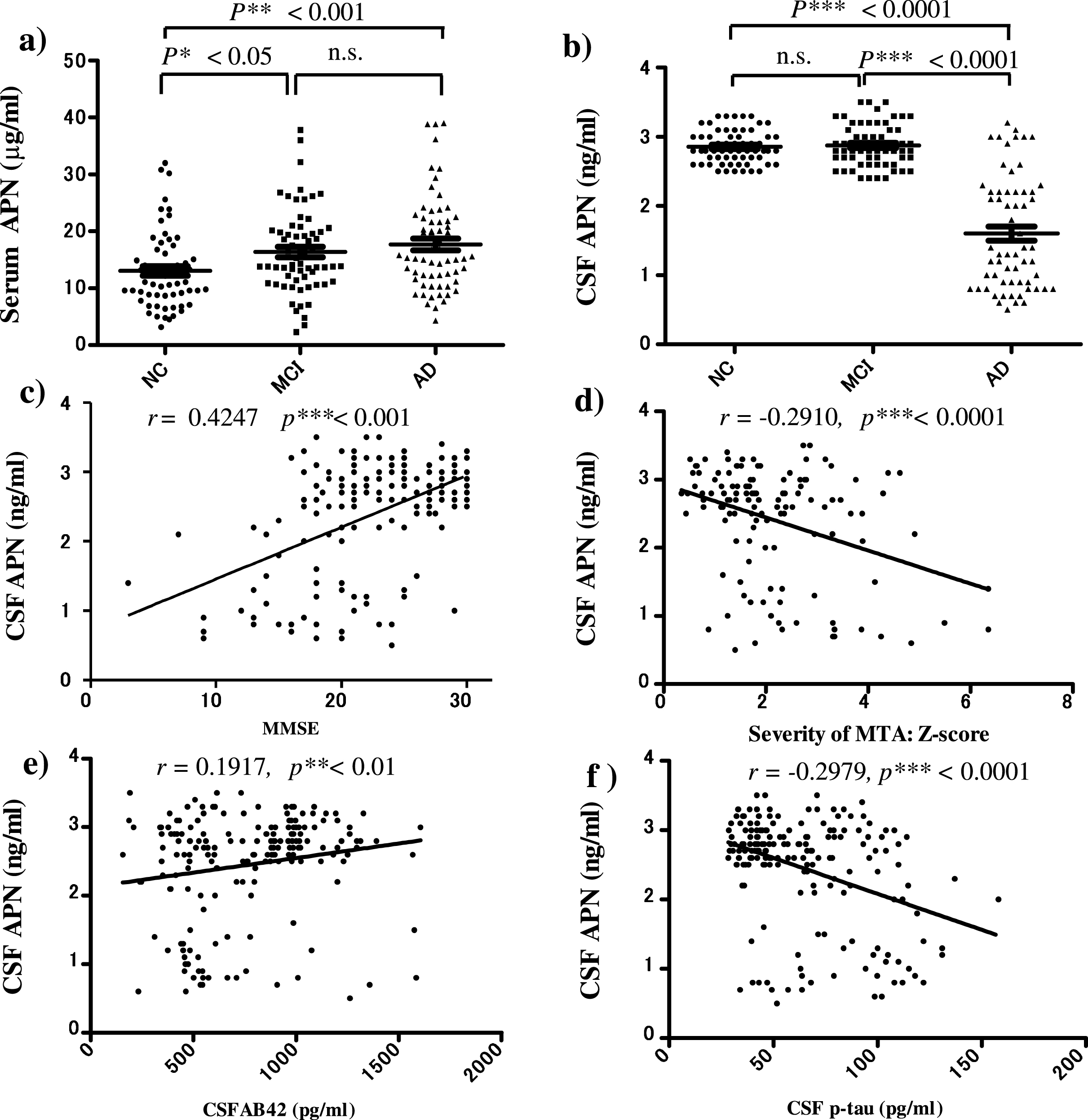
Fig.2
Neurohistochemistry using immunostaining with anti-APN antibody. a) Immunohistochemistry using a polyclonal anti-APN C-terminal antibody in samples from the frontal, parietal and temporal cortex showed strong staining of neurofibrillary tangles (NFTs) in AD brains (middle panel, black arrows), but not in control brains (upper panel). Negative control brain sections treated with secondary antibody without anti-APN antibody showed no staining, indicating the specificity of the anti-APN antibody (lower panel). b) Quantification using Image J showed that the optical density of APN-positive NFTs in (a) was significantly higher in AD brains compared to control brains (p < 0.01). c) Confocal laser scanning microscopy of control and AD brains using anti-tau antibodies AT8 (upper) and PHF (lower), and anti-APN antibody showed that APN co-localized with tau in AD. White arrows indicate NFTs identified by the presence of AT8 or PHF-1 immunoreactive abnormal fibrous inclusions, which were within the perikaryal cytoplasm of neurons. d) Quantification with Image J showed that co-localization of APN with NFTs was significantly higher in AD brains than in control brains (p < 0.01). Scale bar = 10 μm.
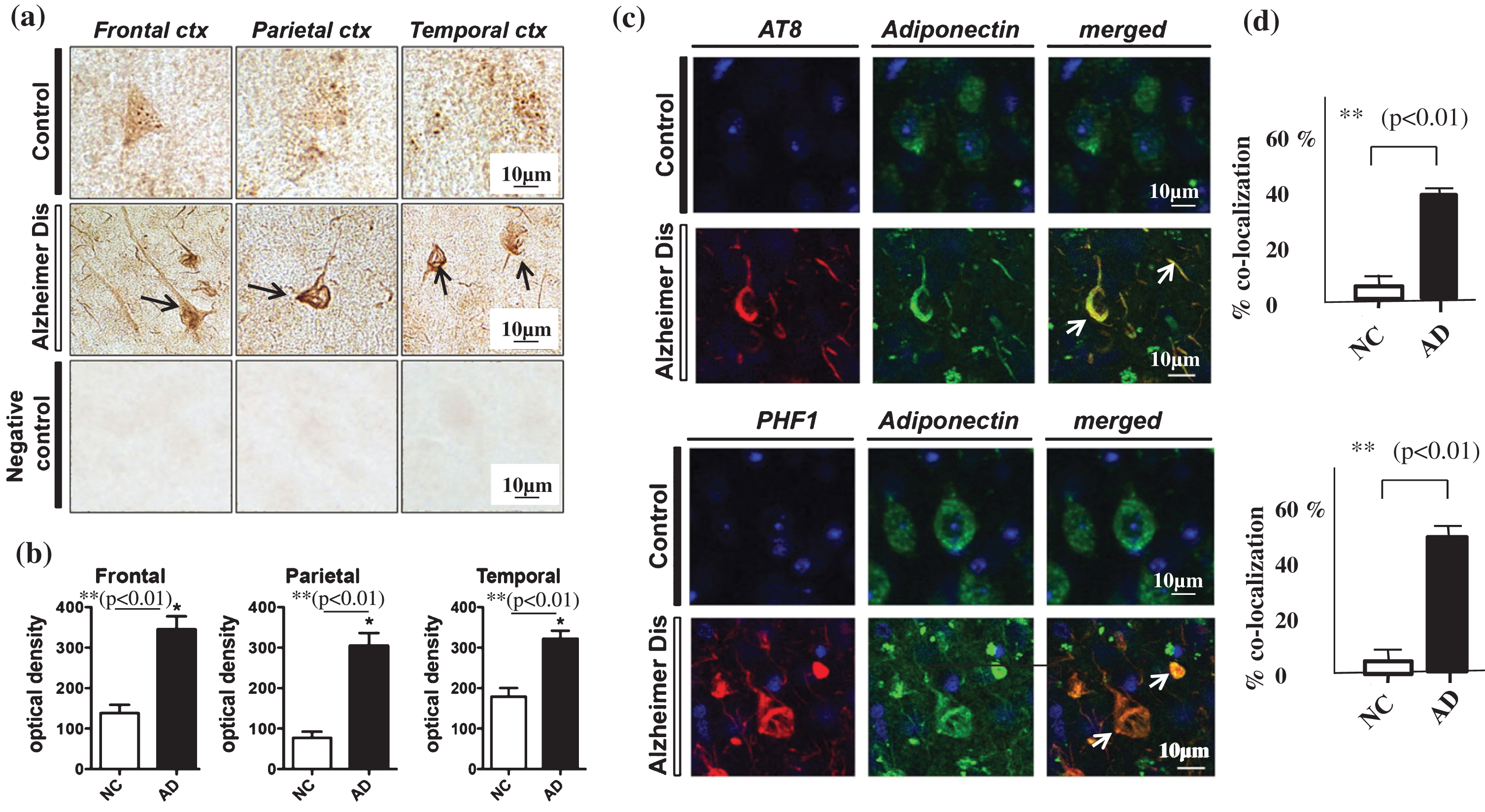
Fig.3
Immunoblot analysis of APN in AD and control brains. Brain extracts were divided into cytosolic and particulate fractions by ultracentrifugation. Bands for APN trimer, dimer and monomer are identified with arrows on the right. a) In cytosolic fractions, the mobilities of the bands corresponding to APN monomer (subunit of APN, 30 kDa) were higher in AD samples compared to those from normal controls (NC). b) In particulate fractions (right), the level of APN trimer (upper arrow: Low molecular weight trimer of APN, 90 kDa) were significantly lower in AD compared to NC, whereas the APN monomer (30 kDa) level was significantly higher in AD. Molecular weight markers (kDa) are shown on the left. Quantification of APN trimer in AD and NC brains (c and d). Bands corresponding to trimeric APN (red square) were quantified using the band signal/beta-actin ratio in ImageJ. The ratio for the trimer band (90-100 kDa) in cytosolic fractions did not differ significantly between AD and NC brains (c), but that in particulate fractions was significantly lower in AD brains (n = 3, p < 0.01) (d).