
Characterization of Amyloid-β Deposits in Bovine Brains
Abstract
Amyloid-β (Aβ) deposits are seen in aged individuals of many mammalian species that possess the same aminoacid sequence as humans. This study describes Aβ deposition in 102 clinically characterized cattle brains from animals aged 0 to 20 years. Extracellular and intracellular Aβ deposition was detected with 4G8 antibody in the cortex, hippocampus, and cerebellum. X-34 staining failed to stain Aβ deposits, indicating the non β-pleated nature of these deposits. Western blot analysis and surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry revealed in Tris, Triton, and formic acid fractions the presence of different Aβ peptides, characterized mainly by C-terminally truncated forms. Exploration of the genetic variability of APOE, PSEN1, and PSEN2 genes involved in Alzheimer’s disease pathogenesis revealed several previously unreported polymorphisms. This study demonstrates certain similarities between Aβ deposition patterns exhibited in cattle brains and those in the human brain in early stages of aging. Furthermore, the identification of the same Aβ peptides reported in humans, but unable to form aggregates, supports the hypothesis that cattle may be protected against amyloid plaque formation.
INTRODUCTION
Amyloid-β (Aβ) is a secreted peptide produced through sequential cleavage of the amyloid-β protein precursor (AβPP), a transmembrane protein widely expressed in the brain. Processed through the amyloidogenic pathway, the majority of cleaved Aβ peptides are 40 residues in length (Aβ40), and a minority is composed of the 42 residue variant (Aβ42), which is more hydrophobic and more prone to fibril formation than Aβ40 [1]. Although these peptides (Aβ40 and Aβ42) have been the dominant focus of research, N- and C-terminally truncated or modified forms of Aβ peptides are also found in the human brain [2] and cerebrospinal fluid [3].
Several genetic factors are known to influence Aβ deposition in the Alzheimer’s disease (AD) brain. Specifically, mutations in APP, presenilin 1 [PSEN1], and presenilin 2 [PSEN2] genes lead with certainty to Aβ aggregation and early onset AD [4]. Moreover, three polymorphisms of APOE strongly alter the likelihood of developing AD and represent a relevant risk factor for late-onset AD [5].
Aβ40 and Aβ42 peptides are the major components of senile plaques that form in the cortex during aging and the neuropathological hallmark of both familiar and sporadic AD. Aβ deposits may also occur in other mammalian species, including non-human primates, domestic carnivores, and wild animals.
The bulk of the literature on Aβ cerebral deposition in animals describes findings in domestic carnivores and wild omnivores, while far fewer reports about domestic and wild large herbivores are available to date. Several studies have been published on sheep [6], elephant [7], horse [8], and camel [9]. Sheep and elephant appear to be spared cerebral Aβ deposition, except for the detection of neurofibrillary tangles in sheep, which are concentrated in certain areas of the neocortex [6]. Methenamine-positive diffuse (preamyloid) plaques sporadically found in the brain of horses are characterized by the accumulation of the N-truncated Aβ42 isoform [8]. Senile plaques detected by histopathological examination in the brain of a 20-year-old camel [9] were mostly of the diffuse type and mainly distributed throughout the cerebral cortex but absent in the hippocampus and the cerebellum. A detailed characterization of Aβ deposition in the central nervous system (CNS) in cattle has never been reported before, except in one study [10] describing Aβ40 and Aβ42 peptides in bovine aqueous and vitreous humors. Since the amino acid sequences of Aβ-protein are identical in bovines and humans, the detection of senile plaques in cattle might be expected [9, 11] and Aβ formation might result from similar molecular mechanisms. The aims of the present study were to characterize Aβ deposition in cattle brain and correlate Aβ fragment patterns with age, health status, and PSEN1, PSEN2, and APOE gene profiles.
MATERIALS AND METHODS
Animals and tissue collection
Brain sections of the frontal cortex, hippocampus, cerebellum, and brainstem samples obtained at necropsy from 102 cattle of various breeds (Piedmontese, Podolica, Friesian, and mixed breed), ranging in age from 0 to 240 months, from the Italian National Reference Center for Animal Encephalopathies (CEA, Turin, Italy) archive, were investigated with different methods (Supplementary Figure 1). Fifty cattle were healthy at death and 52 had shown neurological signs (gait abnormalities, weakness, and decreased mental status) in vivo and undergone neuropathological examination. Twenty-three animals in this latter group did not display any brain abnormalities and 29 presented neuropathological features attributable to different diseases: The majority were classified as neuroinflammatory diseases and the remaining as toxic-metabolic or other diseases (food poisoning, nutritional deficiencies, foreign body syndrome, etc.). At necropsy, the brain was removed and divided into two parts by a sagittal paramedian cut. The small part was frozen at –80°C until immune proteomic analysis, and the other was fixed in 10% buffered formaldehyde solution for immunohistochemical analysis.
Single-label immunohistochemistry (IHC)
Following formaldehyde fixation, sections of the whole brain from each animal were cut coronally, embedded in paraffin wax, sectioned at a thickness of 5 μm, and mounted on glass slides. Slides were dewaxed and rehydrated by routine methods and then immersed in 98% formic acid for 10 min. To enhance Aβ immunoreactivity, sections were washed in distilled water and then boiled in citrate buffer (pH 6.1) for 10 min. Tissues were then incubated overnight at 4°C with mouse monoclonal antibody 4G8 (1:500 dilution; Signet-Covance, Emeryville, CA). After rinsing, a biotinylated secondary antibody (1:200 dilution; Vector Laboratories, Burlingame, CA) was applied to tissue sections for 30 min at room temperature (RT), followed by the avidin-biotinperoxidase complex (Vectastain ABC kit; Vector Laboratories) according to the manufacturer’s protocol. Immunoreactivity was visualized using 3, 3’-diaminobenzidine (DakoCytomation, Carpinteria, CA) as a chromogen; sections were then counterstained with Meyer’s hematoxylin. To test the specificity of staining, primary antibodies were omitted. Furthermore, to simultaneously localize lipofuscin and Aβ in the same tissue sections from older cattle, a combined IHC: Histochemical (IHC: HC) staining protocol was performed, incubating sections with 1% periodic acid for 10 min and Schiff’s reagent (Carlo Erba Reagents, Cornaredo, Italy) for 15 min before 4G8 immunostaining [12]. Human AD brain tissue was used as positive control. Aβ deposition in the four brain regions was graded according to the severity of immunoreactive lesions visualized by 4G8 antibody. The intensity grade of deposition was scored as none (0), slight (1), moderate (2), and marked (3).
Immunofluorescence
X34 staining
Several brain areas with greater amounts of Aβ deposition were selected for X34 staining, a highly fluorescent derivative of Congo red. Fixed brain tissues were stained with methoxy-X34 diluted 1:250 in 40% ethanol for 10 min at RT and analyzed under confocal laser scanning microscopy (SP8, Leica Instruments, Heidelberg, Germany).
Amyloid-β and glia detection
Brain sections staining positive for Aβ by IHC were selected for immunofluorescence studies performed according to the protocol described above. Double labeling to evaluate the relationship between astrocytes and microglia cells with Aβ deposits was performed. Tissue sections from frontal cortexes were dewaxed, rehydrated, formic-acid treated, and boiled in citrate buffer (pH 6.1), as in the IHC protocol described above. Primary antibodies 4G8 (1:100 dilution; Signet-Covance) and rabbit polyclonal Iba-1(1:100 dilution; Wako Chemicals, Richmond, VA) to visualize microglial cells or rabbit polyclonal glial fibrillary acidic protein (GFAP) (1:100 dilution; DakoCytomation) to evidence astrocytes were applied for 1 h at RT. Tissue sections were then incubated at RT with either Alexa fluor 555 or Alexa fluor 488 secondary antibodies (1:250 dilution; Invitrogen, Life Technologies, Carlsbad, CA), respectively, for 15 min. Sections were examined under confocal laser scanning microscopy (SP8, Leica Instruments). Secondary antibody specificity was tested by applying these antisera without the primary antibodies. Glia deposits were considered to be associated if their cell bodies or processes were confined within the area of amyloid deposits.
Immunoblotting for Iba-1 and GFAP
Thirty cortex pools were collected from cattle with low (n = 15) and high presence (n = 15) of Aβ detected by IHC. Lysis buffer 1X was added to each sample and centrifuged at 14000 rpm at 4°C for 15 min. The supernatant was retained, loaded with 2% Laemmli buffer, and boiled at 99°C for 5 min; samples were then loaded onto polyacrylamide gel and separated by electrophoresis. Proteins were transferred to PVDF membranes (GE Healthcare/Amersham Biosciences, Little Chafont, UK) using Wet Blot at 150V for 1 h. Membranes were blocked at RT for 1 h and incubated with primary antibodies (Iba-1 for microglia 1:100 and GFAP for astrocytes 1:10000, Millipore, Darmstadt, Germany). After rinsing, membranes were incubated at RT for 30 min with secondary antibodies (diluted 1:12000). Blots were developed using chemiluminescent substrate (Invitrogen) for 5 min, and proteins were visualized on autoradiographic films.
Biochemical compartmentalization
Strips of tissue (300 mg) were dissected from the frozen cortex, hippocampus, cerebellum, and brainstem of each animal. A 4-step extraction protocol modified from Steinerman et al. was used [13]. Tissue was mechanically homogenized in Tris extraction buffer and then sonicated. After centrifugation (100,000×g, 4°C, 1 h), the supernatant was retained as the Tris-soluble fraction. The pellet was homogenized again in Triton extraction buffer, sonicated, and centrifuged (100,000×g, 4°C, 1 h), and the supernatant was retained as the Triton-soluble fraction. The remaining pellet was homogenized in sodium dodecyl sulfate (SDS) extraction buffer, spun, and the supernatant was saved as the SDS-soluble fraction. The remaining pellet was homogenized in 70% formic acid (FA) to obtain the FA-extracted fraction, and neutralized in 20 volumes 1M Tris. These fractions are defined by their biochemical properties; however, they are predicted to be enriched with proteins from distinct cellular compartments: Extracellular soluble (Tris), intracellular soluble (Triton), membrane-associated (SDS), and insoluble (FA) proteins. Because each fraction may also contain proteins from other cellular compartments and Aβ may spill over between compartments during the extraction procedure, these fractions do not correspond precisely to cellular compartments.
Immunoblotting for Aβ
Forty-seven samples (30 from frontal cortex, 10 from cerebellum, and 7 from brainstem) were obtained from 34 cattle. Sample buffer 3X was added to Tris, Triton, and SDS brain fractions, sonicated, and boiled for 5 min. Each sample and Aβ peptides 1–38, 1–40, and 1–42 were loaded onto urea gels and separated by electrophoresis. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (GE Healthcare/Amersham Biosciences) using a Trans-Blot Semi Dry for 1 h. Membranes were boiled in a microwave for 3 min in TBS and incubated with primary monoclonal antibody (6E10 antibody, diluted 1:500, Covance) at 4°C overnight. After rinsing, membranes were incubated at RT for 30 min with AP-conjugated anti-mouse secondary antibody (diluted 1:12000, Invitrogen). Blots were developed using chemiluminescent substrate (Invitrogen) for 5 min and proteins were visualized on autoradiographic films.
Surface-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) mass spectrometry
Thirty brain samples (Tris, Triton, and formic-acid fractions for each brain) were analysed by SELDI-TOF-MS. Three μl of the specific monoclonal antibodies (mAbs) (6E10 + 4G8) (Covance) at a total mAbs concentration of 0.125 mg/ml (concentration of each mAb was 0.0625 mg/ml) were incubated for 2 h at RT to allow covalent binding to the PS20 ProteinChip Array (Bio-Rad, Hercules, CA, USA). Unreacted sites were blocked with Tris-HCl 0.5 M, pH 8 in a humid chamber at RT for 30 min. Each spot was washed three times with PBS containing 0.5% v/v Triton X-100 and then twice with PBS. Spots were coated with 5 μl of sample and incubated in a humid chamber overnight. Each spot was washed first three times with PBS containing 0.1% v/v Triton X-100, twice with PBS, and finally with deionized water. One μl of α-cyano-4- hydroxyl cinnamic acid (Bio-Rad) was added to the coated spots (Bio-Rad). Mass identification was performed using a ProteinChip SELDI System, Enterprise Edition (Bio-Rad).
Genetic analyses
APOE
DNA was isolated using a NucleoSpin Tissue kit (Macherey-Nagel, Düren, Germany) from frozen brain tissue of 21 cattle. A genomic region of ∼1.8 Kb, encompassing exon 2 to exon 4 of the bovine APOE gene was PCR amplified. The selected region included the entire APOE open reading frame (ORF) (Fig. 1). PCR primers were designed using the Primer3 application (http://primer3.ut.ee/); their sequences were: APOE_Bt_Ex2_F (5’ CCAATCGCAAGCCAGAAG 3’) and APOE_Bt_Ex4a_R (5’ GAGACTCGGGGTGGGAGTA 3’). Thermocycling parameters were an initial denaturation step (95°C, 10 min) followed by 40 cycles of denaturation (94°C, 1 min), annealing (57°C, 1 min), and extension (72°C, 2 min). APOE sequences were determined by direct DNA sequencing of PCR products on an ABI 3130 Genetic Analyser (Life Technologies) by Big Dye terminator v. 3.1 cycle-sequencing using the amplification primer pairs and two internal primers, APOE_Bt_Ex3_F (5’ GAGGAGCCCCTGACTACCC 3’) and APOE_Bt_Ex4b_R (5’ ACACCCAGGTCATTCAGGAA 3’). The sequence reactions were prepared as follows: 2 μl sequencing buffer 10x, 2 μl of Big Dye Terminator v3.1, 3.2 pmol of the sequencing primer (PCR primers), 50–100 ng of template DNA in a final volume of 20 μl. All APOE sequences were assembled using the SeqMan II program (Lasergene package, DNASTAR Inc., Madison, WI) to obtain a consensus sequence for each sample. Polymorphic nucleotides were annotated and the final consensus sequences were assembled into a single dataset. Each variable site was numbered based on the corresponding position in the bovine APOE sequence (GenBank acc. no. NC_007316.5). Finally, allele frequencies of detected polymorphisms were calculated.
PSEN1 and PSEN2
To amplify the entire ORF of PSEN1 and PSEN2 genes, an approach based on amplification and sequencing of RNA transcripts was selected from 19 brain tissues for PSEN1 and from 18 brain tissues for PSEN2. This was done to avoid having to set up several PCR reactions to cover all coding exons of the two genes, since PSEN1 and PSEN2 loci comprise 11 and 10 short coding exons, respectively, separated by long introns. Total RNA was isolated using Trizol (Life Technologies) and then DNase-treated employing Baseline-ZERO Dnase (Epicentre, Madison, WI). One μg of total RNA was reverse transcribed using a high-capacity cDNA Reverse Transcription kit (Life Technologies) and cDNA was used as a template for amplification of the entire ORFs of PSEN1 and PSEN2. PCR primers for PSEN1 were designed using Primer3 to amplify the complete ORF in a single reaction. Their sequences were: PSEN1_PCR_1F (5’ GCAGCCTGTGAGGTCCTTAG 3’) and PSEN1_PCR_4 R (5’ CACCGGAAAATCACCTTTGT 3’) giving a PCR product of 1653 bp. PCR primers for PSEN2 were designed using Primer3 to produce two overlapping PCR amplicons covering the gene ORF. Their sequences were: PSEN2_PCR_1F (5’ AGCAGGTGTGCTAAGGCACT 3’) coupled with PSEN2_PCR_3 R (5’ ACAGCACAGCCACAAGATCA 3’), giving a PCR product of 843 bp, and PSEN2_PCR_2F (5’ CAGGAGGCCTACCTCATCAT 3’) coupled with PSEN2_PCR_4 R (5’ TACCGCTTCCTACAGCTTCC 3’), giving a PCR product of 708 bp. Thermocycling parameters were an initial denaturation step (95°C, 10 min) followed by 40 cycles of denaturation (94°C, 1 min), annealing (56°C, 1 min), and extension (72°C, 2 min). Cycles were raised to 45 for PSEN2_PCR_2F/PSEN2_PCR_4 R primer pair. Sequences were obtained by direct DNA sequencing of the PCR products on an ABI 3130 Genetic Analyser (Life Technologies) by Big Dye terminator v. 3.1 cycle-sequencing using the amplification primer pairs and the addition of two internal sequencing primers for PSEN1. Their sequences were: PSEN1_SEQ_2F (5’ CCTCATGGCCCTGGTATTTA 3’) and PSEN1_SEQ_3 R (5’ CGGTCCATTCTGGGAGGTA 3’). The sequence reactions were prepared as follows: 2 μl sequencing buffer 10x, 2 μl Big Dye Terminator v3.1, 3.2 pmol of the sequencing primer, and 50–100 ng of template DNA in a final volume of 20 μl. All sequences were assembled using the SeqMan II program (Lasergene package, DNASTAR Inc.) to obtain a consensus sequence for each sample. Polymorphic nucleotides were annotated, and the final consensus sequences were assembled into a single dataset for each PSEN gene. Each variable site was enumerated based on the corresponding position in the bovine PSEN1 and PSEN2 sequences (GenBank acc. nos. BC151458 and NM_174440.4). Finally, allele frequencies of detected polymorphisms were calculated.
Statistical analysis
Several logistic regression models were set up to assess the relationship between the animal’s health status and the genetic, immunoproteomic, or immunohistochemical patterns. A logistic regression model was also used to investigate the relationship between the proteomic pattern, genetic profile, and animal age. Sex and breed were entered in the model as confounding variables. The distribution of IBA and GFAP values for different grades of Aβ deposition was described by boxplots. The association of IBA and GFAP values with Aβ deposition grading was analysed using a Kruskal-Wallis test. Statistical data analysis was performed using STATA 11 (StataCorp. 2009. Stata Statistical Software: Release 11. College Station, TX, StataCorp LP).
RESULTS
Single-label immunohistochemistry
IHC analysis for Aβ revealed 4G8-immunoreactivity in 59 out of 102 (57%) cattle brains. Immunopositive deposits presented two different patterns classified as intracellular and extracellular. The intracellular pattern was identified in 2 out of 59 (3%) cattle and was characterized by fine, randomly dispersed immunoreactive granules around the nucleus. The extracellular pattern was identified in 16 out of 59 (27%) cattle and was characterized by aggregates frequently associated with glial cells. The coexistence of intracellular and extracellular patterns was seen in 41 out of 59 IHC-positive cases. The remaining 43 cases resulted completely negative for Aβ immunoreactivity (Table 1). Immunostaining of samples from the frontal cortex was slight to marked; intracellular Aβ deposition patterns were observed in the neurons of the external pyramidal and granular layers and extracellular patterns in the molecular layer. Aβ immunolabeling in the hippocampus appeared slight to moderate, with extracellular Aβ granules scattered within the neurons of the dentate gyrus. Both intracellular and extracellular patterns were identified in the cerebellum, Purkinje cell bodies, and the molecular layer. No Aβ deposits were observed in the brainstem; no Aβ immunoreactive plaques or congophilic angiopathy were found in any of the tissue sections (Fig. 1A–E). Lipofuscin was predominantly present in the cytoplasmic regions of neurons exhibiting prominent 4G8 immunolabelling. Lipofuscin deposits were not co-localized with Aβ in the neurons but were distributed in distinct cellular compartments (Fig. 1F). The relationship between the presence of Aβ and animal age was investigated using a logistic model: via IHC, both intracellular and extracellular Aβ deposition in cattle brain increased with age (p < 0.000). The model also highlighted a close relationship between the animal’s health status and 4G8 immunoreactivity. In particular, the IHC-positive animals had a 14-fold higher risk to develop neurological disease (95% confidence interval [CI] 2.7 to 73.8), and Aβ accumulation occurred earlier (starting from 12 months) in the diseased animals as compared with the healthy ones (120 months).
Immunofluorescence
Aβ deposits and glia detection
All the examined brain sections were negative to X34 staining, indicating the non β-pleated nature of Aβ deposits. Double immuno-labelling with GFAP and 4G8 of frontal cortexes showed hypertrophic astrocytes that were occasionally observed wrapping Aβ deposits inside neuronal cell bodies or in the neuropil (data not shown). Dual immunofluorescence with Iba-1 and 4G8 confirmed microglia involvement in the extracellular Aβ deposition pattern. Activated microglial cells were localized in proximity to Aβ deposits. Some microglia and cell processes were distributed around the periphery of the aggregates, revealing active phagocytic activity (Fig. 2). No Aβ or glia immunolabeling was observed by omitting the primary antibodies.
Immunoblotting for Iba-1 and GFAP
To quantify the extent of glial cell expression, western blot analysis (WB) of GFAP in the whole cortex homogenates of cattle with low or high grade Aβ deposition revealed by IHC showed no differences in GFAP protein expression (Supplementary Figure 2). WB analysis of IBA-1 in the whole cortex homogenates showed a marked increase in IBA-1 protein expression in animals with high amounts of Aβ protein (Supplementary Figure 3). The boxplots in Fig. 3 show the distribution of GFAP and IBA (relative) values for low and high grade Aβ deposition, respectively. There was no statistically significant difference in the GFAP values between the two groups (Kruskal-Wallis, p < 0.09), whereas IBA increased with Aβ deposition and there was a significant difference in its concentration between the low and high-grade groups (Kruskal-Wallis, p < 0.0001).
Biochemical characterization of Aβ deposits
Frozen brain specimens were analyzed by immunoblot analysis. Aβ 1–38, 1–40, 1–42, and 3–42 were detected in 10 out of the 34 animals examined. Specifically, Aβ 1–38 was found in the cortex in 4 out of 10 positive animals, Aβ 1–40 in the cortex of 7 out of 10 positive animals, and Aβ 1–42 in the cortex of 7 out of 10 positive animals. Aβ 3–42 was found in the cortex of only 1 out of 10 positive animals; 2 cerebella out of 8 were positive for Aβ peptides: Aβ 1–42 in 2 cerebella out of 2 positive cases and Aβ 1–40 in only 1 cerebellum out of 2; no Aβ peptides were detected by WB analysis in the 7 brainstems. In all cases, the signal intensity was weaker than the signal detected in human AD brain (Fig. 4). Statistical analysis showed a correlation between WB-positive cattle for the presence of Aβ peptides with general 4G8 immunoreactivity, although this datum was not statistically significant (odds ratio [OR] 4.7, 95% CI 0.7 to 29.8).
SELDI-TOF MS immunoproteomic analyses
Immunoproteomic analysis with two different antibodies directed against Aβ (6E10 and 4G8) in 30 brain samples revealed, in the three fractions (Tris, Triton, and formic acid), the presence of 12 different Aβ peptides (10 C-terminally truncated and 2 N-terminally truncated), the smallest being Aβ1–18 (2166 Da) and the largest Aβ 1–42 (4515 Da). The relative amount of C-terminally truncated Aβ peptides was highest in 1–37 (4074 Da) and progressively decreased in 1–40 (4329 Da) and 1–42 in the Tris fractions, while in the Triton fractions the highest amount was observed in Aβ 1–40 > 1–37 > 1–34 (3787 Da). Finally, in the formic-acid fractions the highest amount was observed in Aβ 1–34 > 1–42 > 1–37 (Table 2). Aβ 1–42 was present in the insoluble fractions (Triton and formic acid) and N-terminally truncated Aβ peptide 10–40 was present in the formic-acid treated fractions. Aβ 1–37 and Aβ 1–40 were predominant in both the Tris and Triton fractions in the young animals (<36 months), whereas Aβ 1–34 and 1–42 were prevalent in the formic-acid fractions, and Aβ 1–40 in the Triton fractions in the aged animals (Fig. 5A, B). Aβ peptide 1–34 was expressed in all three fractions in the healthy animals, with a relevant presence of Aβ 1–40 (Triton) and 1–42 (Tris and formic acid) (Fig. 5C). In the diseased cattle, there was a large variety of Aβ peptides, with Aβ 1–37 and 1–34 present in all three fractions, Aβ 1–42 mostly in the formic-acid fraction, Aβ 1–40 in the Tris and Triton fractions, and Aβ 5–42 (4050 Da) in the Triton and formic-acid fractions (Fig. 5D). The SDS fraction was not analyzed because of the analytical interference of SDS. Statistical analysis showed that Aβ 10–40 and Aβ 1–42 correlated (albeit not statistically significant) with extracellular IHC-positive results in the cortex (OR 2, 95% CI 0.3 to 15.4; OR 2.2, 95% CI 0.1 to 42.7, respectively). In the frontal cortex, the presence of Aβ10–40 also correlated with the IHC intracellular- positive result.
Genetic analyses
APOE
Sequence analysis of the APOE gene of 21 cattle resulted in the identification of nine single nucleotide polymorphisms (SNPs). Table 3 presents the polymorphisms, their position in the sequence, and relative allele frequencies (numbering refers to NCBI reference sequence acc. no. NC_007316.5 starting from the coding sequence in exon 2). The three polymorphisms not reported in the NCBI Gene reference database were one non-synonymous mutation in exon 2 (22 G/T, codon 8, Val⟶Leu) and two SNPs in intron 2 (54 G/A) and intron 3 (725 G/T). Two further mutations located in exons 3 and 4 were synonymous (exon 3, 575 C/G, codon 32, Thr⟶Thr and in exon 4, 1215 T/C, codon 146, Ser⟶Ser). SNPs at positions 824 (intron 3) and 1215 (exon 4) were in complete linkage disequilibrium. A statistically significant association was found between the APOE allele 725-T in intron 3 and the presence at WB of Aβ peptides 1–38 (p = 0.000), 1–40 (p = 0.005), and 1–42 (p = 0.016) in the cortex, whereas the absence in the WB profile of peptides 1–38 in the cortex was associated with allele133-G in intron 2 and 824-G in intron 3.
PSEN1 and PSEN2
Sequence analysis of PSEN1 of 19 cattle revealed conservation of this gene since no SNPs were detected in the ORF. Having sequenced RNA transcripts, we were able to detect two different length variants differing by 12 nucleotides and corresponding to computationally predicted bovine PSEN1 transcript variants (GenBank acc. nos. XM_005211931-XM_005211937). This difference in length between transcripts relies on a polymorphism located in the coding DNA sequence (CDS) of the gene (290indelGTACGTAGCCAG; numbering relative to NCBI reference sequence acc. no. BC151458), resulting in the insertion/deletion of a tetrapeptide (ValArgSerGln) after codon 25 of the PSEN1 protein (Table 4). Sequence analysis of PSEN2 of 18 cattle identified five mutations in the ORF: Four SNPs and one indel polymorphism. Table 4 presents the polymorphisms, their position in the reference sequence, and allele frequencies (GenBank acc. nos. NM_174440.4; numbering relative to the ORF). The two not reported in the NCBI Gene reference database were one non-synonymous mutation at position 85 (Arg⟶Cys) and one indel polymorphism determining the deletion of an AGA triplet at position 973–975. This deletion causes the loss of one glutamic acid (Glu) residue in the resulting PSEN2 protein and was detected at 36% allele frequency. The indel was in complete linkage disequilibrium with the synonymous polymorphism at position 849 (G/A). Statistical analysis highlighted a significant association between allele 290_302ins GTACGTAGCCAG of PSEN1 and the absence of peptide 1–33 (p = 0.02) in the formic-acid fraction of the cortex. Full analysis of the Aβ peptides in the three fractions showed a correlation between the presence of Aβ peptide 1–40 (OR 6.2, 95% CI 0.12 to 311.7) in the cortex and the presence of 290_302insGTACGTAGCCAG, but the difference was not statistically significant. Allele 973_975delAGA of PSEN2 was associated with the absence of Aβ peptide 1–34 (p = 0.02) in the formic-acid fraction of the cortex. This allele also showed a correlation with a positive result for the presence of intracellular Aβ deposition in IHC (OR 3.8, 95% CI 0.4 to 39.9), but the difference was not statistically significant.
DISCUSSION
Aging entails morphological, neurochemical, and functional modifications of the brain caused by detrimental factors such as oxidative stress and altered cell metabolism; however, the molecular mechanisms underlying aging remain elusive. To date, animal models, including rodents, dogs, and non-human primates, provide only partial clues to the progressive accumulation of toxic proteins during aging.
Here we demonstrated an age-related progression of Aβ deposition in cattle brain as occurs in humans. Granular aggregates of Aβ peptides, without the presence of plaques, were observed in the cattle brains. Although a similar Aβ deposition pattern has been described in aged domestic cats, cheetahs, and leopards, the presence of an alternative N-terminal epitope of Aβ peptides is thought to be responsible for their low propensity to aggregate in these felines. An alternative explanation for this phenomenon can be given for cattle, where N-terminal modification has not been reported and where the Aβ sequence is similar to that in humans who naturally develop Aβ plaques during aging. The deposits observed in the cattle brains were constituted by Aβ protein that was not organized in fibrils and that lacked β-sheet secondary protein structure. It is conceivable, therefore, that amyloid deposits are intermediate cleavage products of AβPP detectable in the earlier stages of Aβ deposition. Similar deposits are frequently detectable in some forms of human dementias [14] and in non-demented elderly [15].
Aβ deposition seen in both the intracellular and extracellular patterns increased with advancing age. Similar results are described in healthy humans, where postmortem studies have shown that Aβ accumulation appears in the first year of life, increases in childhood, and remains high throughout adulthood, indicating that Aβ immunoreactive material in the cell body reflects normal neuronal metabolism and is not a neuronal pathology [16, 17]. This age-dependent Aβ deposition is similar in other animal species such as dogs, cats, and monkeys [18–21]. In the cattle, Aβ deposition seemed to start with an extracellular pattern and then evolve toward coexisting patterns of intracellular and extracellular deposits prevalently seen in the older animals. According to LaFerla, previously secreted Aβ in the form of an extracellular pool may be taken up by cells and internalized through receptors or transporters into intracellular compartments over time[1].
Although 52 out of 102 cattle displayed signs of neurological disease, a complete evaluation of cognitive status was not possible. This is a limitation of our retrospective study. Of note is that cognitive decline, as described in other animal species [22], has not been reported in cattle. Since these animals demonstrated gait abnormalities, brainstem or spinal cord seem to be more involved than hippocampal or frontal cortex, the two areas implicated in cognitive impairment. Moreover, we observed a strong relation between health status and Aβ deposition: IHC-positive animals had a 14-fold higher risk to develop neurological pathology probably due to the impairement of the phagocytic abilities of microglial cells [23]. Furthermore, the fact that diseased cattle have an increased risk to accumulate Aβ in the brain provides evidence that the presence of neuropathological disease induces early accumulation of Aβ. It is conceivable, therefore, that an underlying neuropathological process may speed up cerebral Aβ accumulation.
The extracellular deposits were composed of many fine granules frequently associated with microglial cells, as confirmed by double immunofluorescence and WB with antibody directed to activated microglia. Otherwise, reactive astrocytes were only rarely associated with Aβ deposits. These observations are shared by previous research in non-demented and AD patients. In AD, the accumulation of Aβ and other neuronal debris establishes chronic inflammation. Sustained exposure to Aβ and other inflammatory mediators seems to be responsible for the functional impairment of microglial cells seen around Aβ deposits. There are two plausible explanations for the association between Aβ and microglial cells: i) Aβ in glial cells may originate from an inefficient clearance of high levels of soluble Aβ present in the brain [24]; ii) Aβ secreted from neurons may be phagocytosed by glial cells in its early, non fibrous form and then degraded [25]. The presence of amyloid deposits with a non-fibrillar conformation of the protein associated with a lack of immunoreactivity to X34 in neurons and glial cells supports the latter hypothesis and suggests microglial involvement at a very early stage of Aβ deposition.
Notably, this is the first report to demonstrate the presence of Aβ peptides in cattle brain. Although a small number of samples was tested by WB analysis, the same Aβ isoforms characterizing AD brains were found in cattle brain, albeit consistently below the Aβ levels detected in human brains and whose distribution, starting from frontal to caudal brain regions, was similar to that observed in AD patients [26]. Biochemical cellular fractionation analyzed by SELDI TOF-MS revealed the presence of 12 different Aβ peptides, the majority of which are also abundantly expressed in cognitively healthy humans, schizophrenics, and AD patients [27]. Although in our samples the Tris fraction was poorly indicative in general, the Triton fraction (intracellular soluble) showed a greater expression of Aβ 1–37 and 1–40, peptides that have also been found in the cerebrospinal fluid of AD patients and controls [28]. FA-insoluble fraction showed a greater expression of Aβ 1–34 and 1–42 that increase with the age of the animals, as described in dogs [29], AD and healthy aged humans, AβPP-transgenic mice and primates [30, 31], although non statistically significant. Owing to the small sample size, a consistent correlation could not be found between the presence of Aβ peptides and IHC deposition patterns. In particular, the presence of Aβ 10–40 and Aβ42 was associated with the IHC extracellular deposition pattern in the frontal cortex. The association with the Aβ42 isoform was not surprising because it is well described in human AD brains and other animal species, including dogs [32], cats [33], and monkeys [34]. More peculiar was the association with Aβ 10–40, a peptide that contains all the structure-forming peptide segments of the full-length sequence, except for the N-terminal segment previously determined to be unstructured in Aβ 1–40 fibrillar assemblies. A study by Paravastu et al. showed that omission of residues 1–9 did not prevent the peptide from forming amyloid fibrils or eliminate fibril polymorphism [35], whereby, expressing such peptides, it is important not to underestimate the potential of bovine species to form amyloid plaques.
The genetic variability of three genes involved in AD pathogenesis was explored to unravel possible associations with Aβ deposition in cattle. We sequenced the complete ORF of the bovine APOE gene and identified nine SNPs, none involving positions 112 and 158 of the homologous human gene. Only one mutation determines a change in the amino acid composition of the resulting APOE protein, but it is located in the N-terminus of the protein and was detected at very low frequency. Interestingly, statistical analysis revealed a significant association between APOE allele 725-T and the presence at WB of Aβ peptides 1–38, 1–40, and 1–42 in the cortex, whereas the absence in the WB profile of peptide Aβ 1–38 in the cortex was associated with alleles 133-G and 824-G. These polymorphisms were not identified in the coding region, but they were located in introns 2 and 3. Given the limited number of animals involved in this study, caution is warranted. Should this association be confirmed by future studies involving a larger number of animals, these alleles might be hypothesized to contribute to the control of gene expression and, consequently, of APOE dosage. Many genes have conserved regulatory regions located within intron sequences; the enhancer effects of these control regions are so essential that some genes will not be expressed in their absence [36].
We analyzed the complete PSEN1 ORF of 19 cattle and found no SNPs. Nevertheless, statistical analysis showed a significant association between allele 290_302ins GTACGTAGCCAG and the absence of peptide 1–33 in the formic-acid fraction of the cortex. This indel polymorphism determines the presence/absence of four additional amino acid residues in the PSEN1 protein and might influence the ability of γ-secretase to cleave AβPP. The limited genetic variability of bovine PSEN1, as compared with the homologous human gene, seems to suggest that PSEN1 does not play a primary role in modulating Aβ deposition in cattle brain. The bovine PSEN2 gene is located on chromosome 16 and consists of 14 total exons (exons 5–14 coding). We identified five mutations by analysing 18 PSEN2 ORFs. Interestingly, the deletion of one glutamic acid (Glu) residue at codon 325 (973_975delAGA), in complete linkage disequilibrium with a synonymous SNP at codon 283 (849 G/A), was significantly associated with the absence of Aβ peptide 1–34 in the formic-acid fraction of the cortex. This allele also showed a trend for correlation with the presence of intracellular Aβ deposition on IHC analysis. Here, we report this novel mutation, which does not correspond to any human PSEN2 mutation recorded in the Alzheimer Disease & Frontotemporal Dementia Mutation Database (http://www.molgen.ua.ac.be/ADMutations/). The glu deletion occurs in the hydrophilic loop (HL) domain of PSEN2, located between transmembrane domains 6 and 7. Hydrophobic domains of both presenilins are highly evolutionarily conserved, with limited variability in amino acid sequences for structural and functional reasons. In particular, the HL domain between transmembrane domains 6 and 7 is thought to contain sites for phosphorylation, caspase cleavage, and sequences that bind several presenilin-interacting proteins.
In conclusion, this is the first study that fully characterizes Aβ deposition in cattle brain. Since the bovine species possesses the same amino acid sequence of AβPP as humans and displays the same Aβ fragments found in human brain, it provides an interesting model to study the lack of aggregability of Aβ peptides and the role of glial cells in the early stages of Aβ deposition. Further studies are needed to establish the involvement of other genes or epigenetic factors able to modulate Aβ fibril formation in cattle brain.
ACKNOWLEDGMENTS
The authors thank Sabrina Nodari (IZS of Turin) for technical assistance in sequencing.
This study was funded by the Italian Ministry of Health with IZS PLV 05/11 RC and IZS PLV 04/13 RC to Cristiano Corona.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1007r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-151007
REFERENCES
[1] | LaFerla FM , Green KN , Oddo S ((2007) ) Intracellular amyloid-β in Alzheimer’s disease, Nat Rev Neurosci 8: , 499–509. |
[2] | Wang R , Sweeney D , Gandy SE , Sisodia SS ((1996) ) The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry, J Biol Chem 271: , 31894–31902. |
[3] | Ghidoni R , Benussi L , Paterlini A , Albertini V , Binetti G , Emanuele E ((2011) ) Cerebrospinal fluid biomarkers for Alzheimer’s disease: The present and the future, Neurodegener Dis 8: , 413–420. |
[4] | Reitz C , Mayeux R ((2014) ) Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers, Biochem Pharmacol 88: , 640–651. |
[5] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thorton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin CF , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau MT , Choi SH , Reitz C , Pasquier F , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Morón FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fiévet N , Huentelman MW , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuiness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossú P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Deniz Naranjo MC , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F , European Alzheimer’s Disease Initiative (EADI), Genetic, Environmental Risk in Alzheimer’s Disease, Alzheimer’s Disease Genetic Consortium, Cohorts for Heart, Aging Research in Genomic Epidemiology, Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannefelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O’Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley TH Jr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RF , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JS , Boerwinkle E , Riemenschneider M , Boada M , Hiltuenen M , Martin ER , Schmidt R , Rujescu D , Wang LS , Dartigues JF , Mayeux R , Tzourio C , Hofman A , Nöthen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease, Nat Genet 45: , 1452–1458. |
[6] | Nelson PT , Greenberg SG , Saper CB ((1994) ) Neurofibrillary tangles in the cerebral cortex of sheep, Neurosci Lett 170: , 187–190. |
[7] | Cole G , Neal JW ((1990) ) The brain in aged elephants, J Neuropathol Exp Neurol 49: , 190–192. |
[8] | Capucchio MT , Márquez M , Pregel P , Foradada L , Bravo M , Mattutino G , Torre C , Schiffer D , Catalano D , Valenza F , Guarda F , Pumarola M ((2010) ) Parenchymal and vascular lesions in ageing equine brains: Histological and immunohistochemical studies, J Comp Pathol 142: , 61–73. |
[9] | Nakamura S , Nakayama H , Uetsuka K , Sasaki N , Uchida K , Goto N ((1995) ) Senile plaques in an aged two-humped (Bactrian) camel (Camelus bactrianus), Acta Neuropathol 90: , 415–418. |
[10] | Prakasam A , Muthuswamy A , Ablonczy Z , Greig NH , Fauq A , Rao KJ , Pappolla MA , Sambamurti K ((2010) ) Differential accumulation of secreted AβPP metabolites in ocular fluids, J Alzheimers Dis 20: , 1243–1253. |
[11] | Johnstone EM , Chaney MO , Norris FH , Pascual R , Little SP ((1991) ) Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis, Brain Res Mol Brain Res 10: , 299–305. |
[12] | Bancher C , Grundke-Iqbal I , Iqbal K , Kim KS , Wisniewski HM ((1989) ) Immunoreactivity of neuronal lipofuscin with monoclonal antibodies to the amyloid beta-protein, Neurobiol Aging 10: , 125–132. |
[13] | Steinerman JR , Irizarry M , Scarmeas N , Raju S , Brandt J , Albert M , Blacker D , Hyman B , Stern Y ((2008) ) Distinct pools of beta-amyloid in Alzheimer disease affected brain: A clinicopathologic study. Arch Neurol 65: , 906–912. |
[14] | Rostagno A , Ghiso J ((2008) ) Preamyloid lesions and cerebrovascular deposits in the mechanism of dementia: Lessons from non-beta-amyloid cerebral amyloidosis, Neurodegener Dis 5: , 173–175. |
[15] | Delaère P , Duyckaerts C , Masters C , Beyreuther K , Piette F , Hauw JJ ((1990) ) Large amounts of neocortical beta A4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person, Neurosci Lett 116: , 87–93. |
[16] | Wegiel J , Kuchna I , Nowicki K , Frackowiak J , Mazur-Kolecka B , Imaki H , Wegiel J , Mehta PD , Silverman WP , Reisberg B , Deleon M , Wisniewski T , Pirttilla T , Frey H , Lehtimäki T , Kivimäki T , Visser FE , Kamphorst W , Potempska A , Bolton D , Currie JR , Miller DL ((2007) ) Intraneuronal Abeta immunoreactivity is not a predictor of brain amyloidosis-beta or neurofibrillary degeneration, Acta Neuropathol 113: , 389–402. |
[17] | Kok E , Haikonen S , Luoto T , Huhtala H , Goebeler S , Haapasalo H , Karhunen PJ ((2009) ) Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age, Ann Neurol 65: , 650–657. |
[18] | Cummings BJ , Head E , Ruehl W , Milgram NW , Cotman CW ((1996) ) The canine as an animal model of human aging and dementia, Neurobiol Aging 17: , 259–268. |
[19] | Vite CH , Head E ((2014) ) Aging in the canine and feline brain, Vet Clin North Am Small Anim Pract 44: , 1113–1129. |
[20] | Gunn-Moore DA , McVee J , Bradshaw JM , Pearson GR , Head E , Gunn-Moore FJ ((2006) ) Ageing changes in cat brains demonstrated by beta-amyloid and AT8-immunoreactive phosphorylated tau deposits, J Feline Med Surg 8: , 234–842. |
[21] | Cork LC ((1993) ) Plaques in prefrontal cortex of aged, behaviorally-tested rhesus monkeys: Incidence, distribution, and relationship to task performance, Neurobiol Aging 14: , 675–676. |
[22] | Landsberg GM , Nichol J , Araujo JA ((2012) ) Cognitive dysfunction syndrome: A disease of canine and feline brain aging, Vet Clin North Am Small Anim Pract 42: , 749–768. |
[23] | Solito E , Sastre M ((2012) ) Microglia function in Alzheimer’s disease, Front Pharmacol 3: , 14. |
[24] | Funato H , Yoshimura M , Yamazaki T , Saido TC , Ito Y , Yokofujita J , Okeda R , Ihara Y ((1998) ) Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain, Am J Pathol 152: , 983–992. |
[25] | Akiyama H , Mori H , Saido T , Kondo H , Ikeda K , McGeer PL ((1999) ) Occurrence of the diffuse amyloid beta-protein (Abeta) deposits with numerous Abeta-containing glial cells in the cerebral cortex of patients with Alzheimer’s disease, Glia 25: , 324–331. |
[26] | Rapoport M , van Reekum R , Mayberg H ((2000) ) The role of the cerebellum in cognition and behavior: A selective review, J Neuropsychiatry Clin Neurosci 12: , 193–198. |
[27] | Albertini V , Benussi L , Paterlini A , Glionna M , Prestia A , Bocchio-Chiavetto L ((2012) ) Distinct cerebrospinal fluid amyloid-beta peptide signatures in cognitive decline associated with Alzheimer’s disease and schizophrenia, Electrophoresis 33: , 3738–3744. |
[28] | Maddalena AS , Papassotiropoulos A , Gonzalez-Agosti C , Signorell A , Hegi T , Pasch T , Nitsch RM , Hock C ((2004) ) Cerebrospinal fluid profile of amyloid beta peptides in patients with Alzheimer’s disease determined by protein biochip technology, Neurodegener Dis 1: , 231–235. |
[29] | Head E , Pop V , Sarsoza F , Kayed R , Beckett TL , Studzinski CM , Tomic JL , Glabe CG , Murphy MP ((2010) ) Amyloid-beta peptide and oligomers in the brain and cerebrospinal fluid of aged canines, J Alzheimers Dis 20: , 637–646. |
[30] | Van Helmond Z , Miners JS , Kehoe PG , Love S ((2010) ) Higher soluble amyloid beta concentration in frontal cortex of young adults than in normal elderly or Alzheimer’s disease, Brain Pathol 20: , 787–793. |
[31] | Miners JS , Jones R , Love S ((2014) ) Differential changes in Aβ42 and Aβ40 with age, J Alzheimers Dis 40: , 727–735. |
[32] | Cummings BJ , Satou T , Head E , Milgram NW , Cole GM , Savage MJ , Podlisny MB , Selkoe DJ , Siman R , Greenberg BD , Cotman CW ((1996) ) Diffuse plaques contain C-terminal A beta 42 and not A beta 40: Evidence from cats and dogs, Neurobiol Aging 17: , 653–659. |
[33] | Head E , Moffat K , Das P , Sarsoza F , Poon WW , Landsberg G , Cotman CW , Murphy MP ((2005) ) Beta-amyloid deposition and tau phosphorylation in clinically characterized aged cats, Neurobiol Aging 26: , 749–763. |
[34] | Kimura N, Nakamura SI, Honda T, Takashima A, Nakayama H, Ono F, Sakakibara I, Doi K, Kawamura S, Yoshikawa Y ((2001) ) Age-related changes in the localization of presenilin-1 in cynomolgus monkey brain. Brain Res 922: , 30–41. |
[35] | Paravastu AK , Petkova AT , Tycko R ((2006) ) Polymorphic fibril formation by residues 10-40 of the Alzheimer’s beta-amyloid peptide, Biophys J 90: , 4618–4629. |
[36] | Haigh CL , Brown DR ((2006) ) Regulation of prion protein expression: A potential site for therapeutic intervention in the transmissible spongiform encephalopathies, Int J Biomed Sci 2: , 315–323. |
Figures and Tables
Fig.1
IHC and IHC: HC immunoreactive deposits in different brain areas. A) 4G8-deposits in the frontal cortex. Bar = 250 μm. B) 4G8-deposits in the frontal cortex (Bar = 50 μm) with magnification of intraneuronal localization (100x). C) 4G8-deposits in the hippocampus. Bar = 50 μm. D) 4G8-deposits in the cerebellum. Bar = 50 μm. E) Absence of 4G8-deposits in the brainstem. Bar = 50 μm. F) IHC: HC, 4G8-deposits (arrow) and lipofuscin (arrowhead) in the frontal cortex. Bar = 20 μm.
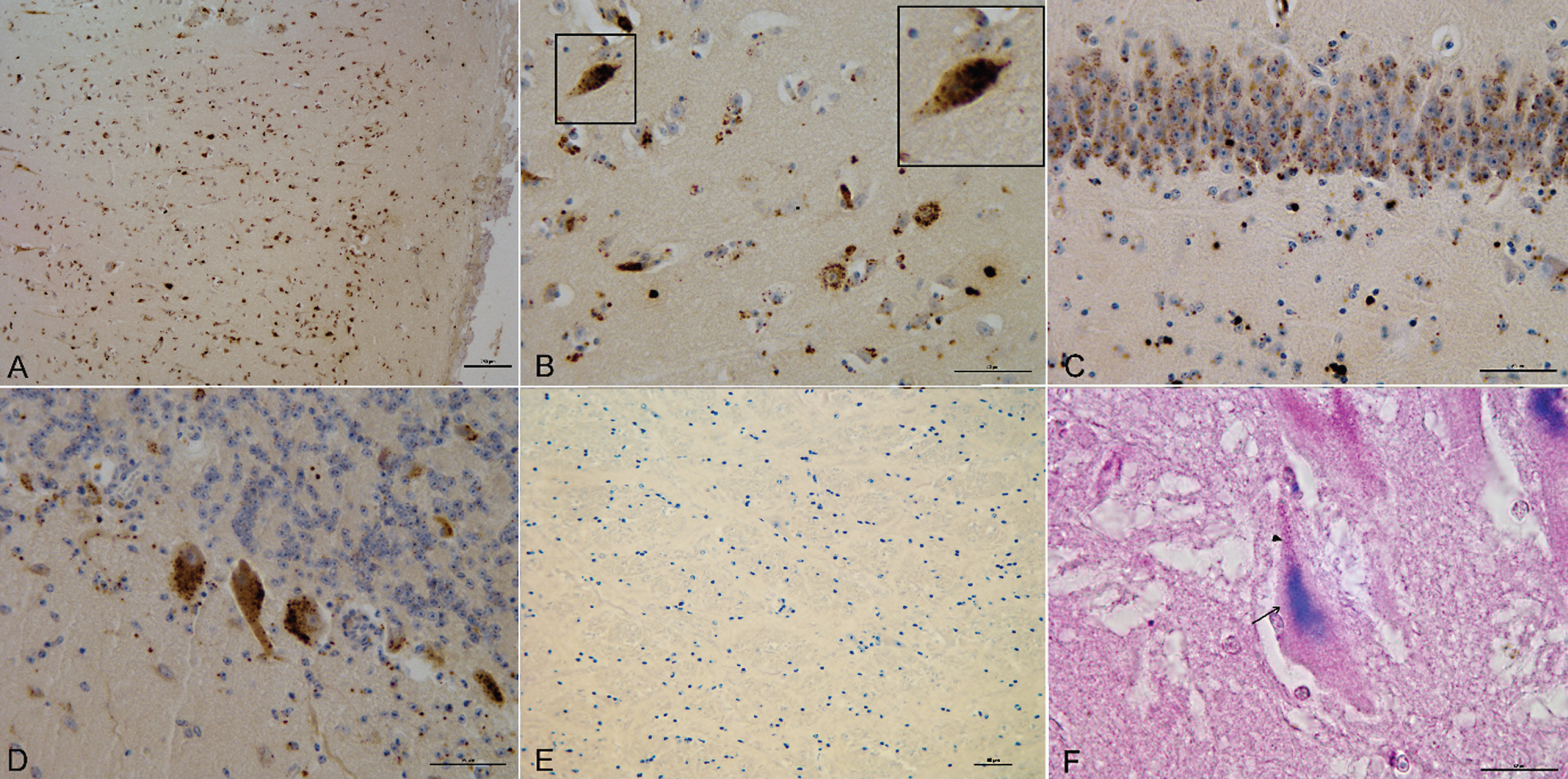
Fig.2
Double immunofluorescence staining with IBA-1/4G8. Immunofluorescent Aβ deposits associated with microglial inflammation: A) Activated microglia (green, Iba1) and (B) Aβ deposits (red, 4G8) in the frontal cortex of brain cattle. C) Merge. Bar = 13.1 μm.
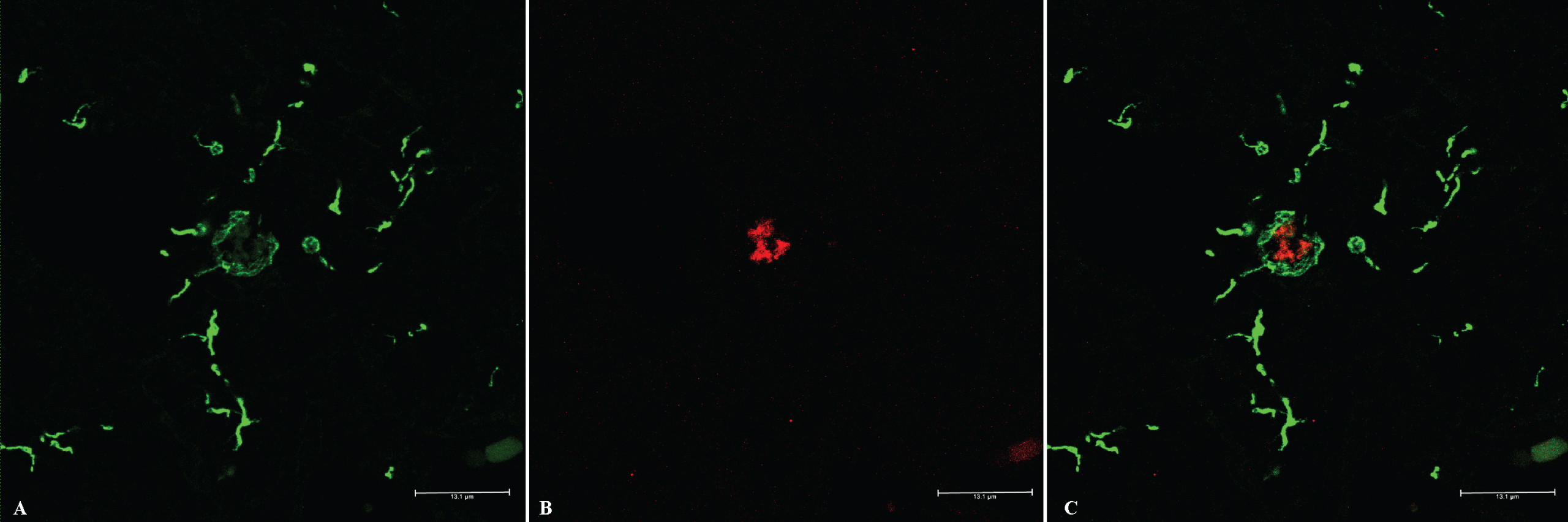
Fig.3
Boxplots of western blot analysis using IBA-1 and GFAP. A) IBA-1 values for low and high-grade Aβ deposition (total n = 32).B) GFAP values for low and high-grade Aβ deposition (total n = 29).
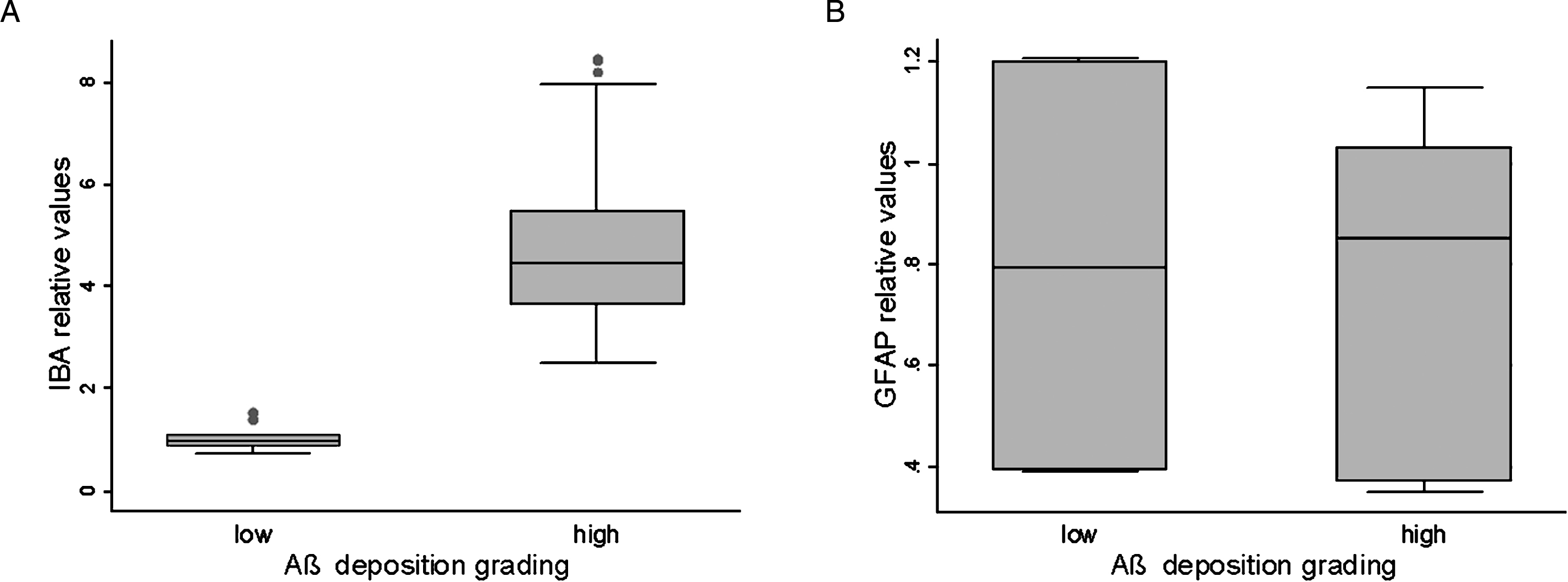
Fig.4
Western blot analysis of Aβ using 6E10 antibody. Aβ peptides 1–38, 1–40, and 1–42 in Tris, Triton, and SDS fractions of cattle cortex pools. Synthetic Aβ peptides were used as controls.
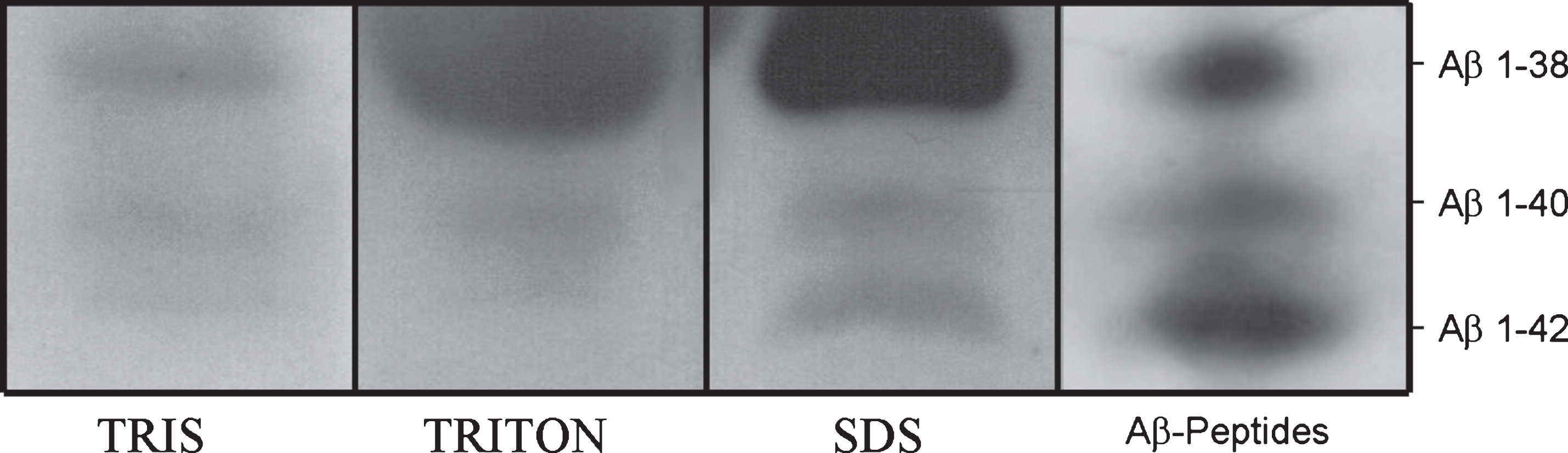
Fig.5
Mean peak intensity of Aβ peptides ( μA±SE) measured in Tris, Triton, and formic-acid fractions of cattle brains. A) Cattle less than 36 months old. B) Cattle more than 36 months old. C) Healthy cattle. D) Diseased cattle.
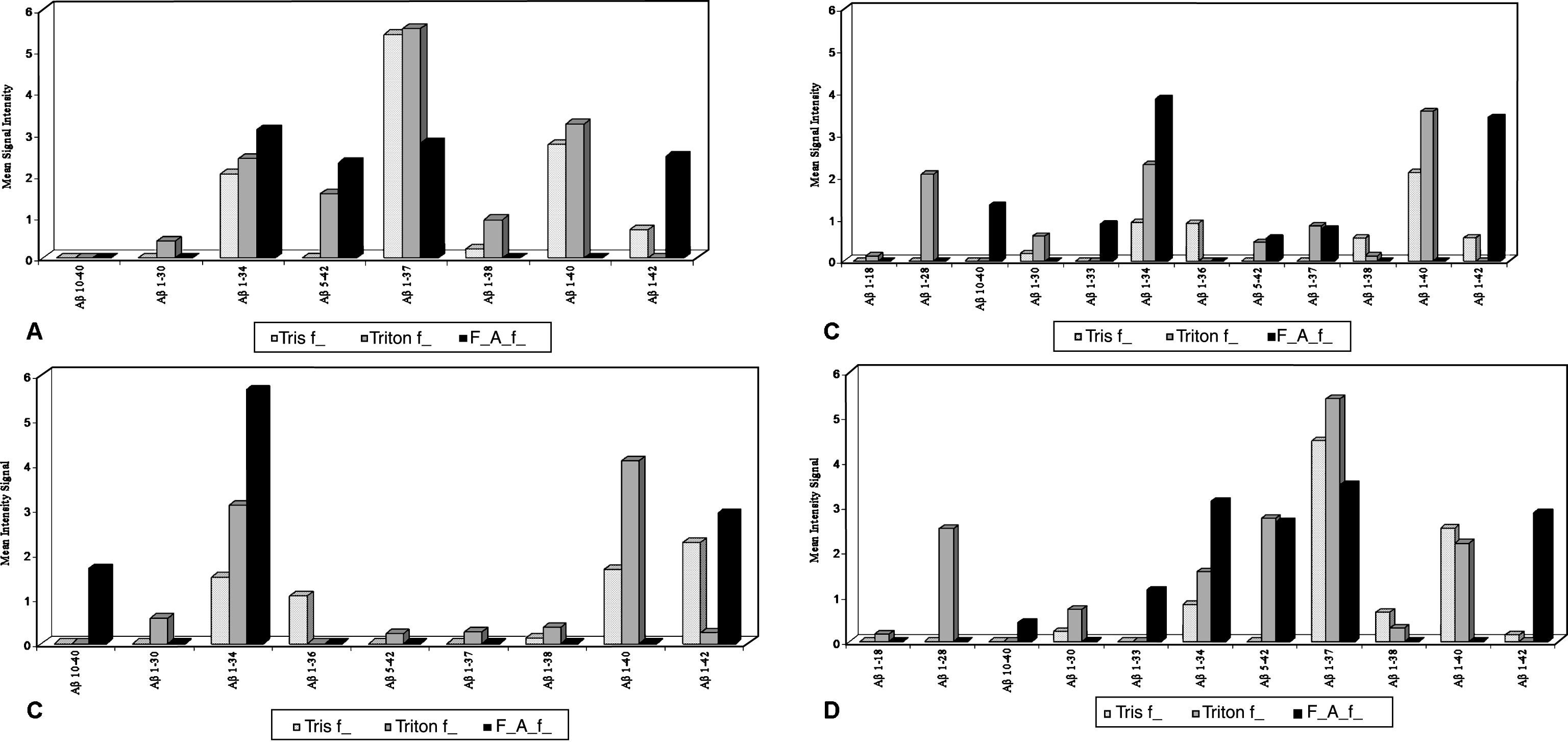
Table 1
| Aβ deposition patterns | ||||||||
| Intracellular Total | Extracellular Total | Coexistence Total | Negative Total | |||||
| animals = 2 | animals = 16 | animals = 41 | animals = 43 | |||||
| No. of animals | 1 | 1 | 11 | 5 | 17 | 24 | 24 | 19 |
| Age range (months) | 36 | 60 | 8–120 | 120–199 | 36–120 | 120–199 | 0–120 | 8–112 |
| Neurological signs | no | decubitus | decubitus | no | decubitus | no | tremors | no |
| paresis | paresis | paresis | paresis | |||||
| d.m.s. | d.m.s. | ataxia | ||||||
| d.m.s. | ||||||||
| Main affected areas | Frontal, parietal | Hippocampus | Frontal cortex | Brainstem | ||||
| cortexes | ||||||||
| Intensity IHC signal | Slight | Slight to moderate | Slight to marked | None | ||||
β deposition patterns based on the localization of 4G8 immunoreactivity. (d.m.s.: Decreased mental status)
Table 2
SELDI-TOF MS signal intensities (int.) and relative percentages (%) of Aβ peptides in cattle brains in Tris, Triton and formic-acid fractions
| Peptide | Tris int. (%) | Triton int. (%) | Formic Acid int. (%) |
| Aβ 1–18 | 0 | 0.08±0.4 (0,6) | 0 |
| Aβ 1–28 | 0 | 1.35±6.7 (10,8) | 0 |
| Aβ 10–40 | 0 | 0 | 1.05±1.5 (8,70) |
| Aβ 1–30 | 0.11±0.4 (1,63) | 0.65±1.6 (5,20) | 0 |
| Aβ 1–33 | 0 | 0 | 0.57±1.7 (4,7) |
| Aβ 1–34 | 0.15±2.0 (2,2) | 2.26±1.7 (18,1) | 4.4±6.2 (36,7) |
| Aβ 1–36 | 0.53±1.4 (7,8) | 0 | 0 |
| Aβ 5–42 | 0 | 1.58±4.2 (12,6) | 1.33±3.1 (11,1) |
| Aβ 1–37 | 2.23±4.6 (33,2) | 3.03±6.9 (24,3) | 1.74±4.1 (14,5) |
| Aβ 1–38 | 0.39±1.4 (5,8) | 0.33±1.0 (2,6) | 0 |
| Aβ 1–40 | 2.09±2.0 (31,1) | 3.06±2.2 (24,5) | 0 |
| Aβ 1–42 | 1.21±3.0 (18) | 0.11±0.5 (0,8) | 2.89±2.1 (24,1) |
Table 3
Polymorphisms, their position in the APOE sequence, and relative allele frequencies
| Polymorphism location | APOE gene | Minor allele |
| (Ref. Seq. NC_007316.5) | region | frequency |
| 22 G/T (codon 8, Val⟶Leu) | exon 2 | 0.08 |
| 54 G/A | intron 2 | 0.06 |
| 57 C/G | intron 2 | 0.24 |
| 75 T/C | intron 2 | 0.26 |
| 133 C/G | intron 2 | 0.37 |
| 575 C/G (codon 32, Thr) | exon 3 | 0.61 |
| 725 G/T | intron 3 | 0.09 |
| 824 A/G | intron 3 | 0.38 |
| T1215 T/C (codon 146, Ser) | exon 4 | 0.38 |
Table 4
Polymorphisms, their position in the PSEN1 and PSEN2 sequences, and allele frequencies
| Polymorphism location | |
| PSEN1 (Ref.Seq. BC151458) | Minor allele frequency |
| 290_302ins GTACGTAGCCAG | 0.40 |
| (codon 25, ValArgSerGln insertion) | |
| PSEN2 (Ref.Seq. NM_174440.4) | Minor allele frequency |
| 85 C/T (codon 29, Arg⟶Cys) | 0.03 |
| 198 C/T | 0.08 |
| 264 T/C | 0.03 |
| 849 G/A | 0.36 |
| 973_975delAGA (codon 325, Glu deletion) | 0.36 |


