
AZD3293: A Novel, Orally Active BACE1 Inhibitor with High Potency and Permeability and Markedly Slow Off-Rate Kinetics
Abstract
A growing body of pathological, biomarker, genetic, and mechanistic data suggests that amyloid accumulation, as a result of changes in production, processing, and/or clearance of brain amyloid-β peptide (Aβ) concentrations, plays a key role in the pathogenesis of Alzheimer’s disease (AD). Beta-secretase 1 (BACE1) mediates the first step in the processing of amyloid-β protein precursor (AβPP) to Aβ peptides, with the soluble N terminal fragment of AβPP (sAβPPβ) as a direct product, and BACE1 inhibition is an attractive target for therapeutic intervention to reduce the production of Aβ. Here, we report the in vitro and in vivo pharmacological profile of AZD3293, a potent, highly permeable, orally active, blood-brain barrier (BBB) penetrating, BACE1 inhibitor with unique slow off-rate kinetics. The in vitro potency of AZD3293 was demonstrated in several cellular models, including primary cortical neurons. In vivo in mice, guinea pigs, and dogs, AZD3293 displayed significant dose- and time-dependent reductions in plasma, cerebrospinal fluid, and brain concentrations of Aβ40, Aβ42, and sAβPPβ. The in vitro potency of AZD3293 in mouse and guinea pig primary cortical neuronal cells was correlated to the in vivo potency expressed as free AZD3293 concentrations in mouse and guinea pig brains. In mice and dogs, the slow off-rate from BACE1 may have translated into a prolongation of the observed effect beyond the turnover rate of Aβ. The preclinical data strongly support the clinical development of AZD3293, and patients with AD are currently being recruited into a combined Phase 2/3 study to test the disease-modifying properties of AZD3293.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disease that affects an ever-growing number of people worldwide. At the present time, there are no effective disease-modifying treatments for AD, although symptomatic treatments are available. Thus, there is an urgent medical need for treatments that can halt or delay the progress of the disease.
Amyloid-β (Aβ) pathology (i.e., plaque deposits) is a hallmark of AD, and the overproduction and/or the reduced clearance of brain Aβ are believed to be key elements in the pathogenesis of both sporadic and early-onset AD. Aβ species of different length, such as Aβ40 and the more pathogenic Aβ42, are produced by sequential cleavage of amyloid-β protein precursor (AβPP) to Aβ peptides via beta-secretase 1 (BACE1) and γ-secretase [1–5], with the soluble N terminal fragment of AβPP (sAβPPβ) as a direct product of BACE1 processing. Modulation of Aβ production or clearance is, therefore, considered one of the highest priority targets for disease modification in AD [6].
Genetic evidence, including causative and protective mutations in AβPP expression around the BACE1 cleavage site, points to a key role of BACE1 in the pathophysiology of AD [7]. The Swedish AβPP mutation, K670N/M671L, has a higher affinity for BACE1, resulting in increased Aβ production and earlier onset of AD in individuals expressing the mutation than in those without the mutation [8]. Conversely, individuals who express AβPP with the A673T substitution are protected against AD and cognitive decline; the substitution reduces Aβ production [9, 10]. Mice that express mutant AβPP [11] show progressive Aβ deposition, and reducing BACE1 activity yields large decreases in amyloid plaque load [12].
Numerous studies have demonstrated that BACE1 inhibitors can reduce brain Aβ concentrations in primates and other animals [13–18]. However, poor blood-brain barrier (BBB) penetration has been an issue with many of these compounds, leading to low brain:plasma concentration ratios [19]. Thus, high doses and high peripheral exposures were required to achieve central effect, because reduction of peripheral Aβ concentration alone does not lower brain Aβ concentration [20, 21].
The promising preclinical data regarding the role of BACE1 inhibitors in reducing amyloid load and possibly modifying the progression of AD have spurred a quest for compounds with more favorable brain:plasma exposure ratios and/or higher potency, as exemplified by AZD3839 and AZ-4217 [17, 18]. While several BACE1 inhibitor compounds have entered into clinical studies [22], some of these compounds have encountered safety issues unrelated to BACE1 inhibition and have thus been terminated [23, 24].
The onset of BACE1 inhibitory effect on Aβ concentration in vivo typically depends on the Aβ turnover rate in the respective effect compartment and is maintained as long as the BACE1 inhibitor concentration is sustained [25]. Typically, as the BACE1 inhibitor plasma and brain concentration decreases, the plasma and brain Aβ concentrations return to baseline as new Aβ is produced. However, a BACE1 inhibitor with a slow off-rate might display prolonged Aβ reduction that is more dictated by the turnover rate of the BACE1 enzyme than to the Aβ recovery rate. Here, we report the in vitro and in vivo pharmacological characteristics of AZD3293, a novel, very potent, highly permeable, orally active BACE1 inhibitor with BBB penetration, a slow off-rate, and excellent in vivo efficacy with prolonged on-target effect. Based on these findings, AZD3293 has been progressed into clinical development as a potential disease-modifying treatment for AD, and the recruitment of patients with AD into a combined Phase 2/3 study (NCT02245737; AMARANTH) is in progress.
MATERIALS AND METHODS
Compound
The AZD3293 compound, (1r,1′R,4r)-4-Methoxy-5″-methyl-6′-[5-(prop-1-yn-1-yl)pyridin-3-yl]-3′H-dispiro[cyclohexane-1,2′- inden-1′,2″-imidazol]-4″-amine, was designed and synthesized by AstraZeneca R&D.
Human BACE1 and BACE2 (hBACE1 and hBACE2) TR-FRET assays
The affinity of AZD3293 was evaluated using hBACE1 and hBACE2 TR-FRET assays. The soluble part of the human β-secretase (recombinant hBACE1 enzyme, aa1–aa460, or hBACE2 enzyme, aa1–aa473) was mixed with compound in reaction buffer (NaOAc, chaps, triton X-100, EDTA pH 4.5) and preincubated for 10 min. Substrate (Europium) CEVNLDAEFK(Qsy7) (PerkinElmer) was added, and the reaction was allowed to proceed for 6.5 h in darkness at 22°C until stopped with the addition of NaOAc pH 9. The fluorescence of the product was measured on a Victor II 1420 multilabel counter plate reader (Wallac) with an excitation wavelength of 340 nm and an emission wavelength of 615 nm. Additional detail about the assay procedure is available in the literature [26].
Cellular amyloid assays
The potency of AZD3293 was evaluated in a number of amyloid assays using cells of various origins: SH-SY5Y, SH-SY5Y overexpressing wild type AβPP, HEK293 cells overexpressing AβPP with the Swedish mutation (K595N/M596L), N2A cells, and primary cortical neurons isolated from fetal C57BL/6 mice (E16) or Dunkin-Hartley guinea pigs (E25-27). The assay methods have been described extensively in the literature [17, 18, 27]. In brief, the cells were incubated with different AZD3293 concentrations for 5 to 16 h, and the release of sAβPPβ, Aβ1 - 40, Aβ1 - 42, or sAβPPα into the medium was analyzed using specific commercial ELISA or kits from Meso Scale Discovery. Any cytotoxic effect of AZD3293 noted in a cellular assay was directly evaluated in the cell plates using the ViaLighttrademark Plus cell proliferation/cytotoxicity kit (Cambrex BioScience, New Jersey, USA), according to the manufacturer’s instructions.
hCathepsin D FRET, Notch, and human ether-a-go-go-related gene (hERG) assays
The specificity of AZD3293 was evaluated using hCathepsin D FRET, Notch, and hERG assays. In the hCathepsin D FRET assay, cathepsin D enzyme (Calbiochem) and substrate (Ac-Glu-Asp(EDANS)-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Gly-Lys(DABCYL)-Glu-NH2) (Bachem) were separately diluted in glycine-HCl buffer. The enzyme was mixed with compound dissolved in DMSO and preincubated for 10 min. Substrate was added, and the reaction mixture was incubated in darkness for 15 min at 22°C. The fluorescent signal was measured on a Victor II 1420 multilabel plate reader (Wallac) with an excitation wavelength of 355 nm and an emission wavelength of 460 nm.
The Notch assay was performed according to previously described methodology [27]. HEK_PS1 and HEK_PS2 cells [28] were transiently transfected with extracellular truncated Notch (Notch1ΔE). The cells were then incubated with different concentrations of AZD3293 for 12 h. The γ-secretase-liberated Notch1 intracellular domain was analyzed using an Image-Xpress scanner (Molecular Devices) and specific, labeled antibodies. The affinity of AZD3293 on hERG was investigated using the patch clamp technique and hERG-expressing CHO cells in a whole-cell configuration. Recordings were made at room temperature. For each cell, following addition of vehicle solution (0.1% DMSO), AZD3293 was tested at six concentrations: 0.3, 1, 3, 10, 30, and 100μM. Following application of the highest test concentration, vehicle solution was re-applied to test the reversibility of the effect. The positive control (3μM cisapride) was then applied. For each cell recorded, data obtained in the presence of the test compound were expressed as a percentage of those obtained during the first application of vehicle solution.
Biacore direct binding assay (DBA)
The binding properties of AZD3293 were evaluated using a Biacore direct binding assay. A CM7 sensor chip was prepared by immobilizing 30μg/mL purified hBACE1 on Spots 1 and 5, to 10000 response units (RU) in Flow Cell 4 of a BC4000 instrument (GE Healthcare, Bio-Sciences AB, Uppsala, Sweden). The standard Biacore amine coupling procedure, with 10-min activation, was followed. Spot 3 was used as a reference surface (unmodified). The running buffer for immobilization was 100 mM NaOAc pH 4.5, 150 mM NaCl, 0.005% P20; the coupling buffer was 10 mM Na-acetate pH 4.5; contact time was 16 min; and the temperature was 25°C. The surface was left to stabilize overnight. The sample association time was 60 seconds, and the dissociation time was 30 min. AZD3293 and AZD3839 were prepared from a 10 mM DMSO stock to 20 μM compound in assay buffer to a final concentration of 2% DMSO. The Biacore data were generally analyzed with the Biacore 4000 evaluation software using the Kinetics and Affinity evaluation method (reference- and blank-subtracted data). The dissociation-rate constant (koff) was calculated using the 1:1 dissociation model and then recalculated to a half-life (t1/2 = ln 2/koff) for the complex. This method has been suitable for comparative experiments with AZD3839 [17, 26] and other similarly fast dissociating compounds. However, for slowly dissociating compounds like AZD3293, dissociation could not be distinguished from the background drift in the signal. Therefore, the t1/2 of AZD3293 was also estimated with single-cycle kinetics from an experiment using a Biacore T200 instrument in which hBACE1 was immobilized with amine coupling on a CM7 chip to 3000 RU, and five pulses of compound at increasing concentrations were injected in the same cycle, followed by a long, 30 min dissociation phase.
The reversibility of AZD3293 binding was also studied in a displacement experiment with AZD3839 using the same immobilization conditions as described above. After six start-up cycles with running buffer, AZD3839 was injected twice at 20μM to determine the initial binding capacity of the surface. The high-affinity compound AZD3293 was then injected twice at 20μM to occupy the binding pocket. AZD3839 was then injected every 60 min over a 20 h period to compete with AZD3293 for available binding sites. Each injection of compound was followed by an injection of running buffer as a negative control.
In vitro drug metabolism and pharmacokinetic (DMPK) assays
AZD3293 has been evaluated in a range of in vitro DMPK assays to determine plasma protein binding, brain tissue binding, brain permeability, efflux ratio, and solubility. The plasma protein binding of AZD3293 was analyzed by equilibrium dialysis using radio-labeled AZD3293 at nominal concentrations of 0.2, 5, and 100μmol/L in mouse, rat, guinea pig, dog, and human plasma. Spectra/Por 2 dialysis membranes (Spectrum Medical Industries Inc.) of molecular weight cut-off 12 to 14 kilodaltons were pre-soaked and assembled within Teflon dialysis cells. For each concentration, triplicate aliquots of spiked plasma solution were dialyzed against Dulbecco’s phosphate-buffered saline (pH 7.4) for 6 h at 37°C. Duplicate aliquots of the dialyzed plasma and buffer were taken for radioactivity measurement by liquid scintillation counting (LCS). The plasma protein binding of AZD3293 metabolite M1 was determined at a concentration of 10μmol/L using cold compound. The unbound fraction in plasma was calculated from the ratio of the area under the peak obtained by mass spectrometry of M1 in buffer divided by sum of the areas of M1 in buffer and plasma [29]. For plasma and blood cell partitioning, triplicate whole blood samples from a single male human subject were spiked with radio-labeled AZD3293 at a concentration of 5μmol/L, mixed gently, and incubated at 37°C for 0, 0.5, and 2 h. Duplicate aliquots were centrifuged to obtain plasma for radioactivity measurement by LCS.
The in vitro metabolism of AZD3293 was studied in human and rat hepatocytes according to the methods described by Floby et al. [30].
To determine brain tissue binding, 300 μm thick coronal rat brain slices were incubated for 5 h in 10 mL of an artificial interstitial fluid buffer containing 1μM of compound. Following the incubation, the slices were weighed and homogenized, proteins were removed, and liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed. The unbound fraction of AZD3293 in brain was calculated as described by Fridén et al. [31]. Free drug concentrations in mouse, guinea pig, and dog brains were estimated by multiplying the total brain analyte concentrations by the unbound fraction determined from the rat brain, as it has been shown that this unbound fraction in brain is interchangeable among species [32].
Brain permeability and efflux ratio were determined in the Caco-2 and MDCK-MDR1 cell assays as described by Gravenfors et al. [29]. Solubility was measured using the dried-DMSO method as described by Alelyunas et al. [33].
Animals and animal handling
Animal experimentation (using mice, rats, guinea pigs, and dogs) was performed in accordance with relevant guidelines and regulations provided by the Swedish Board of Agriculture. Ethical permission was provided by an ethical board that specializes in animal experimentation (Stockholm North Animal Research Ethical Board).
Female 7- to 14-week-old C57BL/6 mice (n = 6 per treatment group and timepoint; Harlan Laboratories, The Netherlands) received vehicle or AZD3293 solution at 50, 100, or 200μmol/kg (20, 41, or 82 mg/kg) as a single dose via oral gavage.
Male 5-week old Dunkin-Hartley guinea pigs (n = 6 to 8 per treatment group and timepoint; HB Lidköping Kaninfarm, Sweden), received vehicle (5% dimethylacetamide in 0.3 M gluconic acid, pH 3) or AZD3293 solution at 10, 30, or 100μmol/kg (4, 12, or 41 mg/kg) as a single dose via oral gavage. Mice and guinea pigs were anesthetized 1.5, 2, 3, 4, 6, 8, 16, 24, or 48 h after the (last) administration of vehicle or drug and were then kept under isoflurane anesthesia. Cerebrospinal fluid (CSF) was aspirated from the cisterna magna, and plasma was isolated from blood collected by cardiac puncture into EDTA tubes. The animals were then sacrificed by decapitation, and the brains were dissected into hemispheres.
Plasma AZD3293 concentration and its effect on plasma Aβ40 were studied in the standard regulatory toxicology studies in male and female beagle dogs aged 14.7 to 19.0 months (from Rååhöjden, Sweden) or 8.5 months (from Charles River Laboratories, Canada) at the start of the experiments. AZD3293 was formulated in a 0.3 M gluconic acid, pH 3 solution. In one male dog and one female dog, AZD3293 was administered orally in a repeated, ascending-dose regimen at doses of 9.7, 29, 87, 240, and 9.7μmol/kg (4, 12, 36, 100, and 4 mg/kg) on Days 1, 3, 8, 10, and 16. Six male and six female dogs were given daily oral AZD3293 doses of 0.48, 3.4, and 24μmol/kg (0.2, 1.4, and 10 mg/kg) for 14 or 15 days. In addition, data were collected for daily oral doses of 2.4, 7.2, and 36μmol/kg (1, 3, and 15 mg/kg; three female and three male dogs per dose) over 91 days. The dynamics of plasma Aβ40 were studied in more detail following a single oral dose of 12, 36, or 60μmol/kg (5, 15, or 25 mg/kg; one male and one female dog per dose). Bioavailability was determined from two 3-μmol/kg (1.2 mg/kg) oral and two 1.5-μmol/kg (0.62 mg/kg) intravenous single-dose AZD3293 concentration-time profiles in male dogs. Blood samples were taken via jugular vein; CSF and hippocampus samples were collected from anesthetized animals in connection with necropsy.
Extraction and analysis of Aβ and sAβPPβ from animal samples
The extraction of brain tissue and the analysis of Aβ and sAβPPβ species were performed as described in detail by Eketjäll et al. [18]. Brain tissue was sonicated in 0.2% dietylamin with 50 mM NaCl, then subjected to ultracentrifugation. Dietylamin brain extracts, plasma, and CSF were analyzed for concentrations of Aβ1 - 40, Aβ1 - 42, and sAβPPβ using commercial and specific immunoassays. Concentrations of sAβPPβ in guinea pig brain extracts, plasma, and CSF were analyzed using a commercial high-sensitivity sAβPPβ ELISA (#JP27732, IBL International, Hamburg, Germany). In dog plasma, CSF, and brain (hippocampus), Aβ1 - 40 was measured using a commercial ELISA (#KHB3482, Invitrogen, Camarillo, CA, USA). The lower limit of quantification (LLOQ) was determined for each immunoassay plate based on the lowest standard point with a coefficient of variation (CV) <20% and an accuracy (back-calculated concentrations) of 80% to 120% . Log-transformed data were analyzed with pair-wise t-tests within a 1-way ANOVA model, using Prism 4 (GraphPad, USA). The level of significance was p < 0.05 (2-tailed).
Bioanalysis of brain, plasma, and CSF samples
The bioanalysis of brain, plasma, and CSF samples has previously been described in detail [34]. Brain samples from mice, guinea pigs, and dogs were analyzed for concentrations of AZD3293. The right brain hemisphere was homogenized in two volumes (weight/volume) of Ringer solution. Aliquots of plasma (25μL) and brain homogenate (50μL) were precipitated with 150μL acetonitrile containing 200 nmol/L warfarin as an internal standard. Samples were mixed, centrifuged, diluted with mobile phase, and analyzed on an LC-MS/MS system. CSF aliquots (15μL) were directly diluted and injected. A correction for blood content in brain was made by subtracting 1.3% of the plasma analyte concentration from the total brain analyte concentration.
Pharmacokinetic/pharmacodynamic analysis
Mouse and guinea pig pharmacokinetic (PK) and pharmacodynamic (PD; Aβ40) data were analyzed using a population modeling approach (NONMEM Version 7.2.0, ICON, Hanover, MD), fitting the data simultaneously using the Monte Carlo Importance Sampling Expectation Maximization for estimation. Model selection was based on the minimum value of the objective function, defined as –2 * log likelihood. A visual predictive check [35] was used to evaluate whether the model was able to predict the observed variability for 80% of the data population used for model identification. Monte Carlo methods were employed to simulate 1000 hypothetical datasets, using the estimated model parameters and variability. Differences in, and overlap of, the simulated (median 10th percentile and 90th percentile) and original distributions indicated the accuracy of the identified model.
The above exercise resulted in different mathematical models for the two animal species. In mice, a model with a central and two separate peripheral PK compartments provided the best fit to the PK/PD data. Plasma AZD3293 concentration was linked to plasma or brain Aβ40 concentration using an indirect/turnover model: dR/dt = kin*(1 –effect) –kout*R, in which R = plasma or brain Aβ40 concentration, kin = zero order Aβ40 production rate, kout = first order turnover rate (kin was set to kout), effect = concentration-effect relationship with effect = Imax * CP/(IC50 + CP) where CP is the AZD3293 concentration in plasma, Imax = maximum response and IC50 = the AZD3293 concentration at which a 50% reduction in the biomarker level from baseline is achieved in vivo. In addition, a constant brain:plasma ratio was assumed, and brain Aβ40 concentration was also fitted. In guinea pigs, the PK model included a central and two peripheral PK compartments, one of which is linked to brain AZD3293 concentration. The second peripheral PK compartment is linked to two additional peripheral compartments, a brain-binding compartment and a CSF compartment. The total brain AZD3293 concentration is the sum of the concentrations in the brain and the brain-binding compartments, with these values corrected for brain:plasma ratio. CSF AZD3293 concentration is directly linked to the brain AZD3293 concentration, excluding the bound-to-brain compartment and corrected with a CSF:brain ratio. Plasma AZD3293 clearance (CL) was non-linear and fitted by actual clearance = CL*(1-CP*CLEmax/(CLEC50+CP)), where CLEmax is the maximum clearance and CLEC50 the half maximum concentration at which the clearance is reduced by 50% from baseline.
Data from 119 mice and 123 guinea pigs were included in the population analysis originating from the PK and PK/PD studies. Biomarkers Aβ42 and sAβPPβ in mice and guinea pig were analyzed in Phoenix WinNonlin 6.4 using the NMLE 1.3 module in which the PK models were adopted from the models obtained above.
In dogs, dose-response exposure for brain and CSF Aβ40 concentrations and dose- and time-response exposure data and plasma Aβ40 concentrations were available, including a total of 18 plasma AZD3293 concentration-time profiles. In dog plasma, predose samples were available, and in brain and CSF reference values for Aβ40 were used. A population analysis using Phoenix WinNonlin evaluated the goodness of fit. AZD3293 and Aβ40 concentration-time profiles were fitted with an indirect/turnover model in which kout was estimated separately from kin but linked by baseline. Bioavailability was calculated using non-compartmental analysis.
RESULTS
In vitro potency and selectivity
AZD3293 was identified as a potent (inhibitory constant [Ki] = 0.4 nM) inhibitor of hBACE1 using an enzymatic TR-FRET assay (Table 1). When the potency of AZD3293 with respect to secretion of Aβ40 and sAβPPβ was studied in a range of cellular models, the compound displayed pM potency in primary neuron cultures from mice and guinea pigs and in SH-SY5Y cells over-expressing AβPP (IC50 = 610 pM, 310 pM, and 80 pM, respectively). AZD3293 selectivity was evaluated against the two aspartyl proteases, hBACE2 and cathepsin D, as well as Notch processing, and the compound showed near equipotency against hBACE2 and a >25,000- and >41,000-fold selectivity against cathepsin D and Notch IC50 values, respectively. Important Ki and IC50 values are summarized in Table 1.
AZD3293 was also tested in a panel of more than 350 in vitro radioligand binding and enzyme activity assays, covering a diverse range of receptors, ion channels, transporters, kinases, and enzymes, up to a concentration of 10μM of AZD3293. A few significant responses were observed, but these had at least a 1,000-fold selectivity against BACE1, thus indicating specificity to BACE1.
Binding characteristics
AZD3293 acts as a full inhibitor of BACE1 in vitro, with a competitive and reversible mechanism of action towards the hBACE1 active site. The mode of action for AZD3293 was determined by characterizing the binding properties in three different Biacore assays (direct binding, single cycle kinetics, and displacement). AZD3293 displayed a very high target affinity and a markedly slow target off-rate, also relative to the potent BACE inhibitor AZD3839 (described in Jeppsson et al. [17]). The off-rate of AZD3293 had an estimated t1/2 of approximately 9 h (Fig. 1A, C). The reversibility of AZD3293 binding was confirmed in a displacement experiment with AZD3839 (Fig. 1B); the time to complete displacement was estimated to be approximately 15 h.
DMPK
AZD3293 is a compound with high permeability, a high apical-to-basolateral apparent permeability coefficient in Caco-2 cells, and medium solubility (Table 1). The in vitro plasma protein binding of AZD3293 was determined by equilibrium dialysis using mouse, rat, guinea pig, dog, and human plasma. The compound was stable in the plasma of these species for at least the duration of the in vitro incubation period. The unbound fractions were 1.3% to 1.8% for mice, 4.2% to 5.9% for rats, 8.3% to 10.3% for guinea pigs, 9.4% to 10.3% for dogs, and 7.7% to 9.4% for human plasma (Table 1). The mean blood:plasma ratio of 0.7 in human blood indicated no significant association with red blood cells. The free fraction in the brain tissue binding assay was 4.5% .
In vitro in hepatocytes and in vivo in mice and dogs, formation of an active metabolite (M1) was observed. While the metabolite M1 was 10-fold less potent than the parent compound in human in vitro cellular systems, in mouse primary neurons, the metabolite was equipotent relative to the parent compound (Tables 1 and 2).
In C57BL/6 mice, from 45 min until 24 h after a 100 μmol/kg oral dose of AZD3293, the ratio between total parent and metabolite plasma concentrations was relatively constant, with the parent concentration approximately 13-fold higher than the metabolite concentration. When brain exposure of parent and compound was tested at 0.5, 1, and 2 h after dosing, the minimum unbound brain: plasma ratio was approximately 30-fold lower for the metabolite relative to the parent compound, and in vitro cellular systems showed higher efflux ratios for the metabolite than for the parent (Tables 1 and 2).
In the dog PK study, the bioavailability of AZD3293 was determined to be 80% (F = 0.8). The time-concentration relationships of AZD3293 and its metabolite M1 were studied in vivo in a repeated daily oral-dose regimen. On Day 1 and Day 91, blood (plasma) samples were obtained before dosing and after dosing at 30 min and 1, 2, 4, 8, 12, and 24 h at doses ranging from 2.4 to 36μmol/kg. The area under the plasma concentration-time curve (AUC) of the parent compound was approximately 8-fold higher than the AUC of the metabolite M1.
AZD3293 concentration-time-effect relationships in mice
When C57BL/6 mice were treated with a single oral administration of AZD3293 (50, 100, or 200μmol/kg), plasma Aβ40 concentration was reduced (88%) down to the LLOQ of the assay and was still significantly reduced (20% to 25%) at 16 h after the dose (Fig. 2C). The concentrations of brain Aβ40 and Aβ42 displayed similar patterns, with statistically significant reductions (20% to 40%) between 1.5 and 8 h after the dose and a return to baseline 16 h after AZD3293 administration (Fig. 2E, F). For these peptides the maximum effect level in brain was already achieved at 3 h after the dose. The maximum effect on brain sAβPPβ (21%) was observed 8 h after AZD3293 administration (Fig. 2D). Free plasma (Fig. 2A) and brain (Fig. 2B) AZD3293 maximum concentrations were reached at 1.5 and 1.5 to 3 h, respectively. The inhibitory effect of AZD3293 on Aβ generation was also investigated in C57BL/6 mice after 7 days of twice-daily administration (100μmol/kg) and found to be equipotent to a single administration of AZD3293, with no accumulation of compound. In mice, the IC50 for brain Aβ40 reduction was estimated to be 0.6 nM (CV 8%) based on free AZD3293 concentration in brain, with an apparent Aβ40 turnover rate of approximately 2.3 h (CV 9%), similar to the rate for Aβ42. In contrast, the turnover rate for sAβPPβ was determined to be 2-fold slower.
AZD3293 concentration-time-effect relationships in guinea pigs
In guinea pigs treated with AZD3293 as a single oral administration (10, 30, or 100, μmol/kg), plasma Aβ40 appeared to reach a plateau reduction of 50% to 56% relative to vehicle-treated animals, regardless of dose, at 2 h after administration (Fig. 3B). This phenomenon has been described previously for BACE inhibitors [18]. In contrast, plasma Aβ42 was reduced (>90%) down to the LLOQ of the assay (Fig. 3C). Plasma concentrations of sAβPPβ, Aβ40, and Aβ42 were profoundly and significantly reduced over time and only returned to baseline concentrations 48 h after AZD3293 administration (Fig. 3D–F). Brain Aβ40 and Aβ42 were reduced in dose-dependent fashion 2 h after administration (Fig. 4B, C) and displayed maximum reductions (70% to 90%) at about 4 h, with concentrations returning toward baseline 24 h after dosing. Brain sAβPPβ concentrations showed time-dependent reductions after AZD3293 treatment, with maximum effect (59%) 8 h after administration (Fig. 4D). Interestingly, the brain sAβPPβ concentration was still significantly reduced (>25%) 24 h after the oral dose. In CSF, Aβ40 concentration demonstrated a dose-dependent reduction at 2 h after dosing (Fig. 5B), and the maximum effects on sAβPPβ (65%), Aβ40 (88%), and Aβ42 (90%) concentrations appeared 8 h after AZD3293 administration (Fig. 5C–E). Plasma, brain, and CSF AZD3293 concentrations were quantified at all doses and timepoints (Figs. 3A, 4A, 5A), and the mean maximum AZD3293 concentrations were reached 2 h after dosing in plasma and CSF and 2 to 4 h after dosing in the brain. The IC50 for brain Aβ40 reduction was calculated to be 0.9 nmol/L (CV 4%) in guinea pigs, based on the free AZD3293 concentration and an apparent Aβ40 turnover rate of approximately 50 min (CV 51%).
AZD3293 concentration-time-effect relationships in dogs
In dogs, after 2 weeks of repeated oral doses of 0.48, 3.4, or 24μmol/kg, maximum plasma AZD3293 concentrations were observed 2 h after dosing (Fig. 6A). Dose-dependent reductions in plasma Aβ40 concentrations were observed, with the highest dose showing a reduction of >50% (from predose) at 2 h after dosing and a reduction of >50% observed at 4 h after dosing for all doses tested. No return to baseline plasma concentration had occurred at 24 h after dosing (Fig. 6B). At 24 h after the last dose, there was a dose-dependent difference in brain (Fig. 6C) and CSF (Fig. 6E) AZD3293 concentrations. The daily-dose regimen resulted in dose-dependent reductions in Aβ40 concentrations in brain (Fig. 6D) and CSF (Fig. 6F) at 24 h after the last oral AZD3293 administration. The IC50 for Aβ40 reduction treated as direct effect was estimated to be 0.8 nmol/L (CV 9%) in the brain, based on free AZD3293 concentration in that compartment, and 3.8 nmol/L (CV 31%) in CSF.
To explore the maximum effect of AZD3293 on plasma Aβ40 concentrations and the return of this biomarker to baseline, a time-response study at doses of 12, 36, and 60μmol/kg was performed. At 2 weeks after the single dose, but not at 1 week, Aβ40 concentrations had returned to pre-dose levels (Fig. 6H). Maximum effect was observed 24 to 72 h after the dose, and the maximum reduction was irrespective of dose, indicating that the maximum effect was reached already at the lowest dose. An apparent turnover half-life of Aβ40 in plasma was estimated to be approximately 8 h (CV 13%). The free plasma AZD3293 IC50 was estimated to be approximately 0.1 nmol/L.
DISCUSSION
The in vitro and in vivo data available to date show that AZD3293 is a potent BACE1 inhibitor, with an extremely slow off-rate from hBACE1. AZD3293 has displayed time- and dose-dependent reductions of brain Aβ40 in mice, guinea pigs, and dogs as well as in other tissue types and for other markers in the Aβ pathway, i.e., plasma, brain, and CSF sAβPPβ and Aβ42. The impact of the slow off-rate from BACE1 in vitro was further studied in vivo by a prolongation of AZD3293 effect beyond its plasma and CSF exposure as well as the turnover rate of Aβ. This high-potency compound also displayed high gastrointestinal permeability, resulting in a favorable total bioavailability in dogs. These, and other desirable properties, have supported the progression of AZD3293 into a Phase 2/3 clinical study as a potential disease-modifying treatment for AD.
The very slow off-rate of AZD3293 from hBACE1 (estimated t1/2 of 9 h) may have had an impact on the observed duration of effect in vivo. In theory, a slow off-rate could result in a prolonged reduction of Aβ-pathway biomarker concentrations, now mainly driven by the turnover rate of the BACE1 protein in addition to the turnover rates of sAβPPβ, Aβ40, and Aβ42 alone. As could be estimated from the time- and dose-response studies in vivo, this phenomenon resulted in slower apparent turnover rates and slower return to baseline of sAβPPβ, Aβ40, or Aβ42 as would be predicted by the known turnover rate of these peptides. In mice, the apparent turnover rate of brain Aβ40 as determined in the time- and dose-response studies with AZD3293, was considerably longer than the turnover rate previously reported using a population modeling approach on a collection of BACE1 inhibitors [25]; 2.3 h versus 0.5 h, respectively. However, brain Aβ40 in guinea pigs had an apparent turnover rate similar to that established with AZD3293, and in the population modeling approach, approximately 50 min versus 60 min, respectively. The population modeling approach reported by Janson et al. [25] was applied to reduce the impact of individual compounds on the turnover rate estimates of the biomarkers. The estimated turnover rate in the current report is based on a single compound, and the deviation in Aβ40 turnover rate in mice but not in guinea pigs might be due to individual variation in this parameter estimate. However, in dogs, the prolonged Aβ lowering effect beyond the turnover rate of plasma Aβ40 strongly suggest additional contribution of the BACE1 enzyme turnover rate to duration of the overall Aβ suppression effect; the apparent turnover half-life of Aβ40 in plasma was estimated to be approximately 8 h, while a reduction in Aβ40 in plasma of >50% was already observed at 2 h after administration of the highest dose. May et al. [24] also reported a rapid decline in plasma Aβ concentration in dogs following oral administration of a BACE1 inhibitor, but, unfortunately, no turnover rate for Aβ in dog plasma was reported. In cellular systems, the turnover half-life of BACE1 has been determined to be >9 h [36, 37], which is longer than observed in any of the preclinical species studied in this current report. However, species differences in the off-rates of compounds from BACE1 and turnover rates of BACE1 might explain the deviations from previously estimated turnover rates of Aβ40 and Aβ42 in mice and guinea pigs. Thus, in mice and dogs, BACE1 turnover rate might contribute to the effect observed in vivo. How the very slow off-rate of AZD3293 from BACE1 affects the apparent turnover rate of Aβ and sAβPPβ in man remains to be assessed, but it is considered likely that the prolonged effect seen with AZD3293 in mice and dogs will translate to humans. One could thus speculate that, with a once-daily dosing regimen, a single missed dose in man would have a negligible effect on maintaining the reduction of Aβ concentration in the brain, which might prove important in a population of cognitively impaired patients with potential unintentional compliance issues. However, it remains to be elucidated if the magnitude of reduction in brain Aβ concentration will translate to humans and if this will result in improved cognitive function in AD patients.
In dogs, AZD3293 administration produced dose-dependent reductions in plasma, CSF, and (reported for the first time) brain Aβ40 concentrations. Due to the absence of time-resolved data in brain and CSF, only direct effects could be calculated. The resulting IC50 estimates for inhibition of Aβ production matched reasonably well between the free brain and CSF concentrations, supporting the translational value of CSF for brain Aβ40 in dogs. BACE1 inhibitors have previously been shown to reduce CSF Aβ in dogs in a time- and dose-dependent manner [24, 38, 39]. The independent reports of an effect of BACE1 inhibitors on CSF Aβ concentrations generated against human BACE1 enzyme are consistent with the highly conserved BACE1 protein homology between humans and dogs, evident from amino acid sequence alignment (not shown).
In mice and dogs, the concentrations of the main AZD3293 metabolite M1 were studied alongside those of the parent compound. In mouse plasma, the parent concentration was approximately 13-fold higher than the metabolite concentration. In dog plasma, the AUC of the parent was 8-fold higher than the AUC of the metabolite. In vitro data indicated that, in human cells, M1 was 10-fold less potent than AZD3293, but in mouse primary neurons the M1 metabolite and AZD3293 appeared equipotent. It has previously been shown that in vivo potency correlates well with in vitro potency as determined in primary neuron cells [25]. Indeed, in mice and guinea pigs, the potency estimated relative to the parent concentration in vivo (IC50 0.6 in mice and 0.9 nM in guinea pigs) correlated well with the potency determined in vitro (IC50 0.6 and 0.3 nM, respectively). Taken together, the possible involvement of the metabolite on the in vivo Aβ reduction was considered to be of only minor impact.
In addition to the drugability and slow off-rate of AZD3293, sAβPPβ was explored as an early biomarker of BACE1 inhibition by establishing time- and dose-dependent reduction of sAβPPβ after treatment with AZD3293. In the amyloid pathway, sAβPPβ is the direct product of BACE1 activity, upstream of Aβ; thus, given the technical challenges of determining Aβ concentration, sAβPPβ could be an attractive biomarker in the clinical setting. Although sAβPPβ is the direct product of BACE1 activity, in mouse and guinea pig brains, the maximum effect of AZD3293 on the biomarkers Aβ40 and Aβ42 occurred before the maximum effect on sAβPPβ, which has not been reported previously. This delay in effect relative to AZD3293 concentration was also evident from the estimated apparent turnover rates of Aβ40 and Aβ42 versus sAβPPβ, respectively. Time-response studies of CSF sAβPPβ concentrations in non-human primates [17, 40] support, in part, the concept that reduction in Aβ precedes the effect on sAβPPβ. This delay in effect on sAβPPβ relative to Aβ has an impact on future clinical study design, if sAβPPβ is to be used as a biomarker; target engagement of a BACE inhibitor on a biomarker in the Aβ pathway can only be observed when time is allowed for its degradation or secretion. However, a decreased turnover rate also results in a reduction of maximum observed effect at the same dose. Indeed, the effect on sAβPPβ is of smaller amplitude relative to the effect on brain Aβ40 and Aβ42 in mice and guinea pigs and on CSF Aβ40 and Aβ42 in guinea pigs. This phenomenon was also evident in non-human primate CSF [40]. Thus, both sampling time and dose may need to be adjusted when sAβPPβ, rather than Aβ, is to be used as a study outcome. CSF sAβPPβ concentrations can still be used as a translational compartment of effect on sAβPPβ in brain, as was established for AZD3293 in guinea pigs, with a similar pattern of turnover rates for Aβ40 and Aβ42 versus sAβPPβ in brain and CSF.
In conclusion, AZD3293 is a novel, very potent, highly permeable, orally active BACE1 inhibitor with good BBB penetration and a markedly slow off-rate from hBACE1. The slow off-rate may have had an impact on the duration of effect observed on brain sAβPPβ, Aβ40, and Aβ42 concentrations in mice and on plasma Aβ40 concentration in dogs. Although sAβPPβ is upstream of Aβ in the amyloid pathway, the effect of AZD3293 on sAβPPβ was delayed relative to the effects on Aβ40 and Aβ42 in guinea pig plasma, brain, and CSF and in mouse brain. Translation of effect from CSF to brain was further confirmed for Aβ40 and Aβ42 in guinea pigs and for sAβPPβ and Aβ40 in dogs. The in vitro and in vivo data for AZD3293 reported here increase our knowledge of both the potency and the duration of effect of this novel compound, enhance our understanding of target engagement and biomarker changes, and aid study design as AZD3293 progresses in its clinical development as a potential disease-modifying treatment for AD.
ACKNOWLEDGMENTS
The authors would like to acknowledge the talented and dedicated work done by past and present members of the BACE program, especially former colleagues from AstraZeneca R&D Södertälje. Medical editing services were provided by Pamela A. Spencer from Audubon PM Associates, Inc. and Emma Bryant, Macclesfield UK, and were funded by AstraZeneca. This research was sponsored by AstraZeneca. All authors were employees of AstraZeneca when this research was conducted. In addition, AK, GC, JJ, KK and SE report ownership of shares in AstraZeneca.
Authors’ disclosures available online (http://www.j-alz.com/manuscript-disclosures/15-0834r1).
REFERENCES
[1] | Younkin SG ((1995) ) Evidence that A beta 42 is the real culprit in Alzheimer’s disease. Ann Neurol 37: , 287–288. |
[2] | Steiner H , Capell A , Leimer U , Haass C ((1999) ) Genes and mechanisms involved in beta-amyloid generation and Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci 249: , 266–270. |
[3] | Iwatsubo T , Odaka A , Suzuki N , Mizusawa H , Nukina N , Ihara Y ((1994) ) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 13: , 45–53. |
[4] | Vassar R , Bennett BD , Babu-Khan S , Kahn S , Mendiaz EA , Denis P , Teplow DB , Ross S , Amarante P , Loeloff R , Luo Y , Fisher S , Fuller J , Edenson S , Lile J , Jarosinski MA , Biere AL , Curran E , Burgess T , Louis JC , Collins F , Treanor J , Rogers G , Citron M ((1999) ) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: , 735–741. |
[5] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[6] | Karran E , Mercken M , De Strooper B ((2011) ) The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov 10: , 698–712. |
[7] | Menting KW , Claassen JA ((2014) ) β-secretase inhibitor: A promising novel therapeutic drug in Alzheimer’s disease. Front Aging Neurosci 6: , 165–. |
[8] | Citron M , Oltersdorf T , Haass C , McConlogue L , Hung AY , Seubert P , Vigo-Pelfrey C , Lieberburg I , Selkoe DJ ((1992) ) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360: , 672–674. |
[9] | Jonsson T , Atwal JK , Steinberg S , Snaedal J , Jonsson PV , Bjornsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jönsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488: , 96–99. |
[10] | Maloney JA , Bainbridge T , Gustafson A , Zhang S , Kyauk R , Steiner P , van der Brug M , Liu Y , Ernst JA , Watts RJ , Atwal JK ((2014) ) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 289: , 30990–31000. |
[11] | Kawarabayashi T , Younkin LH , Saido TC , Shoji M , Ashe KH , Younkin SG ((2001) ) Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21: , 372–381. |
[12] | McConlogue L , Buttini M , Anderson JP , Brigham EF , Chen KS , Freedman SB , Games D , Johnson-Wood K , Lee M , Zeller M , Liu W , Motter R , Sinha S ((2007) ) Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem 282: , 26326–26334. |
[13] | Stachel SJ , Coburn CA , Sankaranarayanan S , Price EA , Wu G , Crouthamel M , Pietrak BL , Huang Q , Lineberger J , Espeseth AS , Jin L , Ellis J , Holloway MK , Munshi S , Allison T , Hazuda D , Simon AJ , Graham SL , Vacca JP ((2006) ) Macrocyclic inhibitors of beta-secretase: Functional activity in an animal model. J Med Chem 49: , 6147–6150. |
[14] | Sankaranarayanan S , Holahan MA , Colussi D , Crouthamel MC , Devanarayan V , Ellis J , Espeseth A , Gates AT , Graham SL , Gregro AR , Hazuda D , Hochman JH , Holloway K , Jin L , Kahana J , Lai MT , Lineberger J , McGaughey G , Moore KP , Nantermet P , Pietrak B , Price EA , Rajapakse H , Stauffer S , Steinbeiser MA , Seabrook G , Selnick HG , Shi XP , Stanton MG , Swestock J , Tugusheva K , Tyler KX , Vacca JP , Wong J , Wu G , Xu M , Cook JJ , Simon AJ ((2009) ) First demonstration of cerebrospinal fluid and plasma A beta lowering with oral administration of a beta-site amyloid precursor protein-cleaving enzyme 1 inhibitor in nonhuman primates. J Pharmacol Exp Ther 328: , 131–140. |
[15] | May PC , Dean RA , Lowe SL , Martenyi F , Sheehan SM , Boggs LN , Monk SA , Mathes BM , Mergott DJ , Watson BM , Stout SL , Timm DE , Smith Labell E , Gonzales CR , Nakano M , Jhee SS , Yen M , Ereshefsky L , Lindstrom TD , Calligaro DO , Cocke PJ , Greg Hall D , Friedrich S , Citron M , Audia JE ((2011) ) Robust central reduction of amyloid-beta in humans with an orally available, non-peptidic beta-secretase inhibitor. J Neurosci 31: , 16507–16516. |
[16] | Huang H , La DS , Cheng AC , Whittington DA , Patel VF , Chen K , Dineen TA , Epstein O , Graceffa R , Hickman D , Kiang YH , Louie S , Luo Y , Wahl RC , Wen PH , Wood S , Fremeau RT Jr ((2012) ) Structure- and property-based design of aminooxazoline xanthenes as selective, orally efficacious, and CNS penetrable BACE inhibitors for the treatment of Alzheimer’s disease. J Med Chem 55: , 9156–9169. |
[17] | Jeppsson F , Eketjäll S , Janson J , Karlström S , Gustavsson S , Olsson LL , Radesäter AC , Ploeger B , Cebers G , Kolmodin K , Swahn BM , von Berg S , Bueters T , Fälting J ((2012) ) Discovery of AZD3839, a potent and selective BACE1 inhibitor clinical candidate for the treatment of Alzheimer disease. J Biol Chem 287: , 41245–41257. |
[18] | Eketjäll S , Janson J , Jeppsson F , Svanhagen A , Kolmodin K , Gustavsson S , Radesäter AC , Eliason K , Briem S , Appelkvist P , Niva C , Berg AL , Karlström S , Swahn BM , Fälting J ((2013) ) AZ-4217: A high potency BACE inhibitor displaying acute central efficacy in different in vivo models and reduced amyloid deposition in Tg2576 mice. J Neurosci 33: , 10075–10084. |
[19] | Butini S , Brogi S , Novellino E , Campiani G , Ghosh AK , Brindisi M , Gemma S ((2013) ) The structural evolution of beta-secretase inhibitors: A focus on the development of small-molecule inhibitors. Curr Top Med Chem 13: , 1787–1807. |
[20] | Henderson SJ , Andersson C , Narwal R , Janson J , Goldschmidt TJ , Appelkvist P , Bogstedt A , Steffen AC , Haupts U , Tebbe J , Freskgård PO , Jermutus L , Burrell M , Fowler SB , Webster CI ((2014) ) Sustained peripheral depletion of amyloid-beta with a novel form of neprilysin does not affect central levels of amyloid-beta. Brain 137: , 553–564. |
[21] | Georgievska B , Gustavsson S , Lundkvist J , Neelissen J , Eketjäll S , Ramberg V , Bueters T , Agerman K , Juréus A , Svensson S , Berg S , Fälting J , Lendahl U ((2015) ) Revisiting the peripheral sink hypothesis: Inhibiting BACE1 activity in the periphery does not alter beta-amyloid levels in the CNS. J Neurochem 132: , 477–486. |
[22] | Vassar R ((2014) ) BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther 6: , 89. |
[23] | Sparve E , Quartino AL , Lüttgen M , Tunblad K , Gårdlund AT , Fälting J , Alexander R , Kågström J , Sjödin L , Bulgak A , Al-Saffar A , Bridgland-Taylor M , Pollard C , Swedberg MD , Vik T , Paulsson B ((2014) ) Prediction and modeling of effects on the QTc interval for clinical safety margin assessment, based on single-ascending-dose study data with AZD3839. J Pharmacol Exp Ther 350: , 469–478. |
[24] | May PC , Willis BA , Lowe SL , Dean RA , Monk SA , Cocke PJ , Audia JE , Boggs LN , Borders AR , Brier RA , Calligaro DO , Day TA , Ereshefsky L , Erickson JA , Gevorkyan H , Gonzales CR , James DE , Jhee SS , Komjathy SF , Li L , Lindstrom TD , Mathes BM , Martenyi F , Sheehan SM , Stout SL , Timm DE , Vaught GM , Watson BM , Winneroski LL , Yang Z , Mergott DJ ((2015) ) The potent BACE1 inhibitor LY2886721 elicits robust central Abeta pharmacodynamic responses in mice, dogs, and humans. J Neurosci 35: , 1199–1210. |
[25] | Janson J , Eketjäll S , Tunblad K , Jeppsson F , Von Berg S , Niva C , Radesäter AC , Fälting J , Visser SA ((2014) ) Population PKPD modeling of BACE1 inhibitor-induced reduction in Abeta levels in vivo and correlation to in vitro potency in primary cortical neurons from mouse and guinea pig. Pharm Res 31: , 670–683. |
[26] | Swahn BM , Kolmodin K , Karlström S , von Berg S , Soderman P , Holenz J , Berg S , Lindström J , Sundström M , Turek D , Kihlström J , Slivo C , Andersson L , Pyring D , Rotticci D , Öhberg L , Kers A , Bogar K , von Kieseritzky F , Bergh M , Olsson LL , Janson J , Eketjall S , Georgievska B , Jeppsson F , Fälting J ((2012) ) Design and synthesis of beta-site amyloid precursor protein cleaving enzyme (BACE1) inhibitors with in vivo brain reduction of beta-amyloid peptides. J Med Chem 55: , 9346–9361. |
[27] | Borgegård T , Gustavsson S , Nilsson C , Parpal S , Klintenberg R , Berg AL , Rosqvist S , Serneels L , Svensson S , Olsson F , Jin S , Yan H , Wanngren J , Juréus A , Ridderstad-Wollberg A , Wollberg P , Stockling K , Karlström H , Malmberg A , Lund J , Arvidsson PI , De Strooper B , Lendahl U , Lundkvist J ((2012) ) Alzheimer’s disease: Presenilin 2-sparing gamma-secretase inhibition is a tolerable Abeta peptide-lowering strategy. J Neurosci 32: , 17297–17305. |
[28] | Strömberg K , Hansson EM , Laudon H , Bergstedt S , Naslund J , Lundkvist J , Lendahl U ((2005) ) gamma-Secretase complexes containing N- and C-terminal fragments of different presenilin origin retain normal gamma-secretase activity. J Neurochem 95: , 880–890. |
[29] | Gravenfors Y , Viklund J , Blid J , Ginman T , Karlström S , Kihlström J , Kolmodin K , Lindström J , von Berg S , von Kieseritzky F , Bogar K , Slivo C , Swahn BM , Olsson LL , Johansson P , Eketjäll S , Fälting J , Jeppsson F , Strömberg K , Janson J , Rahm F ((2012) ) New aminoimidazoles as beta-secretase (BACE-1) inhibitors showing amyloid-beta (Abeta) lowering in brain. J Med Chem 55: , 9297–9311. |
[30] | Floby E , Briem S , Terelius Y , Sohlenius-Sternbeck AK ((2004) ) Use of a cocktail of probe substrates for drug-metabolizing enzymes for the assessment of the metabolic capacity of hepatocyte preparations. Xenobiotica 34: , 949–959. |
[31] | Fridén M , Ducrozet F , Middleton B , Antonsson M , Bredberg U , Hammarlund-Udenaes M ((2009) ) Development of a high-throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab Dispos 37: , 1226–1233. |
[32] | Di L , Umland JP , Chang G , Huang Y , Lin Z , Scott DO , Troutman MD , Liston TE ((2011) ) Species independence in brain tissue binding using brain homogenates. Drug Metab Dispos 39: , 1270–1277. |
[33] | Alelyunas YW , Liu R , Pelosi-Kilby L , Shen C ((2009) ) Application of a Dried-DMSO rapid throughput 24-h equilibrium solubility in advancing discovery candidates. Eur J Pharm Sci 37: , 172–182. |
[34] | Borgegård T , Minidis A , Jureus A , Malmborg J , Rosqvist R , Gruber S , Almqvist H , Yan H , Bogstedt A , Olsson F , Dahlström J , Ray C , Närhi K , Malinowsky D , Hagström E , Jin S , Malm Å ((2011) ) In vivo analysis using a presenilin-1-specific inhibitor: Presenilin1-containingγ-secretase complexes mediate the majority of CNS Aβ production in the mouse. Alzheimers Dis Res J 3: , 29–45. |
[35] | Post TM , Freijer JI , Ploeger BA , Danhof M ((2008) ) Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn 35: , 185–202. |
[36] | Puglielli L , Ellis BC , Saunders AJ , Kovacs DM ((2003) ) Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J Biol Chem 278: , 19777–19783. |
[37] | Cole SL , Vassar R ((2007) ) The Alzheimer’s disease beta-secretase enzyme, BACE1.22. Mol Neurodegener 2: –. |
[38] | Borghys H , Jacobs T , Van Broeck B , Dillen L , Dhuyvetter D , Gijsen H , Mercken M ((2014) ) Comparison of two different methods for measurement of amyloid-beta peptides in cerebrospinal fluid after BACE1 inhibition in a dog model. J Alzheimers Dis 38: , 39–48. |
[39] | Butler CR , Brodney MA , Beck EM , Barreiro G , Nolan CE , Pan F , Vajdos F , Parris K , Varghese AH , Helal CJ , Lira R , Doran SD , Riddell DR , Buzon LM , Dutra JK , Martinez-Alsina LA , Ogilvie K , Murray JC , Young JM , Atchison K , Robshaw A , Gonzales C , Wang J , Zhang Y , O’Neill BT ((2015) ) Discovery of a series of efficient, centrally efficacious BACE1 inhibitors through structure-based drug design. J Med Chem 58: , 2678–2702. |
[40] | Dobrowolska JA , Michener MS , Wu G , Patterson BW , Chott R , Ovod V , Pyatkivskyy Y , Wildsmith KR , Kasten T , Mathers P , Dancho M , Lennox C , Smith BE , Gilberto D , McLoughlin D , Holder DJ , Stamford AW , Yarasheski KE , Kennedy ME , Savage MJ , Bateman RJ ((2014) ) CNS amyloid-beta, soluble APP-alpha and -beta kinetics during BACE inhibition. J Neurosci 34: , 8336–8346. |
Figures and Tables
Fig.1
hBACE1 binding kinetics of AZD3293. The binding of AZD3293 was evaluated in a direct binding assay (A) and in a displacement assay (B) in a Biacore 4000. AZD3293 was further characterized with single-cycle kinetics in a Biacore T200 (C). AZD3293 dissociated significantly slower than AZD3839 (A). The reversibility of AZD3293 binding was confirmed in the displacement experiment with AZD3839 (B). After 6 start-up cycles (not shown), AZD3839 was injected twice to determine the initial binding capacity of the surface (1) followed by AZD3293 which was injected twice to fully occupy the binding pocket (2). AZD3839 was then injected every 60 min over a 20 h period to gradually replace AZD3293 (3). Each injection of compound (cycle time 30 min) was followed by an injection of running buffer as negative control (cycle time 30min). The time to displace AZD3293, monitored as the time for AZD3839 to regain a stable binding level, was estimated to be approximately 15 h (30 cycles).
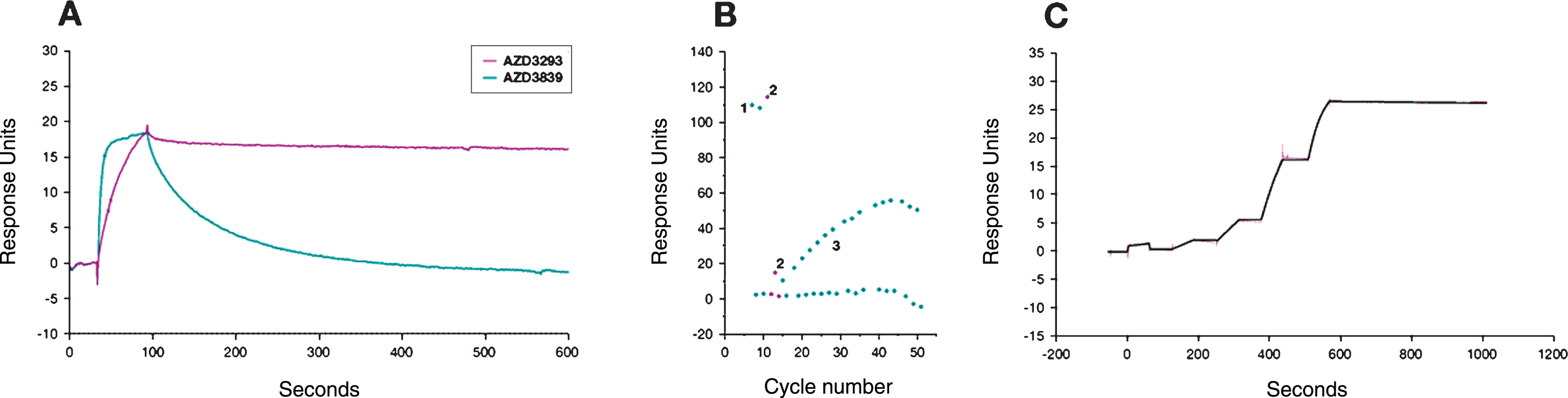
Fig.2
Time- and dose-dependent inhibition of Aβ generation in C57BL/6 mice. After a single oral administration of AZD3293, free plasma AZD3293 concentration (Cp free) peaked around 1.5 h after dosing (A). Free brain AZD3293 concentrations (Cbr free) peaked between 1.5 and 3 h after administration; all samples were above the lower limit of quantification (LLOQ) of the assay only at 1.5 h after the highest dose (100μmol/kg; B). All doses tested (50, 100, and 200μmol/kg [not shown]) reduced plasma Aβ40 to the LLOQ of the assay at 1.5 h after administration. Time-response data showed that the plasma Aβ40 concentrations were reduced ≥40% 1.5 to 8 h after AZD3293 administration. At 16 h after the dose, concentrations were returning to baseline but were still significantly reduced (C). At the high dose (100μmol/kg), brain sAβPPβ was significantly reduced from 3 to 8 h after administration (D). Brain Aβ40 and Aβ42 displayed dose-dependent (50, 100, and 200μmol/kg [not shown]) reductions 1.5 h after dosing (E and F). Brain Aβ40 and Aβ42 concentrations were significantly reduced at all timepoints studied, except at 16 h where the concentrations had returned to baseline (E and F). Data are presented as Mean±SEM.
![Time- and dose-dependent inhibition of Aβ generation in C57BL/6 mice. After a single oral administration of AZD3293, free plasma AZD3293 concentration (Cp free) peaked around 1.5 h after dosing (A). Free brain AZD3293 concentrations (Cbr free) peaked between 1.5 and 3 h after administration; all samples were above the lower limit of quantification (LLOQ) of the assay only at 1.5 h after the highest dose (100μmol/kg; B). All doses tested (50, 100, and 200μmol/kg [not shown]) reduced plasma Aβ40 to the LLOQ of the assay at 1.5 h after administration. Time-response data showed that the plasma Aβ40 concentrations were reduced ≥40% 1.5 to 8 h after AZD3293 administration. At 16 h after the dose, concentrations were returning to baseline but were still significantly reduced (C). At the high dose (100μmol/kg), brain sAβPPβ was significantly reduced from 3 to 8 h after administration (D). Brain Aβ40 and Aβ42 displayed dose-dependent (50, 100, and 200μmol/kg [not shown]) reductions 1.5 h after dosing (E and F). Brain Aβ40 and Aβ42 concentrations were significantly reduced at all timepoints studied, except at 16 h where the concentrations had returned to baseline (E and F). Data are presented as Mean±SEM.](https://content.iospress.com:443/media/jad/2016/50-4/jad-50-4-jad150834/jad-50-jad150834-g002.jpg)
Fig.3
Time- and dose-dependent inhibition of Aβ generation in guinea pig plasma after a single oral AZD3293 dose. Plasma AZD3293 concentrations (Cp) peaked around 2 h after administration (A), and all doses tested (10, 30, and 100μmol/kg) significantly reduced both Aβ40 and Aβ42, with the Aβ42 concentrations diminished below the lower limit of quantification (LLOQ) of the assay (B and C). The maximum effects on plasma sAβPPβ (D), Aβ40 (E), and Aβ42 (F) were observed at around 8 h after dosing. The effects were sustained, with concentrations not returning to baseline until more than 48 h after dosing. Data are presented as Mean±SEM ( *p < 0.05; **p < 0.01, ***p < 0.001, compared with vehicle).
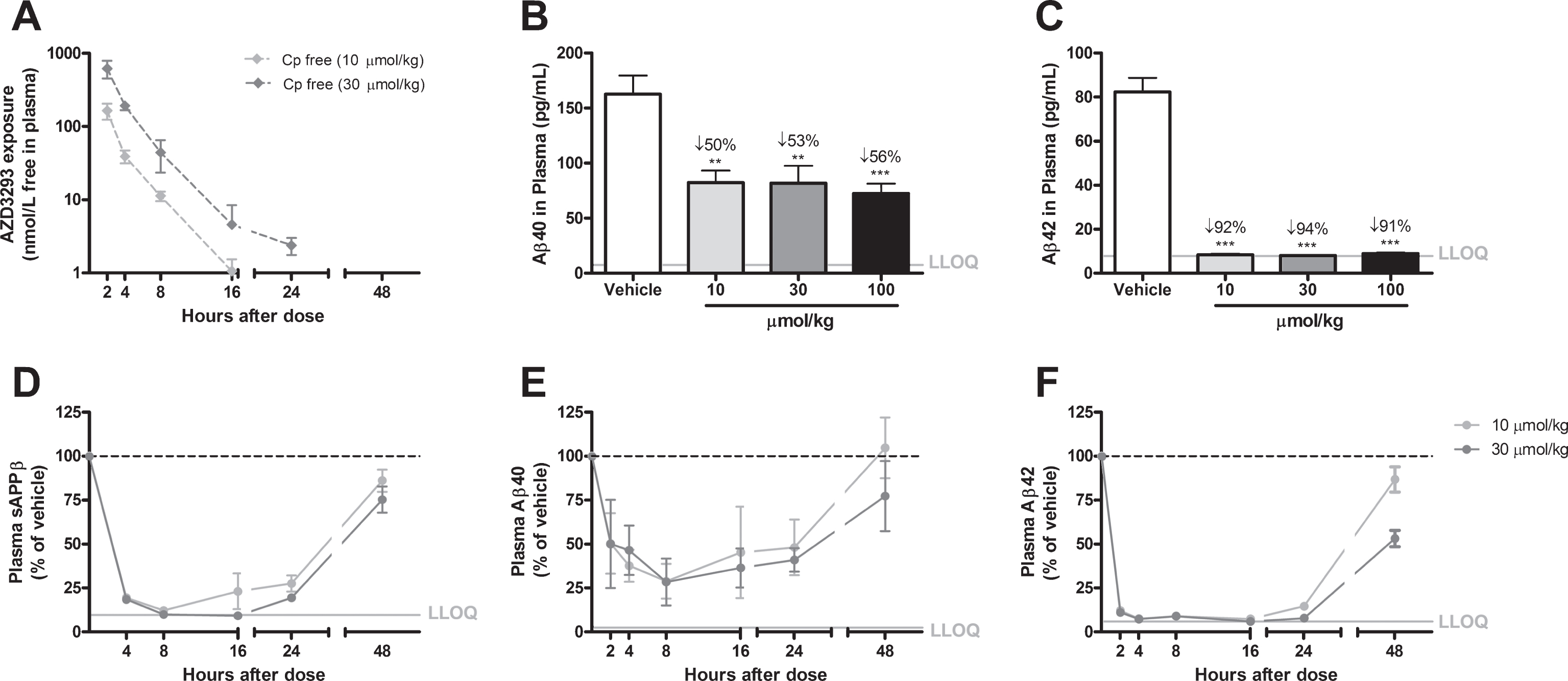
Fig.4
Time- and dose-dependent inhibition of Aβ generation in guinea pig brain after a single oral AZD3293 dose. Free brain AZD3293 concentration (Cbr free) peaked around 2 h after a single administration of AZD3293 (A). Dose-dependent reductions in Aβ40 and Aβ42 were observed at 2 h after administration (10, 30, or 100μmol/kg) (B and C for Aβ40 and Aβ42, respectively). The time-dependent reduction in brain sAβPPβ reached maximum effect at around 8 h after dosing and, after a small increase, appeared to be almost stable for up to 24 h after the single administration (D). The time-dependent reductions in Aβ40 (E) and Aβ42 (F) reached maximum effect between 4 and 8 h after dosing, and concentrations appeared to have returned to baseline at 24 h for the lower dose. Data are presented as Mean±SEM ( *p < 0.05; **p < 0.01, ***p < 0.001, compared with vehicle). Lower limit of quantification (LLOQ).
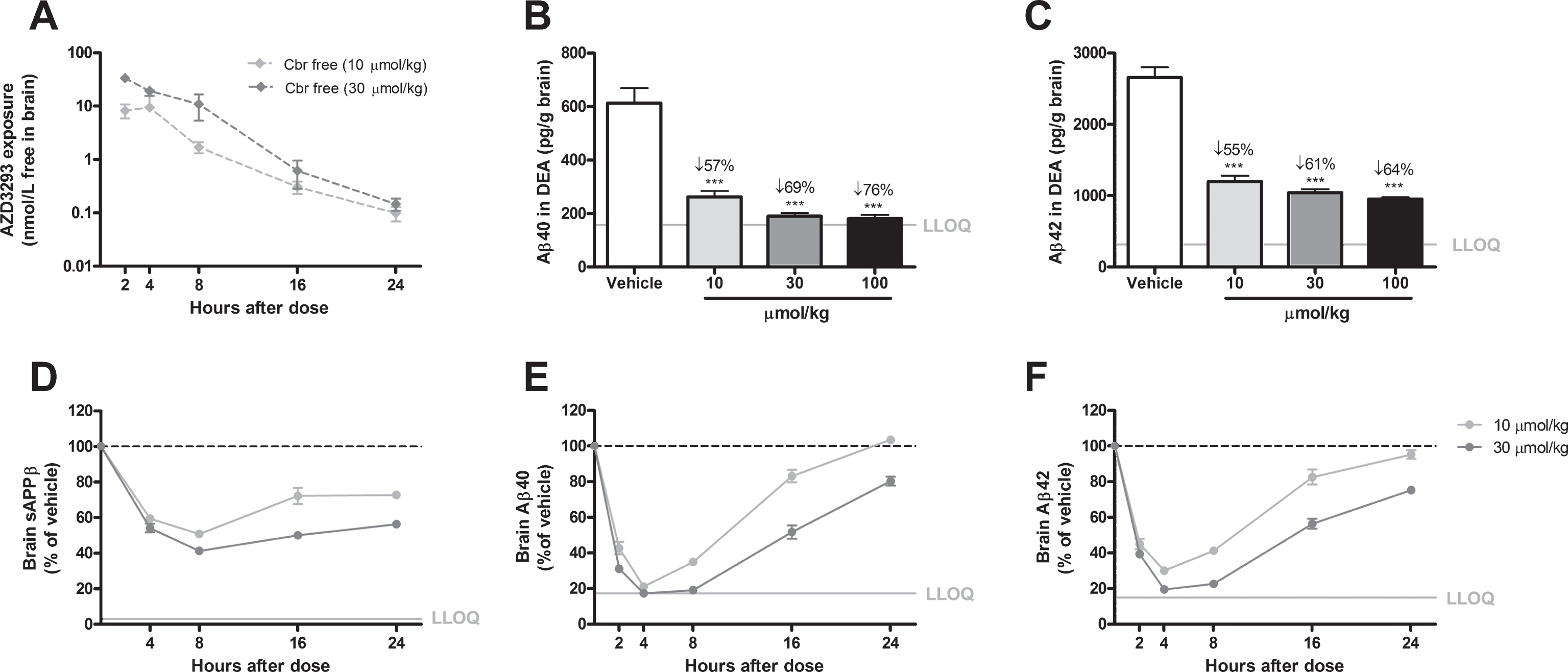
Fig.5
Time- and dose-dependent inhibition of CSF Aβ in guinea pigs after a single administration of AZD3293. Maximum CSF AZD3293 concentration was observed at 2 h after dosing (A). Dose-dependent reduction of Aβ40 was studied at 2 h after a single administration of AZD3293, with the 2 highest doses (30 and 100μmol/kg) resulting in significant effect (B). Maximum effect on CSF sAβPPβ was observed at around 8 h after the dose, and the effect was sustained for up to 24 h (C). CSF Aβ40 (D) and Aβ42 (E) displayed maximum concentrations between 4 and 8 h after AZD3293 administration. CSF Aβ40 appeared to have returned to baseline at 24 h. Data are presented as Mean±SEM ( *p < 0.05; **p < 0.01, ***p < 0.001, compared with vehicle). Lower limit of quantification(LLOQ).
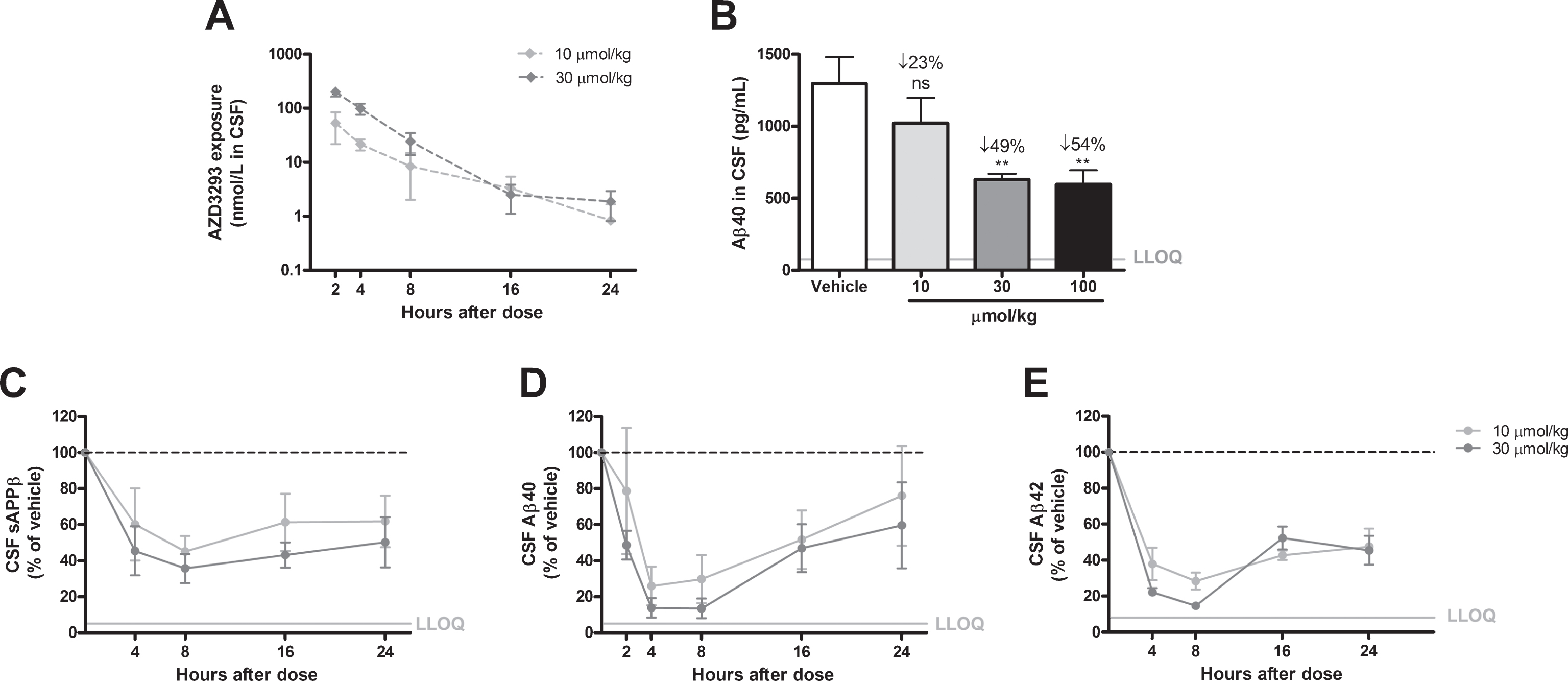
Fig.6
Time- and dose-dependent inhibition of plasma, CSF, and brain Aβ in dogs after oral administration of AZD3293. The dose regimens included 0.48, 3.4, or 24μmol/kg (0.2, 1.4, or 10 mg/kg) daily for 2 weeks (A–F) and a single dose of 12, 36, or 60μmol/kg (5, 15, or 25 mg/kg) (G, H). Maximum plasma AZD3293 concentration was observed at 2 h after dosing (A). AZD3293 resulted in time- and dose-dependent effects on plasma Aβ40 (B). A dose-dependent increase in free brain AZD3293 concentration (hippocampus, C) and reduction in hippocampus Aβ40 (D) were observed at 24 h after the last dose. A dose-dependent increase in CSF exposure (E) and a reduction in brain Aβ40 (G) at 24 h after the last dose were established. At doses of 12, 36, or 60μmol/kg, plasma AZD3293 concentrations increased with dose (G), while the increase in dose did not result in increased effect with Aβ40 concentrations in plasma as read-out (H). Data are presented as Mean±SEM ( *p < 0.05; **p < 0.01, ***p < 0.001, compared with low dose). Lower limit of quantification (LLOQ).
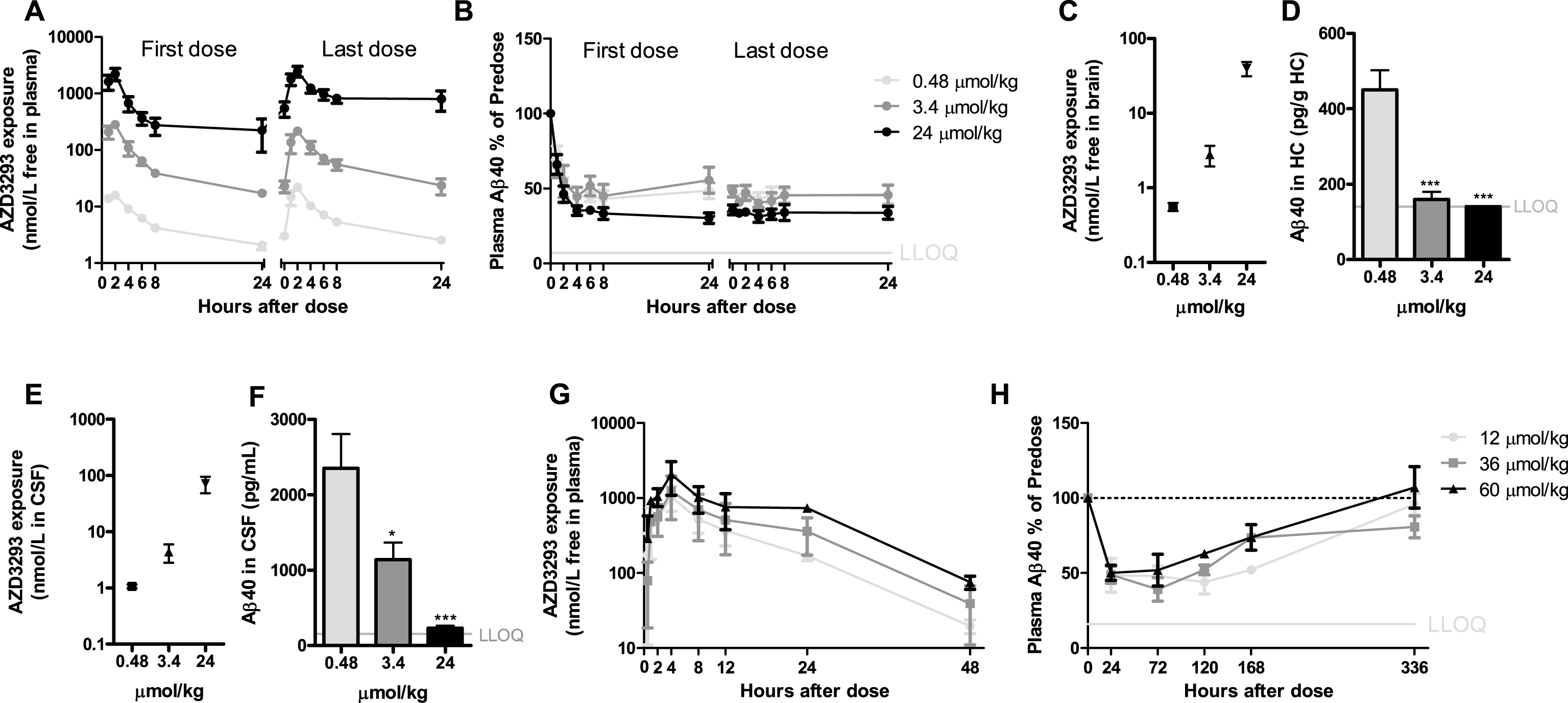
Table 1
AZD3293 in vitro BACE1 potency, selectivity, plasma protein and blood cell binding, solubility, and permeability
| Study | Results |
| Target potency, nM±SEM | |
| hBACE1 affinity, TR-FRET (Ki / IC50) | 0.4±0.03 / 0.6±0.04 (n = 4) |
| SH-SY5Y/AβPP, Aβ40 readout (IC50) | 0.08±0.004 (n = 12) |
| SH-SY5Y, sAβPPβ readout (IC50) | 0.1±0.01 (n = 6) |
| HEK293/AβPPswe, Aβ42 readout (IC50) | 0.37±0.04 (n = 5) |
| N2A, Aβ40 readout (IC50) | 0.29±0.02 (n = 4) |
| Mouse primary neurons, Aβ40 readout (IC50) | 0.61±0.09 (n = 6) |
| Tg2576 primary neurons, hAβ40 readout (IC50) | 78.0±3.2 (n = 3) |
| Guinea pig primary neurons, Aβ40 readout (IC50) | 0.31±0.03 (n = 9) |
| Inhibition mechanism | Competitive &Reversible (Off-rate, t1/2 ∼ 9h) |
| sAβPPα/β profile | sAβPPα increase, sAβPPβ decrease |
| Selectivity, nM±SEM | |
| hBACE2 affinity, TR-FRET (Ki / IC50) | 0.8 / 0.9 (n = 2) |
| hCathepsin D affinity, TR-FRET (Ki / IC50) | 3797±17 / 16100±767 (n = 10) |
| γ-secretase Notch cleavage (IC50) | >25,000 (n = 2) |
| hERG (IC50) | 4700±0.25 (n = 4) |
| Plasma protein and blood cell binding | |
| Mouse, % free | 1.6±0.1 (n = 9) |
| Rat, % free | 5.0±0.3 (n = 9) |
| Guinea pig, % free | 9.2±0.3 (n = 9) |
| Dog, % free | 9.9±0.2 (n = 8) |
| Human, % free | 8.7±0.3 (n = 9) |
| Plasma/blood cell partitioning, ratio in human | 0.7 (n = 9) |
| Solubility, μM | 35.3±6.2 (n = 3) |
| Permeability | |
| Caco-2, apical-to-basolateral Papp (10–6 cm/s) | 34.8±6.8 (n = 6) |
| Caco-2, efflux ratio | 0.7 (n = 2) |
| MDCK, efflux ratio | 10 (n = 1) |
Table 2
AZD3293 metabolite M1 potency, plasma protein binding, and permeability in a human and mouse cellular AβPP processing assay.
| Study | Results |
| Target potency, nM | |
| SH-SY5Y, sAβPPβ readout (IC50) | 1.0 (n = 2) |
| Mouse primary neurons, Aβ40 readout (IC50) | 0.8 (n = 2) |
| Selectivity, nM | |
| hERG | >33,000 (n = 1) |
| Plasma protein binding, % free | |
| Mouse | 34 (n = 1) |
| Rat | 19 (n = 1) |
| Guinea pig | 40 (n = 1) |
| Dog | 30 (n = 1) |
| Human | 37 (n = 2) |
| Permeability | |
| CaCo-2, efflux ratio | 7.9 (n = 1) |
| MDCK, efflux ratio | 32 (n = 2) |

