
Diabetic Ketoacidosis Induces Tau Hyperphosphorylation in Rat Brain
Abstract
Background:
Diabetes mellitus (DM) increases the risk for cognitive impairment and Alzheimer’s disease (AD). Diabetic ketoacidosis (DKA), a serious complication of DM, may also cause brain damage and further AD, but the underlying molecular mechanisms remain unclear.
Objective:
Our objective was to understand how DKA can promote neurodegeneration in AD.
Methods:
We induced DKA in rats through intraperitoneal injection of streptozotocin, followed by starvation for 48 hours and investigated AD-related brain alterations focusing on tau phosphorylation.
Results:
We found that DKA induced hyperphosphorylation of tau protein at multiple sites associated with AD. Studies of tau kinases and phosphatases suggest that the DKA-induced hyperphosphorylation of tau was mainly mediated through activation of c-Jun N-terminal kinase and downregulation of protein phosphatase 2A. Disruption of the mTOR-AKT (the mechanistic target of rapamycin−protein kinase B) signaling pathway and increased levels of synaptic proteins were also observed in the brains of rats with DKA.
Conclusions:
These results shed some light on the mechanisms by which DKA may increase the risk for AD.
INTRODUCTION
Diabetic ketoacidosis (DKA) is a serious complication of diabetes mellitus (DM) that can lead to diabetic coma. DKA is common in type 1 diabetes mellitus (T1DM) and is primarily caused by a severe lack of insulin, which results in high glucose levels and accumulation of ketones in the blood. In addition to numerous metabolic disorders, recurrent DKA in DM can lead to morphological and functional brain impairments that are associated with neurocognitive outcomes [1]. Substantial evidence suggests that DKA can cause different types of brain damage. Memory deficits are observed in children with T1DM plus DKA diagnosis as compared to those with only T1DM [2]. The cause of these deficits remains unclear, in part because T1DM often has other complications that may pose multiple risks for brain damage and cognitive impairment. Although the effects of T1DM on memory have been investigated previously [3–6], the underlying molecular mechanism is not understood.
DKA is also reported to increase the risk for Alzheimer’s disease (AD) in patients with type 2 diabetes mellitus (T2DM) [7]. It has been demonstrated in diabetic rats that DKA can induce deficits in learning and memory associated with astrogliosis [8, 9]. There is increasing evidence to support that DM is associated with neurological disorders. Patients with AD have shown a reduction of insulin in cerebrospinal fluid, impairment in insulin signaling, and reduction in glucose metabolism in the brain [10–15]. The impairment of brain insulin signaling appears to contribute to neurodegeneration and cognitive impairment by promoting abnormal hyperphosphorylation of tau, which may underlie neurodegeneration and cognitive deficits in AD and other tauopathies[16–20].
Under diabetic conditions, especially with DKA, hyperglycemia, free fatty acids, and cytokines produce an excess of reactive oxygen species [21], which in turn can induce impairment in synapses, synaptic plasticity, hippocampus-dependent learning and memory, and adult neurogenesis. The brain damage is associated with structural lesions, including axonal loss and neuronal death [21–23]. In addition, DKA is associated with brain changes that are also observed in hypoxic and ischemic brains, such as a decrease in cerebral blood flow, high levels of energy phosphates, and low pH [24–26]. Cognitive impairment with poorer performance on executive function was observed in older adults with T1DM with recurrent DKA [27]. Therefore, multiple mechanisms are likely involved in the increased risk for cognitive impairment and AD in DM, and DKA might add further insult to the brain.
In the present study, we induced DKA and investigated the subsequent brain alterations, focusing on tau phosphorylation and other changes involved in AD neurodegeneration and cognitive impairment in rats. We found that DKA resulted in hyperphosphorylation of tau and activation of several tau kinases. These findings provide some insights into the molecular mechanisms by which DKA may promote neurodegeneration and cognitiveimpairment.
MATERIALS AND METHODS
Animals and induction of DKA
This study was conducted according to the protocol approved by the Institutional Animal Care and Use Committee of the New York State Institute for Basic Research in Developmental Disabilities and was in accordance with the Guidelines by the NIH Office of Laboratory Animal Welfare. DKA was induced in female Wistar rats (weight approximately 300 g) by a single intraperitoneal injection of 150 mg/kg streptozotocin (STZ) followed by starvation for 48 h according to an established method previously described [28]. STZ was dissolved in a 100-mM citrate buffer, pH 4.5. Control rats were injected with a citrate buffer only. DKA in rats was confirmed by measuring ketoacids (acetoacetate) and pH in urine with Siemens Multistix 7 (Fisher Scientific, Santa Clara, CA) and tail blood glucose levels with a commercial glucometer. Only female rats were employed for this initial study due to the limited resources. Similar studies in male rats will be carried out in thefuture.
Rats were sacrificed by decapitation 48 h after STZ or control injection, and the hippocampi and the whole cerebral cortex tissue were dissected from the rat brains immediately and stored at –80°C until being used. The brain tissue was homogenized in cold homogenization buffer containing 50 mM Tris-HCl, pH 7.4, 8.5% sucrose, 2 mM EDTA, 2 mM EGTA, 10 mM β-mercaptoethanol, 5 mM benzamidine, 0.5 mM 4-(2-aminoethyl) benzenesulfonyl fluoride, 4 μg/ml pepstatin A, and 10 μg/ml each of aprotinin and leupeptin, 20 mM β-glycerolphosphate, 100 mM sodium fluoride, 1 mM sodium vanadate, and 100 nM okadaic acid. The homogenates 10% (w/v) were stored at –80°C in aliquots, and the protein concentrations of the homogenate samples were determined by using Pierce™ 660 nm Protein Assay Reagent (Thermo Scientific, Rockford, IL), according to the manufacturer’s instructions.
Western blots
The homogenate samples were resolved in 10 or 15% SDS-PAGE, followed by electro-transfer to PVDF membrane of 0.45 μm pore size. After initial blocking of the membrane with 5% skim milk, the blots were incubated with primary antibodies (Table 1) diluted in 5% skim milk at 4°C overnight. The blots were then probed with the corresponding anti-mouse or anti-rabbit (1 : 5000; Jackson ImmunoResearch, West Grove, PA) or anti-sheep HRP secondary antibodies (1 : 1000; Invitrogen, Waltham, MA), and finally detected by using enhanced chemiluminescence (ECL) reagents (Thermo Scientific). Multigauge V3 software (Fuji Photo Film, Tokyo, Japan) was used to quantify the specific protein bands on western blots.
Table 1
Primary antibodies used in this study
| Antibody | Type | MW | Specificity | Source | Catalog # |
| CAMK II | Mouse | 50KD | CaMKII | Santa Cruz | sc-5306 |
| CAMK II α-Subunit | Mouse | 50KD | p-CaMKII (Thr286) | Calbiochem | NB13 |
| GSK-3β | Rabbit | 46KD | GSK-3β | Cell signaling | 27c10 |
| P-GSK-3β | Rabbit | 46KD | p-GSK-3β (S9) | Cell signaling | 9336 |
| MAPK (Erk1/2) | Rabbit | 42/44 | Total p44/42 MAP Kinase (Erk1/Erk2) | Cell signaling | 9102 |
| P-MAPK (Erk1/2) (197G2) | Rabbit | 42/44 | p-p44/p42 MAP Kinase (Thr202/Tyr204) | Cell signaling | 4377 |
| Cdk5 (DC17) | Mouse | 34 | Cdk5 | Santa Cruz | sc-249 |
| P-Cdk5 (C7) | Mouse | 34/37 | p-Cdk5 (Ser159) | Santa Cruz | sc-377558 |
| p35 (4G11) | Mouse | 35 | Cdk5 regulatory subunit, p35 | Santa Cruz | sc-293184 |
| AMPK | Rabbit | 62 | Total AMPK | Cell signaling | 2532 |
| P-AMPK (40H9) | Rabbit | 62 | p-AMPK (Thr172) | Cell signaling | 2535 |
| AKT (pan) (11E7) | Rabbit | 60 | Total AKT | Cell signaling | 4685 |
| P-AKT (193H12) | Rabbit | 60 | p-AKT (Ser473) | Cell signaling | 4058 |
| JNK2 (56G8) | Rabbit | 46/54 | Total JNK2 | Cell signaling | 9258 |
| P-SAPK/JNK | Rabbit | 46/55 | p-JNK (Thr183/Tyr185) | Cell signaling | 9251 |
| P13 Kinase p85 | Rabbit | 85 | P13 Kinase p85 not p50 or p55 | Cell signaling | 4292 |
| P-P13 kinase p85 | Rabbit | 85 | p-p85/p55 (Tyr458/TYr199) | Cell signaling | 4228 |
| PDK | Rabbit | 58/68 | Total PDK | Cell signaling | 3062 |
| P-PDK | Rabbit | 58/69 | p-PDK (S241) | Cell signaling | 3061 |
| mTOR (7C10) | Rabbit | 289 | Total mTOR | Cell signaling | 2983 |
| P-mTOR (D9C2) | Rabbit | 289 | p-mTOR (Ser2448) | Cell signaling | 5536 |
| PP2A (46/PP2A catalytic α) | Mouse | 36 | PP2Ac | BD Trans. Lab | 610555 |
| Demethylated PP2A-C (4i57) | Mouse | 36 | Unmethylated PP2Ac subunit | Santa Cruz | sc-80990 |
| Synpsin-1 | Rabbit | 80 | Synpsin-1 | Stressgen | VAP-SV060 |
| PSD 95 | Rabbit | 95 | PSD 95 | Cell signaling | 3450 |
| NMDA-R2B | Rabbit | 190 | NMDA-R2B | Cell signaling | 4207 |
| R92e | Rabbit | 50–75 | pan tau | Pei et al. (1998) | |
| anti pS199 | Rabbit | 50–75 | p-tau (S199) | Invitrogen | 44734G |
| anti-pT205 | Rabbit | 50–75 | p-tau (T205) | Invitrogen | 44738G |
| anti-pT212 | Rabbit | 50–75 | p-tau (T212) | Invitrogen | 44740G |
| anti-pS214 | Rabbit | 50–75 | p-tau (S214) | Invitrogen | 44742G |
| anti-p262 | Rabbit | 50–75 | p-tau (S262) | Invitrogen | 44750G |
| PHF-1 | Mouse | 50–75 | p-tau (S396/404) | Dr. P. Davis | |
| anti-PS409 | Rabbit | 50–75 | p-tau (S409) | Biosource | 44-760G |
| GAPDH | Rabbit | 37 | GAPDH | Sigma | G9545 |
Immuno-dot blots
The cerebral cortical homogenates were centrifuged at 5,000 g at 4°C for 5 min, and the supernatants were collected and adjusted to the same protein concentration with the homogenate buffer, followed by dilution to 1.0 μg protein/μl with Tris-buffered saline (TBS). The amount of 5.0 μl each sample was doted in duplicates onto nitrocellulose paper. After drying at 37°C, the blots were blocked with 5% Carnation milk in TBS solution for 30 min. The blots were then probed with rabbit anti-4-HNE (Abcam, Cat# AB46545) or, as a loading control, rabbit anti-GAPDH (Sigma, Cat # G9545). The dot-blots were finally developed with ECL reagents (PierceThermal Scientific) and quantified by densitometry using ImageJ 1.54 g.
Statistical analysis
To analyze the densitometric data from Western blots, a Student’s t-test was performed for the statistical analysis between the two groups. Correlation analysis was performed by the Spearman correlation test. All data were analyzed by employing Prisma 7 statistical software (GraphPad Software, Inc., Boston, MA). All data were expressed as means±SEM. Differences between the means were treated as statistically significant if p < 0.05.
RESULTS
Verification of DKA in rats
To verify the success of DKA induction with STZ injection and starvation, we tested the levels of blood glucose, urine ketoacids (acetoacetate), and urine pH values at various time points post–STZ injection. The results (Table 2) confirmed that the rats had DKA 48 h post–STZ injection and starvation.
Table 2
Rat blood glucose levels, urine ketone levels, and pH values
| Rat # | Time (h) post STZ injection | ||||||||
| Blood glucose (mg/dL) | Urine ketones* (mg/dL) | Urine pH value* | |||||||
| 0 | 24 | 48 | 0 | 24 | 48 | 0 | 24 | 48 | |
| 1 | 122 | 203 | 410 | Neg | Neg | 40 | 7 | 7 | 6 |
| 2 | 123 | 129 | 363 | Neg | Neg | ≥160 | 7 | 7 | 6 |
| 3 | 128 | 201 | 382 | Neg | Neg | ≥160 | 7 | 7 | 6 |
| 4 | 139 | 281 | 287 | Neg | Neg | ≥160 | 7 | 7 | 6 |
| 5 | 131 | 222 | 395 | Neg | Neg | ≥160 | 7 | 6 | 6 |
| 6 | 134 | 202 | 370 | Neg | Neg | ≥160 | 7 | 6 | 6 |
| 7 | 122 | 232 | 366 | Neg | Neg | ≥160 | 7 | 7 | 6 |
*Values were taken from Multistix as indicated by manufacturer’s scale.
Tau hyperphosphorylation induced by DKA
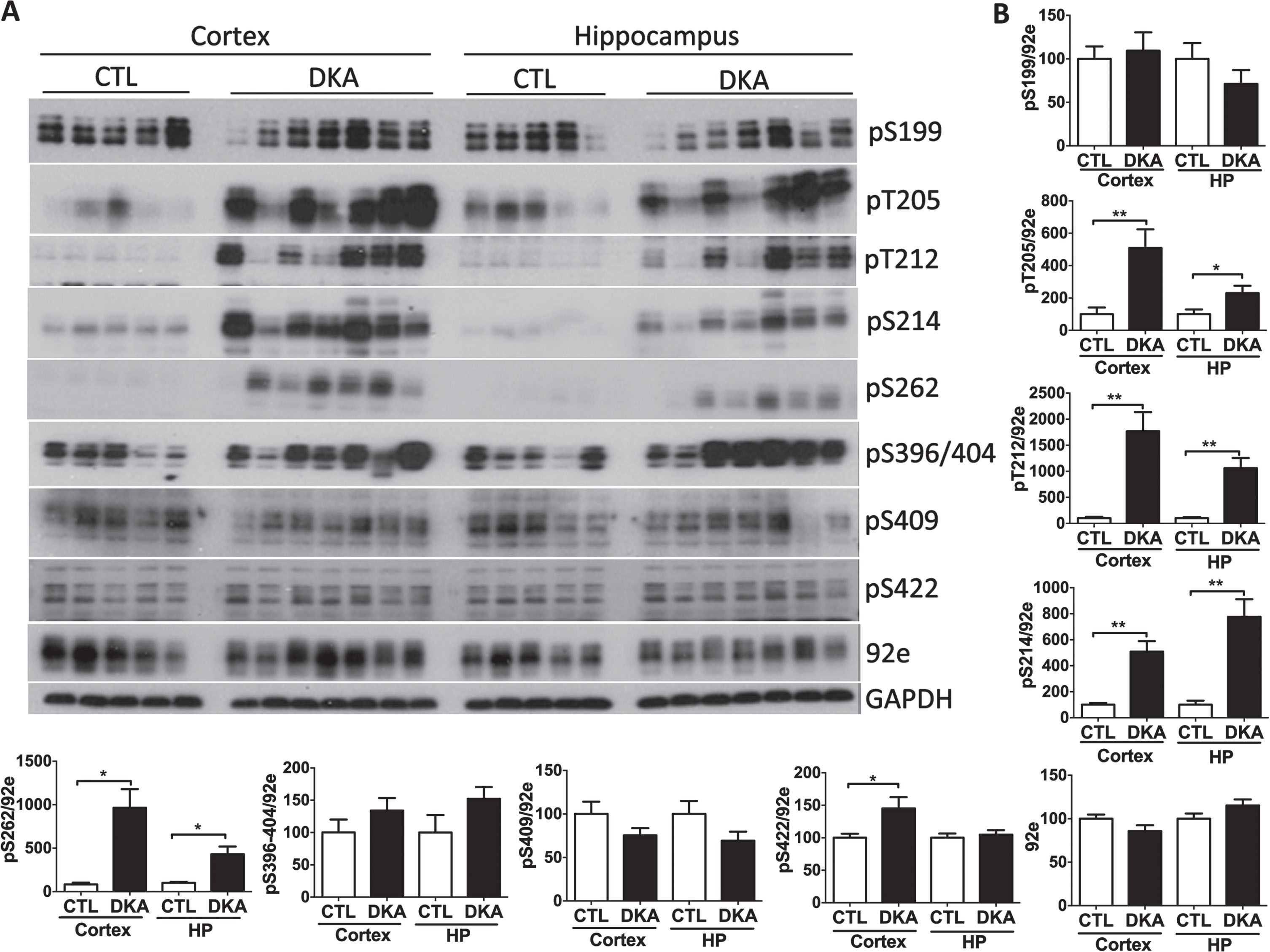
Abnormal hyperphosphorylation of tau is believed to be the initial step of neurofibrillary pathology and is crucial to neurodegeneration [29]. We, therefore, studied whether DKA can induce hyperphosphorylation of tau at the phosphorylation sites observed in AD brain. Western blot analyses using several phosphorylation-dependent and site-specific tau antibodies indicated that tau in the brains of rats with DKA became markedly hyperphosphorylated at pT205, pT212, pS214, and pS262 in both the cerebral cortex and the hippocampus and at pS422 in the cerebral cortex (Fig. 1). All these sites are the abnormal hyperphosphorylation sites seen in AD brain. The total tau protein level, as determined by using the phosphorylation-independent tau antibody 92e, was not changed with DKA, because no significant differences were seen between the DKA and the control groups. These results indicate that DKA can induce hyperphosphorylation of tau at multiple phosphorylation sites relevant to neurofibrillary degeneration.
Fig. 1
Tau hyperphosphorylation in the brains of rats with DKA. Brain homogenates derived from the rat hippocampal and cortical homogenates were subjected to western blots developed with the indicated antibodies (A). The blots were then quantified by densitometry, and the relative levels of tau phosphorylation at individual phosphorylation sites were normalized with the total tau level (determined with 92e) (B). GAPDH blots were included as a loading control. Data shown in the graphs are presented as mean±SEM (n = 5−7/group), where * and ** indicate p < 0.05 and 0.01, respectively.
Alterations of tau kinases in the brains of rats with DKA
Hyperphosphorylation of tau at multiple sites in the brains of rats with DKA, as observed above, suggests dysregulation of tau protein kinases and/or phosphatases in the brain during DKA, because the phosphorylation level of tau at individual sites results from a balance of tau kinase and phosphatase activities acting at the site. Therefore, we investigated the levels and activities (evidenced by their phosphorylation status that regulates the kinase activity) by using quantitative western blots. We first determined the levels and the activity-determining phosphorylation of glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (CDK5), the brain protein kinases implied in the regulation of tau phosphorylation at multiple sites [30]. GSK3β activity is mainly regulated negatively with its phosphorylation at S9 [31]. Surprisingly, we found marked phosphorylation of GSK3β at S9 in both the cerebral cortex and the hippocampus of rats during DKA (Fig. 2), suggesting downregulation of this kinase in the brains of rats with DKA as compared to controls. CDK5 activity requires both phosphorylation at its Ser159 and an activator p35. Whereas the CDK5 phosphorylation was found to be slightly increased, the p35 level was markedly decreased in the brains of rats with DKA as compared to the controls, suggesting no activation of this kinase in the brain during DKA.
Fig. 2
Western blots of tau protein kinases in the brains of rats with DKA. Brain homogenates derived from the rat hippocampal and cortical tissue were subjected to western blots developed with the indicated antibodies (A). The blots were then quantified by densitometry, and the relative levels of phosphorylated kinase were normalized with the total kinase level (B). GAPDH blots were included as a loading control. Data shown in the graphs are presented as mean±SEM (n = 5−7/group), where * and ** indicate p < 0.05 and 0.01, respectively.
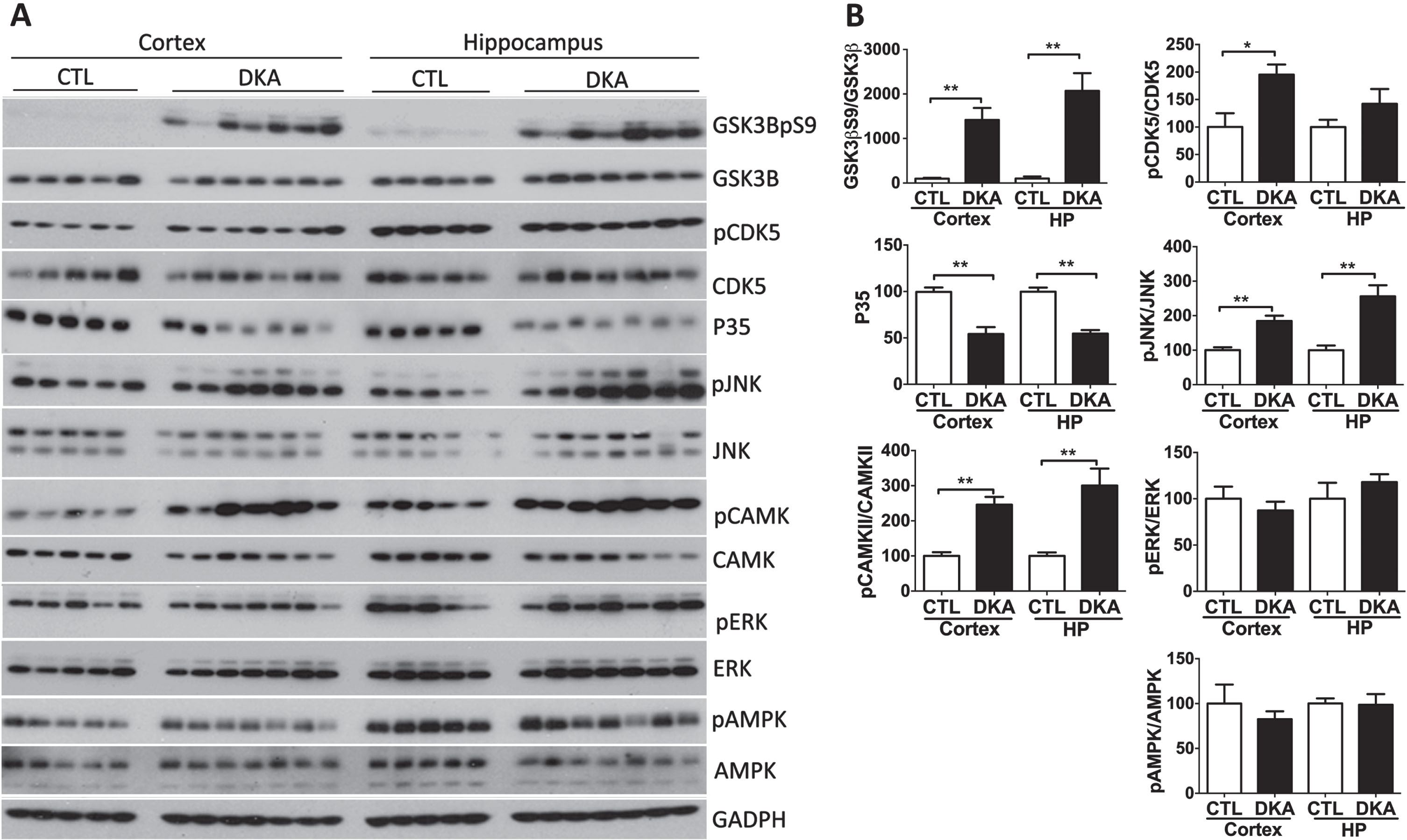
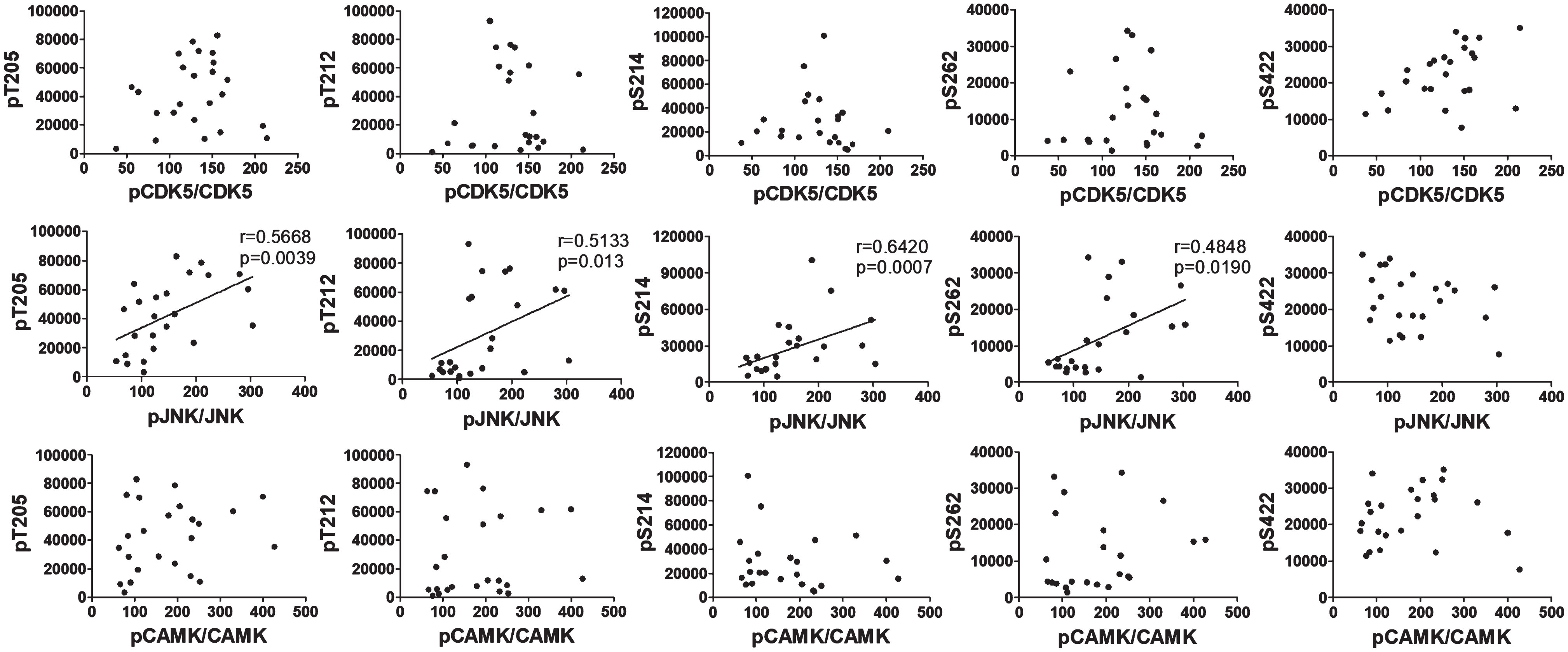
The above results indicate that the alterations of GSK3β and CDK5 did not contribute to the hyperphosphorylation of tau observed in the brains of rats with DKA. Otherwise, decreased phosphorylation, instead of hyperphosphorylation, of tau would have been found. To learn what protein kinases contribute to tau hyperphosphorylation in the brain during DKA, we further studied c-Jun N-terminal kinase (JNK), calcium/calmodulin-dependent protein kinase II (CAMK-II), extracellular signal–regulated kinase (ERK), and AMP-activated protein kinase (AMPK), because these kinases are also reported to regulate tau phosphorylation in the brain [32]. Quantitative western blot analyses found increased phosphorylation/activation of JNK and CAMK-II in the rat brains with DKA as compared to controls, but the expression levels of these kinases were not altered with DKA (Fig. 2). No significant differences in the level and phosphorylation/activation of ERK or AMPK were seen between the DKA and the control groups. These results suggest that DKA may have led to activation of JNK and CAMK-II that in turn resulted in hyperphosphorylation of tau in the rat brains. To verify these possibilities, we performed a Spearman correlation between the phosphorylation of tau at the sites elevated during DKA and the phosphorylation/activation of JNK and CAMK-II. We found a positive correlation of tau phosphorylation at pT205, pT212, pS214, and pS262 with phosphorylation/activation of JNK but not of CAMK-II (Fig. 3). No significant correlation was found between tau phosphorylation at any site studied and phosphorylation/activation of CDK5, which was included as references. These results suggest that the increased phosphorylation of tau was mainly caused by activated JNK in the rat brains during DKA.
Fig. 3
Correlation analysis between phosphorylation of tau and phosphorylation/activation of CDK5, JNK, and CAMK-II. The phosphorylation levels of tau at the indicated sites of brain samples were plotted against the phosphorylation/activation of CDK5, JNK, and CAMK-II and analyzed using Spearman correlation. The linear correlation lines, correlation coefficients (r values), and p values are included in the graphs only if they are statistically significant.
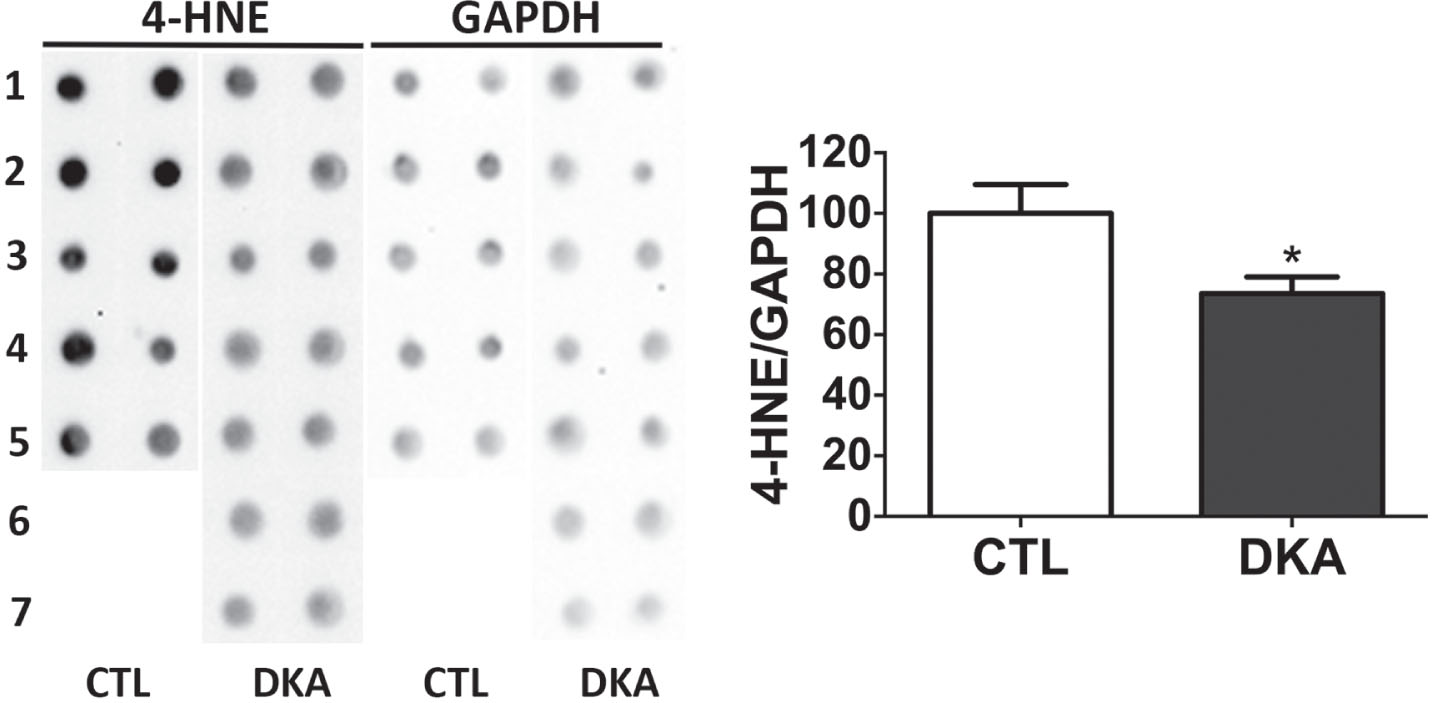
Because STZ injection could induce oxidative stress in the brain, which might in turn promote JNK activation. To learn whether there was significant oxidative stress that could explain the JNK activation we observed in the DKA rat brains, we determined the level of a commonly used oxidative stress marker, 4-hydroxy-2-nonenal (4-HNE), by using immune-dot blots. We found that the rat brain tissue of the DKA group had a reduced level of 4-HNE instead (Fig. 4). These results exclude the brain oxidative stress status at this time point, which could otherwise activate JNK.
Fig. 4
Immuno-dot blots of 4-HNE in cerebral cortical tissue of rats with DKA. Crute extracts of the cerebral cortical homogenates were subjected to immuno-dot blot assays developed with anti-4-HNE against oxidated protein adducts. The blots were then quantified by densitometry, and the relative levels of 4-HNE are shown the graph (mean±SEM, n = 5−7/group).
Alterations of tau phosphatases in the brains of rats with DKA
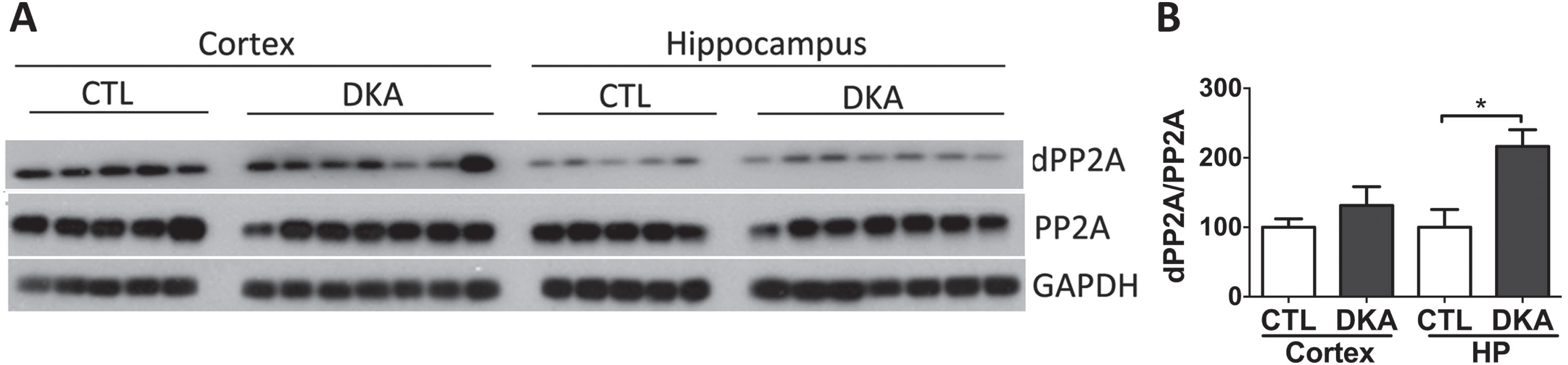
Protein phosphatase 2A (PP2A) is the major phosphatase that regulates tau phosphorylation in the mammalian brain [33]. Thus, we also studied PP2A in the rat brains during DKA. Because PP2A activity is promoted through methylation of the C-terminus of the PP2A catalytic subunit (PP2Ac), and its demethylation inhibits phosphatase activity [34], we determined the levels of total PP2A and demethylated PP2A (dPP2A) in the rat brains by using western blots. We found that while the total level of PP2A was not altered, the dPP2A level was significantly increased in the hippocampus of rats with DKA as compared to the controls (Fig. 5). These results suggest a downregulation of PP2A through its demethylation at least in the hippocampus of rats with DKA, which can also contribute to the hyperphosphorylation of tau that we found in the brains of rats with DKA.
Fig. 5
Western blots of PP2A in the brains of rats with DKA. Brain homogenates derived from the rat hippocampal and cortical homogenates were subjected to western blots developed with antibodies against the catalytic subunit of PP2A or demethylated PP2A (dPP2A) (A). The blots were then quantified by densitometry, and the relative levels of dPP2A/PP2A are shown in (B) (mean±SEM, n = 5−7/group).
Disruption of brain AKT signaling in rats with DKA
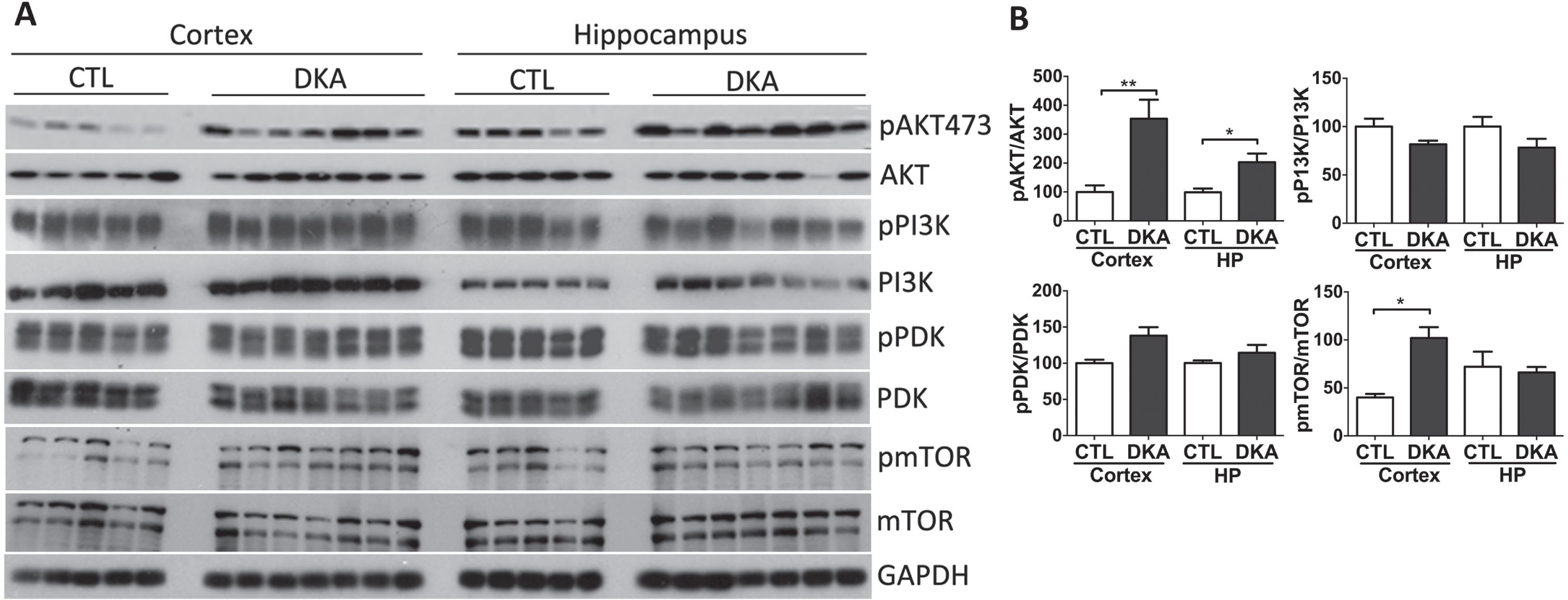
AKT (protein kinase B) signaling is the key molecular signal mediating insulin’s function and is also involved in neurofibrillary degeneration [14, 35]. We thus investigated AKT and its upstream signaling molecules, phosphatidylinositol 3-kinase (PI3K), phosphoinositide-dependent kinase-1 (PDK1), and the mechanistic target of rapamycin (mTOR). These signaling molecules are protein kinases in nature, and their kinase activities are initiated/promoted by phosphorylation. Therefore, we determined the phosphorylation of these kinases in the brains of rats with DKA by western blots developed with the specific phosphorylation-dependent antibodies. We found a marked increase in the phosphorylation of AKT at S473 in the rat brains with DKA, suggesting activation of this kinase in the brain during DKA (Fig. 6). AKT is mainly activated via phosphorylation at T308 by PI3K and PDK1 or at S473 by mTOR [36, 37]. Our quantitative analysis of western blots indicated significant activation/phosphorylation of mTOR in the cerebral cortex but no significant alterations of PI3K or PDK1 in the brains of rats with DKA (Fig. 6). These results revealed disruption of mTOR-AKT signaling in the brains of rats with DKA.
Fig. 6
Western blots of AKT, PI3K, PDK1, and mTOR from brains of rats with DKA. Brain homogenates derived from the rat hippocampal and cortical homogenates were subjected to western blot developed with the antibodies indicated at the right side of (A). The blots were then quantified by densitometry, and the net phosphorylation levels of each kinase were plotted (mean±SEM, n = 5−7/group) (B).
Increase in neuronal plasticity markers at the early stage of DKA
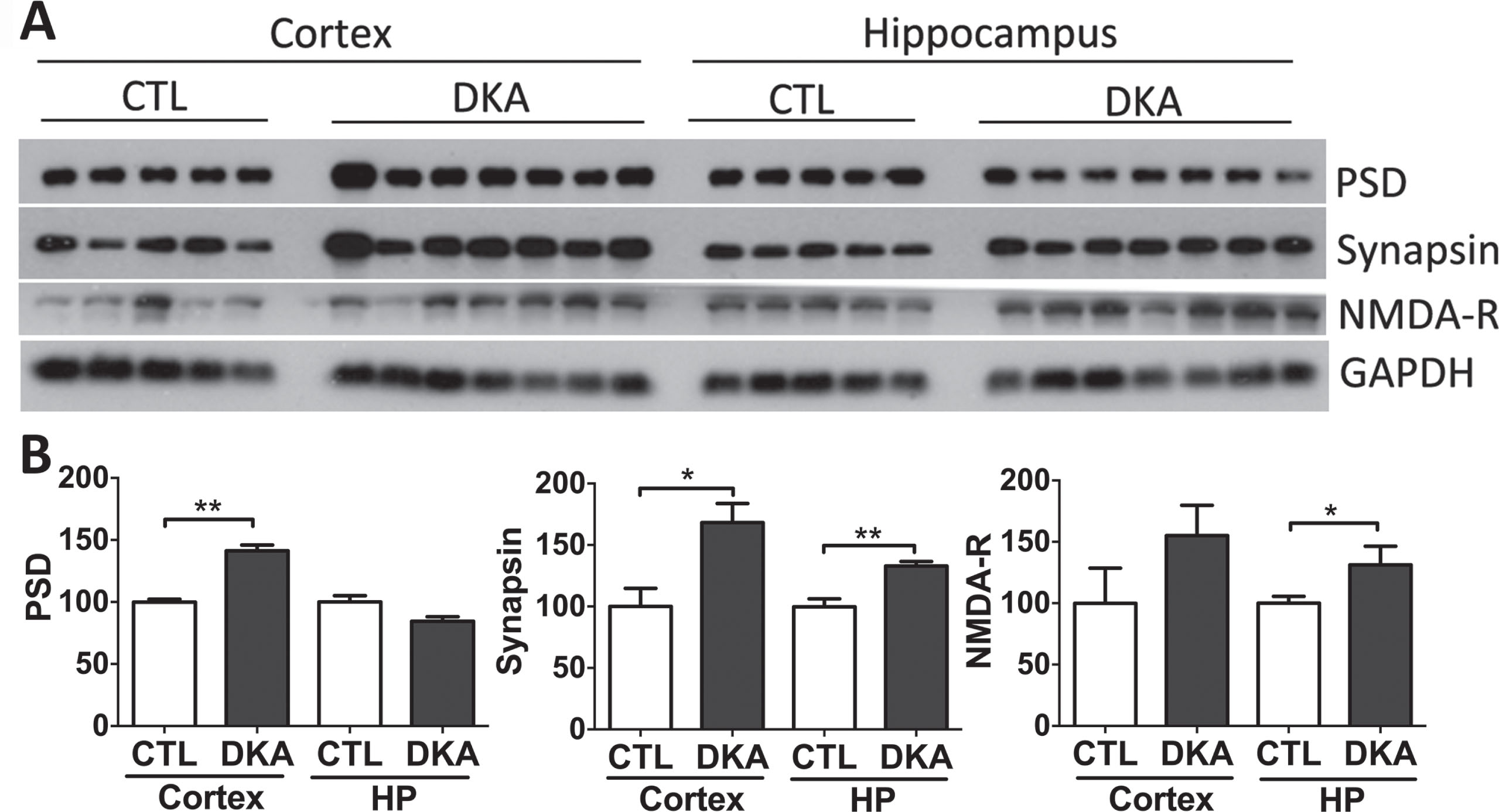
Neuronal synapses connecting and regulating neural networks in the brain are responsible for learning, memory, and cognition. This mechanism is regulated by excitatory synaptic transmission when pre-synapses release neurotransmitters that activate receptors on the postsynaptic neuron. In this study, we investigated the alteration of neuronal plasticity markers in the hippocampus and cerebral cortex of rats under DKA conditions. The levels of synapsin 1 and PSD95, a pre- and post-synaptic protein, respectively, were found to be significantly higher in rats with DKA than in the control group. Likewise, the same response was found in NMDA receptors, which can trigger lethal injury, inducing calcium influx and subsequently cellular calcium overload (Fig. 7).
Fig. 7
Western blots of synaptic proteins in the brains of rats with DKA. Brain homogenates derived from the rat hippocampal and cortical homogenates were subjected to western blots developed with antibodies indicated at the right side of (A). The blots were then quantified by densitometry, and the levels, after being normalized with GAPDH levels, are shown (mean±SEM, n = 5−7/group) (B).
DISCUSSION
DKA is a serious acute complication of DM that occurs in patients with uncontrolled hyperglycemia or other complications. Clinical studies have demonstrated that DKA can increase the risk of, or cause, long-term cognitive impairment. Significant memory impairment was observed in children with T1DM complicated with a history of DKA as compared to those without DKA, and the memory impairment is associated with subtle cerebral injury [2]. Increased white matter volume with decreased grey matter and poor mental score have been observed in children with T1DM with recurrent DKA [1, 38]. A longevity study found that older adults with T1DM, specifically those with recurrent DKA, had lower global cognitive function and reduced scores on psychomotor activity and executive functions [27]. In a large population study that evaluated the relationship between T2DM-DKA and the risk of dementia in adults, DKA was found to increase the risk for AD by 1.86-fold [7]. However, how DKA promotes cognitive impairment and AD remains unclear. Several possible mechanisms, such as hyperglycemic state associated with oxidative stress [39], blood-brain barrier disruption [40, 41], neuroinflammation [42], and brain insulin deficiency [43], have been proposed. In the present study, we found that DKA can induce AD-like hyperphosphorylation of tau, which is crucial to neurofibrillary degeneration and the consequent cognitive impairment in AD. It should be noted that DKA itself is an acute, short-time condition/complication of the chronic disease diabetes, and it is life-threatening if not corrected timely. Our results of the substantial changes in the brains of rats with DKA indicate that the brain is a sensitive organ responding to insults. Our findings suggest that recurrent DKA can probably increase the risk for AD by promoting abnormal hyperphosphorylation of tau in the brain. However, it is unlikely that young diabetes patients with a DKA episode could develop long-lasting tau pathology in the brain, but repeated DKA attacks can induce recurrent abnormal hyperphosphorylation of tau in the brain, which in turn could promote neurodegeneration and thus increase the risk for AD.
A common pathological feature of AD and other tauopathies is the formation of intraneuronal aggregation of tau protein as neurofibrillary tangles. Abnormal hyperphosphorylation of tau is believed to be crucial to neurofibrillary degeneration in these neurodegenerative diseases that all lead to memory loss and dementia [44]. To learn whether DKA promotes neurodegeneration and cognitive impairment through inducing AD-like hyperphosphorylation of tau in the brain, we determined the level of tau phosphorylation at several sites that are seen in AD brain and found that DKA induced significant hyperphosphorylation of tau at T205, T212, S214, S262, S396/404, and S422 in rat brains. Considering that abnormal tau hyperphosphorylation is an early and crucial event of neurofibrillary degeneration, our results suggest that repeated DKA may promote or induce cognitive impairment by promoting abnormal hyperphosphorylation of tau and, consequently, neurofibrillary degeneration in individuals with DM.
Abnormal hyperphosphorylation is an early stage of tau pathology that eventually leads to the formation of neurofibrillary tangles in AD brain. Acute tau phosphorylation does not necessarily indicate a path toward tauopathies such as AD. This initial stage is reversible and can be reversed through inhibition of tau kinases and/or activation of tau phosphatases [29]. In addition, brain insulin signaling is downregulated in AD brain [14], which may also contribute to tau-mediated neurofibrillary degeneration in AD [15]. We previously found that restoration of brain insulin signaling through either insulin sensitizers or intranasal insulin administration can reverse the AD-like tau hyperphosphorylation and improve cognitive function in mice [45–47]. Thus, timely management of DKA for diabetes patients may help prevent the DKA-induced increase in AD risk in these individuals.
The level of tau phosphorylation is a net outcome of the activities of tau protein kinases and phosphatases. To determine which tau kinases mediate hyperphosphorylation of tau in DKA, we investigated the major known tau kinases, including GSK3β, CDK5, JNK, CAMK-II, ERK, and AMPK [30]. Surprisingly, we found downregulation of both GSK3β and probably CDK5 activities, as evidenced by increased S9 phosphorylation and decreased CDK5 activator p35, respectively, in the brains of rats with DKA. Downregulation of these two kinases would induce decreased phosphorylation, rather than hyperphosphorylation, of tau if other modulators were not involved. Therefore, the fact that tau was hyperphosphorylated in the brains of rats during DKA indicates that tau hyperphosphorylation was not mediated by these two protein kinases under this condition. Significant alterations of S9 phosphorylation of GSK3β are not seen in human AD brain either [14]. Further studies of other tau kinases suggested activation of JNK that appears to contribute to tau hyperphosphorylation during DKA, which was further supported by a positive correlation between tau phosphorylation at multiple sites and phosphorylation/activation of JNK. Thus, it appears that JNK in the brain may be activated as a stress response to DKA, which in turn induces hyperphosphorylation of tau during DKA. The type of stress that activates JNK under this condition is unlikely to be oxidative stress because the total oxidative stress level in the rat brains at the time point of investigation was lower with DKA, as determined by our immuno-dot blot assay.
Under both biological and pathological conditions, tau phosphorylation is usually regulated by the combined actions of several tau kinases and phosphatases. In addition to JNK activation, we found that the major tau phosphate in the mammalian brain, PP2A [33], was downregulated through increased demethylation of the PP2A catalytic subunit. Downregulation of PP2A also occurs in the AD brain and may contribute to abnormal hyperphosphorylation of tau in AD [33, 48–50]. We thus speculate that during DKA, activation of JNK and downregulation of PP2A collectively induce tau phosphorylation, which outweighs the counteraction of downregulation of GSK3β and probably also of CDK5, resulting in hyperphosphorylation of tau in the rat brains. A similar phenomenon was previously reported in the brains of starved mice [51].
AKT is a key mediator of both insulin signaling and mTOR signaling, as well as the major upstream protein kinase of GSK3β [52]. We found that mTOR-AKT signaling was activated in rat brains during DKA. These results are consistent with the downregulation of GSK3β activity through its phosphorylation at S9 that we found because the S9 phosphorylation of GSK3β is catalyzed by AKT. The activation of mTOR-AKT signaling appears to be an acute response to DKA, which may contribute to the increased level of synaptic proteins, such as synapsin I, PSD95, and NMDA-R, that we found in the brains of rats during DKA.
In conclusion, we found that DKA induced hyperphosphorylation of tau protein at multiple phosphorylation sites associated with female AD and disruption of the mTOR-AKT signaling and increased levels of synaptic proteins in rat brains. The DKA-induced hyperphosphorylation of tau was mainly mediated through activation of JNK and downregulation of PP2A. Future studies will need to confirm whether these findings can be replicated in male animals. Nevertheless, the present study shed some light on how DKA may increase the risk for AD.
AUTHOR CONTRIBUTIONS
Gustavo Basurto-Islas (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Validation; Visualization; Writing – original draft; Writing – review & editing); Yunn Chyn Tung (Data curation; Methodology; Writing – original draft); Chun-ling Dai (Data curation; Investigation; Methodology; Writing – review & editing); Khalid Iqbal (Conceptualization; Investigation; Project administration; Resources; Supervision; Writing – review & editing); Cheng-Xin Gong (Conceptualization; Formal analysis; Funding acquisition; Investigation; Project administration; Resources; Supervision; Writing – review & editing).
ACKNOWLEDGMENTS
We thank Maureen Marlow of the New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA, for copy-editing of the manuscript.
FUNDING
This study was supported in part by Mexico’s National Science and Technology Board (CONAHCyT) (Grant A1-S-29906 to G.B.-I.), Convocatoria de Apoyo a Profesoras y Profesores 2024, University of Guanajuato, and the New York State Office for People With Developmental Disabilities.
CONFLICT OF INTEREST
KI and CXG are Editorial Board Members of this journal but were not involved in the peer-review process for this article and did not have access to any information regarding its peer review. KI is cofounder and Chief Scientific Officer of Phanes Biotech, Inc. This company is in the process of developing biomarkers and therapeutics of Alzheimer’s disease and related disorders. CXG serves in the Scientific Advisory Board of Alectos Therapeutics.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article.
REFERENCES
[1] | Cameron FJ , Scratch SE , Nadebaum C , Northam EA , Koves I , Jennings J , Finney K , Neil JJ , Wellard RM , Mackay M , Inder TE , DKA Brain Injury Study Group ((2014) ) Neurological consequences of diabetic ketoacidosis at initial presentation of type 1 diabetes in a prospective cohort study of children. Diabetes Care 37: , 1554–1562. |
[2] | Ghetti S , Lee JK , Sims CE , Demaster DM , Glaser NS ((2010) ) Diabetic ketoacidosis and memory dysfunction in children with type 1 diabetes. J Pediatr 156: , 109–114. |
[3] | Bober E , Buyukgebiz A ((2005) ) Hypoglycemia and its effects on the brain in children with type 1 diabetes mellitus. Pediatr Endocrinol Rev 2: , 378–382. |
[4] | Perantie DC , Lim A , Wu J , Weaver P , Warren SL , Sadler M , White NH , Hershey T ((2008) ) Effects of prior hypoglycemia and hyperglycemia on cognition in children with type 1 diabetes mellitus. Pediatr Diabetes 9: , 87–95. |
[5] | Schwartz DD , Wasserman R , Powell PW , Axelrad ME ((2014) ) Neurocognitive outcomes in pediatric diabetes: A developmental perspective. Curr Diab Rep 14: , 533. |
[6] | Strudwick SK , Carne C , Gardiner J , Foster JK , Davis EA , Jones TW ((2005) ) Cognitive functioning in children with early onset type 1 diabetes and severe hypoglycemia. J Pediatr 147: , 680–685. |
[7] | Chen YL , Weng SF , Yang CY , Wang JJ , Tien KJ ((2019) ) Diabetic ketoacidosis further increases risk of Alzheimer’s disease in patients with type 2 diabetes. Diabetes Res Clin Pract 147: , 55–61. |
[8] | Glaser N , Anderson S , Leong W , Tancredi D , O’Donnell M ((2012) ) Cognitive dysfunction associated with diabetic ketoacidosis in rats. Neurosci Lett 510: , 110–114. |
[9] | Glaser N , Sasaki-Russell J , Cohen M , Little C , O’Donnell M , Sall J ((2017) ) Histological and cognitive alterations in adult diabetic rats following an episode of juvenile diabetic ketoacidosis: Evidence of permanent cerebral injury. Neurosci Lett 650: , 161–167. |
[10] | Gasparini L , Netzer WJ , Greengard P , Xu H ((2002) ) Does insulin dysfunction play a role in Alzheimer’s disease? Trends Pharmacol Sci 23: , 288–293. |
[11] | Frolich L , Blum-Degen D , Riederer P , Hoyer S ((1999) ) A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer’s disease. Ann N Y Acad Sci 893: , 290–293. |
[12] | Rivera EJ , Goldin A , Fulmer N , Tavares R , Wands JR , de la Monte SM ((2005) ) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J Alzheimers Dis 8: , 247–268. |
[13] | Mosconi L ((2005) ) Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging 32: , 486–510. |
[14] | Liu Y , Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2011) ) Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol 225: , 54–62. |
[15] | Chen Y , Deng Y , Zhang B , Gong CX ((2014) ) Deregulation of brain insulin signaling in Alzheimer’s disease. Neurosci Bull 30: , 282–294. |
[16] | Planel E , Tatebayashi Y , Miyasaka T , Liu L , Wang L , Herman M , Yu WH , Luchsinger JA , Wadzinski B , Duff KE , Takashima A ((2007) ) Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci 27: , 13635–13648. |
[17] | Ke YD , Delerue F , Gladbach A , Gotz J , Ittner LM ((2009) ) Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One 4: , e7917. |
[18] | Papon MA , El Khoury NB , Marcouiller F , Julien C , Morin F , Bretteville A , Petry FR , Gaudreau S , Amrani A , Mathews PM , Hebert SS , Planel E ((2013) ) Deregulation of protein phosphatase 2A and hyperphosphorylation of tau protein following onset of diabetes in NOD mice. Diabetes 62: , 609–617. |
[19] | Morales-Corraliza J , Wong H , Mazzella MJ , Che S , Lee SH , Petkova E , Wagner JD , Hemby SE , Ginsberg SD , Mathews PM ((2016) ) Brain-wide insulin resistance, tau phosphorylation changes, and hippocampal neprilysin and amyloid-beta alterations in a monkey model of type 1 diabetes. J Neurosci 36: , 4248–4258. |
[20] | Clodfelder-Miller BJ , Zmijewska AA , Johnson GV , Jope RS ((2006) ) Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 55: , 3320–3325. |
[21] | Yan W , Pang M , Yu Y , Gou X , Si P , Zhawatibai A , Zhang Y , Zhang M , Guo T , Yi X , Chen L ((2019) ) The neuroprotection of liraglutide on diabetic cognitive deficits is associated with improved hippocampal synapses and inhibited neuronal apoptosis. Life Sci 231: , 116566. |
[22] | Hamed SA ((2017) ) Brain injury with diabetes mellitus: Evidence, mechanisms and treatment implications. Expert Rev Clin Pharmacol 10: , 409–428. |
[23] | Stranahan AM , Arumugam TV , Cutler RG , Lee K , Egan JM , Mattson MP ((2008) ) Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11: , 309–317. |
[24] | Glaser N , Yuen N , Anderson SE , Tancredi DJ , O’Donnell ME ((2010) ) Cerebral metabolic alterations in rats with diabetic ketoacidosis: Effects of treatment with insulin and intravenous fluids and effects of bumetanide. Diabetes 59: , 702–709. |
[25] | Yuen N , Anderson SE , Glaser N , Tancredi DJ , O’Donnell ME ((2008) ) Cerebral blood flow and cerebral edema in rats with diabetic ketoacidosis. Diabetes 57: , 2588–2594. |
[26] | Glaser N , Ngo C , Anderson S , Yuen N , Trifu A , O’Donnell M ((2012) ) Effects of hyperglycemia and effects of ketosis on cerebral perfusion, cerebral water distribution, and cerebral metabolism. Diabetes 61: , 1831–1837. |
[27] | Lacy ME , Gilsanz P , Eng CW , Beeri MS , Karter AJ , Whitmer RA ((2020) ) Recurrent diabetic ketoacidosis and cognitive function among older adults with type 1 diabetes: Findings from the Study of Longevity in Diabetes. BMJ Open Diabetes Res Care 8: , e001173. |
[28] | Beech JS , Williams SC , Iles RA , Cohen RD , Nolan KM , Evans SJ , Going TC ((1995) ) Haemodynamic and metabolic effects in diabetic ketoacidosis in rats of treatment with sodium bicarbonate or a mixture of sodium bicarbonate and sodium carbonate. Diabetologia 38: , 889–898. |
[29] | Gong CX , Iqbal K ((2008) ) Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr Med Chem 15: , 2321–2328. |
[30] | Wang JZ , Grundke-Iqbal I , Iqbal K ((2007) ) Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 25: , 59–68. |
[31] | Beurel E , Grieco SF , Jope RS ((2015) ) Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol Ther 148: , 114–131. |
[32] | Martin L , Latypova X , Wilson CM , Magnaudeix A , Perrin ML , Yardin C , Terro F ((2013) ) Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res Rev 12: , 289–309. |
[33] | Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2005) ) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22: , 1942–1950. |
[34] | Xing Y , Li Z , Chen Y , Stock JB , Jeffrey PD , Shi Y ((2008) ) Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell 133: , 154–163. |
[35] | Deng Y , Li B , Liu Y , Iqbal K , Grundke-Iqbal I , Gong CX ((2009) ) Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer’s disease. Am J Pathol 175: , 2089–2098. |
[36] | Wei Y , Zhou J , Yu H , Jin X ((2019) ) AKT phosphorylation sites of Ser473 and Thr308 regulate AKT degradation. Biosci Biotechnol Biochem 83: , 429–435. |
[37] | Zeng Z , Sarbassov dD , Samudio IJ , Yee KW , Munsell MF , Ellen Jackson C , Giles FJ , Sabatini DM , Andreeff M , Konopleva M ((2007) ) Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 109: , 3509–3512. |
[38] | Wootton-Gorges SL , Buonocore MH , Caltagirone RA , Kuppermann N , Glaser NS ((2010) ) Progressive decrease in N-acetylaspartate/Creatine ratio in a teenager with type 1 diabetes and repeated episodes of ketoacidosis without clinically apparent cerebral edema: Evidence for permanent brain injury. AJNR Am J Neuroradiol 31: , 780–781. |
[39] | Rosales-Corral S , Tan DX , Manchester L , Reiter RJ ((2015) ) Diabetes and Alzheimer disease, two overlapping pathologies with the same background: Oxidative stress. Oxid Med Cell Longev 2015: , 985845. |
[40] | Biessels GJ , Staekenborg S , Brunner E , Brayne C , Scheltens P ((2006) ) Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol 5: , 64–74. |
[41] | Strachan MW ((2011) ) R D Lawrence Lecture 2010. The brain as a target organ in Type 2 diabetes: Exploring the links with cognitive impairment and dementia. Diabet Med 28: , 141–147. |
[42] | Dalton RR , Hoffman WH , Passmore GG , Martin SL ((2003) ) Plasma C-reactive protein levels in severe diabetic ketoacidosis. Ann Clin Lab Sci 33: , 435–442. |
[43] | Li X , Song D , Leng SX ((2015) ) Link between type 2 diabetes and Alzheimer’s disease: From epidemiology to mechanism and treatment. Clin Interv Aging 10: , 549–560. |
[44] | Iqbal K , Liu F , Gong CX ((2016) ) Tau and neurodegenerative disease: The story so far. Nat Rev Neurol 12: , 15–27. |
[45] | Chen Y , Zhao Y , Dai CL , Liang Z , Run X , Iqbal K , Liu F , Gong CX ((2014) ) Intranasal insulin restores insulin signaling, increases synaptic proteins, and reduces Aβ level and microglia activation in the brains of 3xTg-AD mice. Exp Neurol 261: , 610–619. |
[46] | Zhang Y , Dai CL , Chen Y , Iqbal K , Liu F , Gong CX ((2016) ) Intranasal insulin prevents anesthesia-induced spatial learning and memory deficit in mice. Sci Rep 6: , 21186. |
[47] | Chen Y , Dai CL , Wu Z , Iqbal K , Liu F , Zhang B , Gong CX ((2017) ) Intranasal insulin prevents anesthesia-induced cognitive impairment and chronic neurobehavioral changes. Front Aging Neurosci 9: , 136. |
[48] | Gong CX , Singh TJ , Grundke-Iqbal I , Iqbal K ((1993) ) Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 61: , 921–927. |
[49] | Gong CX , Shaikh S , Wang JZ , Zaidi T , Grundke-Iqbal I , Iqbal K ((1995) ) Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J Neurochem 65: , 732–738. |
[50] | Sontag JM , Sontag E ((2014) ) Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front Mol Neurosci 7: , 16. |
[51] | Planel E , Yasutake K , Fujita SC , Ishiguro K ((2001) ) Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 beta and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem 276: , 34298–34306. |
[52] | Manning BD , Toker A ((2017) ) AKT/PKB signaling: Navigating the network. Cell 169: , 381–405. |


