
Episodic Memory Loss: When Alzheimer’s Disease Is Not the Answer
Abstract
A 60-year-old man presented to a Neurology Clinic specialized in cognitive disorders to evaluate memory complaints. A comprehensive neuropsychological examination detected an isolated and severe hippocampal memory deficit. Laboratory tests, brain magnetic resonance imaging (MRI), and cerebrospinal fluid (CSF) tests, including Alzheimer’s disease (AD) biomarkers, did not show remarkable results. Due to family history of cognitive impairment, we extended the study to non-Alzheimer monogenic mutations (Next Generation Sequencing) detecting a pathogenic variant of the progranulin (PGRN) gene (c.1414-1 G > T) which has been previously associated with the same phenotype. These results should be considered in patients with an Alzheimer-like presentation, negative AD biomarkers’ results, and family history of dementia.
INTRODUCTION
The increasing availability of genetic testing for neurodegenerative diseases has led to a growing recognition of the phenotypic diversity associated with various genetic conditions. Here we report the case of 60-year-old man who presented with episodic memory deficits and negative Alzheimer’s disease (AD) biomarkers, who was found to have a loss of function variant in the GRN (granulin) gene. There has been an increase in disease modifying clinical trials targeting familial frontotemporal dementia (FTD) caused by genetic mutations, especially in progranulin, suggesting that this mutation may be one of the first to benefit [1]. GRN mutations usually present with phenotypes within the FTD spectrum, with some distinctive characteristics as parietal atrophy extension or asymmetrical atrophy on neuroimaging. However, AD-like presentations can take place. Thus, genetic testing for possible familial FTD mutations should encompass a wide variety of possible phenotypes. Here we describe a well-documented AD-like case and discuss its potential diagnostic and therapeutic implications.
CASE REPORT
A 60-year-old man presented to a Neurology Clinic specialized in cognitive disorders to evaluate memory complaints. He reported progressive impairment of episodic memory over the preceding 2 years, resulting in the need for supervision in his professional occupation. Neither the patient nor his wife endorsed any changes in orientation, language, object handling, behavior or motor functions. Despite the difficulties with memory, the patient led an independent life. Past medical history was only notable for hyperlipidemia. In terms of family history, the patient’s father and his paternal aunt suffered from dementia.
Neurologic examination was unremarkable. A comprehensive neuropsychological assessment was performed; details and results are shown in Table 1. Compared to normative Spanish population data, adjusted by sex, age, and educational level [2, 3], memory domain (TMA-93 and FCSRT + IR) was severely impaired, whereas the rest of cognitive and behavioral domains were preserved. These findings were consistent with a hippocampal amnestic syndrome.
Table 1
Initial neuropsychological battery of the patient. The patient only failed in those with respect to the cognitive memory domain
| Neuropsychological test | Pathologic | Normal | |
| MMSE | 29/30 (1 error in Recall Memory) | ||
| TMA-93 | 16/30 (5 intrusions) | ||
| FCSRT + IR | Total Free recall: 6; Total Cued | ||
| Recall: 16; Cued Index: 0.26 | |||
| Boston Naming Test | 53/60 | ||
| Semantic fluency | 23 | ||
| Phonetic fluency | 11 | ||
| Stroop Test | Words: 97 | ||
| Colors: 79 | |||
| Words and Colors: 52 | |||
| Stroop’s effect: 0.19 | |||
| Digit Span | Direct: 7/16 | ||
| (number 5/8) | |||
| Indirect: 6/16 | |||
| (number 3/8) | |||
| RBANS figure | 20/20 | ||
| VOSP (Number Location) | 10/10 | ||
| Barcelona’s test figure | 20/20 | ||
| Goldberg Anxiety and Depression Scale | Anxiety 1/9 | ||
| Depression 0/9 | |||
| MBI-C | Decreased Motivation, 2/18 | ||
| Emotional dysregulation, 0/18 | |||
| Impulse dyscontrol, 3/36 | |||
| Social inappropriateness, 0/15 | |||
| Abnormal perception, 0/15 | |||
| Total, 5/102 | |||
| FAQ | 2/33 | ||
| AD8 | 2/8 |
MMSE, Mini Mental State Examination; TMA-93, Memory Associative Test of the district of Seine-Saint-Denis; FCSRT, Free and Cued Selective Reminding Test; RBANS, Repeatable Battery for the Assessment of Neuropsychological Status; VOSP, Visual Object and Space Perception; MBI-C, Mild Behavioral Impairment Checklist; FAQ, Functional Activities Questionnaire; AD8, Eight-item informant Interview to Differentiate Aging and Dementia.
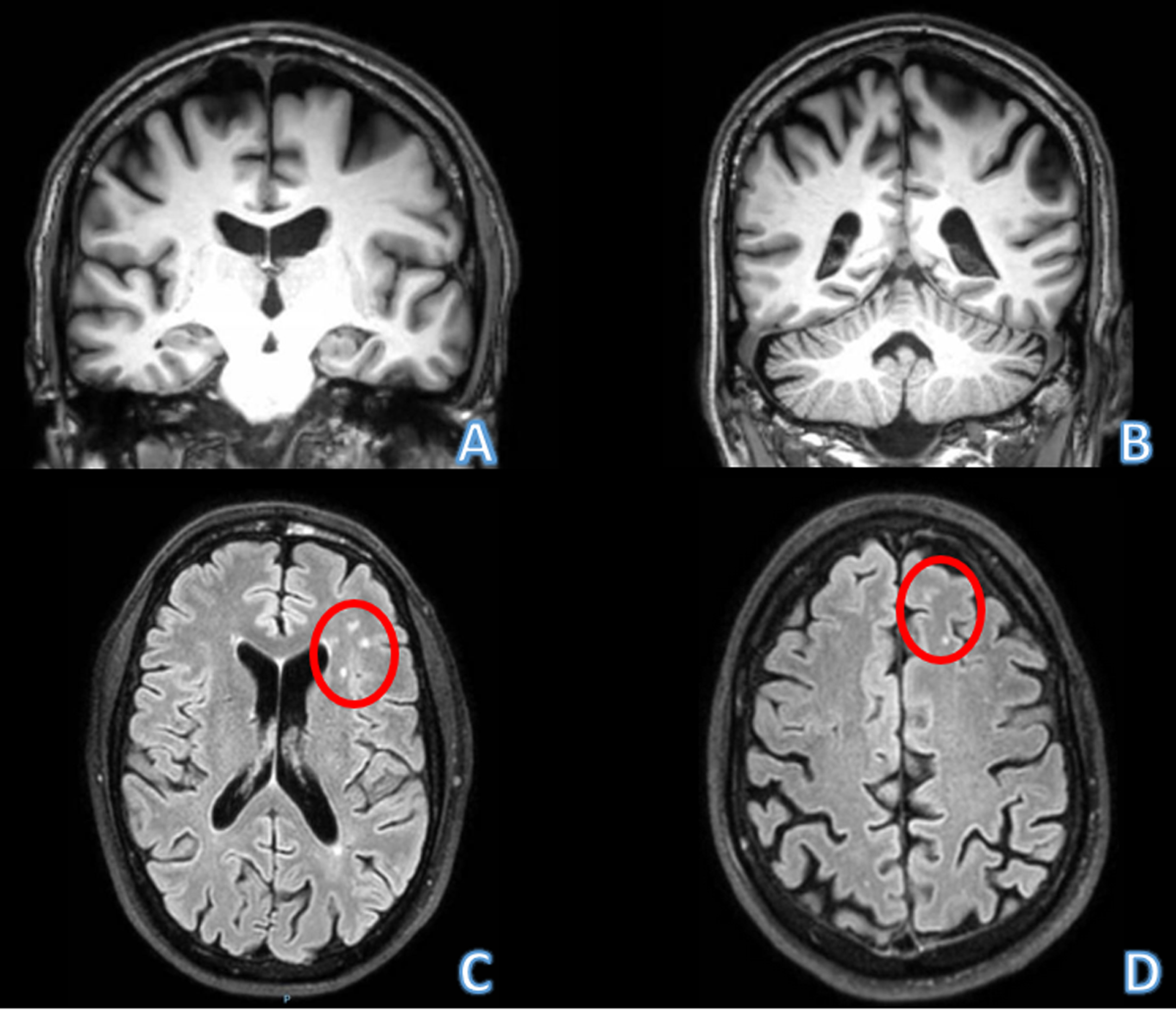
Routine laboratory testing ruled out reversible causes of cognitive impairment, including hypothyroidism, B12 and folate deficiency, syphilis, and HIV. Testing for autoimmune and inflammatory encephalitis, including CSF cell count, protein, and oligoclonal bands; CSF CASPR, DPPX, LGI1, AMPA receptor, GABA-B receptor, and NMDA receptor antibodies; and serum anti-Hu, anti-Ri, and anti-Yo antibodies, was also negative. Brain MRI showed mild parietal atrophy, no hippocampal atrophy, and nonspecific focal white matter hyperintensities (Fig. 1). CSF AD biomarkers were negative (Euroimmun method): Aβ42, 615 pg/ml (pathological, < 570), Ratio Aβ42/Aβ40, 0,1380 (pathological, < 0.095), Total-Tau, 116 pg/ml (pathological, > 412 pg/ml), and phosphorylated-Tau (pTau (181), 8 pg/ml (pathological, > 61). Due to the family history, and negative AD biomarkers, the patient participated in genetic prescreening for C9orf72, MAPT and GRN mutations as part of a clinical trial evaluating the brain penetrant recombinant PGRN TAK-594/DNL593 (NCT05262023). Genetic testing was performed at a commercial laboratory (Prevention Genetics) via standard methodology for short-read next-generation sequencing (NGS). For all assayed genes, read depth was at least 20× for all coding exons and +/– 10 base pairs of flanking DNA. Copy number variant detection was also performed using standard approaches comparing read depth and distribution of each target in the test sample to multiple matched controls. Any differences from the reference sequence were assigned to various degrees of pathogenicity (“Pathogenic”, “Likely Pathogenic”, “Uncertain Significance”, “Benign”, etc.) based on American College of Medical Genetics criteria [4]. Results yielded a heterozygous variant in GRN (c.1414-1G>T) assigned as “Pathogenic” owing to it not having been previously reported in any population databases and its sequence change disrupting a consensus splice acceptor site in GRN. No other pathogenic variants were detected in the screened list of genes. In the two years since initial examination, the patient has maintained these symptoms with little decline in memory tests and no development of signs or symptoms typical of FTD.
Fig. 1
Brain MRI of the patient. A, B) Coronal sections in the T1 sequence. Note predominantly parietal atrophy, but relatively spare of both hippocampi. C, D) Axial section in FLAIR sequence. Note the hyperintensities in white matter possibly related with microglial activation described in patients with progranulin mutation (red circles).
DISCUSSION
GRN mutations were first described as the cause of familial autosomal dominant FTD [5, 6]. All known pathogenic mutations have an effect of loss of function. The biological function of GRN seems to be part of processes such as neurite outgrowth, cytokine release, and synaptic transmission. There is growing evidence that these may be mediated by its lysosomal actions. In the brain, PGRN is expressed in neurons and microglia, and chronic PGRN deficiency results in lysosomal dysfunction, which in turn leads to aberrant microglial activation, neuroinflammation, and TDP-43 proteinopathy, culminating in neurodegeneration [7– 9]. Therefore, replacement or boosting of PGRN levels may be a promising approach to treat the disease [10, 11]. Phenotypic variability is widely known and includes the behavioral variant of FTD and nonfluent/agrammatic primary progressive aphasia (PPA) as the most common clinical syndromes at onset. Unlike most other genetic or sporadic FTD presentations, parietal cortex degeneration is frequently observed and may be associated with corticobasal syndrome presentations [12]. In addition, these mutations should be considered when, in patients with logopenic PPA, AD biomarkers are negative [13]. In terms of neuroimaging, two features may be distinctive for FTD-GRN: striking asymmetry on atrophy patterns, and the presence of white matter lesions predominantly in the frontal and parietal lobes [14], as seen in this case and that do not seem to be related with small vessel cerebral disease (SVCD), but with hyperactivation of microglia (Fig. 1).
In this new era of more generalized use of biomarkers to confirm AD, a percentage of cases presenting with typical amnestic hippocampal syndrome are found to test negative. Excluding, in appropriate settings, rare symptomatic causes such as immune or infectious limbic encephalitis, some neurodegenerative entities that mimic AD as Hippocampal Sclerosis, Argyrophilic Grain Disease, Primary Age-Related Tauopathy, and Limbic Predominant Age-Related TDP-43 Encephalopathy (LATE), should be taken in consideration [15]. However, certain features in this case, such as the relatively young age of onset, the absence of hippocampal atrophy, and the notable family history, prompted an expansion of the investigation to study monogenic FTD mutations. Therefore, not all cases that test negative for AD have to be diagnosed as probable LATE or other age-related entities with similar hippocampal involvement, particularly when atypical data are present. Moreover, patients who test positive for GRN mutations, exhibit an amnestic hippocampal onset, lack dementia, show minimal hippocampal atrophy, have limited behavioral symptoms, and are able to comply with procedures, could be optimal candidates for emergent trials targeting the restoration of PGRN levels.
Another interesting aspect that emerged from the patient’s diagnosis is related to genetics. Indeed, the variant found in this patient, c.1414-1 G>T, has already been documented in seven families, four of which were referred for genetic testing partly due to a strong family history of dementia [16, 17]. Furthermore, all patients and their families had Hispanic origins. In the study by Cruchaga et al. (2012), although the two families described were residents of the United States, both had Hispanic ancestry [16]. Moreover, in Menéndez-González et al. (2022), the same mutation was detected in five families of Spanish origin, suggesting a potential foundereffect [17].
In conclusion, in the new era of widespread use of AD biomarkers, GRN and other FTD mutations should be considered as a diagnostic possibility in presentations with a hippocampal amnestic syndrome, positive family history, and negativity for AD biomarkers. In this setting, and particularly in Hispanic families, the variant c.1414-1 G>T should be considered. This discernment is increasingly relevant as new PGRN targeted treatments become available.
AUTHOR CONTRIBUTIONS
Ernesto García Roldán (Conceptualization; Supervision; Writing – original draft; Writing – review & editing); Richard Tsai (Supervision; Writing – review & editing); Amy Berger (Writing – review & editing); Emilio Franco-Macías (Conceptualization; Supervision; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
The authors wish to thank the patient and his wife.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
Amy Berger and Richard Tsai are employees of Denali Therapeutics.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
[1] | Boeve BF , Boxer AL , Kumfor F , Pijnenburg Y , Rohrer JD ((2022) ) Advances and controversies in frontotemporal dementia: Diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol 21: , 258–272. |
[2] | Rodrigo-Herrero S , Sánchez-Benavides G , Ainz-Gómez L , Luque-Tirado A , Graciani-Cantisán E , Sánchez-Arjona MB , Maillet D , Jiménez-Hernández MD , Franco-Macías E ((2020) ) Norms for testing visual binding using the Memory Associative Test (TMA-93) in older educationally diverse adults. J Alzheimers Dis 75: , 871–878. |
[3] | Peña-Casanova J , Gramunt-Fombuena N , Quiñones-Ubeda S , Sánchez-Benavides G , Aguilar M , Badenes D , Molinuevo JL , Robles A , Barquero MS , Payno M , Antúnez C , Martínez-Parra C , Frank-García A , Fernández M , Alfonso V , Sol JM , Blesa R ; NEURONORMA Study Team ((2009) ) Spanish Multicenter Normative Studies (NEURONORMA Project): Norms for the Rey-Osterrieth complex figure (copy and memory), and free and cued selective reminding test. Arch Clin Neuropsychol 24: , 371–393. |
[4] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL ; ACMG Laboratory Quality Assurance Committee ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[5] | Baker M , Mackenzie IR , Pickering-Brown SM , Gass J , Rademakers R , Lindholm C , Snowden J , Adamson J , Sadovnick AD , Rollinson S , Cannon A , Dwosh E , Neary D , Melquist S , Richardson A , Dickson D , Berger Z , Eriksen J , Robinson T , Zehr C , Dickey CA , Crook R , McGowan E , Mann D , Boeve B , Feldman H , Hutton M ((2006) ) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442: , 916–919. |
[6] | Cruts M , Gijselinck I , van der Zee J , Engelborghs S , Wils H , Pirici D , Rademakers R , Vandenberghe R , Dermaut B , Martin JJ , van Duijn C , Peeters K , Sciot R , Santens P , De Pooter T , Mattheijssens M , Van den Broeck M , Cuijt I , Vennekens K , De Deyn PP , Kumar-Singh S , Van Broeckhoven C ((2006) ) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442: , 920–924. |
[7] | Mendsaikhan A , Tooyama I , Walker DG ((2019) ) Microglial progranulin: Involvement in Alzheimer’s disease and neurodegenerative diseases. Cells 11: , 230. |
[8] | Wu Y , Shao W , Todd TW , Tong J , Yue M , Koga S , Castanedes-Casey M , Librero AL , Lee CW , Mackenzie IR , Dickson DW , Zhang YJ , Petrucelli L , Prudencio M ((2019) ) Microglial lysosome dysfunction contributes to white matter pathology and TDP-43 proteinopathy in GRN-associated FTD. Cell Rep 24: , 109581. |
[9] | Zhang J , Velmeshev D , Hashimoto K , Huang YH , Hofmann JW , Shi X , Chen J , Leidal AM , Dishart JG , Cahill MK , Kelley KW , Liddelow SA , Seeley WW , Miller BL , Walther TC , Farese RV Jr , Taylor JP , Ullian EM , Huang B , Debnath J , Wittmann T , Kriegstein AR , Huang EJ ((2020) ) Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature 588: , 459–465. |
[10] | Davis SE , Roth JR , Aljabi Q , Hakim AR , Savell KE , Day JJ , Arrant AE ((2021) ) Delivering progranulin to neuronal lysosomes protects against excitotoxicity. J Biol Chem 297: , 100993. |
[11] | Logan T , Simon MJ , Rana A , Cherf GM , Srivastava A , Davis SS , Low RLY , Chiu CL , Fang M , Huang F , Bhalla A , Llapashtica C , Prorok R , Pizzo ME , Calvert MEK , Sun EW , Hsiao-Nakamoto J , Rajendra Y , Lexa KW , Srivastava DB , van Lengerich B , Wang J , Robles-Colmenares Y , Kim DJ , Duque J , Lenser M , Earr TK , Nguyen H , Chau R , Tsogtbaatar B , Ravi R , Skuja LL , Solanoy H , Rosen HJ , Boeve BF , Boxer AL , Heuer HW , Dennis MS , Kariolis MS , Monroe KM , Przybyla L , Sanchez PE , Meisner R , Diaz D , Henne KR , Watts RJ , Henry AG , Gunasekaran K , Astarita G , Suh JH , Lewcock JW , DeVos SL , Di Paolo G ((2021) ) Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic. Cell 2: , 4651–4668.e25. |
[12] | Le Ber I , Camuzat A , Hannequin D , Pasquier F , Guedj E , Rovelet-Lecrux A , Hahn-Barma V , van der Zee J , Clot F , Bakchine S , Puel M , Ghanim M , Lacomblez L , Mikol J , Deramecourt V , Lejeune P , de la Sayette V , Belliard S , Vercelletto M , Meyrignac C , Van Broeckhoven C , Lambert JC , Verpillat P , Campion D , Habert MO , Dubois B , Brice A ; French research network on FTD/FTD-MND ((2008) ) Phenotype variability in progranulin mutation carriers: A clinical, neuropsychological, imaging and genetic study. Brain 131: , 732–746. |
[13] | Saracino D , Ferrieux S , Noguès-Lassiaille M , Houot M , Funkiewiez A , Sellami L , Deramecourt V , Pasquier F , Couratier P , Pariente J , Géraudie A , Epelbaum S , Wallon D , Hannequin D , Martinaud O , Clot F , Camuzat A , Bottani S , Rinaldi D , Auriacombe S , Sarazin M , Didic M , Boutoleau-Bretonnière C , Thauvin-Robinet C , Lagarde J , Roué-Jagot C , Sellal F , Gabelle A , Etcharry-Bouyx F , Morin A , Coppola C , Levy R , Dubois B , Brice A , Colliot O , Gorno-Tempini ML , Teichmann M , Migliaccio R , Le Ber I ; French Research Network on FTD/FTD-ALS ((2021) ) Primary progressive aphasia associated with mutations: New insights into the nonamyloid logopenic variant. Neurology 6: , e88–e102. |
[14] | Caroppo P , Le Ber I , Camuzat A , Clot F , Naccache L , Lamari F , De Septenville A , Bertrand A , Belliard S , Hannequin D , Colliot O , Brice A ((2014) ) Extensive white matter involvement in patients with frontotemporal lobar degeneration: Think progranulin. JAMA Neurol 71: , 1562–1566. |
[15] | Jicha GA , Nelson PT ((2019) ) Hippocampal sclerosis, argyrophilic grain disease, and primary age-related tauopathy. Continuum (Minneap Minn) 25: , 208–233. |
[16] | Cruchaga C , Haller G , Chakraverty S , Mayo K , Vallania FL , Mitra RD , Faber K , Williamson J , Bird T , Diaz-Arrastia R , Foroud TM , Boeve BF , Graff-Radford NR , St Jean P , Lawson M , Ehm MG , Mayeux R , Goate AM ; NIA-LOAD/NCRAD Family Study Consortium ((2012) ) Rare variants in APP, PSEN1, and PSEN2 increase risk of AD in late-onset Alzheimer’s disease families. PLoS One 7: , e31039. |
[17] | Menéndez-González M , García-Martínez A , Fernández-Vega I , Pitiot A , Álvarez V ((2022) ) A variant in GRN of Spanish origin presenting with heterogeneous phenotypes., Neurologia (Engl Ed), 10.1016/j.nrleng.2022.10.001. |


