
FUS G559A Mutation in a Patient with a Frontotemporal Dementia-Motor Neuron Disease Compatible Syndrome: A Case Report
Abstract
Fused in sarcoma (FUS) mutations cause frontotemporal dementia (FTD) and motor neuron disease (MND). Here, we describe a 43-year-old man with progressive behavioral and cognitive change, myelopathy, clinical and electrophysiologic evidence of MND, and a FUS variant of unknown significance (VUS). This VUS, a heterozygous G559A transition (Gly187Ser), was previously reported in a patient with sporadic MND and affects important FUS biophysical properties. While this rare variant’s presence in a second patient with a related neurodegenerative syndrome does not establish pathogenicity, it raises the question of whether its association with our patient is coincidental and increases the possibility that FUS G559A is pathogenic.
INTRODUCTION
Genetic testing panels report variants of unknown significance (VUS), which can confuse clinicians and cause patient and family anxiety. The detection of VUS in patients with sporadic disease presents a particular challenge. Here, we report a frontotemporal dementia (FTD) and motor neuron disease (MND) syndrome in a patient with a fused in sarcoma (FUS) gene VUS. We discuss the case for changing the status of this designation from VUS to pathogenic.
CASE REPORT
A 43-year-old, diabetic man of white European descent with 2.5 years of fluctuating yet overall progressive behavioral and cognitive change was evaluated at the University of Kansas Memory Clinic. In spring, 2019 he presented to a hospital emergency department following a near-drowning event. The next day he phoned friends with no apparent purpose, sounded confused, and was returned to that hospital where he was diagnosed with diabetic ketoacidosis and told a head CT revealed remote “ministrokes.” He moved in with his mother and took leave from his job, improved over the next several months, and that autumn returned to independent living and his job.
Over the next year he uncharacteristically did not initiate family contact. In October 2020, friends told his mother he was acting strange. At the local hospital, he was again diagnosed with diabetic ketoacidosis, and contributions from alcohol and possibly recreational drug use (urine toxicology was positive for amphetamines) were considered. His mother assumed his diabetes management and eliminated alcohol and recreational drug access. He initially improved, but not to baseline, and then subsequently manifested progressive behavioral and cognitive decline. He ceased to socialize and developed profound apathy, abulia, and amotivation; his mother reported “when he is awake, he just stares straight ahead.” His memory declined. He crashed his car and lost his driving privileges. He developed intermittent bladder and bowel incontinence.
Past medical history included insulin-dependent diabetes since the first half of his third decade and hypertension. Medications included insulin, blood pressure medications, atorvastatin, daily aspirin, thiamine, and escitalopram. He was born at 29 weeks; a twin sibling died several months after birth. Mother and sister were in good health, and his father died from esophageal cancer at age 62. There was no family history of neurodegenerative disease. He graduated high school, where he excelled in athletics. He worked in construction and operated construction equipment. Throughout adulthood, he consumed alcohol but was not diagnosed with alcoholism. He was not known to seek or consume recreational drugs.
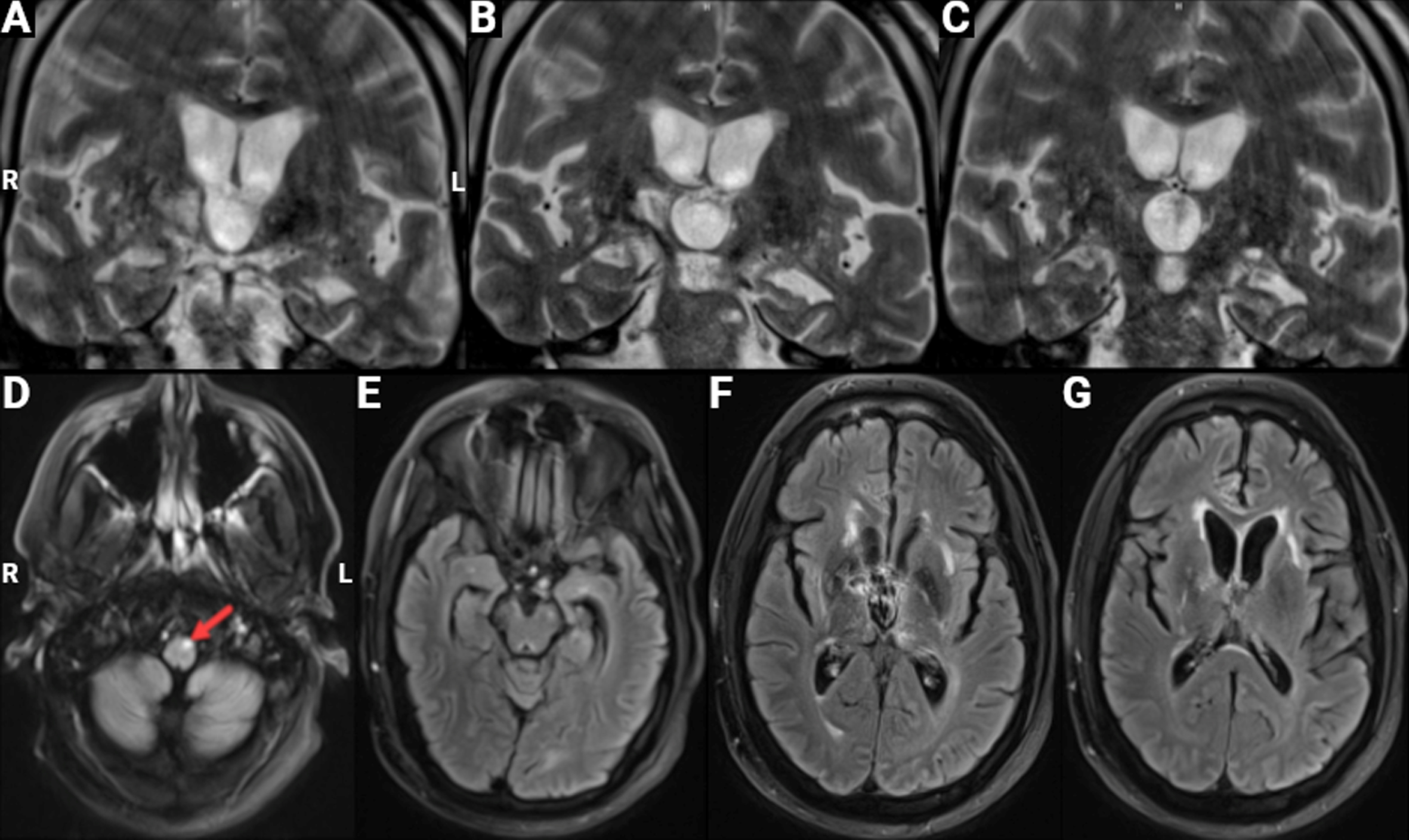
Two brain MRIs showed stable signal abnormalities in the right putamen and thalamus, which were felt to represent remote infarcts or possibly sequelae of a Wernicke’s encephalopathy event (Fig. 1). An EEG was unremarkable. Cerebrospinal fluid revealed a very minor protein elevation.
Fig. 1
Brain MRI. Images from a February 2022 MRI scan are shown; the obtained images were mildly degraded by motion artifact. A-C) Coronal T2 high-resolution 3MM sequence depicting left greater than right hippocampal volume loss in the anterior, medial, and posterior hippocampus regions. There is also significant ex vacuo dilatation of the third ventricle. D-G) Axial T2 FLAIR BLADE sequence showing focal signal hyperintensity (arrow) in the pyramidal tract on the left (D), potentially reflective of Wallerian degeneration consequent to a past stroke or selective primary degeneration of upper motor neurons. There are small, chronic, bilateral signal changes thought to represent sequelae of subcortical infarcts in the anterior temporal (E), basal ganglia/thalamus (F), and internal capsule (G) regions.
At his initial Memory Care Clinic evaluation (October 2021), the AD8 [1] was pan-positive except for the repetitive questioning item, as the patient never initiated conversation. He no longer shopped, cooked, or managed his medications. General neurologic exam showed reduced hand intrinsic muscle bulk, hypoactive upper extremity reflexes, hyperactive lower extremity reflexes, and suspected extension plantar responses. There were occasional non-purposeful movements of the right upper extremity. He had a normal gait, walked tandem, and the Romberg sign was absent.
His Mini-Mental State Exam score was 18 of 30. He had difficulty encoding information, was rapidly amnestic on verbal and visual recall testing, and did not improve with cueing. He scored 6/7 on a seven-point clock drawing scale [2] as digit placement was uneven. He named 3 animals over one minute. He calculated one dollar contained 25 nickels and showed persistent ideomotor apraxia. Anomia was not present. He spoke only in response to direct questions.
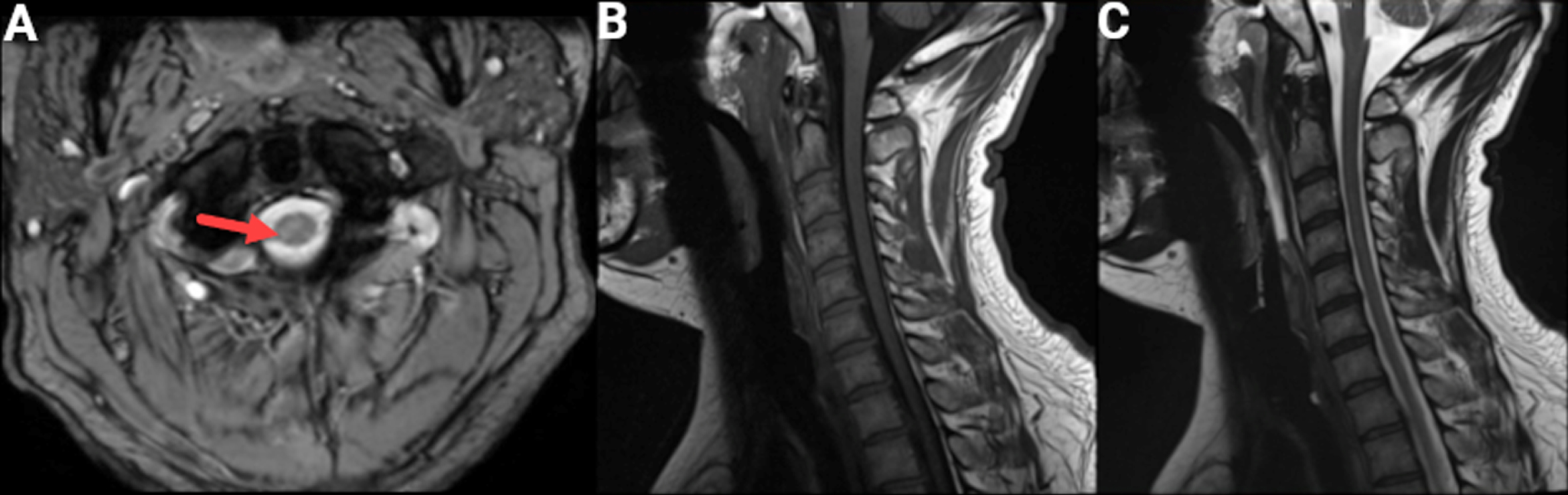
He was diagnosed with a frontal lobe syndrome of unclear etiology. Cervical MRI ruled out structural-induced myelopathy (Fig. 2) and revealed neither cervical cord nor nerve root compression. The axial images showed subtle bilateral corticospinal tract hyperintensities, greater on the right, which were not felt by the radiologist to represent pathologic signal changes.
Fig. 2
Cervical MRI. An MRI of the cervical spine and cord was obtained because of concomitant signs of central and peripheral motor system dysfunction at a cervical level. A) Axial GRE T2 sequence showing subtle T2 hyperintensity in the right (arrow) greater than left corticospinal tract. Sagittal T1 (B) and sagittal T2 (C) sequences without evidence of cord compression.
Over the next year his mother managed his diabetes. There was no alcohol or recreational drug use, and no focal or stepwise changes. Independent function continued to progressively decline. By October 2022 he rarely left his bed, wore a diaper due to continuous bowel and bladder incontinence, could no longer bathe himself, and occasionally gagged when eating. Cranial nerves were intact. There was again profound atrophy of the intrinsic hand muscles but no obvious fasciculations. Deep tendon reflexes were pervasively suppressed despite bilateral Babinskis. He walked with a slightly widened base and subtle athetoid hand movements. His Mini-Mental State Exam score declined to 14, but otherwise cognitive exam performance was comparable to the previous year. Comprehension was again intact, but he did not initiate conversation. When asked to describe the Western Aphasia Battery picnic picture, he stated “a guy reading a book, a house, kids are playing, I don’t know.”
Nerve conduction study (NCS)-electromyography (EMG) testing revealed severe sensorimotor axonal neuropathy, felt to partially, at least, represent a consequence of longstanding diabetes. There were diffuse, multilevel active denervation changes with fasciculations in lumbar and cervical myotomes and chronic reinnervation changes in lumbar myotomes (Table 1) that suggested motor neuron disease. C9orf72 genotyping by Invitae did not reveal repeat expansion. The Invitae FTD/ALS/Alzheimer’s disease panel noted a heterozygous FUS G559A transition that causes a glycine to serine substitution at amino acid 187 and is reported as a VUS.
Table 1
EMG Summary Table. The EMG study revealed multilevel active denervation changes with fasciculations in lumbar and cervical myotomes and chronic reinnervation changes in lumbar myotomes, which suggest the presence of motor neuron disease that differs in etiology from that of an also noted sensorimotor axonal neuropathy
| Insertional | Spontaneous Activity | Volitional MUAPs | |||||||
| Muscle | Activity | Fibs | PSW | Fasc | Poly | Amp | Dur | Recruit | Other |
| R. Tibialis anterior | ++ | None | None | Many | ++ | ++ | ++ | MOD dec | None |
| R. Gastrocnemius | ++ | 1+ | 1+ | Few | + | + | + | MOD dec | None |
| R. Vastus lateralis | + | 1+ | 1+ | Few | + | + | + | MLD dec | None |
| R. First dorsal interosseous | ++ | 2+ | 2+ | Few | unable to abduct | None | |||
| R. Pronator teres | + | 1+ | 1+ | Few | + | Normal | Normal | Normal | None |
| R. Biceps brachii | + | None | None | None | None | Normal | Normal | Normal | None |
| R. Triceps brachii | Normal | None | None | None | None | Normal | Normal | Normal | None |
| R. Deltoid | + | 1+ | 1+ | None | None | Normal | Normal | Normal | None |
| R. Lumbar paraspinals (mid) | ++ | None | None | None | None | ||||
| R. T7 paraspinal | Normal | None | None | None | None | ||||
Fibs, fibrillations; PSW, positive sharp waves; Fasc, fasciculations; Poly, polyphasic motor units; Amp, amplitude, Dur, duration; Recruit = recruitment.
The gene panel report cited results from three algorithms developed to assess functional impact: PolyPhen-2, SIFT, and Align GVGD. PolyPhen-2 returned a result of “not available”; the SIFT prediction was “tolerated” (the probability value was 0.06, with probabilities of < 0.05 predicted to be deleterious); and the Align GVD prediction was “Class C0,” which presumes a functional consequence is unlikely. We performed an additional analysis using SNAP2 [3], which predicted the variant as “neutral” (score =-24, predicted accuracy 61%).
The patient’s adult relatives declined genetic testing. Following his final neurologic assessment, the patient developed progressive weakness and his ambulation declined, he fell and sustained a humeral fracture, and he exhibited slowly worsening uremia. He was transitioned to hospice care. He died at home 5.5 months after his genetic testing. The cause of death was attributed to his chronic conditions. An autopsy was not requested and, therefore, not performed.
DISCUSSION
Apathy, abulia, amotivation, mutism, and bladder/bowel incontinence suggests frontal lobe dysfunction. We cannot exclude contributions from brain injury due to stroke, vitamin B1 deficiency (Wernicke’s encephalopathy), alcohol and recreational drug consumption, hypoxia from water submersion, and hypo and hyperglycemia. Progressive decline in the absence of ongoing such insults, though, raises the possibility of a neurodegenerative disorder and suggests these factors were to some extent potentially consequences, not causes, of his frontal lobe syndrome.
Strokes could not explain the lower motor neuron clinical or electrophysiology signs. Denervation and reinnervation changes on EMG raise suspicion for a neurodegenerative MND. FTD and MND genetics overlap; genes that cause FTD also cause MND [4, 5].
Our case lacks a neuropathologic assessment. An absence of FUS pathology in the patient’s spinal cord and brain, if found, would argue against FUS G559A pathogenicity. While the presence of FUS histopathology in these tissues would be consistent with FUS G559A pathogenicity, it would still fall short of establishing pathogenicity, since FUS histopathology in FTD-MND cases frequently occurs in the absence of FUS mutation.
The FUS gene, also referred to as translated in liposarcoma (TLS), locates to chromosome 16p11.2. It encodes a DNA/RNA-binding protein that forms part of the heterogeneous nuclear ribonucleoprotein (hnRNP) complex and participates in transcription regulation, RNA splicing, RNA transport, and DNA repair [6]. Variants cause hereditary essential tremor type 4 (ETM4) and amyotrophic lateral sclerosis (ALS), specifically the ALS6 subtype [7, 8]. ALS6 can present as an isolated MND syndrome, or with concurrent FTD [9]. FUS mutation, potentially alone or in conjunction with variation in other genes, can also cause FTD [10, 11].
The GnomAD browser quotes a G559A allele frequency of 0.000008. Algorithms that speculate biological impact do not predict functional consequences, although these algorithms have a demonstrated need for improved reliability [12–14]. As such, it is appropriate that Invitae continues to cite G559A as a VUS. Interestingly, the recent study of Niaki et al. did report that the resulting Gly187Ser substitution alters FUS biophysical properties [15]. Compared to wild-type FUS, Gly187Ser FUS showed altered dwell-times for RNA binding and Gly187Ser complexes with RNA show altered phase separation dynamics. The latter phenomenon is emerging as a critical process for a variety of intracellular functions, and its dysregulation may play a role in protein aggregation diseases [16].
Rademakers et al. previously reported FUS G559A heterozygosity in a 79-year-old woman with ALS [17]. The authors could neither establish a pattern of Mendelian inheritance nor ascertain a family history of neurologic disease. An autopsy was not performed, and it is unknown whether FUS histology was altered. Based on the rarity of the variant and its localization to a gene region where familial ALS mutations reside, the authors speculated this variant was likely pathogenic.
Our report, therefore, notes the presence of FUS G559A in a second patient with an apparent neurodegenerative syndrome. In this case, the syndrome initially presented as a pure frontal lobe syndrome, or FTD, that appears to have evolved into a broader FTD-MND disorder. The patient’s co-morbidities may have influenced his overall course by compounding nervous system stress, enhancing penetrance, and predisposing to a sporadic presentation. It is also possible that FUS G559A disease-relevance in this case was enabled by concomitant, undetected variation in another gene or genes. As we were unable to genotype other family members, we cannot say whether the variant was inherited or arose de novo. Regardless, while the presence of the rare FUS G559A variant in a second patient with a related neurodegenerative syndrome does not establish pathogenicity, it raises the question of whether its presence in our patient is coincidental and increases the possibility that FUS G559A can indeed confer pathogenic consequences.
ACKNOWLEDGMENTS
The authors wish to thank the patient and his mother.
FUNDING
RHS and RT are supported by the University of Kansas Alzheimer’s Disease Center (NIA P30 AG 072973).
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
DATA AVAILABILITY
Upon reasonable request, the data described in this study are available from the corresponding author.
REFERENCES
[1] | Galvin JE , Roe CM , Powlishta KK , Coats MA , Muich SJ , Grant E , Miller JP , Storandt M , Morris JC ((2005) ) The AD8: A brief informant interview to detect dementia. Neurology 65: , 559–564. |
[2] | Solomon PR , Hirschoff A , Kelly B , Relin M , Brush M , DeVeaux RD , Pendlebury WW ((1998) ) A 7 minute neurocognitive screening battery highly sensitive to Alzheimer’s disease. Arch Neurol 55: , 349–355. |
[3] | Hecht M , Bromberg Y , Rost B ((2015) ) Better prediction of functional effects for sequence variants. BMC Genomics 16 Suppl 8: , S1. |
[4] | Crook A , Williams K , Adams L , Blair I , Rowe DB ((2017) ) Predictive genetic testing for amyotrophic lateral sclerosis and frontotemporal dementia: Genetic counselling considerations. Amyotroph Lateral Scler Frontotemporal Degener 18: , 475–485. |
[5] | Gelon PA , Dutchak PA , Sephton CF ((2022) ) Synaptic dysfunction in ALS and FTD: Anatomical and molecular changes provide insights into mechanisms of disease. Front Mol Neurosci 15: , 1000183. |
[6] | Dormann D , Haass C ((2013) ) Fused in sarcoma (FUS): An oncogene goes awry in neurodegeneration. Mol Cell Neurosci 56: , 475–486. |
[7] | Merner ND , Girard SL , Catoire H , Bourassa CV , Belzil VV , Rivière JB , Hince P , Levert A , Dionne-Laporte A , Spiegelman D , Noreau A , Diab S , Szuto A , Fournier H , Raelson J , Belouchi M , Panisset M , Cossette P , Duprée N , Bernard G , Chouinard S , Dion PA , Rouleau GA ((2012) ) Exome sequencing identifies FUS mutations as a cause of essential tremor. Am J Hum Genet 91: , 313–319. |
[8] | Kwiatkowski TJ , Jr. , Bosco DA , Leclerc AL , Tamrazian E , Vanderburg CR , Russ C , Davis A , Gilchrist J , Kasarskis EJ , Munsat T , Valdmanis P , Rouleau GA , Hosler BA , Cortelli P , de Jong PJ , Yoshinaga Y , Haines JL , Pericak-Vance MA , Yan J , Ticozzi N , Siddique T , McKenna-Yasek D , Sapp PC , Horvitz HR , Landers JE , Brown RH , Jr. ((2009) ) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: , 1205–1208. |
[9] | Yan J , Deng HX , Siddique N , Fecto F , Chen W , Yang Y , Liu E , Donkervoort S , Zheng JG , Shi Y , Ahmeti KB , Brooks B , Engel WK , Siddique T ((2010) ) Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75: , 807–814. |
[10] | Brenner D , Müller K , Lattante S , Yilmaz R , Knehr A , Freischmidt A , Ludolph AC , Andersen PM , Weishaupt JH ((2022) ) FUS mutations dominate TBK1 mutations in FUS/TBK1 double-mutant ALS/FTD pedigrees. Neurogenetics 23: , 59–65. |
[11] | Wagner M , Lorenz G , Volk AE , Brunet T , Edbauer D , Berutti R , Zhao C , Anderl-Straub S , Bertram L , Danek A , Deschauer M , Dill V , Fassbender K , Fliessbach K , Götze KS , Jahn H , Kornhuber J , Landwehrmeyer B , Lauer M , Obrig H , Prudlo J , Schneider A , Schroeter ML , Uttner I , Vukovich R , Wiltfang J , Winkler AS , Zhou Q , Ludolph AC , Oexle K , Otto M , Diehl-Schmid J , Winkelmann J Clinico-genetic findings in 509 frontotemporal dementia patients. . Mol Psychiatry ((2021) ) 26: , 5824–5832. |
[12] | Miller M , Bromberg Y , Swint-Kruse L ((2017) ) Computational predictors fail to identify amino acid substitution effects at rheostat positions. Sci Rep 7: , 41329. |
[13] | Andreoletti G , Pal LR , Moult J , Brenner SE ((2019) ) Reports from the fifth edition of CAGI: The Critical Assessment of Genome Interpretation. Hum Mutat 40: , 1197–1201. |
[14] | Pal LR , Kundu K , Yin Y , Moult J ((2020) ) Matching whole genomes to rare genetic disorders: Identification of potential causative variants using phenotype-weighted knowledge in the CAGI SickKids5 clinical genomes challenge. Hum Mutat 41: , 347–362. |
[15] | Niaki AG , Sarkar J , Cai X , Rhine K , Vidaurre V , Guy B , Hurst M , Lee JC , Koh HR , Guo L , Fare CM , Shorter J , Myong S ((2020) ) Loss of dynamic RNA interaction and aberrant phase separation induced by two distinct types of ALS/FTD-linked FUS mutations. Mol Cell 77: , 82–94.e84. |
[16] | Boeynaems S , Alberti S , Fawzi NL , Mittag T , Polymenidou M , Rousseau F , Schymkowitz J , Shorter J , Wolozin B , Van Den Bosch L , Tompa P , Fuxreiter M ((2018) ) Protein phase separation: A new phase in cell biology. Trends Cell Biol 28: , 420–435. |
[17] | Rademakers R , Stewart H , Dejesus-Hernandez M , Krieger C , Graff-Radford N , Fabros M , Briemberg H , Cashman N , Eisen A , Mackenzie IR ((2010) ) Fus gene mutations in familial and sporadic amyotrophic lateral sclerosis. Muscle Nerve 42: , 170–176. |


