
Subtypes of Dementia with Lewy Bodies: Clinical Features, Survival, and Apolipoprotein E Effect
Abstract
Background:
Dementia with Lewy bodies (DLB) is a progressive neurodegenerative disease with various clinical symptoms. Limited data have described the clinical subtypes of DLB.
Objective:
We aimed to compare clinical subtypes of DLB according to initial symptoms and to study the effect of Apolipoprotein E (APOE) gene in DLB.
Methods:
We included DLB patients classified into three groups based on initial symptoms: non-motor onset (cognitive and/or psychiatric) (NMO-DLB), motor onset (parkinsonism and/or gait disorders) (MO-DLB), and mixed onset (non-motor and motor symptoms) (MXO-DLB). Clinical and APOE genotype associations and survival were analyzed.
Results:
A total of 268 patients were included (NMO-DLB = 75%, MXO-DLB = 15.3%, MO-DLB = 9.7%). Visual hallucinations were more frequent (p = 0.025), and attention was less commonly impaired in MXO-DLB (p = 0.047). When adjusting with APOE ɛ4 status (APOE genotype performed in 155 patients), earlier falls and frontal lobe syndrome were more common in MXO-DLB (p = 0.044 and p = 0.023, respectively). The median MMSE decline was 2.1 points/year and the median FAB decline was 1.9 points/year, with no effect of clinical subtypes. Median survival was 6 years. It was similar in DLB subtypes (p = 0.62), but shorter for patients with memory symptoms at onset (p = 0.04) and for males (p = 0.0058).
Conclusions:
Our study revealed a few differences between DLB clinical subtypes. APOE ɛ4 appears to be associated with earlier falls and a higher prevalence of frontal syndrome in MXO-DLB. However, DLB clinical subtypes did not impact on survival. Nevertheless, survival analysis identified other poor prognosis factors, notably inaugural memory impairment and male gender.
INTRODUCTION
Dementia with Lewy bodies (DLB) is a progressive neurodegenerative disorder that belongs to the group of synucleinopathies [1]. It is the second most common cause of neurodegenerative dementia after Alzheimer’s disease (AD) and accounts for 0.3 to 34.4% of all dementia cases [2]. Its classic clinical and neuropathological description was made by Kosaka et al. in 1984 [3] and the “Lewy body dementia” label was coined in 1996 [2]. Clinically, DLB is defined, according to the latest criteria of McKeith et al. (2017), by the presence of dementia associated with a variable combination of core clinical features including visual hallucinations, parkinsonism, cognitive fluctuations, and rapid eye movement sleep behavior disorders (RBD) [1]. A variety of other clinical symptoms are suggestive features and can help with diagnosis. In the prodromal phase of DLB, not all symptoms are present, leading to high clinical heterogeneity. The clinical variability may be related to heterogeneous underlying pathology [4, 5]. From an etiopathogenic point of view, DLB is a multifactorial disease combining environmental and genetic factors. Although the current understanding of the genetic etiology is still limited, several genes seem to play a potential role in DLB such as the Apolipoprotein E (APOE), synuclein (SNCA), and β-glucocerebrosidase (GBA) genes [6].
There are currently limited data on the different DLB clinical subtypes according to initial symptoms. A better understanding of their peculiarities could provide a redesigned insight into the underlying pathophysiology of DLB and the possible factors influencing the disease course.
Thus, the aim of our study was to compare the clinical subtypes of DLB according to initial symptoms and to study the effect of the APOE gene in DLB in a Tunisian cohort.
METHODS
Study subjects
An observational cross-sectional study was carried out in the Department of Neurology at Razi University Hospital, a tertiary referral center in Tunis in North Tunisia, over a period of 18 years (from January 2003 to December 2020). Patients with Major Neurocognitive Disorder (MNCD) according to the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) [7] and with clinically diagnosed probable or possible DLB according to the revised Mc Keith Criteria were included [1]. The latter requires the presence of dementia/MNCD, which is the essential feature, associated with one (possible DLB) or two (probable DLB) core clinical features: fluctuating cognition, recurrent visual hallucinations, RBD and one or more spontaneous cardinal features of parkinsonism. The first 3 typically occur early and may persist throughout the course [1]. We excluded all patients with parkinsonism and/or dementia of other origins.
Clinical and neuropsychological assessment
All patients had a neurological examination performed by a neurologist and a systematic brain imaging (brain CT scan or brain MRI). Demographic, clinical, and neuropsychological data were collected using standardized case-report forms.
Information was obtained from the participants and their caregivers about family and personal medical history and medication. Disease onset was defined as the age of occurrence of either cognitive, psychiatric, or motor symptoms (parkinsonism and/or gait disorders). We conducted a retrospective re-categorization analysis based on initial symptoms to classify patients into three subgroups: non-motor onset (cognitive and/or psychiatric onset) (NMO-DLB), motor onset (parkinsonism and/or gait disorders) (MO-DLB), and mixed onset (non-motor and motor symptoms) (MXO-DLB). We specified the presence and the number of core clinical features, i.e., cognitive fluctuations, recurrent visual hallucination, RBD, and parkinsonism. We also specified the presence of supportive clinical features, including severe sensitivity to antipsychotic agents, postural instability, repeated falls, syncope, severe autonomic dysfunction, hypersomnia, hyposmia, hallucinations in other modalities, systematized delusions, apathy, anxiety, and depression. Unified Parkinson’s disease Rating Scale (UPDRS) section-III was used to rate the severity of extrapyramidal symptoms. The degree of severity of parkinsonian symptoms was measured using the Hoehn & Yahr (H&Y) scale. Other motor signs were assessed on examination including other movement disorders. For the assessment of levodopa responsiveness, we used an acute pharmacological test, namely the Acute levodopa challenge (ALC), which is routinely performed in our department. We used a standard protocol by administering a single dose of levodopa/carbidopa 250/25 mg. Motor response was quantified using the MDS- UPDRS-III. During the ALC, motor examination was performed immediately before and every 30 min after levodopa intake until the motor conditions returned to the motor baseline status. We calculated the percentage of motor response as the ratio of the difference between the baseline and the peak-of-dose motor scores by the baseline motor score.
Levodopa-responsiveness was defined as an improvement rate ≥30% of MDS- UPDRS-III [8].
Besides, each patient underwent a neuropsychological examination at first consultation comprising the 30-item Mini-Mental State Examination (MMSE) standardized and validated in Tunisia and adjusted for age and education [9] to assess overall cognitive efficiency. The validated Arab version of the frontal assessment battery (FAB) [10] was used to evaluate executive functions and a score less than 16 was considered abnormal.
If patients had a second neuropsychological assessment after 6 months or more, the cognitive progression was evaluated for Global cognitive function using the annual decline of MMSE and for executive function using the annual decline of FAB. We calculated the annual decline of MMSE and FAB according to the formula:
- Annual decline of MMSE = (MMSE1-MMSE2)/(Time between MMSE1 and MMSE2 (years))
- Annual decline of FAB = (FAB1-FAB2)/(Time between FAB1 and FAB2 (years))
MMSE1 and FAB1 corresponded to the scores at first evaluation and MMSE2 and FAB2 corresponded to the scores at second evaluation.
The different cognitive domains evaluated by neuropsychological assessment included orientation, attention, episodic memory, language, apraxia, agnosia, visuospatial functions, judgment, and reasoning. Beck’s Depression Inventory (BDI) (if the age <65 years) and Geriatric Depression Scale (GDS) (if the age >65 years) were used to evaluate mood disturbances and detect depression. Behavioral disorders were identified by the Neuropsychiatric Inventory (NPI).
Genetic study
Genotyping of APOE was performed using the Restriction Fragment Length Polymorphism Polymerase Chain Reaction (RFLP-PCR). APOE genotypes were determined by scoring for a unique combination of fragment sizes, as depicted by Hixon et al. In fact, digestion by HhaI restriction enzyme gives various combinations of fragment sizes for each genotype as pursued: ɛ2/ɛ2, 91 and 83 bp; ɛ3/ɛ3, 91 and 48 bp; ɛ4/ɛ4, 72 and 48 bp and a mixed genotype: ɛ2/ɛ3, 91, 83, and 48 bp; ɛ3/ɛ4, 91, 72, and 48 bp; ɛ2/ɛ4, 91, 83, 72, and 48 bp.
Statistical analysis
Statistical analyses were performed using R software for Windows using the “multinom”, “SNPassoc”, “Hmisc”, and “ggplot” packages. Categorical variables were expressed as counts and percentages. For continuous variables, mean and standard deviations (SD) or median and Interquartile range (IQR) were used when appropriate. A chi-square exact test and a Fisher’s exact test were used to calculate differences in categorical data as appropriate. To analyze the continuous variables, ANOVA test or nonparametric tests were used according to the distribution of data. Multinomial logistic regression was used to model outcome variables according to APOE ɛ4 carrying status. Corrections for multiple comparisons were employed with a Bonferroni correction. Analysis with a value of p < 0.05 were considered statistically significant. Survival was explored using Kaplan Meier analysis. Demographic, clinical, neuropsychological features, and frequency of APOE ɛ4 allele were analyzed in the survivalanalysis.
Ethics
All subject investigations conformed to the principles outlined in the Declaration of Helsinki and have been performed with permission of the Razi hospital ethic committee. All subjects were informed about the purposes of the study and gave written consent (patients themselves or caregivers) to participate in the study.
RESULTS
General study population characteristics
We screened 4,132 patients with MNCD and 464 patients with atypical parkinsonian syndrome. A total of 268 patients meeting the McKeith et al. DLB criteria [1] were included in this study: 250 probable DLB and 18 possible DLB. Median time to meet probable DLB criteria was 1.0 year (0.5–2.0). Male predominance was noted with a sex-ratio equal to 1.4. The mean age at disease onset was 75.4±7.9 years and ranged from 52 to 92 years and the mean age of onset of MNCD was 75.7±7.9 years. The mean disease duration at 1st evaluation was 2.8±1.8 years. Parental consanguinity was found in 35.5% of cases, with a family history of MNCD and parkinsonism in 43.7% and 16.4% of cases respectively. Clinical subtypes based on initial symptoms showed a non-motor onset in 75.0 %, a motor onset in 9.7%, and a mixed onset in 15.3% of cases (Fig. 1). There were no significant differences in age at disease onset, disease duration, sex ratio and family history between the 3 groups. Detailed demographic characteristics and family history of total DLB patients as well as stratified according to clinical subtypes were summarized in Table 1.
Fig. 1
Clinical subtypes of DLB according to initial symptoms.
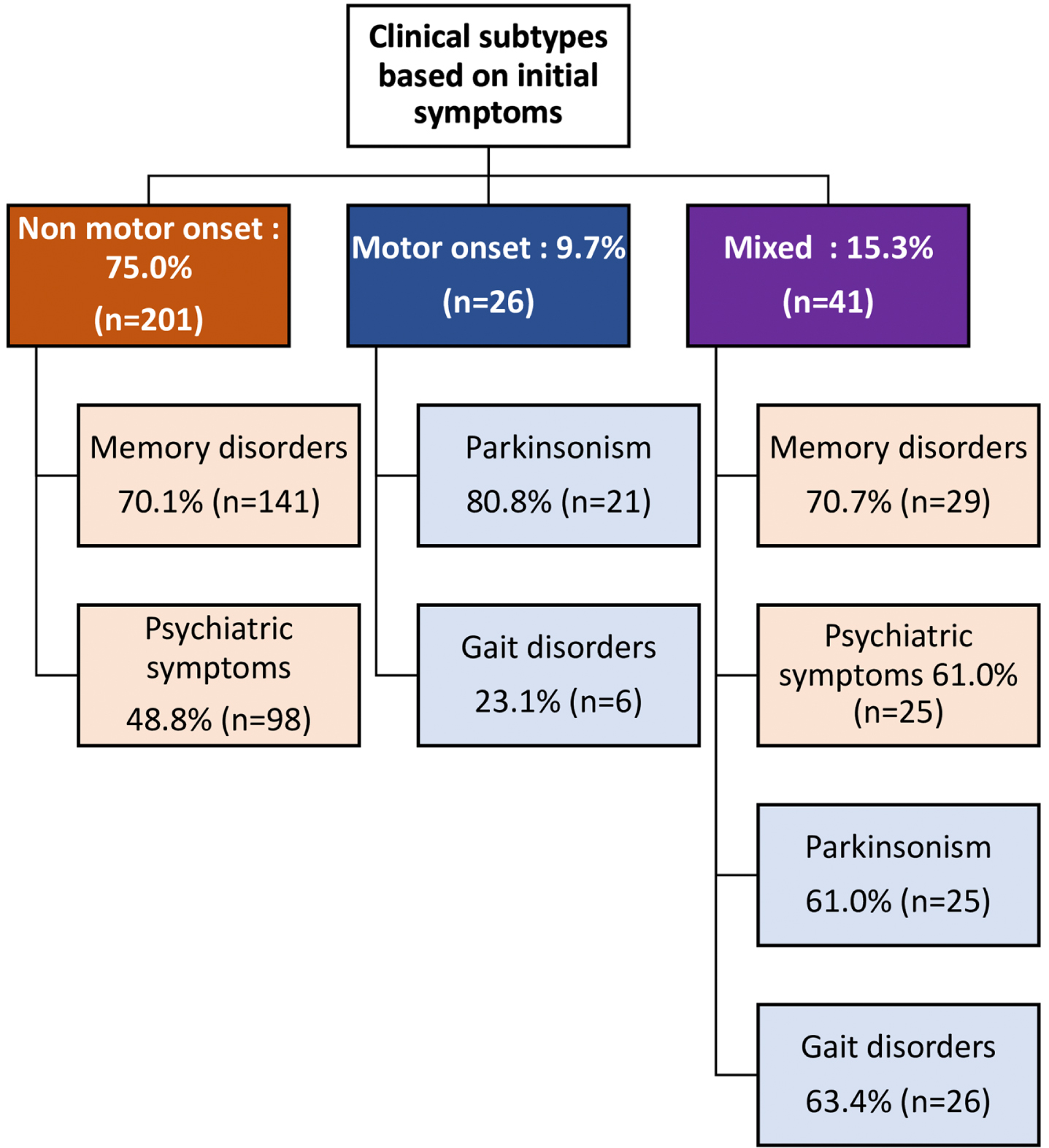
Table 1
Demographic characteristics and family history of DLB patients
| Overall N = 268 | Clinical subtypes | ||||
| NMO-DLB, N = 201 | MO-DLB, N = 26 | MXO-DLB, N = 41 | p | ||
| Gender (M/F), n (%) | 155/113 (57.8/42.2) | 117/84 (58.2/41.8) | 14/12 (53.8/46.2) | 24/17 (58.5/41.5) | 0.743 |
| Age at 1st evaluation, mean±SD (y) | 78.16±7.98 | 78.25±8.25 | 79.9±6.15 | 76.61±7.49 | 0.091 |
| Disease duration at 1st evaluation, mean±SD (y) | 2.75±1.78 | 2.85±1.9 | 2.88±1.34 | 2.20±1.32 | 0.072 |
| Age at disease onset, mean±SD (y) | 75.4±7.94 | 75.39±8.28 | 76.96±6.1 | 74.44±7.25 | 0.216 |
| Educational level, n (%) | |||||
| Illiterate | 184 (68.66) | 138 (68.66) | 17 (65.38) | 29 (70.73) | 0.412 |
| Primary school | 47 (17.54) | 33 (16.42) | 4 (15.38) | 10 (24.39) | |
| High school | 19 (7.09) | 15 (7.46) | 3 (11.54) | 1 (2.44) | |
| University | 12 (4.48) | 10 (4.98) | 1 (3.85) | 1 (2.44) | |
| Not specified | 6 (2.24) | 5 (2.49) | 1 (3.85) | 0 | |
| Parental consanguinity, n (%) | 95 (35.45) | 65 (32.34) | 10 (38.46) | 20 (48.78) | 0.112 |
| Family history of MNCD, n (%) | 117 (43.7) | 92 (45.77) | 10 (38.46) | 15 (36.59) | 0.702 |
| 1st degree relative | 56 (20.9) | 47 (23.38) | 3 (11.54) | 6 (14.63) | 0.967 |
| 2nd degree relative | 57(21.3) | 43 (21.39) | 6 (23.08) | 8 (19.51) | 0.721 |
| 3rd degree relative | 25 (9.33) | 19 (9.45) | 1 (3.85) | 5 (12.2) | 0.273 |
| Family history of Parkinsonism, n (%) | 44 (16.42) | 28 (13.93) | 8 (30.77) | 8 (19.51) | 0.416 |
| 1st degree relative | 12 (4.48) | 9 (4.48) | 1 (3.85) | 2 (4.88) | 0.845 |
| 2nd degree relative | 20 (7.46) | 14 (6.97) | 4 (15.38) | 2 (4.88) | 0.146 |
| 3rd degree relative | 10 (3.73) | 7 (3.48) | 1 (3.85) | 2 (4.88) | 0.273 |
NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies.
At first neuropsychological assessment, the most impaired cognitive domains were executive functions (99.1%), memory (98.8%), and attention (98.7%) followed by and visuo-spatial functions (92.7%). Language was impaired in 83.7%, apraxia was noted in 81.0% and judgment and reasoning were impaired respectively in 63.6 % and 66.5% while agnosia was found in only 17.7% of cases and was the least affected cognitive domain. Mean MMSE and FAB scores at first evaluation were respectively 15.5/30±6.2 and 5.9/18±3.4.
APOE genotype in DLB patients
The genetic study was performed in 155 patients. The APOE ɛ4 allele was found in 19.68% of cases and APOE ɛ2 in 5.16% of cases. The APOE ɛ3/ɛ3 genotype was the most frequent (54.83%) in the total DLB population as well as in the different clinical subtypes, followed by APOE ɛ3/ɛ4 (32.25%). The APOE ɛ4/ɛ4 genotype was found in only 3.25% of cases. There were no significant differences in allelic and genotypic frequencies of APOE gene across the clinical subtypes (Table 2).
Table 2
Allelic and genotypic frequencies of APOE gene in DLB patients
| Clinical subtypes | |||||
| Overall N = 155 | NMO-DLB N = 116 | MO-DLB N = 16 | MXO-DLB N = 23 | p | |
| Allelic frequencies, n (%) | |||||
| APOE ɛ2 | 16 (5.16) | 13 (5.60) | 0 (0.0) | 3 (6.52) | 0.639 |
| APOE ɛ3 | 233 (75.16) | 174 (75.0) | 26 (81.25) | 33 (71.74) | |
| APOE ɛ4 | 61 (19.68) | 45 (19.4) | 6 (18.75) | 10 (21.74) | |
| Genotypic frequencies, n (%) | |||||
| APOE ɛ2/ɛ2 | 1 (0.64) | 1 (0.9) | 0 (0.0) | 0 (0.0) | 0.899 |
| APOE ɛ2/ɛ3 | 13 (8.39) | 10 (8.6) | 0 (0.0) | 3 (13.0) | |
| APOE ɛ2/ɛ4 | 1 (0.64) | 1 (0.9) | 0 (0.0) | 0 (0.0) | |
| APOE ɛ3/ɛ3 | 85 (54.83) | 64 (55.2) | 10 (62.5) | 11 (47.8) | |
| APOE ɛ3/ɛ4 | 50 (32.25) | 36 (31.0) | 6 (37.5) | 8 (34.8) | |
| APOE ɛ4/ɛ4 | 5 (3.25) | 4 (3.4) | 0 (0.0) | 1 (4.3) | |
NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies.
Clinical features and APOE ɛ4 genotype effect in DLB clinical subtypes
Regarding symptoms at disease onset, memory complaints were the most frequent (63.4%), followed by psychiatric symptoms (45.9%), parkinsonism (17.2%), and gait disorders (11.9%). Memory disorders were equally common in both NMO-DLB and MXO-DLB, but psychiatric symptoms were more prevalent in MXO-DLB. Additionally, parkinsonism was more frequent in MO-DLB, while gait disorders were observed more often in MXO-DLB (p < 0.001) (Fig. 1).
At the time of the study, all core clinical features were present in 28.7%, three of them were present in 41.8%, two of them were present in 23.1% and only one core clinical feature was present in 6.3% of the cases. Among core clinical features, visual hallucinations were significantly more frequent in MXO-DLB than in MO-DLB (p = 0.025). Among supportive clinical features, postural instability was significantly more common in MO-DLB when adjusting with APOE ɛ4 carrying status (p = 0.0027) (Table 3).
Table 3
Essential feature, core clinical features and supportive features of DLB across clinical subtypes
| Clinical subtypes | |||||||
| Overall N = 268 (%) | NMO-DLB N = 201 | MO-DLB N = 26 | MXO-DLB N = 41 | p | p1 | ||
| Cognitive complaint | |||||||
| Memory complaint, n (%) | 252 (94.03) | 190 (94.52) | 23 (88.46) | 39 (95.12) | 0.327 | 0.06 | |
| Age of onset of memory disorders, mean±SD (y) | 76.22±7.9 | 75.9±8.38 | 78.9±6.18 | 75.79±6.7 | 0.219 | 0.200 | |
| Core clinical features | |||||||
| Number of core clinical features, mean±SD | 2.93±0.88 | 2.95±0.85 | 2.92±0.93 | 2.83±1.0 | 0.724 | 0.644 | |
| Cognitive fluctuations, n (%) | 181 (67.54) | 138 (68.65) | 16 (61.53) | 27 (65.85) | 0.820 | 0.900 | |
| Visual Hallucinations †, n (%) | 243 (90.67) | 187 (93.03) | 21 (80.76) | 35 (85.36) | 0.025 | 0.037 | |
| Age of onset of VH, mean±SD (y) | 76.52±7.8 | 76.5±8.09 | 79.39±4.96 | 74.80±7.93 | 0.052 | 0.318 | |
| Disease duration at onset of VH, median [IQR] (y) | 1 [0–2] | 0.75 [0.0–2.0] | 2.0 [1.0–4.0] | 0.0 [0.0–1.0] | 0.0007 | 0.044 | |
| Parkinsonism, n (%) | 230 (85.82) | 166 (82.58) | 26 (100.0) | 38 (92.68) | 0.759 | 0.810 | |
| Age of onset of parkinsonism, mean±SD (y) | 76.65±7.9 | 77.12±8.23 | 77.15±6.35 | 74.22±7.19 | 0.095 | 0.653 | |
| Disease duration at onset of parkinsonism, median [IQR] (y) | 2 [0.125–3.0] | 2.0 [1.0–4.0] | 0.0 [0.0–0.0] | 0.0 [0.0–0.0] | <0.001 | <0.001 | |
| RBD, n (%) | 140 (52.24) | 100 (49.75) | 12 (46.15) | 16 (39.02) | 0.438 | 0.396 | |
| Age of onset of RBD, mean±SD (y) | 71.01±14.96 | 70.95±15.46 | 76.25±7.63 | 68.57±14.6 | 0.444 | 0.446 | |
| Supportive clinical features | |||||||
| Severe sensitivity to antipsychotic agents *, n (%) | 44/101 (43.56) | 34/82 (41.46) | 5/8 (62.5) | 5/11 (45.45) | 0.281 | 0.814 | |
| Gait disorders, n (%) | 161 (60.07) | 109 (54.22) | 19 (73.07) | 33 (80.48) | 0.211 | 0.645 | |
| Postural instability ‡, n (%) | 32 (11.94) | 18 (8.95) | 8 (30.76) | 6 (14.63) | 0.103 | 0.0027 | |
| Age of gait disorder, mean±SD (y) | 77.3±7.35 | 77.93±7.52 | 78.18±7.26 | 75.16±6.68 | 0.098 | 0.197 | |
| Repeated falls, n (%) | 123 (45.9) | 87 (43.28) | 16 (61.53) | 20 (48.78) | 0.478 | 0.242 | |
| Age of appearance of repeated falls §, mean±SD (y) | 76.66±7.5 | 77.95±7.72 | 76.73±6.87 | 72.95±6.53 | 0.071 | 0.044 | |
| Syncope, n (%) | 23 (8.58) | 18 (8.95) | 2 (7.69) | 3 (7.31) | 0.900 | 0.239 | |
| Severe autonomic dysfunction, n (%) | 182 (67.91) | 135 (67.16) | 20 (76.92) | 27 (65.85) | 0.403 | 0.496 | |
| Hypersomnia, n (%) | 52 (19.4) | 40 (19.9) | 5 (19.23) | 7 (17.07) | 0.778 | 0.333 | |
| Hyposmia, n (%) | 16 (5.97) | 12 (5.97) | 2 (7.69) | 2 (4.87) | 0.643 | 0.947 | |
| Hallucinations in other modalities, n (%) | 85 (31.72) | 70 (34.82) | 8 (30.77) | 7 (17.07) | 0,219 | 0.701 | |
| Systematized delusions, n (%) | 145 (54.10) | 115 (57.21) | 13 (50.00) | 17 (41.46) | 0,245 | 0.163 | |
| Apathy, n (%) | 45 (16.79) | 38 (18.90) | 1 (3.84) | 6 (14.63) | 0.416 | 0.824 | |
| Anxiety, n (%) | 66 (24.63) | 56 (27.86) | 6 (23.07) | 4 (9.75) | 0.105 | 0.074 | |
| Depression, n (%) | 100 (37.31) | 75 (37.31) | 10 (38.46) | 15 (36.58) | 0.879 | 0.071 | |
†MO-DLB versus MXO-DLB: p = 0.0017. ‡MO-DLB versus NMO-DLB: p = 0.00137 and MO-DLB versus MXO-DLB: p = 0.00163. §MO-DLB versus MXO-DLB: p = 0.021. *Calculated based on the number of patients who received antipsychotics. p1: adjusted for APOE ɛ4 carrying status. NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies.
Frequencies of repeated falls were not significantly different across clinical subtypes; however, they appeared at an earlier age in MXO-DLB subtype when adjusting with APOE ɛ4 genotype (p = 0.044) (Table 3).
Neurological examination showed that the parkinsonian syndrome involved mostly both trunk and limbs (77.83%), was mainly bilateral (94.35%), symmetrical (43.91%), and included mostly the 3 motor features (akinesia, tremor, and rigidity) (55.65%). Mean UPDRS-III score was 41.2±17.1, mean Hoehn and Yahr score was 2.9±0.9 and a positive response to levodopa was present in only 12.2% of cases. The comparison of the clinical subtypes showed no significant differences regarding parkinsonism characteristics. Frontal lobe syndrome was significantly more frequent in MXO-DLB when adjusting with APOE ɛ4 carrying status (p = 0.023) (Table 4).
Table 4
Clinical characteristics of DLB patients across clinical subtypes
| Clinical subtypes | |||||||
| Overall N = 268 (%) | NMO-DLB N = 201 | MO-DLB N = 26 | MXO-DLB N = 41 | p | p1 | ||
| Frontal syndrome †, n (%) | 56 (20.90) | 37 (18.4) | 5 (19.23) | 14 (34.14) | 0.067 | 0.023 | |
| Parkinsonian syndrome, n (%) | 230 (85.82) | 166 (82.58) | 26 (100.0) | 38 (92.68) | 0.759 | 0.810 | |
| Distribution, n (%) | |||||||
| Trunk | 1 (0.43) | 1 (0.60) | 0 (0.0) | 0 (0.0) | 0.517 | 0.892 | |
| Limbs | 38 (16.52) | 28 (16.87) | 5 (19.23) | 5 (13.16) | |||
| Trunk and limbs | 179 (77.83) | 128 (77.11) | 20 (76.92) | 31 (81.58) | |||
| Not specified | 12 (5.22) | 9 (5.42) | 1(3.85) | 2 (5.26) | |||
| Type, n (%) | |||||||
| Tremo-akineto-rigid ‡ | 128 (55.65) | 85 (51.20) | 20 (76.92) | 23 (60.53) | 0.052 | 0.077 | |
| Tremo-akinetic | 7 (3.04) | 6 (3.61) | 0 (0.0) | 1 (2.63) | 0.679 | 0.306 | |
| Akineto-rigid | 89 (38.70) | 70 (42.16) | 5 (19.23) | 14 (36.84) | 0.092 | 0.212 | |
| Not specified | 6 (2.61) | 5 (3.01) | 1(3.85) | 0 (0.0) | – | – | |
| Distribution, n (%) | |||||||
| Unilateral | 4 (1.74) | 2 (1.20) | 1 (3.85) | 1 (2.63) | 0.836 | 0.255 | |
| Bilateral | 217 (94.35) | 157 (94.58) | 24 (92.31) | 36 (94.74) | |||
| Not specified | 9 (3.91) | 7 (4.22) | 1(3.85) | 1 (2.63) | |||
| Symmetry, n (%) | |||||||
| Symmetrical | 101 (43.91) | 74 (44.58) | 11 (42.31) | 16 (42.11) | 0.798 | 0.531 | |
| Asymmetrical | 99 (43.04) | 70 (42.17) | 13 (50.0) | 16 (42.11) | |||
| Not specified | 30 (13.04) | 22 (13.25) | 2 (7.69) | 6 (15.79) | |||
| UPDRS-III, mean±SD | 41.18±17.1 | 39.38±17.15 | 47.53±15.18 | 40.65±17.89 | 0.266 | 0.070 | |
| Sensitivity to Levodopa, n (%) | 28 (12.17) | 17 (10.24) | 5 (19.23) | 6 (15.78) | 0.887 | 0.642 | |
| Levodopa response, mean±SD | 23.65±17.03 | 24.42±19.22 | 22.3±15.43 | 23.19±13.68 | 0.829 | 0.893 | |
| Hoehn and Yahr score, mean±SD | 2.90±0.89 | 2.8±0.84 | 3.19±0.88 | 3.05±1.01 | 0.741 | 0.088 | |
| Dystonia §, n (%) | 33 (12.31) | 25 (12.43) | 3 (11.53) | 5 (12.19) | 0.954 | 0.021 | |
| Myoclonus, n (%) | 52 (19.4) | 40 (19.9) | 3 (11.53) | 9 (21.95) | 0.338 | 0.426 | |
†MO-DLB versus MXO-DLB: p = 0.05. ‡MO-DLB versus NMO-DLB: p = 0.039. §MO-DLB versus MXO-DLB: p = 0.017. p1: adjusted for ApoEɛ4 carrying status. NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies.
There were no significant differences between the different clinical subtypes in neuropsychological assessment except for attention which was less frequently impaired in MXO-DLB (p = 0.047) (Table 5).
Table 5
Neuropsychological characteristics of DLB patients across clinical subtypes
| Clinical subtypes | ||||||
| Overall N = 268 | NMO-DLB N = 201 | MO-DLB N = 26 | MXO-DLB N = 41 | p | p1 | |
| Cognitive impaired domain, % | ||||||
| Attention † | 98.7 | 99.4 | 100 | 94.6 | 0.047 | 0.844 |
| Memory | 98.8 | 98.9 | 100 | 97.4 | 0.651 | 0.823 |
| Visuo-spatial | 92.7 | 93.9 | 100 | 84.2 | 0.252 | 0.572 |
| Executive | 99.1 | 98.8 | 100 | 100 | 0.698 | 0.826 |
| Linguistic | 83.7 | 85.5 | 78.3 | 78.4 | 0.429 | 0.428 |
| Apraxia | 81.0 | 82.7 | 77.3 | 74.3 | 0.459 | 0.692 |
| Agnosia | 17.7 | 20.0 | 9.5 | 10.8 | 0.243 | 0.273 |
| Judgment | 63.6 | 61.9 | 65.2 | 70.3 | 0.626 | 0.752 |
| Reasoning | 66.5 | 65.7 | 72.7 | 66.7 | 0.806 | 0.540 |
| MMSE, mean±SD/median | 15.46±6.21 | 15.34±6.32 | 16.12±6.02 | 15.74±5.88 | 0.861 | 0.652 |
| [IQR] (/30) | 15.5 [12–19.0] | 15 [11.25–19] | 17 [13–19] | 15.7 [12.2–20.5] | ||
| FAB score, mean±SD/median | 5.86±3.36 | 5.68±3.19 | 6.04±3.30 | 6.74±4.19 | 0.416 | 0.857 |
| [IQR] (/18) | 5.0 [4.0–7.0] | 5.0[4.0–7.0] | 5.0 [3.5–9.0] | 6.0 [3.5–8.5] | ||
| Annual MMSE decline, | 3.95±5.22 | 3.86±5.38 | 5.14±4.10 | 3.66±5.31 | 0.604 | 0.992 |
| mean±SD/median [IQR] (/y) | 2.11 [0.0–5.82] | 2.0 [0.0–5.25] | 3.98 [0.64–8.79] | 0.72 [0.0–4.23] | ||
| Annual FAB decline, | 1.92±2.61 | 1.77±2.39 | 2.33±4.17 | 2.63±3.09 | 0.699 | 0.349 |
| mean±SD/median [IQR] (/y) | 1.92 [0.0–2.89] | 0.82 [0.0–2.8] | 0.96 [0.42–7.2] | 1.93 [0.0–3.56] | ||
| Mood evaluation | ||||||
| GDS score, mean±SD/median | 12.82±6.72 | 12.9±6.38 | 14.71±8.01 | 11.38±7.97 | 0.285 | 0.345 |
| [IQR] | 13 [7.0–18.0] | 13 [7.75–17.0] | 18 [8.50–20.0] | 14 [3.0–17.0] | ||
| BECK score, mean±SD/median | 19.6±22.8 | 58.0 | 2.5±3.54 | 17.5±2.12 | 0.589 | – |
| [IQR] | 16 [5.0–19.0] | 58 [58–58] | 2.50 [1.2–3.75] | 17.5 [16.7–18.2] | ||
| Evaluation of psychiatric and behavioral symptoms | ||||||
| NPI score: FxG mean±SD/ | 48.96±28.4 | 50.99±28.4 | 39.56±24.67 | 44.76±30.62 | 0.793 | 0.673 |
| median [IQR] | 47 [26.25–65.7] | 48.5 [33–67.7] | 32 [21.7–55.7] | 45 [19–68] | ||
| NPI score: R ‡ | 23.97±15.5 | 24.08±11.52 | 16.08±9.07 | 27.83±29.64 | 0.0386 ‡a | 0.0028 ‡b |
| mean±SD/median [IQR] | 22 [15.0–31.0] | 23 [16.5–32.0] | 18 [10.0–19.0] | 22 [13.0–28.5] | ||
†MO-DLB versus MXO-DLB: p = 0.027. ‡aMO-DLB versus MXO-DLB: p = 0.029. ‡bMO-DLB versus MXO-DLB: p = 0.0169 and MO-DLB versus NMO-DLB: p = 0.035. p1: adjusted for APOE ɛ4 carrying status. NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies.
On follow-up assessment, 71 patients had a second MMSE and 69 patients had a second FAB. Median MMSE decline was 2.1 points/ year and median FAB decline was 1.9 points/year. Clinical subtypes did not influence the rate of cognitive decline(Table 5).
On brain imaging, we found cerebral atrophy in 91.79% of the cases, mainly moderate and diffuse, and associated vascular lesions in half of the cases, with no significant difference across the clinical subtypes (Table 6).
Table 6
Brain imaging characteristics of DLB patients across clinical subtypes
| Overall N = 268 (%) | Clinical subtypes | p | p1 | |||
| NMO-DLB N = 201 | MO-DLB N = 26 | MXO-DLB N = 41 | ||||
| Atrophy, n (%) | 246 (91.79) | 184 (91.54) | 25(96.15) | 37 (90.24) | 0.361 | 0.495 |
| Site | ||||||
| Localized | 21 (8.54) | 15 (8.15) | 3 (12.00) | 3 (8.11) | 0.650 | 0.841 |
| Diffuse | 220 (89.43) | 167 (90.76) | 21(84.00) | 32 (86.49) | ||
| Not specified | 5 (2.03) | 2 (1.09) | 1 (4.00) | 2 (5.41) | ||
| Degree | ||||||
| Mild | 76 (30.89) | 62 (33.70) | 6 (24.00) | 8 (21.62) | 0,492 | 0.382 |
| Moderate | 82 (33.33) | 59 (32.07) | 8 (32.00) | 15 (40.54) | ||
| Severe | 36 (14.63) | 25 (13.59) | 4 (16.00) | 7 (18.92) | ||
| Not specified | 52 (21.14) | 38 (20.65) | 7 (28.00) | 7 (18.92) | ||
| Vascular leukopathy, n (%) | 154 (57.46) | 107 (53.23) | 18(69.23) | 29 (70.73) | 0.576 | 0.402 |
p1: adjusted for APOE ɛ4 carrying status. NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies.
Survival analysis
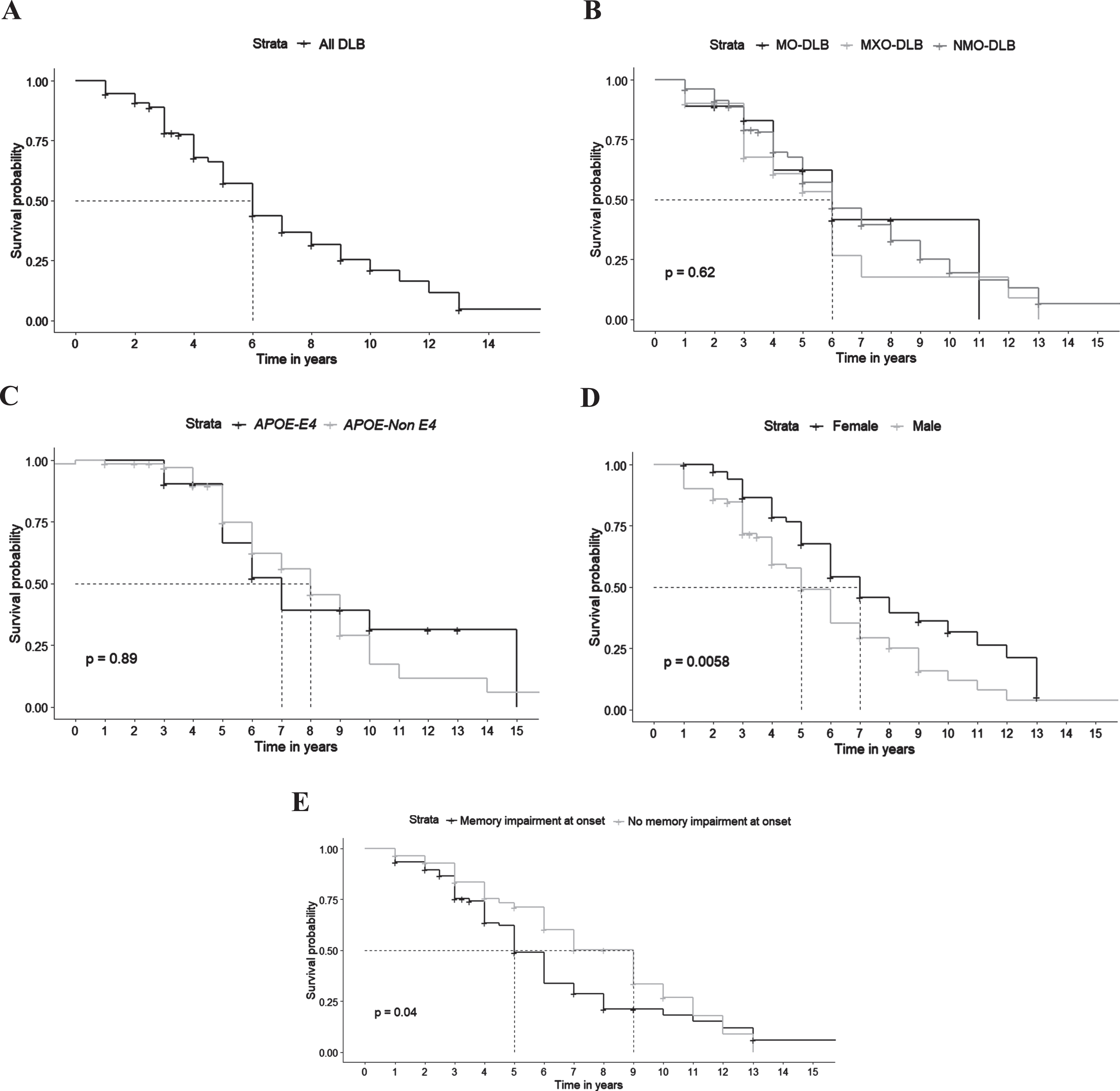
Survival data were available for 151 patients. Median survival from symptoms onset was 6 years. Median survival was significantly shorter for patients with memory symptoms at onset (5 years versus 9 years without, p = 0.04) and for males (5 years versus 7 years for women, p = 0.0058). There was no significant effect of clinical subtypes (p = 0.62) and APOE ɛ4 carrying status (p = 0.89) on survival in our DLB patients (Fig. 2).
Fig. 2
Survival in DLB patients. A) Global survival. B) Survival according to clinical subtypes (NMO-DLB, non-motor onset dementia with Lewy bodies; MO-DLB, motor onset dementia with Lewy bodies; MXO-DLB, mixed onset dementia with Lewy bodies). C) Survival according to APOE status. D) Survival according to gender. E) Survival according to the presence of memory complaint at onset.
DISCUSSION
In the present study, we classified the patients into three clinical subtypes according to initial symptoms and highlighted the characteristics of each subtype. To our knowledge, this large cohort of DLB patients is the first Tunisian and African study describing the clinical and cognitive features, genetic and prognostic factors of DLB according to clinical subtypes [11].
The onset of the disease was non-motor in 75.0%, motor in 9.7%, and mixed in 15.3% of cases. Non-motor onset was more frequent than in other atypical parkinsonian syndromes (APS) reported in our previously reported Tunisian APS series, where non-motor onset symptoms were noted in 6.7% of multiple system atrophy cases, 13.6% of supranuclear palsy cases and 20.7% of corticobasal degeneration cases. Conversely, the motor onset was less frequent than in other APS, where it varied between 62.9 and 66.7% of cases [11]. Hence, clinical subtype at disease onset could be used as an additional distinctive tool for the differential diagnosis of DLB versus other APS. In our study, the most frequent inaugural symptoms were memory disorders, followed by psychiatric manifestations, parkinsonism, and gait disorders. Along with our results, Morenas-Rodriguez et al. also reported, in their series including 81 DLB, the preponderance of a cognitive-predominant onset (56.8%), followed by neuropsychiatric-predominant onset (27.2%), then parkinsonism-predominant onset (16.0%) [12]. This predominance of isolated cognitive symptoms at onset highlights the difficulty in diagnosing DLB and distinguishing it from AD, at prodromal stages. Indeed, misdiagnosis during the initial assessments is common in DLB. In the post-mortem study of Smirnov et al., 71% of DLB confirmed after postmortem pathology were classified as AD at first assessment and only 26% were classified as DLB [13]. The variability of inaugural phenotypes of DLB may be related to pathological heterogeneity and a particular distribution of α-synuclein in the brain. Besides, Fujishiro et al. compared the distribution of amyloid deposition in patients with Lewy body diseases among different clinical phenotypes and noticed a greater amyloid overload in patients whose disease was initiated by cognitive disorders. Thus, the predominance of cognitive disorders could be explained by the associated amyloidopathy which would act “in synergy” with α-synuclein [14].
In our cohort, MXO-DLB patients developed more visual hallucinations, had an earlier age of onset of falls, less frequent attentional deficit, and more frequent frontal lobe syndrome, compared to MO-DLB and NMO-DLB. These two latter subgroups did not exhibit any specific characteristics. The minor clinical differences are likely to be the reflection of α-synuclein distribution. Ferman et al. (2020) noticed indeed that visual hallucinations and cognitive fluctuations were more common and parkinsonism less frequent in DLB patients with diffuse α-synuclein deposition compared to transitional DLB [5].
The median annual decline in MMSE score was 2.1 points in our cohort and clinical subtypes did not influence the rate of decline. This mean progression of decline was comparable to that previously reported [15, 16]. The clinical predictive factors of cognitive decline are currently unknown and of those studied, gender, initial MMSE score and core clinical features did not seem to have an impact on cognitive progression [15]. It appears interesting to mention that cognitive decline according to initial symptoms has not been described. However, Morenas-Rodriguez et al. who also studied the different clinical subtypes of DLB based on the main clinical features during the prodromal phase of the disease using cluster analysis, reported that the cognitive-predominant cluster was characterized by a long prodromal phase [12]. One of the only predictors of cognitive decline described in DLB is the presence of AD concomitant pathology measured in CSF or found in neuropathological studies [17–19]. Furthermore, previous studies also concluded to a faster disease progression in the presence of APOE ɛ4 allele in synucleinopathies without any relation with associated AD pathology. Indeed, Davis et al., who studied the effect of the APOE ɛ4 and APOE ɛ2 alleles in transgenic mice, demonstrated the presence of elevated levels of phosphorylated synuclein and greater gliosis in mice with APOE ɛ4 [20]. Although DLB phenotypes may be related to a particular brain distribution of α-synuclein [5], the brain imaging study of our DLB patients did not show any specific signs, in particular, no specific localized atrophy and there were no significant differences between the clinical subtypes.
The genetic study of our cohort revealed that the APOE ɛ4/ɛ4 genotype was found in only 3.3% of cases and the APOE ɛ3/ɛ4 in 32.3% of DLB. Comparing the APOE genotype distribution in DLB with AD and controls in Tunisia, the frequencies APOE ɛ3/ɛ4 genotype in DLB was higher than in the general population and lower than in AD [21]. Furthermore, the frequency of the APOE ɛ3/ɛ4 genotype in our cohort was comparable to that of this genotype in pure DLB in the Caucasian population where it was 30.8% [22]. The presence of the APOE ɛ4 allele did not influence the clinical phenotype of our DLB patients, including the cognitive profile and cognitive decline.
Finally, the median survival of DLB in our study was 6 years. In agreement with our data, previous studies have reported a median survival in DLB ranging from 2 to 7 years [23, 24]. Clinical subtypes did not appear to influence survival in our study, in contrast to the literature, where inaugural visual hallucinations were correlated with shorter survival and parkinsonism was rather associated with a longer survival [24–27]. Indeed, Jellinger et al. showed that the presence of parkinsonism as the initial symptom in Lewy body dementia (LBD), which encompasses both DLB and Parkinson’s disease with dementia (PDD), can have a significant impact on mortality and is associated with a longer survival rate. However, these patients with inaugural parkinsonism experienced a delayed onset of dementia, suggesting that they may align more closely with the diagnosis of PDD. Consequently, it is expected that this subgroup will show a higher survival rate [26]. Nevertheless, in our cohort, DLB patients with memory symptoms at onset had shorter survival. Moreover, in accordance with previous studies, median survival was significantly shorter for males. As far as genetic factors are concerned, the APOE ɛ4 allele carrying-status, which has been formerly described as a factor of short survival in DLB, did not influence survival in our cohort [24–27].
The findings of our study have to be seen in light of some limitations. Our study is cross-sectional reflecting little about the dynamics of developing non-motor and motor signs in DLB patients and their chronology providing, as a consequence, limited insight about the evolution of clinical subtypes of DLB across time. Besides, the study of cognitive decline and survival as well as APOE genotyping were not performed in all patients. Nonetheless, it still was carried out on a large enough sample to allow statisticalanalysis.
Conclusion
Our study revealed few differences across DLB clinical subtypes, showing more visual hallucinations and less attention deficits in MXO-DLB. APOE ɛ4 seems to play a role in defining earlier falls and common marked frontal syndrome in MXO-DLB. These DLB clinical subtypes appear to have no impact on cognitive decline or survival. Yet, the survival analysis identified other poor prognosis factors in DLB patients, notably male gender and inaugural memory impairment. These findings do call for longitudinal studies assessing the evolving presentation of DLB clinical subtypes and the impact of genetic background on disease prognosis.
ACKNOWLEDGMENTS
We thank all patients who gave their consent and participated in the present study.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
REFERENCES
[1] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor J-P , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG , Bayston A , Beach TG , Blanc F , Bohnen N , Bonanni L , Bras J , Brundin P , Burn D , Chen-Plotkin A , Duda JE , El-Agnaf O , Feldman H , Ferman TJ , ffytche D , Fujishiro H , Galasko D , Goldman JG , Gomperts SN , Graff-Radford NR , Honig LS , Iranzo A , Kantarci K , Kaufer D , Kukull W , Lee VMY , Leverenz JB , Lewis S , Lippa C , Lunde A , Masellis M , Masliah E , McLean P , Mollenhauer B , Montine TJ , Moreno E , Mori E , Murray M , O’Brien JT , Orimo S , Postuma RB , Ramaswamy S , Ross OA , Salmon DP , Singleton A , Taylor A , Thomas A , Tiraboschi P , Toledo JB , Trojanowski JQ , Tsuang D , Walker Z , Yamada M , Kosaka K ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[2] | McKeith IG , Galasko D , Kosaka K , Perry EK , Dickson DW , Hansen LA , Salmon DP , Lowe J , Mirra SS , Byrne EJ , Lennox G , Quinn NP , Edwardson JA , Ince PG , Bergeron C , Burns A , Miller BL , Lovestone S , Collerton D , Jansen ENH , Ballard C , de Vos RAI , Wilcock GK , Jellinger KA , Perry RH ((1996) ) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology 47: , 1113–1124. |
[3] | Kosaka K , Yoshimura M , Ikeda K , Budka H ((1984) ) Diffuse type of Lewy body disease: Progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol 3: , 185–192. |
[4] | Fujishiro H , Iseki E , Nakamura S , Kasanuki K , Chiba Y , Ota K , Murayama N , Sato K ((2013) ) Dementia with Lewy bodies: Early diagnostic challenges: Early diagnostic challenges of DLB. Psychogeriatrics 13: , 128–138. |
[5] | Ferman TJ , Aoki N , Boeve BF , Aakre JA , Kantarci K , Graff-Radford J , Parisi JE , Van Gerpen JA , Graff-Radford NR , Uitti RJ , Pedraza O , Murray ME , Wszolek ZK , Reichard RR , Fields JA , Ross OA , Knopman DS , Petersen RC , Dickson DW ((2020) ) Subtypes of dementia with Lewy bodies are associated with α-synuclein and tau distribution.e155-e. Neurology 95: , 165. |
[6] | Outeiro TF , Koss DJ , Erskine D , Walker L , Kurzawa-Akanbi M , Burn D , Donaghy P , Morris C , Taylor J-P , Thomas A , Attems J , McKeith I ((2019) ) Dementia with Lewy bodies: An update and outlook. Mol Neurodegener 14: , 5. |
[7] | Sachdev PS , Blacker D , Blazer DG , Ganguli M , Jeste DV , Paulsen JS , Petersen RC ((2014) ) Classifying neurocognitive disorders: The DSM-5 approach. Nat Rev Neurol 10: , 634–642. |
[8] | Zhou W , Jiang J , Peng W , Zhou X , Du J , Mo L , Tan C , Liu X , Chen L ((2021) ) Levodopa responsiveness in Parkinson’s disease patients and white matter alterations in diffusion tensor imaging: A cross-sectional tract-based spatial statistics study. Neuroreport 32: , 636–642. |
[9] | Bellaj T , Jemaa SB , Romdhane NA , Dhiffallah M , Ali NB , Bouaziz M , Mrabet A ((2008) ) Version arabe du mini mental state examination (A-MMSE): Fidélité, validité et données normatives. Tunis Med 86: , 10. |
[10] | Jemaa SB , Bellaj T , Romdhane NA , Chérif A , Zakraoui NO , Bouaziz M ((2008) ) La Frontal Assessment Battery (FAB): Fidélité, Validité et Étalonnage d’une forme arabe. Tunis Med 86: (07), 793–800. |
[11] | Nasri A , Ben Djebara M , Sghaier I , Mrabet S , Zidi S , Gargouri A , Kacem I , Gouider R ((2021) ) Atypical parkinsonian syndromes in a North African tertiary referral center. Brain Behav 11: , e01924. |
[12] | Morenas-Rodríguez E , Sala I , Subirana A , Pascual-Goñi E , Sánchez-Saudinós MB , Alcolea D , Illán-Gala I , Carmona-Iragui M , Ribosa-Nogué R , Camacho V , Blesa R , Fortea J , Lleó A ((2018) ) Clinical subtypes of dementia with Lewy bodies based on the initial clinical presentation. J Alzheimers Dis 64: , 505–513. |
[13] | Smirnov DS , Galasko D , Edland SD , Filoteo JV , Hansen LA , Salmon DP ((2020) ) Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology 94: , e2076–e2087. |
[14] | Fujishiro H , Iseki E , Higashi S , Kasanuki K , Murayama N , Togo T , Katsuse O , Uchikado H , Aoki N , Kosaka K , Arai H , Sato K ((2010) ) Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett 486: , 19–23. |
[15] | Kramberger MG , Auestad B , Garcia-Ptacek S , Abdelnour C , Olmo JG , Walker Z , Lemstra AW , Londos E , Blanc F , Bonanni L , McKeith I , Winblad B , de Jong FJ , Nobili F , Stefanova E , Petrova M , Falup-Pecurariu C , Rektorova I , Bostantjopoulou S , Biundo R , Weintraub D , Aarsland D ((2017) ) long-term cognitive decline in DLB in a large multicentre, international cohort. J Alzheimers Dis 57: , 787–795. |
[16] | Rongve A , Soennesyn H , Skogseth R , Oesterhus R , Hortobágyi T , Ballard C , Auestad BH , Aarsland D ((2016) ) Cognitive decline in dementia with Lewy bodies: A 5-year prospective cohort study. BMJ Open 6: , e010357. |
[17] | Ferreira D , Przybelski SA , Lesnick TG , Lemstra AW , Londos E , Blanc F , Nedelska Z , Schwarz CG , Graff-Radford J , Senjem ML , Fields JA , Knopman DS , Savica R , Ferman TJ , Graff-Radford NR , Lowe VJ , Jack CR , Petersen RC , Mollenhauer B , Garcia-Ptacek S , Abdelnour C , Hort J , Bonanni L , Oppedal K , Kramberger MG , Boeve BF , Aarsland D , Westman E , Kantarci K ((2020) ) β-Amyloid and tau biomarkers and clinical phenotype in dementia with Lewy bodies. Neurology 95: , e3257–e3268. |
[18] | Howlett DR , Whitfield D , Johnson M , Attems J , O’Brien JT , Aarsland D , Lai MKP , Lee JH , Chen C , Ballard C , Hortobágyi T , Francis PT ((2015) ) Regional multiple pathology scores are associated with cognitive decline in Lewy body dementias. Brain Pathol 25: , 401–408. |
[19] | Sarro L , Senjem ML , Lundt ES , Przybelski SA , Lesnick TG , Graff-Radford J , Boeve BF , Lowe VJ , Ferman TJ , Knopman DS , Comi G , Filippi M , Petersen RC , Jack CR Jr , Kantarci K ((2016) ) Amyloid-β deposition and regional grey matter atrophy rates in dementia with Lewy bodies. Brain 139: , 2740–2750. |
[20] | Davis AA , Inman CE , Wargel ZM , Dube U , Freeberg BM , Galluppi A , Haines JN , Dhavale DD , Miller R , Choudhury FA , Sullivan PM , Cruchaga C , Perlmutter JS , Ulrich JD , Benitez BA , Kotzbauer PT , Holtzman DM ((2020) ) genotype regulates pathology and disease progression in synucleinopathy. Sci Transl Med 12: , eaay3069. |
[21] | Rassas AA , Mrabet Khiari H , Hadj Fredj S , Sahnoun S , Batti H , Zakraoui NO , Cherif A , Anane N , Ben Ali N , Messaoud T , Mrabet A ((2012) ) High APOE epsilon 4 allele frequencies associated with Alzheimer disease in a Tunisian population. Neurol Sci 33: , 33–37. |
[22] | Tsuang D , Leverenz JB , Lopez OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Trojanowski JQ , Weintraub D , Chen-Plotkin AS , Irwin DJ , Rick J , Schellenberg GD , Watson GS , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Quinn JF , Chung KA , Yearout D , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2013) ) APOE ɛ4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70: , 223–228. |
[23] | Mueller C , Soysal P , Rongve A , Isik AT , Thompson T , Maggi S , Smith L , Basso C , Stewart R , Ballard C , O’Brien JT , Aarsland D , Stubbs B , Veronese N ((2019) ) Survival time and differences between dementia with Lewy bodies and Alzheimer’s disease following diagnosis: A meta-analysis of longitudinal studies. Ageing Res Rev 50: , 72–80. |
[24] | Vergouw LJM , Bosman B , van de Beek M , Salomé M , Hoogers SE , van Steenoven I , Roks G , Bonifati V , van Swieten JC , Lemstra AW , de Jong FJ ((2020) ) Family history is associated with phenotype in dementia with Lewy bodies. J Alzheimers Dis 73: , 269–275. |
[25] | Savica R , Turcano P , Bower JH , Ahlskog JE , Mielke MM ((2019) ) Survival, progression in synucleinopathy phenotypes with parkinsonism: A population-based study. Mayo Clin Proc 94: , 1825–1831. |
[26] | Jellinger KA , Wenning GK , Seppi K ((2007) ) Predictors of survival in dementia with Lewy bodies and Parkinson dementia. Neurodegener Dis 4: , 428–430. |
[27] | Lemstra AW , de Beer MH , Teunissen CE , Schreuder C , Scheltens P , van der Flier WM , Sikkes SAM ((2017) ) Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 88: , 113–118. |


