
A Novel PSEN1 Variant Leading to Posterior Cortical Atrophy: A Case Report
Abstract
We describe a 52-year-old patient with a progressive visuospatial disorder and apraxia. Neuropsychological assessment, neuroradiological findings, and Alzheimer’s disease (AD) core biomarker assay on cerebrospinal fluid led to a diagnosis of posterior cortical atrophy due to AD. We performed a next generation sequencing dementia-gene panel and found the c.1301 C>T p.(Ala434Val) variant in the Presenilin1 (PSEN1) gene. The missense change affects the PAL (Pro433-Ala434-Leu435) motif critical for catalytic activity of the macromolecular γ-secretase complex. Evolutionary and integrated bioinformatic tools predicted a deleterious effect of the variant supporting its role in the AD pathogenesis.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that afflicts millions of people worldwide [1]. The disease is characterized by the deposition in the brain of amyloid-β (Aβ) plaques and neurofibrillary tangles, mainly composed of hyperphosphorylated tau (p-tau), leading to cognitive decline and functional impairment [2]. While most AD cases are sporadic, a small percentage (<0.5%) are inherited with an autosomal dominant pattern, caused by mutations in genes encoding the amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2) [1, 3].
Posterior cortical atrophy (PCA) is a rare variant of AD that mainly affects the posterior regions of the brain, causing visual and spatial deficits, apraxia, and alexia [4]. Although PCA is often associated with the presence of Aβ and tau pathology, different neuropathological features have been reported in some cases [5]. Furthermore, recent studies have identified novel genetic mutations in patients with PCA, highlighting the importance of genetic testing in the diagnosis of atypical AD variants [6, 7].
In this study, we describe the case of a 52-year-old female patient with a clinic-neuroradiological diagnosis of PCA and based on biomarkers, who underwent genetic analysis to investigate the underlying cause of the disease.
Using targeted next-generation sequencing, we identified a novel mutation in the PSEN1 gene c.1301 C>T p.(Ala434Val) affecting a critical protein domain.
MATERIALS AND METHODS
Due to progressive difficulties at work, a 52-year-old woman presented to our Cognitive Disorders Centre of the ‘Rita-Levi Montalcini’ Neuroscience Department of the University Hospital “Città della Salute e della Scienza” of Torino, Italy. An extensive battery of neuropsychological tests, including the Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MoCA), was performed by an experienced neuropsychologist. The patient underwent computed tomography of the brain (CT), magnetic resonance imaging (MRI), fluorodeoxyglucose positron emission tomography (FDG-PET), and Amyloid PET with flutemetamol. The patient also underwent lumbar puncture to determine core AD biomarkers Aβ42, Aβ42/40 ratio, p-tau, and total tau (t-tau) by enzyme-linked immunosorbent assay kits (ELISA, Fujirebio, Ghent, Belgium). The CSF sample was collected at approximately 10 am, after an overnight fast. The sample was centrifuged at 2500 g for 10 min at 4°C and frozen at –80°C until analysis. Genetic testing included APOE genotyping (Thermo Fisher, USA) and the analysis of the coding exons of the following genes related to dementia, by next-generation sequencing (NGS; S15-SureSelect, Agilent Technologies, Santa Clara, CA, USA): APP, CHCHD10, COL4A1, COL4A2, CSF1 R, FUS, GRN, HTRA1, ITM2B, LRRK2, MAPT, NOTCH3, PINK1, PRNP, PSEN1, PSEN2, SERPINI1, SNCA, SQSRM1, TBK1, TARDBP, TREM2, TYROBP, UBQLN2, VCP. Sequencing had a minimum coverage of 30 reads per base and an average coverage of 809 reads per base. Data were analyzed using the SOPHiA DDMtrademark software (Sophia Genetics, Switzerland). PSEN1 reference sequence is NM _000021.4 (MANE transcript). Sanger sequencing validation was performed by a standard procedure amplifying PSEN1 exon 12 from a second genomic DNA aliquot and using ABI BigDye Terminator Sequencing Kit V 3.1 and an ABI 3500 sequencer (Thermo Fisher Scientific, Waltham, MA, USA). An approval for this report was obtained by the local ethics committee. Informed consent was also obtained from the patient and family members.
THE CASE STUDY
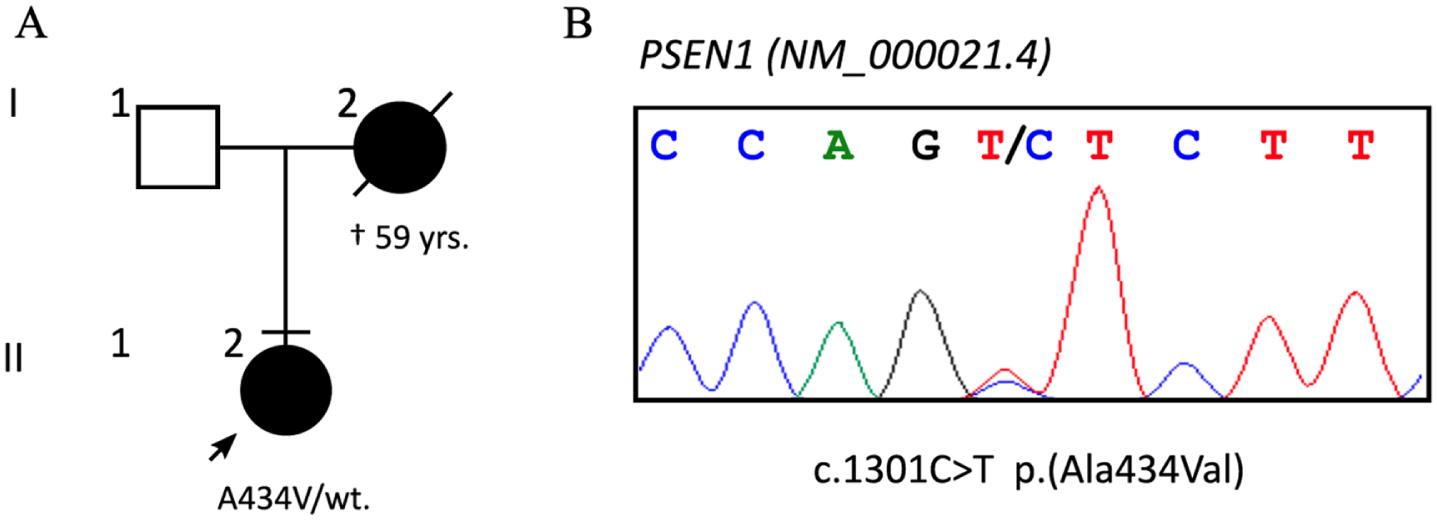
The patient was a 52-year-old right-handed woman, with 23 years of education. For approximately one year prior to her arrival at our memory clinic, the patient, a medical doctor, had been experiencing insidious difficulties performing her job properly due to deficits in spatial perception, simultanagnosia, acalculia, and agraphia. The patient reported these symptoms, which were confirmed by colleagues and family members. The patient underwent a neurological and neuropsychological evaluation, which revealed constructive apraxia and reduced visual-spatial abilities, associated with mild amnestic and attentional deficit. The MMSE and the MoCA scores were 24/30 and 21/30, respectively. The family history revealed the presence of presenile dementia, as the patient’s mother died with severe dementia at the age of 59 years after a clinical history of approximately eight years (Fig. 1A). Since no specific documentation was available, this information was obtained through anamnesis.
Fig. 1
A) Patient’s family tree, with known cases of dementia blacked and deceased barred; the arrow points to the proband. B) Sanger sequencing confirmed the heterozygous c.1301 C>T variant.
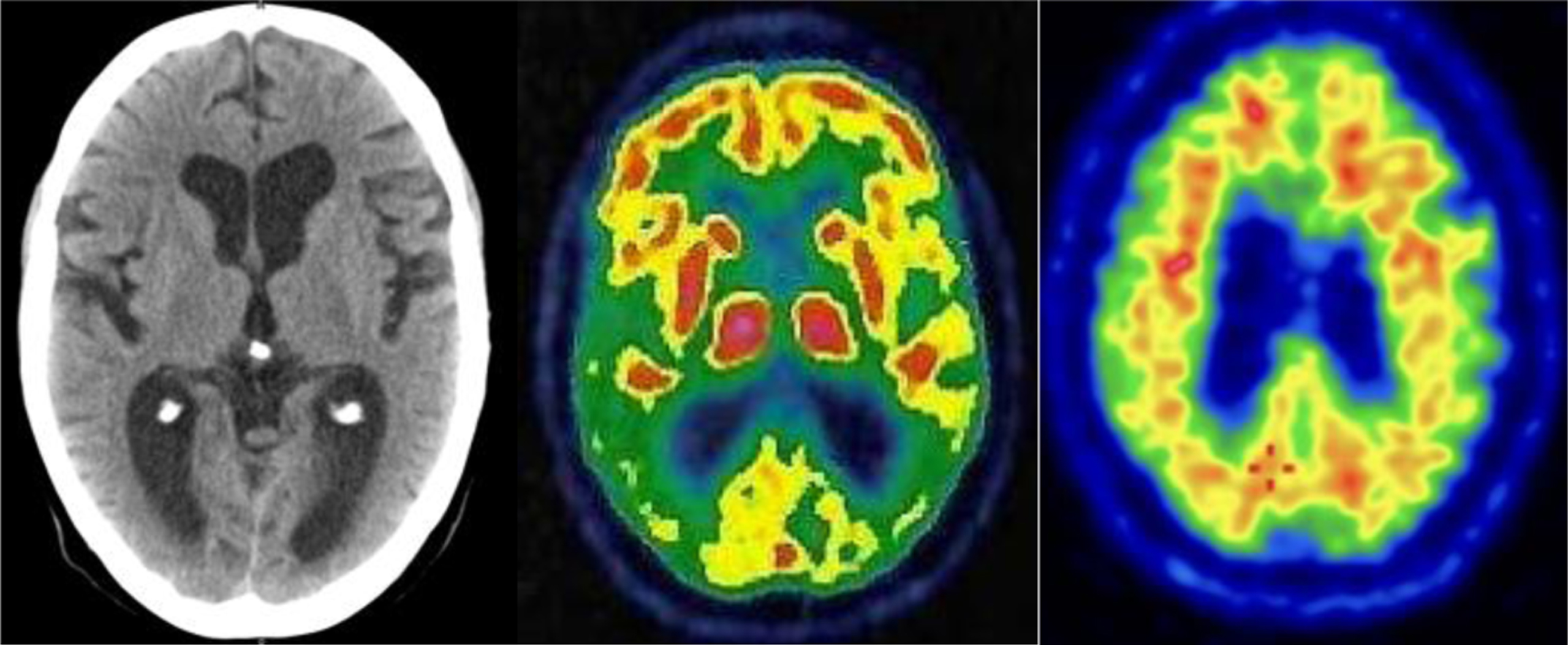
Structural and metabolic neuroimaging examinations of the brain, including CT and FDG-PET, showed predominantly posterior pattern atrophy and marked, left predominant hypometabolism in the occipital, parietal, and temporal regions. A clinical-radiological diagnosis of PCA was then made. The patient then underwent lumbar puncture with the determination of core AD biomarker levels, which showed a reduction of Aβ42 and Aβ42/40 ratio, and increased t-tau. A PET scan with flutemetamol confirmed the presence of cerebral amyloidosis.
APOE genotyping showed the presence of ɛ3/ɛ4 alleles. NGS sequencing of 25 dementia-associated genes identified the c.1301 C>T p.(Ala434Val) variant in the exon 12 of the Presenilin-1 (PSEN1; NM_000021.4) gene, confirmed by Sanger sequencing (Fig. 1B).
The change was not present in control subjects of the GnomAD (ver.2.1.1) and TOPMed BRAVO (freeze 8) genetic databases. The variant hits a highly conserved nucleotide (phyloP: 7.74 (–19.0, 10.9)) and changes an extremely conserved alanine, invariant in vertebrates, D. melanogaster, and C. elegans. The Ala434 is located within the Presenilin, signal peptide peptidase, family domain (PSEN), within a well-known PAL (Pro433-Ala434-Leu435)-motif important for the protein catalytic activity. Almost all bioinformatics tools (97/3 (del|benign); Alamut visual plus ver 1.7) predict a deleterious effect of the variant (CADD (v1.6): Phred: 26.8; Align GVGD (v2007): Class C65 (GV: 0.00 - GD: 65.28); PolyPhen2 (v): HDivPred: probably damaging (score: 0.999); HVarPred: probably damaging (score: 0.992); SIFT (v6.2.0): Deleterious (score: 0.00, median: 3.50); MutationTaster (v2021): Deleterious).
Using the American College of Medical Genetics (ACMG) criteria, we classified the variant as likely pathogenic (PM1, PM2, PP2, and PP3) [8].
A conclusive diagnosis of AD was made. Table 1 and Fig. 2 show the main findings of the investigations conducted.
Table 1
Summary of AD-related investigations
| Findings | Results | Reference range |
| Cognitive Tests | ||
| MMSE | 24/30 | >26 |
| MoCA | 21/30 | >23 |
| CSF Tests | ||
| Aβ42/40 (pg/ml) | 0.041 | 0.068–0.115 |
| Aβ42 (pg/ml) | 218.0 | >500 |
| p-tau 181 (pg/ml) | 27.9 | <61 |
| t-tau (pg/ml) | 519.0 | <500 |
| Neuroimaging | ||
| Brain MRI/CT | P/O atrophy | – |
| FDG-PET | T/P/O hypometabolism | – |
| Amyloid-PET | Positive | Negative |
| Genetic Tests | ||
| APOE | ɛ3/ɛ4 | – |
| NGS | p.(Ala434Val) in PSEN1 | – |
MMSE, Mini-Mental State Examination; MoCA, Montreal Cognitive Assessment; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; CT, computed tomography; FDG-PET, fluorodeoxyglucose positron emission tomography; Amyloid-PET, PET with flutemetamol; T/P/O, temporal/parietal/occipital; APOE, apolipoprotein E; NGS, next-generation sequencing; PSEN1, presenilin 1.
Fig. 2
Neuroimaging findings. From left to right: CT, FDG-PET, and Amyloid PET with flutemetamol of the patient consistent with the diagnosis of PCA due to AD.
The patient has been followed at our Centre for approximately two years. Acetylcholinesterase inhibitor (Donepezil) and Memantine therapy was started with low benefit. The patient has experienced a decline in cognitive abilities on a global scale, affecting most cognitive domains, supporting a diagnosis of dementia.
DISCUSSION
We describe a patient with AD and PCA associated with a novel likely pathogenic variant p.(Ala434Val) in the PSEN1 gene.
Phenotypically, the patient had a disease onset as PCA and in the course of follow-up progressively deteriorated to AD dementia. PCA is often associated with the presence of AD pathology which was demonstrated in vivo in our case study.
Generation of Aβ from the AβPP requires a series of proteolytic processes [9]. The point mutation detected in this case study introduces a variation in the highly conserved PAL motif, which is recognized as critical for the catalytic activity of γ-secretase [9, 10].
In line with our findings, variations in alanine 434 have been described in other cases of AD. The p.(Ala434Cys) variant has been reported in two siblings in their third decade of life and their father affected by autopsy-confirmed, early-onset AD [11]. The clinical course in these three individuals was characterized by cognitive and behavioral impairment accompanied by myoclonus, seizures, and aphasia within 5 years of onset.
The p.(Ala434Thr) mutation was detected in a patient initially misdiagnosed as schizophrenic at 35 years of age due to prevalent psychotic symptoms [12]. The patient developed progressive memory loss associated with other symptoms of cognitive decline over the next three years. Her mother also presented symptoms of cognitive decline and was diagnosed with AD at the age of 55.
Although this study has some limitations, such as the description of a single case and the lack of genetic analysis in other family members, we think important reporting a novel variant affecting Ala434. Moreover, no cases with variants affecting the PAL motif have been associated with AD and PCA.
The present study confirms the importance of genetic analysis in patients with early-onset AD and expands the clinical phenotype associated with PSEN1 mutations.
ACKNOWLEDGMENTS
We thank the patient and her family for being willing to share their clinical history.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author.
REFERENCES
[1] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S , Crutch S , Engelborghs S , Frisoni GB , Fox NC , Galasko D , Habert MO , Jicha GA , Nordberg A , Pasquier F , Rabinovici G , Robert P , Rowe C , Salloway S , Sarazin M , Epelbaum S , de Souza LC , Vellas B , Visser PJ , Schneider L , Stern Y , Scheltens P , Cummings JL ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[2] | Lane CA , Hardy J , Schott JM ((2018) ) Alzheimer’s disease. Eur J Neurol 25: , 59–70. |
[3] | Knopman DS , Amieva H , Petersen RC , Chételat G , Holtzman DM , Hyman BT , Nixon RA , Jones DT ((2021) ) Alzheimer disease. Nat Rev Dis Primers 7: , 33. |
[4] | Crutch SJ , Schott JM , Rabinovici GD , Murray M , Snowden JS , van der Flier WM , Dickerson BC , Vandenberghe R , Ahmed S , Bak TH , Boeve BF , Butler C , Cappa SF , Ceccaldi M , de Souza LC , Dubois B , Felician O , Galasko D , Graff-Radford J , Graff-Radford NR , Hof PR , Krolak-Salmon P , Lehmann M , Magnin E , Mendez MF , Nestor PJ , Onyike CU , Pelak VS , Pijnenburg Y , Primativo S , Rossor MN , Ryan NS , Scheltens P , Shakespeare TJ , Suárez González A , Tang-Wai DF , Yong KXX , Carrillo M , Fox NC; Alzheimer’s Association ISTAART Atypical Alzheimer’s Disease and Associated Syndromes Professional InterestArea ((2017) ) Consensus classification of posterior cortical atrophy. Alzheimers Dement 13: , 870–884. |
[5] | Crutch SJ , Lehmann M , Schott JM , Rabinovici GD , Rossor MN , Fox NC ((2012) ) Posterior cortical atrophy. Lancet Neurol 11: , 170–178. |
[6] | Schott JM , Crutch SJ , Carrasquillo MM , Uphill J , Shakespeare TJ , Ryan NS , Yong KX , Lehmann M , Ertekin-Taner N , Graff-Radford NR , Boeve BF , Murray ME , Khan QU , Petersen RC , Dickson DW , Knopman DS , Rabinovici GD , Miller BL , González AS , Gil-Néciga E , Snowden JS , Harris J , Pickering-Brown SM , Louwersheimer E , van der Flier WM , Scheltens P , Pijnenburg YA , Galasko D , Sarazin M , Dubois B , Magnin E , Galimberti D , Scarpini E , Cappa SF , Hodges JR , Halliday GM , Bartley L , Carrillo MC , Bras JT , Hardy J , Rossor MN , Collinge J , Fox NC , Mead ((2016) ) Genetic risk factors for the posterior cortical atrophy variant of Alzheimer’s disease. Alzheimers Dement 12: , 862–871. |
[7] | Sitek EJ , Narożańska E , Pepłońska B , Filipek S , Barczak A , Styczyńska M , Mlynarczyk K , Brockhuis B , Portelius E , Religa D , Barcikowska M , Sławek J , Żekanowski C ((2013) ) Apatient with posterior cortical atrophy possesses a novel mutation in the presenilin 1 gene. PLoS One 8: , e61074. |
[8] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL , ACMG Laboratory Quality Assurance Committee ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[9] | De Strooper B , Iwatsubo T , Wolfe MS ((2012) ) Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006304. |
[10] | Sato C , Takagi S , Tomita T , Iwatsubo T ((2008) ) The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the gamma-secretase. J Neurosci 28: , 6264–6271. |
[11] | Devi G , Fotiou A , Jyrinji D , Tycko B , DeArmand S , Rogaeva E , Song YQ , Medieros H , Liang Y , Orlacchio A , Williamson J , St George-Hyslop P , Mayeux R ((2000) ) Novel presenilin 1 mutations associated with early onset of dementia in a family with both early-onset and late-onset Alzheimer disease. Arch Neurol 57: , 1454–1457. |
[12] | Jiao B , Tang B , Liu X , Xu J , Wang Y , Zhou L , Zhang F , Yan X , Zhou Y , Shen L ((2014) ) Mutational analysis in early-onset familial Alzheimer’s disease in Mainland China. Neurobiol Aging 35: , 1957.e1–6. |


