
Safety, Tolerability, and Pharmacokinetics of Zagotenemab in Participants with Symptomatic Alzheimer’s Disease: A Phase I Clinical Trial
Abstract
Background:
Zagotenemab (LY3303560), a monoclonal antibody, preferentially binds to extracellular, misfolded, aggregated tau that has been implicated in Alzheimer’s disease (AD).
Objective:
The goal of this study was to assess the safety and pharmacokinetics of multiple doses of zagotenemab in participants with AD.
Methods:
This was a Phase Ib, multi-site, participant- and investigator-blind, placebo-controlled, parallel-group study in participants with mild cognitive impairment due to AD or mild to moderate AD. After screening, participants were randomized to zagotenemab 70 mg, 210 mg, or placebo every 4 weeks for up to 49 weeks and were followed up for 16 weeks.
Results:
A total of 13 males and 9 females, aged 59 to 84 years, were dosed. No deaths occurred during this study. A total of 4 serious adverse events occurred in 2 participants who then discontinued the study. The most commonly reported (3 or more participants) treatment-emergent adverse events were sinus bradycardia, headache, fall, and bronchitis. The pharmacokinetics profile showed generally linear exposures across the dose range studied with a clearance of ~8 mL/h. The half-life of zagotenemab in serum was ~20 days. A dose-dependent increase in plasma tau was observed. No other significant pharmacodynamic differences were observed due to low dose levels and limited treatment duration.
Conclusions:
No dose-limiting adverse events were observed with zagotenemab treatment. Pharmacokinetics of zagotenemab were typical for a monoclonal antibody. Meaningful pharmacodynamic differences were not observed.
Clinicaltrials.gov: NCT03019536
INTRODUCTION
Alzheimer’s disease (AD) is an age-related degenerative brain disorder that is characterized by amyloid-β plaques and neurofibrillary tangles (NFTs). Tau is an axonal microtubule binding protein that normally promotes microtubule assembly and stability. Misfolded, hyperphosphorylated tau is hypothesized to induce tau aggregation, NFT formation, microtubule destabilization, and neuronal toxicity. The capability of tau to spread and generate new aggregates is supported by cell culture and mouse models [1–3].
New disease-modifying therapies aimed at treating the pathogenesis and progressive pathology of AD are needed [4]. Zagotenemab (LY3303560) is a monoclonal antibody being developed for the treatment of AD. It binds preferentially to aggregated misfolded tau (KD:<220 pM) compared with monomer tau (KD: 235 nM) [5] and is hypothesized to block or delay transcellular spread of aggregated tau, NFT formation, and neuronal loss. Therefore, it has the potential to slow the cerebral spread of tau and consequently slow clinical progression of tau-related diseases.
In an initial Phase I study (NCT02754830), healthy participants received a single intravenous (IV) dose of zagotenemab covering an 800-fold range (7 mg to 5,600 mg). Pharmacodynamic (PD) studies showed plasma tau concentrations increased at doses of at least 700 mg. Pharmacokinetic (PK) studies indicated drug performance was consistent with that expected of an IgG4 antibody. No deaths, serious adverse events (AEs), or discontinuations due to AEs were reported when zagotenemab was administered as a single dose up to 5,600 mg. AEs were mild in severity, with headache being the most commonly reported AE deemed related to treatment. Based on the appropriate safety, PD, and PK profile, a subsequent clinical trial of zagotenemab, presented here, was conducted to assess zagotenemab in participants with mild cognitive impairment (MCI) or mild to moderate dementia due to AD (NCT03019536). The intent of this study was to provide an initial exploration of the safety, tolerability, and PK of zagotenemab following multiple doses, before progressing to larger trials.
METHODS
Standard protocol approvals, registrations, and patient consents
The study was conducted in accordance with the protocol approved by local ethical review boards in compliance with principles derived from the international Conference on Harmonization Good Clinical Practice guidelines and the Declaration of Helsinki. The study protocol and statistical analysis plan are available in the Supplementary Material (SAP 1 and SAP 2, respectively). The study was registered at ClinicalTrials.gov (NCT03019536). Patients and/or patients’ legally acceptable representatives provided written informed consent for participation prior to any study-specific procedures.
Patients and study design
This was a Phase Ib, participant- and investigator-blind, placebo-controlled, multiple ascending dose study to assess the safety, tolerability, PK, and PD of zagotenemab. It was planned to recruit 4 multiple-ascending dose cohorts to explore a range of dose levels from approximately 70 to 1400 mg. However, during conduct of the study, it became apparent that the actual effective dose range of zagotenemab could be higher than the doses being tested, so the cohorts that had not yet been conducted (700 and 1,400 mg zagotenemab) were not run.
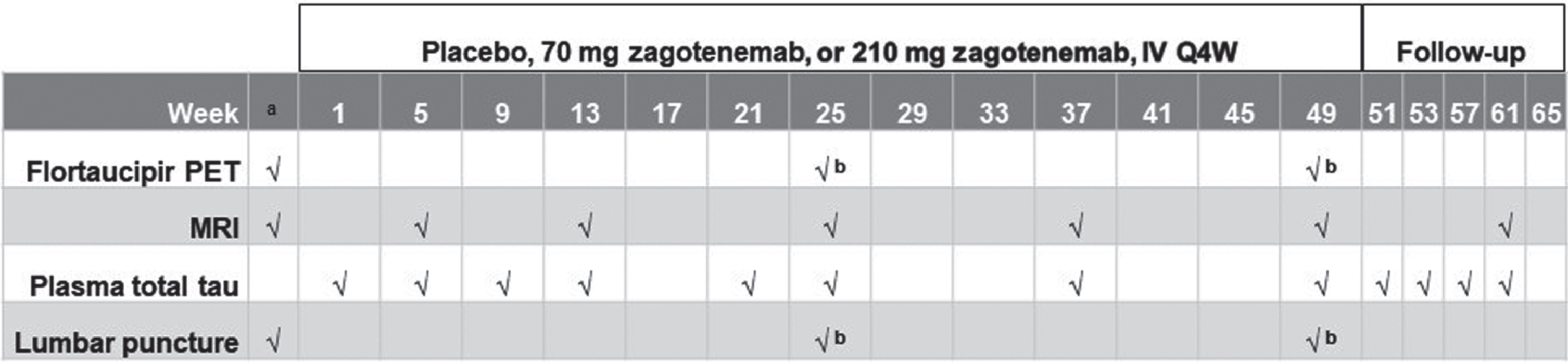
Patients in each dosing cohort (70 mg and 210 mg zagotenemab) were randomized to receive either zagotenemab or placebo (6 LY3303560 : 2 placebo). Male and female participants at least 50 years of age with MCI or mild to moderate dementia due to AD, an amyloid PET scan consistent with amyloid pathology, and a body mass index of 18.0 to 35.0 kg/m2 were eligible for this study. Participants were enrolled from 9 study centers in 3 countries (Japan, UK, and US). The first participant was enrolled on March 2, 2017 and the last participant completed the study on June 5, 2019. Participants attended a screening visit (Visit 1) within 90 days prior to dosing and were randomly assigned to treatment (Visit 2) within 8 days prior to the first dose. Treatment was initially every 4 weeks for a 49-week period. The intent of the 49-week treatment duration was to maximize the opportunity to observe an effect on tau PET at the two highest doses. When the decision was made to remove the two highest doses, a reduced treatment window of 25 weeks was judged adequate to investigate the tolerability of zagotenemab. Accordingly, there was a subsequent protocol amendment in which the higher dose cohorts of the multiple dose escalaton were eliminated from the study and the treatment period was reduced to 25 weeks, after which the participants were given the option of continuing the treatment up to 49 weeks (up to 6 further doses). Participants who completed at least the 25-week treatment period were followed up for 16 weeks after completion of dosing. Dose-escalation decisions were made following a review of the safety data from a minimum of 4 participants having received at least 2 doses of zagotenemab and 1 participant having received at least 2 doses of placebo at the prior dose level. The subsequent cohort did not begin dosing until all participants from the previous cohort had started dosing.
Safety analyses
Safety was assessed throughout the study for each cohort by AE monitoring, electrocardiograms (ECGs), laboratory tests, vital signs (blood pressure and pulse rate), and magnetic resonance imaging (MRI) scans, which were obtained at specified timepoints during the study (Fig. 2). Safety monitoring using MRI images included sequences appropriate for detecting vasogenic edema and microhemorrhage, which were analyzed by BioClinica Inc., Princeton, NJ, USA. Neurological examinations were performed at specified timepoints during the study, and if abnormalities were observed, then additional examinations were performed at daily intervals until the participant had returned to baseline. The Columbia-Suicide Severity Rating Scale (C-SSRS) was used to capture the occurrence, severity, and frequency of suicide-related thoughts and behaviors during the assessment period. As there is no C-SSRS version specifically for cognitively impaired people, the child version of the C-SSRS was used at defined timepoints for this study.
Fig. 2
Illustration of study design. aBefore first dose (including screening). bConducted at Week 25 should the participant follow the 25-week treatment regimen or at Week 49 for the 49-week treatment regimen. Only the 49-week treatment scheme is shown. The 210 mg cohort was initiated following a review of the safety data from the 70 mg cohort. IV, intravenous; MRI, magnetic resonance imaging; PET, positron emission tomography; Q4 W, every four weeks.
Pharmacokinetic analyses
Human serum and cerebrospinal fluid (CSF) samples obtained during this study were analyzed for zagotenemab using validated enzyme-linked immunosorbent assays (ELISA) (Covance Laboratories Inc., Chantilly, VA, USA). PK parameters of zagotenemab in serum at Visit 3 (Week 1) and Visit 19 (Week 49) were determined using standard non-compartmental methods in Phoenix WinNonlin (Version 8.0). The lower limit of quantification in the serum assay was 100.0 ng/mL, and the upper limit of quantification was 3,000.0 ng/mL. For CSF, the lower limit of quantification was 2.5 ng/mL and the upper limit of quantification was 500.0 ng/mL. The CSF:serum zagotenemab concentration percentages were calculated for participants with available CSF data.
Flortaucipir PET imaging
A total of 2 tau-PET scans, utilizing flortaucipir F 18 [6], were performed during the study: 1 prior to first dosing and 1 after the participant’s last dose. At each flortaucipir F 18 PET imaging visit, participants received a single IV administration of approximately 370 MBq (10 mCi) flortaucipir F 18 and a PET scan starting approximately 75 min post injection. An AD-signature weighted neocortical standardized uptake value ratio (SUVr) with respect to a reference signal intensity in white matter (PERSI) and a composite SUVr was calculated to estimate tau signal [7]. The primary comparison was between zagotenemab and placebo arms for change from baseline in composite SUVr at the last flortaucipir F 18 imaging visit.
Immunogenicity methods
Blood samples for the assessment of immunogenicity were taken periodically across all dose cohorts. Serum samples obtained during this study were analyzed at Eurofins Pharma Bioanalytics Services US Inc. (St. Charles, MO, USA). Samples were analyzed using a validated affinity capture elution-bridge ELISA assay method [8, 9] to screen for, confirm, and titer anti-drug antibodies (ADAs) against zagotenemab. The ADA assay was performed with a minimum required dilution (MRD) of 1 : 10, sensitivity of < 2.0 ng/mL, and a drug tolerance of 59.61μg/mL in the presence of 100 ng/mL affinity purified hyper-immunized monkey anti-zagotenemab antibody. If ADAs were detected at baseline, samples from participants considered to have treatment-emergent ADAs (TE ADAs) showed a≥4-fold increase from baseline. If no ADAs were detected at baseline, participants considered to have TE ADAs had samples with a titer 2-fold (1 dilution) greater than the MRD of the ADA assay.
Plasma tau method
Plasma samples obtained during the course of the study were analyzed using an ELISA assay by Eurofins Pharma Bioanalytics Services US Inc.
Statistical analyses
The parameters were summarized using standard descriptive statistics, unless otherwise noted. All p-values were from an exploratory perspective (i.e., hypothesis generating). No adjustments were made for multiplicity.
The composite SUVr data were analyzed using an analysis of covariance model. Treatment dose (zagotenemab and placebo) was fitted as a fixed effect and a baseline covariate adjustment was used.
RESULTS
Participants
A total of 22 participants (13 males and 9 females) between the ages of 59 and 84 years with MCI or mild to moderate dementia due to AD were dosed in this study (Fig. 1). All participants had positive evidence of amyloid at screening. The average age and body mass index were similar across treatments (Table 1). Two participants withdrew their consent: one after 49 weeks of receiving 70 mg zagotenemab, and the other was withdrawn after the Week 21 dose of placebo. Two additional participants withdrew due to serious adverse events (SAEs) (see safety section). Of the 18 participants who completed the initial 25-week treatment period, 17 chose to complete the 49-week treatment period (a total of 13 doses).
Fig. 1
CONSORT diagram.
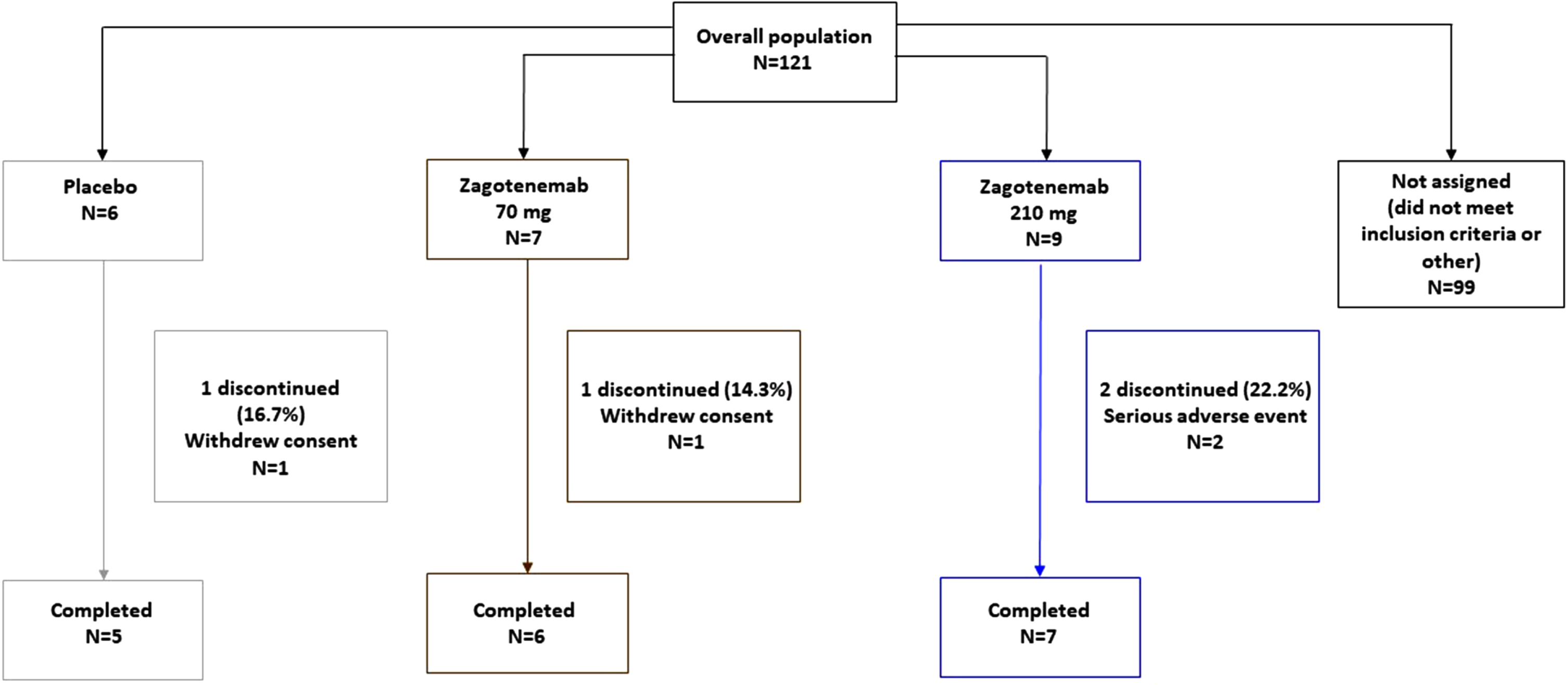
Table 1
Demographics and participant characteristics
| Placebo N = 6 | 70 mg zagotenemab N = 7 | 210 mg zagotenemab N = 9 | Overall N = 22 | |
| Mean years of age (SD) | 72.2 (8.3) | 72.4 (7.7) | 74.3 (6.7) | 73.1 (7.2) |
| Female n (%) | 4 (66.7) | 1 (14.3) | 4 (44.4) | 9 (40.9) |
| Japanese n (%) | 2 (33.3) | 1 (14.3) | 2 (22.2) | 5 (22.7) |
| Mean BMI (SD) | 24.4 (4.8) | 24.9 (3.7) | 23.3 (3.3) | 24.1 (3.7) |
| APOE genotype | ||||
| ɛ2/ɛ3 | 0 (0.0%) | 2 (28.6%) | 0 (0.0%) | 2 (9.1%) |
| ɛ3/ɛ3 | 3 (50.0%) | 3 (42.9%) | 2 (22.2%) | 8 (36.4%) |
| ɛ3/ɛ4 | 3 (50.0%) | 0 (0.0%) | 5 (55.6%) | 8 (36.4%) |
| ɛ4/ɛ3 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| ɛ4/ɛ4 | 0 (0.0%) | 2 (28.6%) | 2 (22.2%) | 4 (18.2%) |
APOE, Apolipoprotein E; BMI, body mass index; n, number of participants in subgroup; N, Number of participants in treatment arm; SD, standard deviation.
Safety
No deaths occurred during the study. A total of 2 participants (both in the 210 mg cohort) reported 4 SAEs during the study. One participant had moderate severity SAEs of pulmonary mass and pneumothorax, and the other had a moderate severity SAE of back pain and subsequently a moderate SAE with a preferred term of metastatic neoplasm. The 2 participants who experienced SAEs discontinued from the study and were the only participants to discontinue the study due to AEs. There were no apparent trends in the clinical laboratory data, vital signs, or ECG data over time nor any notable differences among treatments.
All treatment-emergent adverse events (TEAEs) were mild or moderate in severity; no TEAEs were reported as severe. The most commonly reported TEAEs (incidence of 3 or more) were sinus bradycardia, headache, fall, bronchitis and cerebral microhaemorrhage. A total of 8 sinus bradycardia AEs were reported by 3 participants, after dosing with 70 mg zagotenemab. One participant had sinus bradycardia on Day 225 and another had it on Days 226 and 254 after the first dose of 70 mg zagotenemab. Sinus bradycardia was observed in the third participant prior to dosing with 70 mg zagotenemab. This subject also had 5 events of sinus bradycardia after dosing with 70 mg zagotenemab. None of the sinus bradycardia AEs were associated with any signs or symptoms (Table 2).
Table 2
Treatment-Emergent Adverse Events (Regardless of Causality)
| MedDRA preferred term | Placebo (N = 6) n [n’] | 70 mg zagotenemab (N = 7) n [n’] | 210 mg zagotenemab (N = 9) n [n’] | Overall (N = 22) n [n’] |
| Number of Serious TEAEs, Regardless of Causality | 0 [0] | 0 [0] | 4 [2] | 4 [2] |
| Discontinuation Due to TEAEs, N | 0 | 0 | 2 | 2 |
| Total number of TEAEs, Regardless of Causality | 21 [4] | 39 [7] | 38 [8] | 98 [19] |
| Common TEAEs (≥2 TEAEs in≥2 participants), Regardless of Causality | ||||
| Sinus bradycardia | 0 [0] | 8 [3] | 0 [0] | 8 [3] |
| Headache | 1 [1] | 1 [1] | 2 [2] | 4 [4] |
| Fall | 3 [2] | 1 [1] | 0 [0] | 4 [3] |
| Bronchitis | 1 [1] | 1 [1] | 1 [1] | 3 [3] |
| Cerebral microhaemorrhage (ARIA-H)a | 0 [0] | 1 [1] | 2 [1] | 3 [2] |
| Depression | 0 [0] | 2 [2] | 0 [0] | 2 [2] |
| Diarrhoea | 1 [1] | 0 [0] | 1 [1] | 2 [2] |
| Dizziness | 0 [0] | 0 [0] | 2 [2] | 2 [2] |
| Fatigue | 0 [0] | 1 [1] | 1 [1] | 2 [2] |
| Haematuria | 0 [0] | 1 [1] | 1 [1] | 2 [2] |
| Pruritus generalised | 1 [1] | 1 [1] | 0 [0] | 2 [2] |
| Sinusitis | 0 [0] | 1 [1] | 1 [1] | 2 [2] |
| Urinary tract infection | 1 [1] | 1 [1] | 0 [0] | 2 [2] |
| Vertigo | 1 [1] | 0 [0] | 1 [1] | 2 [2] |
| Vomiting | 1 [1] | 0 [0] | 1 [1] | 2 [2] |
ARIA-H, Amyloid-related imaging abnormalities; MedDRA, Medical Dictionary for Regulatory Activities; n, number of events; N, number of participants; n’, number of participants with event; TEAE, treatment-emergent adverse event. aThere was also a cerebellar microhaemorrhage reported by one participant in the 70 mg zagotenemab dosing group.
A total of 4 TEAEs of cerebellar and cerebral microhaemorrhage (ARIA-H) were detected on MRI in 3 participants following dosing with zagotenemab. Of these, 2 participants were in the 70 mg dosing group. One participant had a left frontal lobe cerebellar microhaemorrhage detected at Day 164 and another had a right parietal lobe cerebral microhaemorrhage detected at the Day 341 MRI scan. The third participant, who received 210 mg zagotenemab, had an occipital lobe cerebral microhaemorrhage detected at the Day 162 MRI scan, and a second occipital lobe cerebral microhaemorrhage detected at the Day 418 MRI scan. This participant reported an AE of worsening headaches on the same day as the first microhemorrhage. The worsening headaches were mild and lasted for 164 days; no additional AEs were noted at the time of the second microhemorrhage. All TEAEs of cerebellar and cerebral microhaemorrhage were deemed mild in severity (Table 2).
The volume of the majority of brain regions was not significantly different when either dose of zagotenemab treatment was compared with placebo. Atrophy was significantly greater in those treated with zagotenemab compared to placebo for 2 bilateral parietal (inferior and lateral) regions.
Among the participants who received at least 1 dose of study treatment, 3 reported suicidal thoughts via the C-SSRS after dosing as follows: one at an isolated timepoint during the dosing period (210 mg treatment group; Day 113), one at 3 timepoints during the follow-up period (210 mg treatment group; Days 351, 431 and 449), and one at 4 timepoints during the dosing period (ranging between Day 85 and Day 337) and 3 timepoints during the follow-up period (70 mg treatment group). None of these subjects reported a history of suicidal thoughts at screening.
Pharmacokinetic evaluations
Serum pharmacokinetics
Mean serum concentrations are shown in Fig. 3a and 3b. At Visit 19 (Week 49), the mean half-life was approximately 20 days and 19 days for the 70 mg and 210 mg dose groups, respectively. The mean clearance was approximately 8 mL/h with relatively low variability (approximately 20%) for both dose groups. The accumulation ratio based on Ctrough at steady state compared with Ctrough prior to the second dose (i.e., Visit 19 predose sample to Visit 8 predose sample) was 1.72 for the 70 mg dose group and 1.36 for the 210 mg dose group (Table 3). Concentration data from 3 Japanese participants appeared consistent with that of non-Japanese participants (data on file).
Fig. 3
Mean serum zagotenemab concentration-time plots following multiple zagotenemab doses administered every 4 weeks: (a) linear scale; (b) semi-log scale. N, number of participants; SD, standard deviation.
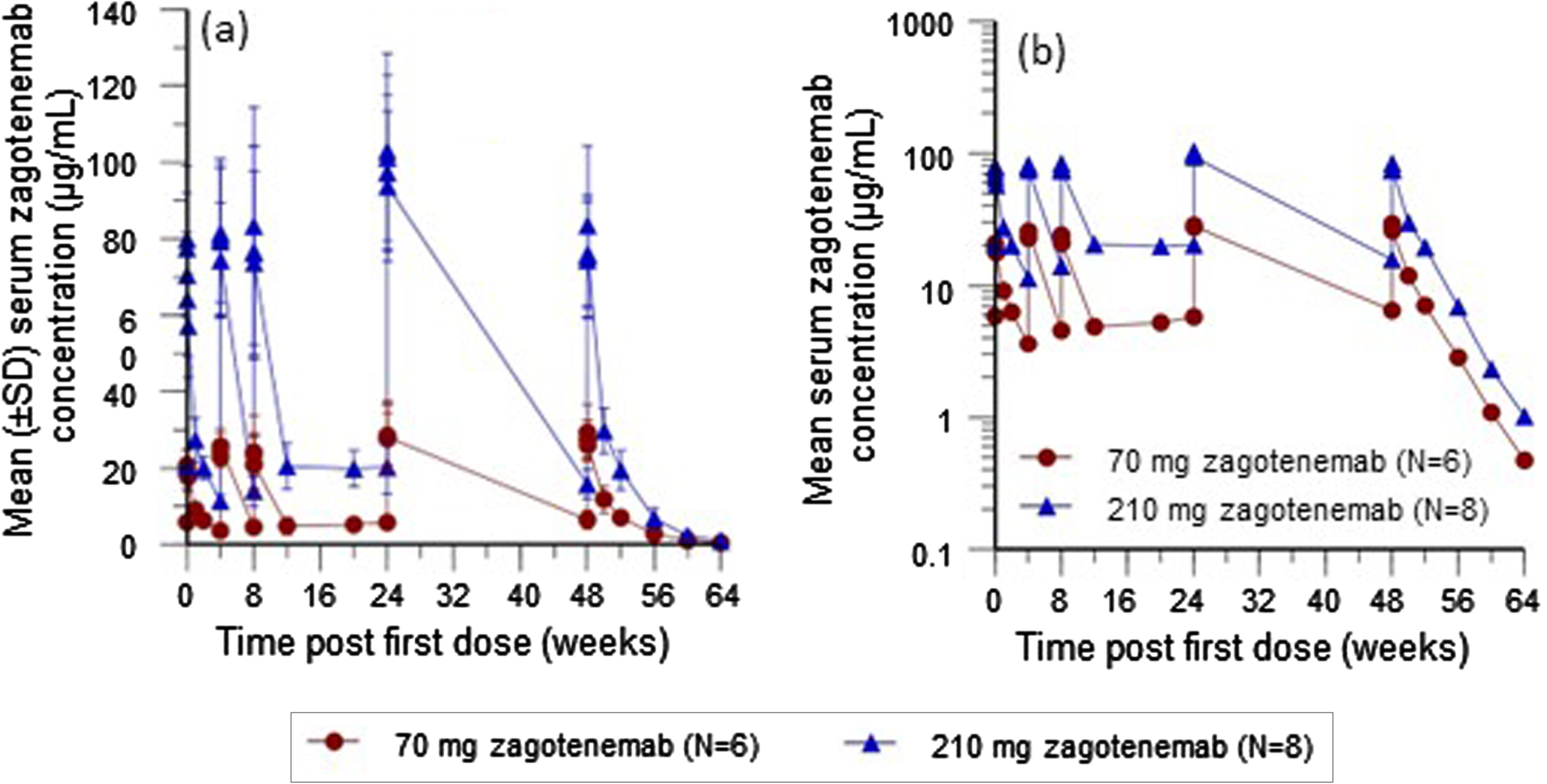
Table 3
Summary of serum pharmacokinetic parameter estimates for zagotenemab after multiple monthly doses
| Geometric Mean (CV%) | ||||
| Serum zagotenemab | ||||
| Zagotenemab 70 mg IV | Zagotenemab 210 mg IV | |||
| Visit/Week | Week 1 | Week 49 | Week 1 | Week 49 |
| N | 7 | 6 | 9 | 8 |
| Cmax (μg/mL) | 21.5 (17%) | 29.2 (23%) | 81.8 (22%) | 83.9 (22%) |
| Ctrough (μg/mL)a | 3.53 (23%) | 6.15 (37%) | 11.2 (16%) | 15.4 (24%) |
| tmaxb (h) | 2.0 (0.6–24) | 1.55 (0.6–4) | 2.5 (0.53–8) | 3.0 (0.53–4.05) |
| t1/2c (day) | 20.2 (14.9–24.7)d | 18.9 (14.8–22.5) | ||
| AUC0 - T (μg*h/mL) | 5180 (15) | 9140 (21)d | 16200 (15) | 24200 (19) |
| CL (mL/h) | 7.66 (21)d | 8.66 (19) | ||
| Vss (mL) | 4640 (25)d | 5120 (17) | ||
| RA (based on Ctrough) | 1.72 (25) | 1.36 (22) | ||
a Ctrough for Visit 3 is the predose concentration at Visit 8. Ctrough for Visit 19 is the predose concentration at Visit 19. b Median (range). cGeometric mean (range). dN = 5. AUC0 - T, area under the concentration versus time curve during the dosing interval; CL, total body clearance of drug; Cmax, maximum observed drug concentration; Ctrough, drug concentration before the next dose; CV, coefficient of variation; IV, intravenous; N, number of participants; RA, accumulation ratio; t1/2, half-life associated with the terminal rate constant in non-compartmental analysis; tmax, time of maximum observed drug concentration; Vss, volume of distribution at steady state.
CSF pharmacokinetics
All available postdose CSF samples (from 4 participants at 70 mg and 6 participants at 210 mg) assayed for zagotenemab concentrations were quantifiable and ranged from 24.8 ng/mL to 228.8 ng/mL. Seven of these 10 quantifiable concentrations (range: 32.9 ng/mL to 228.8 ng/mL) were collected between 3 days to 8 days after the previous dose of zagotenemab. There were 3 participants (all in the 210 mg dose group; range 24.8 ng/mL to 73.8 ng/mL) who had a predose (2 to 5 days before the next dose) CSF sample taken at Week 25. The CSF:serum zagotenemab concentration percentage ranged from 0.147% to 0.282% across these 3 participants in the 210 mg dose group.
Immunogenicity
No TE ADA participants were detected after administration of multiple doses of zagotenemab up to 210 mg.
Flortaucipir PET scans
There were no significant differences in the composite SUVr after treatment with zagotenemab at Week 49 in the participants treated with 70 mg or 210 mg zagotenemab compared with those who received placebo.
Plasma tau
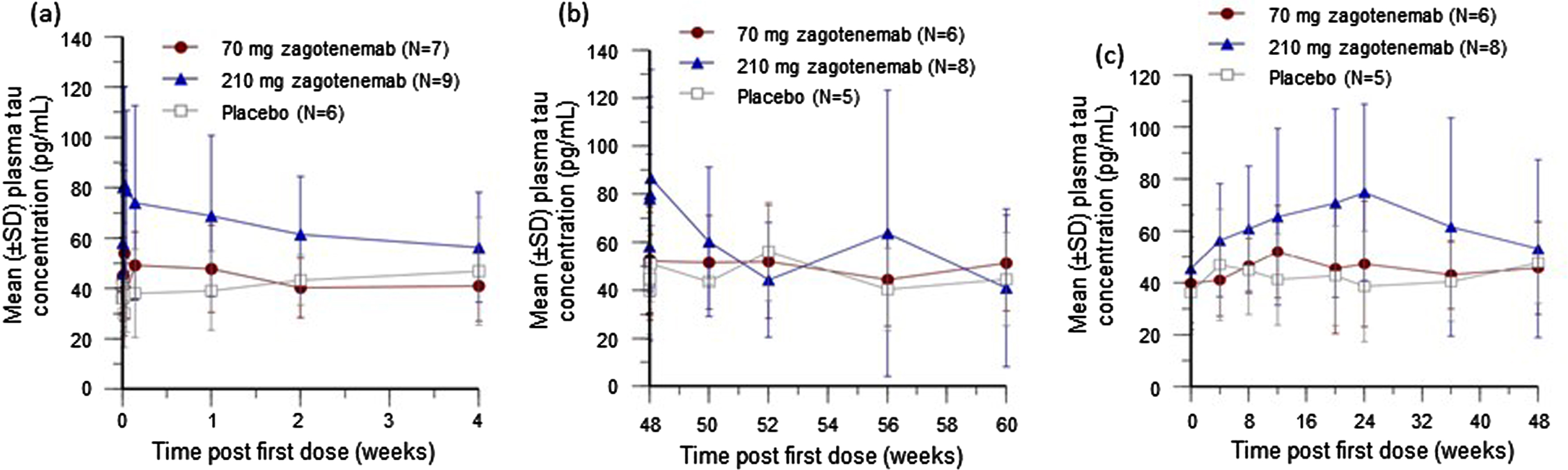
An apparent dose-dependent increase in plasma tau concentrations was observed following the first dose (Fig. 4a) but was similar in all treatment groups by 2 weeks after the last dose (Fig. 4b). Trough concentrations (samples taken just before each dosing) showed an initial dose-dependent trend that peaked at 24 weeks post first dose. All cohorts had similar trough plasma tau concentrations by 48 weeks post first dose (Fig. 4c).
Fig. 4
Mean plasma tau concentration-time plots after zagotenemab following a single dose (a) and completion of multiple doses (b), and mean trough plasma tau concentration-time plot after zagotenemab multiple doses (c). In (b), two participants discontinued the study after the 48-week dose. For 70 mg, N = 6 for Week 48 and N = 5 for Week 50 to Week 60. For 210 mg, N = 8 for Week 48 to Week 56 and N = 7 for Week 60. In (c), samples were obtained prior to next dose (4 weeks after previous dose). N, number of participants; SD, standard deviation.
DISCUSSION
This was the first clinical study to investigate the safety, tolerability, and PK of multiple doses of zagotenemab in participants with MCI or mild to moderate dementia due to AD. No deaths occurred, and while 2 zagotenemab-treated participants reported a total of 4 SAEs, none of the SAEs were deemed to be related to treatment by the investigator. No TEAEs in any of the treatment groups were reported as severe. The most commonly reported TEAEs were events that are common in the participant population recruited to this study, which are cardiac abnormalities such as sinus bradycardia, headache, fall, and bronchitis.
There were no apparent trends in the clinical laboratory, vital signs, ECG, or neurological examination data over time nor any notable differences among treatments in these parameters. De novo microhaemorrhages were detected in 3 participants following their dosing with zagotenemab. However, ARIA-H like microhaemorrhages can occur spontaneously in the elderly or AD population [10] and therefore they may occur spontaneously during a clinical study. For example, the incidence of ARIA-H was 12% over a 2-year period in a memory clinic population [10] and in the placebo arm of aducanamb Phase III studies in early AD patients, the incidence was approximately 7% over 18 months [11].
Immunogenicity was assessed using a validated assay to detect ADAs in the presence of zagotenemab from baseline to Day 287 across all dose cohorts. No TE ADA participants were detected after administration of multiple doses of zagotenemab up to 210 mg.
The secondary objective was to assess steady-state (Week 49) serum zagotenemab PK parameters following multiple IV doses of 70 mg or 210 mg zagotenemab every 4 weeks in participants with MCI or mild to moderate dementia due to AD. The half-life of zagotenemab in serum is approximately 20 days. The CSF:serum zagotenemab concentration ratio of approximately 0.2% was consistent with values reported for other monoclonal antibodies [12–14]. The effects of multiple IV doses of zagotenemab on tau pathology (using flortaucipir F 18) was explored, but no clinically meaningful effect was observed. Although zagotenemab has approximately 1000-fold greater affinity to extracellular aggregated tau, the observed increase in plasma tau after zagotenemab treatment is likely due to its weak affinity for peripheral monomeric tau. The effects of zagotenemab treatment on plasma phosphorylated tau was not conducted because an assay for plasma phosphorylated tau had not been established at the time of this study in 2017. The development of a plasma p-tau181 assay was first published in 2018 [15]. Since our previous work published in 2016 [16] had demonstrated plasma tau to be associated with neurodegeneration and cognitive function, we considered our use of a drug tolerant total tau assay relevant.
The decision to modify the design of the Phase I study was based on an evolving understanding of tau turnover in CSF, which was assumed to be similar to what occurred in the interstitial fluid of the brain. Nonclinical data suggested that this turnover was higher than what had originally be estimated, thus necessitating higher doses than had originally been planned.
As a preliminary exploration of the clinical pharmacology of zagotenemab, this study was subject to several limitations. While the size of the study was appropriate for a Phase I evaluation of safety and PK outcomes, it was not sufficiently powered to detect clinically meaningful effects in the imaging, biomarker or cognitive outcomes. Given the relatively slow rate at which cognitive decline and tau aggregation occur, it would have been unlikely for a disease-modifying treatment to have shown any effect on these outcomes in such a relatively short trial. This study was followed by a larger Phase II study exploring higher doses of zagotenemab to assess its efficacy and effect on AD (NCT03518073). Although this Phase II study did not meet its primary clinical endpoints [17], the data from that study and this Phase I study with zagotenemab add to the Phase I clinical development literature on pursuing anti-tau therapy using an extracellular anti-tau approach. Collectively, along with several other monoclonal antibodies similarly targeted against extracellular tau, tilavonemab [18], semorinemab [19]) have all resulted in negative outcomes in AD, suggesting alternative approaches to tau therapy in AD need to be explored.
Conclusions
There were no dose limiting safety findings after treatment of 210 mg zagotenemab administered every 4 weeks for up to 49 weeks. Zagotenemab was cleared at approximately 8 mL/h with relatively low variability and has a half-life of approximately 20 days. TE ADA were not detected in any participants.
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution and dedication of the patients, their families and caregivers who participated in this study. We are tremendously grateful and thankful to our study sites and staff for their dedication in partnering with us, and the great care taken of patients and families participating in the study.
The authors wish to thank Sinéad Ryan, employee of Eli Lilly and Company, for writing and project management support.
FUNDING
This study was sponsored/funded/supported by Eli Lilly and Company, Indianapolis, IN, USA.
CONFLICT OF INTEREST
Sergey Shcherbinin, Louise Chinchen, Scott W. Andersen, Elizabeth S. LaBell, David G. S. Perahia, Paula M. Hauck, and Stephen L. Lowe are full-time employees and minor stockholders of Eli Lilly and Company. Brian A. Willis, previous employee and minor stockholder of Eli Lilly and Company, is currently at Eisai Inc. Albert C. Lo is a previous employee and minor stockholder of Eli Lilly and Company. Jeffrey L. Dage, previous employee and minor stockholder of Eli Lilly and Company, is currently at Indiana University School of Medicine and has received speaker fees and in kind support for his research from Eli Lilly.
DATA AVAILABILITY
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after primary publication acceptance. For details on submitting a request, see the instructions provided at http://www.vivli.org.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-230012.
REFERENCES
[1] | Vogel JW , Iturria-Medina Y , Strandberg OT , Smith R , Levitis E , Evans AC , Hansson O , Alzheimer’s Disease Neuroimaging Initiative, Swedish BioFinder Study ((2020) ) Spread of pathological tau proteins through communicating neurons in human Alzheimer’s disease. Nat Commun 11: , 2612. |
[2] | Zhang H , Cao Y , Ma L , Wei Y , Li H ((2021) ) Possible mechanisms of tau spread and toxicity in Alzheimer’s disease. Front Cell Dev Biol 9: , 707268. |
[3] | Wu JW , Herman M , Liu L , Simoes S , Acker CM , Figueroa H , Steinberg JI , Margittai M , Kayed R , Zurzolo C , Di Paolo G , Duff KE ((2013) ) Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288: , 1856–1870. |
[4] | Abeysinghe A , Deshapriya R , Udawatte C ((2020) ) Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci 256: , 117996. |
[5] | Alam R , Driver D , Wu S , Lozano E , Key SL , Hole JT , Hayashi ML , Lu J ((2017) ) Preclinical characterization of an antibody [LY3303560] targeting aggregated tau. Alzheimers Dement 13: , P592–P593. |
[6] | Fleisher AS , Pontecorvo MJ , Devous MD Sr., Lu M , Arora AK , Truocchio SP , Aldea P , Flitter M , Locascio T , Devine M , Siderowf A , Beach TG , Montine TJ , Serrano GE , Curtis C , Perrin A , Salloway S , Daniel M , Wellman C , Joshi AD , Irwin DJ , Lowe VJ , Seeley WW , Ikonomovic MD , Masdeu JC , Kennedy I , Harris T , Navitsky M , Southekal S , Mintun MA , A16 Study Investigators ((2020) ) Positron emission tomography imaging with [18F]flortaucipir and postmortem assessment of Alzheimer disease neuropathologic changes. JAMA Neurol 77: , 829–839. |
[7] | Devous MD Sr., Joshi AD , Navitsky M , Southekal S , Pontecorvo MJ , Shen H , Lu M , Shankle WR , Seibyl JP , Marek K , Mintun MA ((2018) ) Test-retest reproducibility for the tau PET imaging agent flortaucipir F 18. J Nucl Med 59: , 937–943. |
[8] | Bourdage JS , Cook CA , Farrington DL , Chain JS , Konrad RJ ((2007) ) An affinity capture elution (ACE) assay for detection of anti-drug antibody to monoclonal antibody therapeutics in the presence of high levels of drug. J Immunol Methods 327: , 10–17. |
[9] | Butterfield AM , Chain JS , Ackermann BL , Konrad RJ ((2010) ) Comparison of assay formats for drug-tolerant immunogenicity testing. Bioanalysis 2: , 1961–1969. |
[10] | Goos JD , Henneman WJ , Sluimer JD , Vrenken H , Sluimer IC , Barkhof F , Blankenstein MA , Scheltens PH , van der Flier WM ((2010) ) Incidence of cerebral microbleeds: A longitudinal study in a memory clinic population. Neurology 74: , 1954–1960. |
[11] | Salloway S , Chalkias S , Barkhof F , Burkett P , Barakos J , Purcell D , Suhy J , Forrestal F , Tian Y , Umans K , Wang G , Singhal P , Budd Haeberlein S , Smirnakis K ((2022) ) Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 79: , 13–21. |
[12] | Curtin F , Vidal V , Bernard C , Kromminga A , Lang AB , Porchet H ((2016) ) Serum pharmacokinetics and cerebrospinal fluid concentration analysis of the new IgG4 monoclonal antibody GNbAC1 to treat multiple sclerosis: A phase 1 study. MAbs 8: , 854–860. |
[13] | Tran JQ , Rana J , Barkhof F , Melamed I , Gevorkyan H , Wattjes MP , de Jong R , Brosofsky K , Ray S , Xu L , Zhao J , Parr E , Cadavid D ((2014) ) Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol Neuroimmunol Neuroinflamm 1: , e18. |
[14] | Willis BA , Sundell K , Lachno DR , Ferguson-Sells LR , Case MG , Holdridge K , DeMattos RB , Raskin J , Siemers ER , Dean RA ((2018) ) Central pharmacodynamic activity of solanezumab in mild Alzheimer’s disease dementia. Alzheimers Dement (N Y) 4: , 652–660. |
[15] | Mielke MM , Hagen CE , Xu J , Chai X , Vemuri P , Lowe VJ , Airey DC , Knopman DS , Roberts RO , Machulda MM , Jack CR Jr., Petersen RC , Dage JL ((2018) ) Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimers Dement 14: , 989–997. |
[16] | Dage JL , Wennberg AMV , Airey DC , Hagen CE , Knopman DS , Machulda MM , Roberts RO , Jack CR Jr., Petersen RC , Mielke MM ((2016) ) Levels of tau protein in plasma are associated with neurodegeneration and cognitive function in a population-based elderly cohort. Alzheimers Dement 12: , 1226–1234. |
[17] | Fleisher A , Munsie L , Perahia D , Andersen S , Higgins I , Lo A , et al. (2022) Assessment of safety, tolerability, and efficacy of zagotenemab: Results from PERISCOPE-ALZ, a phase 2 study in early symptomatic Alzheimer’s disease. International Conference on Alzheimer’s and Parkinson’s Disease - 16th (AD/PD 2022), Barcelona. https://cslide.ctimeetingtech.com/adpd22/attendee/confcal/show/session/191 |
[18] | Florian H , Wang D , Arnold SE , Boada M , Guo Q , Jin Z , Zheng H , Fisseha N , Kalluri HV , Rendenbach-Mueller B , Budur K , Gold M ((2023) ) Tilavonemab in early Alzheimer’s disease: Results from a phase 2, randomized, double-blind study. Brain 146: , 2275–2284. |
[19] | Teng E , Manser PT , Pickthorn K , Brunstein F , Blendstrup M , Sanabria Bohorquez S , Wildsmith KR , Toth B , Dolton M , Ramakrishnan V , Bobbala A , Sikkes SAM , Ward M , Fuji RN , Kerchner GA ((2022) ) Safety and efficacy of semorinemab in individuals with prodromal to mild Alzheimer disease: A randomized clinical trial. JAMA Neurol 79: , 758–767. |


