
How to Prevent and/or Revert Alzheimer’s Disease Continuum During Preclinical Phases
Abstract
The development of Alzheimer’s disease (AD) follows three consecutive phases: namely preclinical, prodromal or mild cognitive impairment (MCI), and dementia. In addition, the preclinical phase can be divided into subphases related to the presence of biomarkers that appear at different points before the onset of MCI. Indeed, an early risk factor could promote the appearance of additional ones through a continuum. The presence of various risk factors may trigger specific biomarkers. In this review, we comment on how modifiable risk factors for AD may be reverted, thus correlating with a possible decrease in the specific biomarkers for the disease. Finally, we discuss the development of a suitable AD prevention strategy by targeting modifiable risk factors, thereby increasing the level of “precision medicine” in healthcare systems worldwide.
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent form of dementia [1]. It is a silent pathology in which symptoms appear before the clinical diagnosis of dementia [1–4]. Indeed, most researchers in the field hold the view that, when the AD diagnosis has been confirmed, it is too late to repair the damage and reverse the disease. AD is characterized by the appearance of aberrant brain structures, whose components are found in fluids such as cerebrospinal fluid and plasma, a decrease in some brain functions, and structural changes in the brain, among others [5]. These features could appear through a continuum before clinical diagnosis. The identification of these markers as early as possible would facilitate research into preventing or slowing down the progression of the disease. These features are related to the presence of non-modifiable or modifiable risk factors [6], and from the perspective of prevention, the major goal is to minimize the former or to potentiate the resilience mechanisms or protective factors. However, this field has not been widely explored [7, 8]. Given that non-modifiable risk factors such as genetics cannot be prevented, here we focus on the modifiable ones and how these may correlate with the presence of specific early biomarkers for the disease [9]. For practical purposes, these kinds of studies should be complemented with thorough clinical (precision medicine) analysis of patients [10].
In summary, here we discuss whether intervention on risk factors could lead to a decrease in a specific biomarker of the AD continuum and thus ultimately result in the prevention or modification of the progression of the disease.
POSSIBLE LINKS BETWEEN EARLY BIOMARKERS AND EARLY RISK FACTORS IN ALZHEIMER’S DISEASE
Early biomarkers and risk factors in AD
AD can be considered as the overlapping of two major pathologies, amyloid-β (Aβ) plaques and neurofibrillary tangles, that could be linked due to changes in glial cells [11, 12]. These two pathologies begin in different regions of the brain, and they overlap as the disease progresses, thereby facilitating the onset of dementia.
The presence and spread of aberrant amyloid or tau [13] aggregates occur through different stages: amyloid deposits spread following the stages reported in [14], whereas tau aggregates expand as described in [13]. Amyloid and tau aggregates appear at different times and in distinct regions of the brain during AD. The presence of both structures in distinct regions, for example, amyloid in the cingulate cortex [15], or tau in the entorhinal cortex [16], may manifest in early negative processes, leading to specific cognitive and functional deficits. However, clinical changes appear mainly after stage 3 of these two pathologies, correlating with the onset of dementia, the main disorder related to AD [17]. Glia (microglial cells) have been proposed to play a critical role in this overlap [18, 19].
Additional preclinical markers
Some studies have examined changes in moral judgment in subjects potentially [20] in the preclinical phase of AD, and the increased tolerance of pain in patients with mild to moderate AD, compared to healthy controls [21, 22]. Also, homocysteine levels in plasma [23], and decreased levels of calcium in bone, which could result in osteoporosis [24], should be taken into account. Thus, given that modifiable risk factors could involve around 40% of the cases of dementia, targeting these factors in the preclinical stage in individuals identified (through longitudinal studies) as having more modifiable risk factors should bring about the prevention or retardation of the condition [9].
Presence of biomarkers for lesions without clinical symptoms: Resilience
There is not always a total correlation between the presence of amyloid or tau aggregates with clinical consequences. Indeed, a small proportion of subjects whose autopsies show a high amount of Aβ or tau deposits may have remained cognitively intact until death. In such cases, these subjects may have been protected by resilience mechanisms [25]. These mechanisms are not fully understood and merit further analysis.
Risk factors
There are risk and protective factors for AD. Here we mainly focus only on the former. Among concepts related with protective modifiable factors; cognitive reserve, brain reserve or maintenance should be included. In this way, a correlation between cognitive reserve and a reduction of risk of developing dementia has been indicated [26]. In addition, the role of brain reserve, cognitive reserve, and maintenance in cognitive disorders like AD, has been widely commented by Dr. Stern [27, 28]. Also, lifestyle (psychical or leisure activity) may supply cognitive reserve [29]. Alternatively, some autoinflammatory agents could act as a possible protective factor for AD [30]. Returning to risk factors, they can be classified into non-modifiable and modifiable. The most relevant non-modifiable ones are aging and genetic factors [31].
Regarding modifiable risk factors, there are discrepancies between the number and incidence of such factors [7–9, 23, 32]. To choose relevant modifiable risk factors, as criteria here we used their prevalence as indicated in the literature [9, 32]. In Fig. 1, we also include factors such as socio-economic status [33], dyslipidemia [34], and nutrition-microbiota changes [35], which were commonly excluded in other reviews. About socio-economic status, we have indicated the impact of individual and neighborhood dimensions of socio-economic status on the presence of mild cognitive impairment (MCI) [33].
Fig. 1
Modifiable risk factors-Kinetics and links between them. Sleep disturbance is related to increasing age and it could be one of the earlier modifiable risk factors for AD (see text). Other modifiable risk factors related to the environment, lifestyle, or damage to the nervous system, peripheral tissues or organs are indicated. Arrows show the links between these modifiable risk factors.
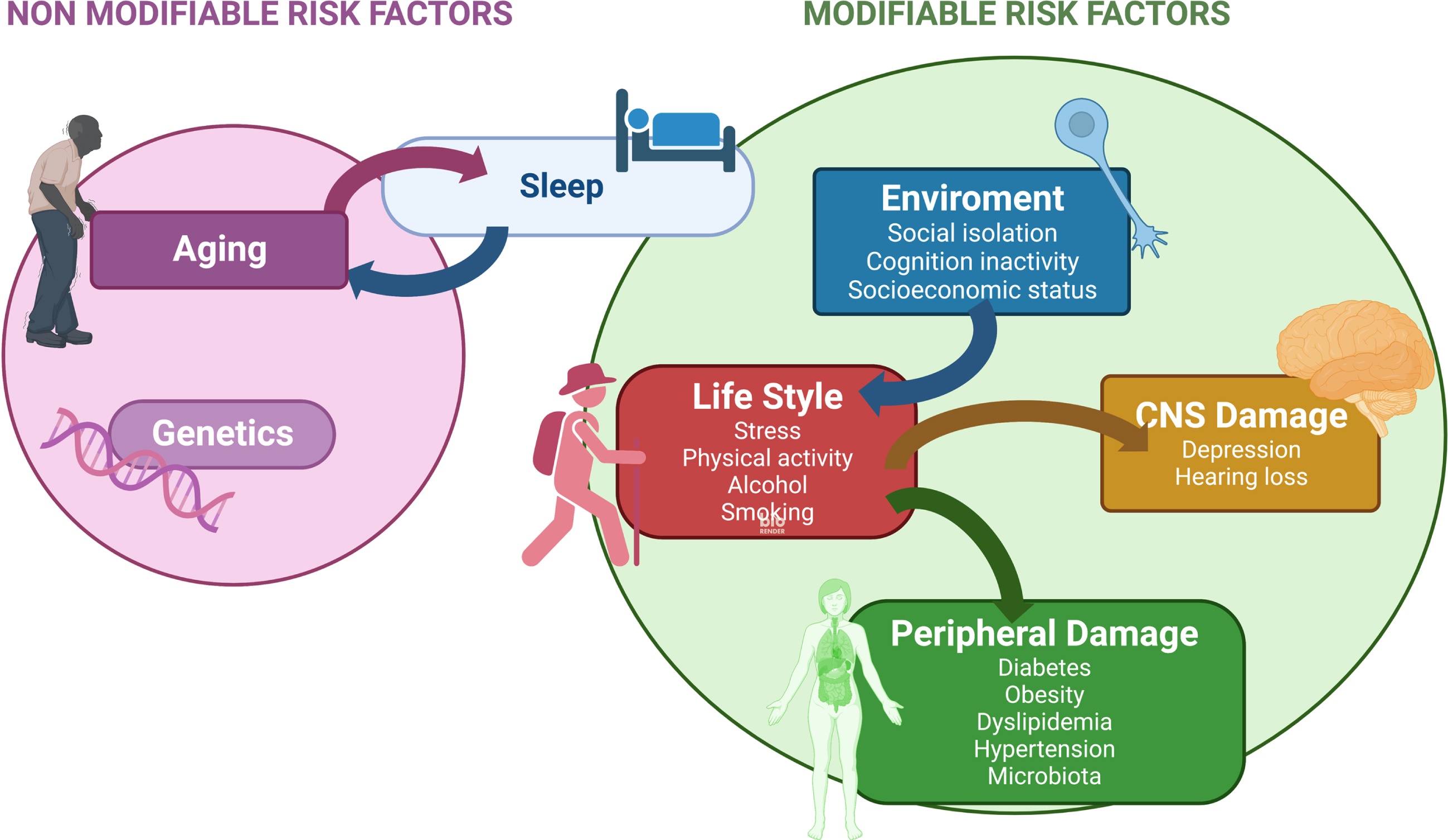
Also it has been described that socio-economic deprivation is a risk to develop dementia compared to people with a better economic status, even with a higher genetic risk [36].
About microbiota changes, the link between gut microbiota and AD has been reported [35]. Thus, gut microbiota modulation, for example, through probiotics or specific diet, has been suggested as a potential therapy in AD [37, 38].
About nutrition, long term high-fat diet consumption may indicate cognitive decline in aging, as shown in mouse models [39]. In addition, low serum vitamin D status could be associated with the appearance of dementia in the oldest old [40].
As previously indicated [6], some modifiable risk factors for AD can be intervened upon in a preventive manner to delay the development of the disease. It has been reported that modifiable risk factors are associated with the development of the disease in about 40% of dementia cases [9]. Modifiable risk factors also include the following: hypertension, obesity, physical inactivity, chronic stress, depression, and social isolation [6, 41, 42]. During the preclinical phase of AD, various risk factors are activated at different times and some factors that act, early on, may promote the emergence of new ones [43]. For example, chronic stress or social isolation can induce later depression [41, 42], and physical inactivity may lead to subsequent obesity. Thus, the risk factors that are activated at different times during the development of the disease may be linked.
Possible kinetics for risk factors
MCI is the (late) stage just before the diagnosis of dementia [44, 45]. Thus, it is currently considered a target phase in which intervention on modifiable risk factors may have positive effects on the further progression of the disease [46]. Indeed, a substantial proportion of subjects with MCI could annually revert to a normal cognitive state [47].
Prior to MCI, there is even a previous stage called subjective cognitive decline [48], during which it is also possible to intervene to prevent disorders associated with AD.
Sleep dysfunction as an early risk factor for Alzheimer’s disease
Some modifiable risk factors for AD are the consequences of life changes that occur during aging. It is sometimes difficult to establish whether the decrease in specific functions is the result of aging or, in fact, a preclinical sign of the disease. Sleep disturbances could be related to aging and AD. Indeed, sleep affects several functions related to aging and AD, such as episodic memory consolidation and executive functions [49, 50].
By using the “Slow Oscillation Switch Model”, the relationship between sleep and these two domains was explained [49]. Episodic memory is replaced during sleep through interactions between the hippocampal and neocortical networks. On the other hand, prefrontal-subcortical networks regulate executive functions and working memory [49]. These sleep functions can be disturbed during aging, but dysfunction can also occur earlier, being a very early marker for AD [51]. However, there are some discrepancies in this regard [52]. Sleep deficits have been reported in subjects during the early preclinical phase of AD [53, 54], around (or before) the appearance of amyloid pathology [2, 5, 55]. On the other hand, it has been described that sleep is bidirectionally modified by Aβ oligomers [51, 53, 54]; in a zebrafish model, Aβ reversibly enhances or suppresses sleep as a function of Aβ oligomer length [54]. Short Aβ oligomers facilitate acute wakefulness while long ones induce sleep [54]. During the progression of AD, sleep can be disrupted [51].
In addition, aging and the presence of Aβ aggregates alter circadian rhythms, including sleep regulation. In this regard, a relation between changes in the circadian rhythm and neurodegenerative disorders has been proposed [56].
Could sleep disturbances precede amyloid accumulation?
Sleep disturbance may be a consequence of claustrum dysfunction [46, 57, 58], which, at the onset of the preclinical phase of familiar AD, could precede amyloid accumulation [58]. Indeed, a link between claustrum deficits and human sleep has recently been described [5, 58]. Therefore, sleep disturbances emerge as a very early risk factor for AD [5, 57] and they may correlate with the presence of Aβ oligomers, which are increased mainly during sleep deprivation [59–61], as also shown by examining these oligomers in soluble fluids (plasma or cerebrospinal fluid) [62]. Although further work is required in this field, treatment of sleep disturbances using medication, psychological strategies, or circadian rhythm treatments could be considered as approaches to prevent the development of AD [63].
Finally, other proteins related to AD, like tau, may play a role in sleep disturbances. In this regard, it has been found that the absence (dysfunction) of tau results in sleep dysregulation [64].
Links between early modifiable risk factors and biomarkers for the preclinical phase of AD
Given that modifiable risk factors related to lifestyle can often be altered, some studies addressed whether lifestyle changes could also decrease the level of some AD biomarkers [65, 66].
Increasing physical activity results in a decrease in the level of biomarkers for AD
An example of a modifiable lifestyle risk factor is physical inactivity. This risk factor can be targeted by doing physical exercise. In this regard, in a mouse model for AD subjected to five months of voluntary exercise showed a decreased level of Aβ deposits in the cortex and hippocampus [67]. Also, long-term treadmill exercise attenuates tau pathology in the P301S-tau transgenic mouse model [68]. In addition, exercise decreases microglia activation in the hippocampus, the levels of cytokines like IL-1β or TNFα being decreased [69]. Thus, there is some evidence that links early modifiable risk factors with a decrease in the level of early markers for the preclinical phase of AD.
In addition, changes in physical activity can also improve functional features like cognitive impairment. Indeed, normal cognitive status has been regained in animal models [66, 70].
In humans, aerobic exercise at early AD stages correlates with an increase in cardiorespiratory fitness that could be linked to an increase in memory function [71]. Also, the effects of different types of physical exercise in humans, have been commented as a possible therapy to prevent neuronal death that could be taken place in neurodegenerative disorders [72].
REVERSION OF RISK FACTORS FOR THE PREVENTION AND REVERSION OF PRECLINICAL PHASES OF ALZHEIMER’S DISEASE
Modifiable risk factors can be targeted when they become activated and, in several cases, the damage they cause, like hypertension, depression, and diabetes, can be decreased by pharmacological therapy, or by psychological strategies that modify lifestyle, for example, by preventing social isolation or depression. Indeed, there is some controversy about the targeting of risk factors to prevent the progression of dementia [7, 8]. Some authors have indicated that such targeting is not a suitable strategy [8]. However, on the other hand, decreasing hypertension by changes in diet could reduce the incidence of AD [71]. Also, analyses performed by the Cochrane Organization examining the effect of hypertension treatments suggest that such strategies retard dementia [73]. In addition, treatment of depression may enhance cognitive functions [74]. To clarify this controversy, additional analyses that include new relevant modifiable risk factors are required. Thus, it is of interest to collect solid clinical data from patients in the preclinical phase of AD. This could be achieved simply by including additional clinical observations to the previously defined risk factors. In other words, healthcare systems should foster an increased level of “precision medicine” [10, 75].
Finally, cognitive stimulation or lifelong learning have been suggested to improve cognitive functions in people with dementia [76]. In this way, cognitive stimulation therapy has been proposed for treatment of AD [77].
CONCLUSIONS
In this review, we have addressed the targeting of modifiable risk factors to delay or prevent dementias like AD. However, this strategy depends on the capacity of healthcare systems to monitor changes in these risk factors over several years before a clinical diagnosis is made. However, as indicated in a recent study, vast improvements worldwide are required before these health providers can undertake this task [78]. That study gave examples of countries needing improvement of their systems. However, we question whether any country in the world takes care of brain health in a regular manner like dental health is.
Kerwin et al. [78] highlighted the problems to be tackled in several countries. In this regard, in China, there is a lack of physician training in dementia. In Brazil and Nigeria, socio-economic inequalities are an issue. In Sweden, there is inconsistent use of biomarkers, and in Spain, patient hesitancy, together with an overloaded health care system, are barriers to a proper diagnosis [78]. In conclusion, for more effective clinical management of dementia, healthcare systems worldwide would need to commit to boosting “precision medicine”.
Around this point, the Alzheimer Precision Medicine Initiative (APMI) was launched with collaborations, at the present, from USA, France, Germany, UK, Ireland, Canada, Ital,y and Switzerland. AMPI is at an early development stage [10].
ACKNOWLEDGMENTS
We thank Universidad Complutense de Madrid and Instituto de Investigación Sanitaria Hospital Clínico San Carlos for the support. We thank Ms. Nuria de la Torre for the edition help. Figure was created with BioRender.com
FUNDING
This work has been done during a Margarita Salas contract founded by Next Generation EU.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Masters CL , Bateman R , Blennow K , Rowe CC , Sperling RA , Cummings JL ((2015) ) Alzheimer’s disease. Nat Rev Dis Primers 1: , 15056. |
[2] | Bateman RJ , Xiong C , Benzinger TL , Fagan AM , Goate A , Fox NC , Marcus DS , Cairns NJ , Xie X , Blazey TM , Holtzman DM , Santacruz A , Buckles V , Oliver A , Moulder K , Aisen PS , Ghetti B , Klunk WE , McDade E , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Schofield PR , Sperling RA , Salloway S , Morris JC , Dominantly Inherited Alzheimer Network ((2012) ) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367: , 795–804. |
[3] | Hampel H , Hardy J , Blennow K , Chen C , Perry G , Kim SH , Villemagne VL , Aisen P , Vendruscolo M , Iwatsubo T , Masters CL , Cho M , Lannfelt L , Cummings JL , Vergallo A ((2021) ) The amyloid-beta pathway in Alzheimer’s disease. Mol Psychiatry 26: , 5481–5503. |
[4] | Prince M , Bryce R , Albanese E , Wimo A , Ribeiro W , Ferri CP ((2013) ) The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement 9: , 63–75 e62. |
[5] | Avila J , Perry G ((2021) ) A multilevel view of the development of Alzheimer’s disease. Neuroscience 457: , 283–293. |
[6] | Edwards Iii GA , Gamez N , Escobedo G Jr., Calderon O , Moreno-Gonzalez I ((2019) ) Modifiable risk factors for Alzheimer’s disease. Front Aging Neurosci 11: , 146. |
[7] | Barnes DE , Yaffe K ((2011) ) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10: , 819–828. |
[8] | Flicker L ((2010) ) Modifiable lifestyle risk factors for Alzheimer’s disease. J Alzheimers Dis 20: , 803–811. |
[9] | Lee M , Whitsel E , Avery C , Hughes TM , Griswold ME , Sedaghat S , Gottesman RF , Mosley TH , Heiss G , Lutsey PL ((2022) ) Variation in population attributable fraction of dementia associated with potentially modifiable risk factors by race and ethnicity in the US. JAMA Netw Open 5: , e2219672. |
[10] | Hampel H , Vergallo A , Perry G , Lista S , Alzheimer Precision Medicine Initiative ((2019) ) The Alzheimer Precision Medicine Initiative. J Alzheimers Dis 68: , 1–24. |
[11] | Hamelin L , Lagarde J , Dorothee G , Leroy C , Labit M , Comley RA , de Souza LC , Corne H , Dauphinot L , Bertoux M , Dubois B , Gervais P , Colliot O , Potier MC , Bottlaender M , Sarazin M , Clinical It ((2016) ) Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 139: , 1252–1264. |
[12] | Martin E , Boucher C , Fontaine B , Delarasse C ((2017) ) Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer’s disease models: Effects of aging and amyloid pathology. Aging Cell 16: , 27–38. |
[13] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[14] | Thal DR , Rub U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[15] | Avila-Villanueva M , Gomez-Ramirez J , Avila J , Fernandez-Blazquez MA ((2021) ) Alzheimer’s disease and empathic abilities: The proposed role of the cingulate cortex. J Alzheimers Dis Rep 5: , 345–352. |
[16] | Maass A , Lockhart SN , Harrison TM , Bell RK , Mellinger T , Swinnerton K , Baker SL , Rabinovici GD , Jagust WJ ((2018) ) Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging. J Neurosci 38: , 530–543. |
[17] | Hojjati SH , Feiz F , Ozoria S , Razlighi QR , Alzheimer’s Disease Neuroimaging Initiative ((2021) ) Topographical overlapping of the amyloid-beta and tau pathologies in the default mode network predicts Alzheimer’s disease with higher specificity. J Alzheimers Dis 83: , 407–421. |
[18] | Hopp SC , Lin Y , Oakley D , Roe AD , DeVos SL , Hanlon D , Hyman BT ((2018) ) The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 15: , 269. |
[19] | Pascoal TA , Benedet AL , Ashton NJ , Kang MS , Therriault J , Chamoun M , Savard M , Lussier FZ , Tissot C , Karikari TK , Ottoy J , Mathotaarachchi S , Stevenson J , Massarweh G , Scholl M , de Leon MJ , Soucy JP , Edison P , Blennow K , Zetterberg H , Gauthier S , Rosa-Neto P ((2021) ) Microglial activation and tau propagate jointly across Braak stages. Nat Med 27: , 1592–1599. |
[20] | van Honk J , Terburg D , Montoya ER , Grafman J , Stein DJ , Morgan B ((2022) ) Breakdown of utilitarian moral judgement after basolateral amygdala damage. Proc Natl Acad Sci U S A 119: , e2119072119. |
[21] | Cao S , Fisher DW , Yu T , Dong H ((2019) ) The link between chronic pain and Alzheimer’s disease. J Neuroinflammation 16: , 204. |
[22] | Jensen-Dahm C , Werner MU , Dahl JB , Jensen TS , Ballegaard M , Hejl AM , Waldemar G ((2014) ) Quantitative sensory testing and pain tolerance in patients with mild to moderate Alzheimer disease compared to healthy control subjects. Pain 155: , 1439–1445. |
[23] | Xu W , Tan L , Wang HF , Jiang T , Tan MS , Tan L , Zhao QF , Li JQ , Wang J , Yu JT ((2015) ) Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry 86: , 1299–1306. |
[24] | Woodman I ((2013) ) Osteoporosis: Linking osteoporosis with Alzheimer disease. Nat Rev Rheumatol 9: , 638. |
[25] | Gomez-Isla T , Frosch MP ((2022) ) Lesions without symptoms: Understanding resilience to Alzheimer disease neuropathological changes. Nat Rev Neurol 18: , 323–332. |
[26] | Panico F , Sagliano L , Magliacano A , Santangelo G , Trojano L ((2022) ) The relationship between cognitive reserve and cognition in healthy adults: A systematic review. Curr Psychol. doi: 10.1007/s12144-022-03523-y. |
[27] | Stern Y ((2012) ) Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol 11: , 1006–1012. |
[28] | Stern Y , Arenaza-Urquijo EM , Bartrés-Faz D , Belleville S , Cantilon M , Chetelat G , Ewers M , Franzmeier N , Kempermann G , Kremen WS , Okonkwo O , Scarmeas N , Soldan A , Udeh-Momoh C , Valenzuela M , Vemuri P , Vuoksimaa E ; the Reserve, Resilience and Protective Factors PIA Empirical Definitions and Conceptual Frameworks Workgroup ((2020) ) Whitepaper: Defining and investigating cognitivereserve, brain reserve, and brain maintenance. Alzheimers Dement 16: , 1305–1311. |
[29] | Song S , Stern Y , Gu Y ((2022) ) Modifiable lifestyle factors and cognitive reserve: A systematic review of current evidence. Ageing Res Rev 74: , 101551. |
[30] | McGeer PL , Schulzer M , McGeer EG ((1996) ) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology 47: , 425–432. |
[31] | Tanzi RE ((2012) ) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006296. |
[32] | Omura JD , McGuire LC , Patel R , Baumgart M , Lamb R , Jeffers EM , Olivari BS , Croft JB , Thomas CW , Hacker K ((2022) ) Modifiable risk factors for Alzheimer disease and related dementias among adults aged >/=45 years - United States, 2019. MMWR Morb Mortal Wkly Rep 71: , 680–685. |
[33] | Fernandez-Blazquez MA , Noriega-Ruiz B , Avila-Villanueva M , Valenti-Soler M , Frades-Payo B , Del Ser T , Gomez-Ramirez J ((2021) ) Impact of individual and neighborhood dimensions of socioeconomic status on the prevalence of mild cognitive impairment over seven-year follow-up. Aging Ment Health 25: , 814–823. |
[34] | Reitz C ((2013) ) Dyslipidemia and the risk of Alzheimer’s disease. Curr Atheroscler Rep 15: , 307. |
[35] | Jiang C , Li G , Huang P , Liu Z , Zhao B ((2017) ) The gut microbiota and Alzheimer’s disease. J Alzheimers Dis 58: , 1–15. |
[36] | Maresova P , Mohelska H , Dolejs J , Kuca K ((2015) ) Socio-economic aspects of Alzheimer’s disease. Curr Alzheimer Res 12: , 903–911. |
[37] | Kesika P , Suganthy N , Sivamaruthi BS , Chaiyasut C ((2021) ) Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci 264: , 118627. |
[38] | Shabbir U , Arshad MS , Sameen A , Oh DH ((2021) ) Crosstalk between gut and brain in Alzheimer’s disease: The role of gut microbiota modulation strategies. Nutrients 13: , 690. |
[39] | Liang Z , Gong X , Ye R , Zhao Y , Yu J , Zhao Y , Bao J ((2023) ) Long-term high-fat diet consumption induces cognitive decline accompanied by tau hyper-phosphorylation and microglial activation in aging. Nutrients 15: , 250. |
[40] | Melo van Lent D , Egert S , Wolfsgruber S , Kleineidam L , Weinhold L , Wagner-Thelen H , Stoffel-Wagner B , Bickel H , Wiese B , Weyerer S , Pentzek M , Jessen F , Schmid M , Maier W , Scherer M , Riedel-Heller SG , Ramirez A , Wagner M ((2022) ) Low serum vitamin D status is associated with incident Alzheimer’s dementia in the oldest old. Nutrients 15: , 61. |
[41] | Avila-Villanueva M , Gomez-Ramirez J , Avila J , Fernandez-Blazquez MA ((2022) ) Loneliness as risk factor for Alzheimer’s disease. Curr Aging Sci 15: , 293–296. |
[42] | Avila-Villanueva M , Gomez-Ramirez J , Maestu F , Venero C , Avila J , Fernandez-Blazquez MA ((2020) ) The role of chronic stress as a trigger for the Alzheimer disease continuum. Front Aging Neurosci 12: , 561504. |
[43] | Aisen PS , Cummings J , Jack CR Jr., Morris JC , Sperling R , Frolich L , Jones RW , Dowsett SA , Matthews BR , Raskin J , Scheltens P , Dubois B ((2017) ) On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimers Res Ther 9: , 60. |
[44] | Gauthier S , Reisberg B , Zaudig M , Petersen RC , Ritchie K , Broich K , Belleville S , Brodaty H , Bennett D , Chertkow H , Cummings JL , de Leon M , Feldman H , Ganguli M , Hampel H , Scheltens P , Tierney MC , Whitehouse P , Winblad B , International Psychogeriatric Association Expert Conference on mild cognitive impairment ((2006) ) Mild cognitive impairment. Lancet 367: , 1262–1270. |
[45] | Petersen RC ((2011) ) Clinical practice. Mild cognitive impairment. N Engl J Med 364: , 2227–2234. |
[46] | Avila-Villanueva M , Marcos Dolado A , Gomez-Ramirez J , Fernandez-Blazquez M ((2022) ) Brain structural and functional changes in cognitive impairment due to Alzheimer’s disease. Front Psychol 13: , 886619. |
[47] | Sanz-Blasco R , Ruiz-Sanchez de Leon JM , Avila-Villanueva M , Valenti-Soler M , Gomez-Ramirez J , Fernandez-Blazquez MA ((2022) ) Transition from mild cognitive impairment to normal cognition: Determining the predictors of reversion with multi-state Markov models. Alzheimers Dement 18: , 1177–1185. |
[48] | Jessen F , Amariglio RE , van Boxtel M , Breteler M , Ceccaldi M , Chetelat G , Dubois B , Dufouil C , Ellis KA , van der Flier WM , Glodzik L , van Harten AC , de Leon MJ , McHugh P , Mielke MM , Molinuevo JL , Mosconi L , Osorio RS , Perrotin A , Petersen RC , Rabin LA , Rami L , Reisberg B , Rentz DM , Sachdev PS , de la Sayette V , Saykin AJ , Scheltens P , Shulman MB , Slavin MJ , Sperling RA , Stewart R , Uspenskaya O , Vellas B , Visser PJ , Wagner M , Subjective Cognitive Decline Initiative Working Group ((2014) ) A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 10: , 844–852. |
[49] | Chen PC , Zhang J , Thayer JF , Mednick SC ((2022) ) Understanding the roles of central and autonomic activity during sleep in the improvement of working memory and episodic memory. Proc Natl Acad Sci U S A 119: , e2123417119. |
[50] | Creery JD , Brang DJ , Arndt JD , Bassard A , Towle VL , Tao JX , Wu S , Rose S , Warnke PC , Issa NP , Paller KA ((2022) ) Electrophysiological markers of memory consolidation in the human brain when memories are reactivated during sleep. Proc Natl Acad Sci U S A 119: , e2123430119. |
[51] | Ju YS , Ooms SJ , Sutphen C , Macauley SL , Zangrilli MA , Jerome G , Fagan AM , Mignot E , Zempel JM , Claassen J , Holtzman DM ((2017) ) Slow wave sleep disruption increases cerebrospinal fluid amyloid-beta levels. Brain 140: , 2104–2111. |
[52] | Olsson M , Arlig J , Hedner J , Blennow K , Zetterberg H ((2018) ) Sleep deprivation and cerebrospinal fluid biomarkers for Alzheimer’s disease. Sleep 41: , zsy025. |
[53] | Roh JH , Huang Y , Bero AW , Kasten T , Stewart FR , Bateman RJ , Holtzman DM ((2012) ) Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med 4: , 150ra122. |
[54] | Ozcan GG , Lim S , Leighton P , Allison WT , Rihel J ((2020) ) Sleep is bi-directionally modified by amyloid beta oligomers. Elife 9: , e53995. |
[55] | Jack CRJr. , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , Shaw LM , Vemuri P , Wiste HJ , Weigand SD , Lesnick TG , Pankratz VS , Donohue MC , Trojanowski JQ ((2013) ) Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: , 207–216. |
[56] | Nassan M , Videnovic A ((2022) ) Circadian rhythms in neurodegenerative disorders. Nat Rev Neurol 18: , 7–24. |
[57] | Atilgan H , Doody M , Oliver DK , McGrath TM , Shelton AM , Echeverria-Altuna I , Tracey I , Vyazovskiy VV , Manohar SG , Packer AM ((2022) ) Human lesions and animal studies link the claustrum to perception, salience, sleep and pain. Brain 145: , 1610–1623. |
[58] | Goutagny R , Gu N , Cavanagh C , Jackson J , Chabot JG , Quirion R , Krantic S , Williams S ((2013) ) Alterations in hippocampal network oscillations and theta-gamma coupling arise before Abeta overproduction in a mouse model of Alzheimer’s disease. Eur J Neurosci 37: , 1896–1902. |
[59] | Allen SR , Seiler WO , Stahelin HB , Spiegel R ((1987) ) Seventy-two hour polygraphic and behavioral recordings of wakefulness and sleep in a hospital geriatric unit: Comparison between demented and nondemented patients. Sleep 10: , 143–159. |
[60] | Moran M , Lynch CA , Walsh C , Coen R , Coakley D , Lawlor BA ((2005) ) Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med 6: , 347–352. |
[61] | Shokri-Kojori E , Wang GJ , Wiers CE , Demiral SB , Guo M , Kim SW , Lindgren E , Ramirez V , Zehra A , Freeman C , Miller G , Manza P , Srivastava T , De Santi S , Tomasi D , Benveniste H , Volkow ND ((2018) ) beta-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci U S A 115: , 4483–4488. |
[62] | Lucey BP ((2020) ) It’s complicated: The relationship between sleep and Alzheimer’s disease in humans. Neurobiol Dis 144: , 105031. |
[63] | McCurry SM , Reynolds CF , Ancoli-Israel S , Teri L , Vitiello MV ((2000) ) Treatment of sleep disturbance in Alzheimer’s disease. Sleep Med Rev 4: , 603–628. |
[64] | Cantero JL , Hita-Yanez E , Moreno-Lopez B , Portillo F , Rubio A , Avila J ((2010) ) Tau protein role in sleep-wake cycle. J Alzheimers Dis 21: , 411–421. |
[65] | Mee-Inta O , Zhao ZW , Kuo YM ((2019) ) Physical exercise inhibits inflammation and microglial activation. Cells 8: , 691. |
[66] | Rashid MH , Zahid MF , Zain S , Kabir A , Hassan SU ((2020) ) The neuroprotective effects of exercise on cognitive decline: A preventive approach to Alzheimer disease. Cureus 12: , e6958. |
[67] | Adlard PA , Perreau VM , Pop V , Cotman CW ((2005) ) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci 25: , 4217–4221. |
[68] | Ohia-Nwoko O , Montazari S , Lau YS , Eriksen JL ((2014) ) Long-term treadmill exercise attenuates tau pathology in P301S tau transgenic mice. Mol Neurodegener 9: , 54. |
[69] | Koga S , Kojima A , Ishikawa C , Kuwabara S , Arai K , Yoshiyama Y ((2014) ) Effects of diet-induced obesity and voluntary exercise in atauopathy mouse model: Implications of persistent hyperleptinemia and enhanced astrocytic leptin receptor expression. Neurobiol Dis 71: , 180–192. |
[70] | Llorens-Martin M , Teixeira CM , Jurado-Arjona J , Rakwal R , Shibato J , Soya H , Avila J ((2016) ) Retroviral induction of GSK-3beta expression blocks the stimulatory action of physical exercise on the maturation of newborn neurons. Cell Mol Life Sci 73: , 3569–3582. |
[71] | Morris MC , Tangney CC , Wang Y , Sacks FM , Bennett DA , Aggarwal NT ((2015) ) MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement 11: , 1007–1014. |
[72] | Andreotti DZ , Silva JDN , Matumoto AM , Orellana AM , de Mello PS , Kawamoto EM ((2020) ) Effects of physical exercise on autophagy and apoptosis in aged brain: Human and animal studies. Front Nutr 7: , 94. |
[73] | McGuinness B , Todd S , Passmore P , Bullock R ((2009) ) Blood pressure lowering in patients without prior cerebrovascular disease for prevention of cognitive impairment and dementia. Cochrane Database Syst Rev CD004034. |
[74] | Butters MA , Becker JT , Nebes RD , Zmuda MD , Mulsant BH , Pollock BG , Reynolds CF 3rd ((2000) ) Changes in cognitive functioning following treatment of late-life depression. Am J Psychiatry 157: , 1949–1954. |
[75] | Toups K , Hathaway A , Gordon D , Chung H , Raji C , Boyd A , Hill BD , Hausman-Cohen S , Attarha M , Chwa WJ , Jarrett M , Bredesen DE ((2022) ) Precision medicine approach to Alzheimer’s disease: Successful pilot project. J Alzheimers Dis 88: , 1411–1421. |
[76] | Woods B , Aguirre E , Spector AE , Orrell M ((2012) ) Cognitive stimulation to improve cognitive functioning in people with dementia. Cochrane Database Syst Rev CD005562. |
[77] | Spector A , Woods B , Orrell M ((2008) ) Cognitive stimulation for the treatment of Alzheimer’s disease. Expert Rev Neurother 8: , 751–757. |
[78] | Kerwin D , Abdelnour C , Caramelli P , Ogunniyi A , Shi J , Zetterberg H , Traber M ((2022) ) Alzheimer’s disease diagnosis and management: Perspectives from around the world. Alzheimers Dement (Amst) 14: , e12334. |


