
Blood Kallikrein-8 and Non-Amnestic Mild Cognitive Impairment: An Exploratory Study
Abstract
Background:
Blood kallikrein-8 is supposed to be a biomarker for mild cognitive impairment (MCI) due to Alzheimer’s disease (AD), a precursor of AD dementia. Little is known about the association of kallikrein-8 and non-AD type dementias.
Objective:
To investigate whether blood kallikrein-8 is elevated in individuals with non-amnestic MCI (naMCI), which has a higher probability to progress to a non-AD type dementia, compared with cognitively unimpaired (CU) controls.
Methods:
We measured blood kallikrein-8 at ten-year follow-up (T2) in 75 cases and 75 controls matched for age and sex who were participants of the population-based Heinz Nixdorf Recall study (baseline: 2000–2003). Cognitive performance was assessed in a standardized manner at five (T1) and ten-year follow-up. Cases were CU or had subjective cognitive decline (SCD) at T1 and had naMCI at T2. Controls were CU at both follow-ups. The association between kallikrein-8 (per 500 pg/ml increase) and naMCI was estimated using conditional logistic regression: odds ratios (OR) and 95% confidence intervals (95% CI) were determined, adjusted for inter-assay variability and freezing duration.
Results:
Valid kallikrein-8 values were measured in 121 participants (45% cases, 54.5% women, 70.5±7.1 years). In cases, the mean kallikrein-8 was higher than in controls (922±797 pg/ml versus 884±782 pg/ml). Kallikrein-8 was not associated with having naMCI compared to being CU (adjusted; OR: 1.03 [95% CI: 0.80–1.32]).
Conclusion:
This is the first population-based study that shows that blood kallikrein-8 tends not to be elevated in individuals with naMCI compared with CU. This adds to the evidence of the possible AD specificity of kallikrein-8.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia, followed by vascular and mixed dementia pathologies [1]. Dementia due to frontotemporal lobar degeneration, Parkinson’s disease, or Lewy bodies are less frequently reported. In order to distinguish between different forms of dementia, there is often a need for invasive and expensive diagnostics [2]. Although there have been no breakthroughs in treatment options to date, intensive research is continuing. There is consensus that future treatments can only be successful if the underlying cause of dementia can be identified at a very early stage. An early treatment might be able to stop or at least slow the progression of the disease and preserve cognitive functioning. An optimal, less invasive, and cost-effective support for early diagnosis could be achieved using blood-based dementia biomarkers. So far, blood-based biomarkers for tau and amyloid-β (Aβ) have been developed, but their diagnostic value in early stage of AD is controversial. They appear to be too insensitive for diagnosis in the preclinical phase of AD [3, 4].
In the course of AD, there is a dysregulation of the kallikrein-kinin system (KKS), which is controlled by kallikreins. Low and high molecular weight kininogen (LMWK and HMWK) is processed by tissue kallikrein (KLK1) and plasma kallikrein (KLKB1) to generate the peptide hormone bradykinin (BK), which is rapidly transformed to des-Arg9-BK (DA9BK). BK and DA9BK activate the bradykinin 1 and 2 receptors (B1 R and B2 R) and so induce vasodilation, vessel permeability, and inflammation [5]. In the blood of AD patients, KKS hyperactivity [6] and elevated BK levels [7] are found. In previous studies, enhanced hippocampal B1 R immuno-reactivity in AD mice [8] are demonstrated, while B1 R blockade results in cognitive and cerebrovascular improvements [8]. Kallikrein-8 (KLK8, alias neuropsin) is one of the 15 serine proteases of the kallikrein family [9, 10]. Biochemically, it is a 228 amino acid long enzyme with a catalytic triad of histidine-aspartate-serine [11]. KLK8 is expressed cerebrally, mainly in the hippocampus [12], but also in extracerebral tissues [13]. As a secreted enzyme, it can be detected in biological body fluids such as blood serum and cerebrospinal fluid (CSF). Recently, KLK8 is thought to be involved in the development of AD [14–19]. Whether this pathological alteration contributes to KKS hyperactivity is unclear and requires further investigation.
Our recent studies suggest KLK8 to be a potential early AD blood and CSF biomarker [14–19]. KLK8 is a dose-dependent modulator of neuroplasticity and memory [20–23]. It appears to regulate several signaling pathways associated with neuroplasticity [22, 24–26] by processing the substrates neuregulin-1 [27], neuronal cell adhesion molecule L1 [28], fibronectin [29], and ephrin receptor B2 [30]. KLK8 inhibition and knockdown impedes amyloidogenic amyloid-β protein precursor processing, facilitates Aβ clearance, boosts autophagy, reduces Aβ load and tau pathology, enhances neuroplasticity, improves memory, and unfolds anxiolytic effects [16–18]. A clinical study of MCI and AD patients showed that KLK8 levels were elevated in blood and CSF of patients with MCI due to AD and early AD [14]. The diagnostic accuracy of KLK8 in CSF was as good as that of the major CSF biomarkers (Aβ, phosphorylated tau, and total tau) for AD and, in the case of MCI, even better than Aβ42 in CSF. KLK8 in blood was a similarly strong discriminator for MCI due to AD but slightly weaker for AD [14]. Furthermore, our recently published results of a population-based study showed that blood KLK8 was elevated in individuals with amnestic MCI compared with cognitively healthy individuals [19]. In different regions of transgenic murine [18] and AD-affected human brain [18, 31] pathologically high levels of KLK8 mRNA and protein were shown long before the clinical signs of AD appear [18]. This exceedingly early (in mice prior to onset of amyloid pathology and in patients in CERAD A/ Braak I-II stage) and multifocal rise of KLK8 (i.e., in the frontal cortex and hippocampus and even in regions such as cerebellum that are not that affected by ADs pathology) [18] is suggestive of a causal role in the cascade of events leading to AD. This hypothesis is supported further by the fact that short-term inhibition of KLK8 by an anti-KLK8-antibody [16, 18] or its long-term downregulation by genetic knockdown [17] exerted considerable therapeutic effect in transgenic mice. Thus, KLK8 might be a very early biomarker of AD and a therapeutic target as well.
It was hypothesized that blood and CSF KLK8 is elevated only in individuals who are on the AD pathway and not in individuals with other dementias. There is only one study regarding other dementia causes and KLK8 by Li et al. (2021) that found blood KLK8 levels to be significantly higher in participants with vascular dementia (defined using the Mini-Mental State Examination only) compared to controls [32]. However, no study is published on the association of KLK8 and prodromal stages of non-AD type dementia. Therefore, it is still not known if blood KLK8 is elevated in early stages of non-AD type dementia. Looking at the dementia continuum, MCI is a condition in which individuals demonstrate mild cognitive symptoms that do not interfere with everyday activities but are noticeable to the individual [33]. Participants with MCI have a higher probability to progress to dementia in the further course [33]. When memory dysfunction predominates, MCI is described by the term “amnestic”. When impairment of other cognitive features is more prominent, MCI is described as “non-amnestic” [33]. Whereas amnestic MCI (aMCI) has a higher probability to progress to AD (MCI due to AD) [34], non-amnestic MCI (naMCI) has been shown to be more likely to progress to non-AD type dementias (like vascular dementia, dementia with Lewy bodies or frontotemporal dementia) [33, 35–37]. The aim of this present study is to examine if blood KLK8 levels of participants with naMCI are elevated compared to cognitively unimpaired (CU) participants using a population-based case-control sample.
MATERIALS AND METHODS
Study design and study population
We analyzed data of our population-based Heinz Nixdorf Recall (Risk Factors, Evaluation of Coronary Calcification, and Lifestyle) study (HNR study). The details of the cohort and study design have been described previously [38]. Briefly, the participants for the HNR study were randomly selected inhabitants of the Ruhr area living in Bochum (371,582 residents), Essen (589,676 residents), and Mülheim/Ruhr (172,759 residents). A total of 4,814 men and women aged 45 to 75 participated in the baseline survey from 2000 to 2003. The overall recruitment efficacy proportion was 55.8% [39]. After five (T1) and ten years (T2), participants were followed up and they received annual questionnaires. The standardized assessment of cognitive performance described below was introduced at T1 and extended at T2.
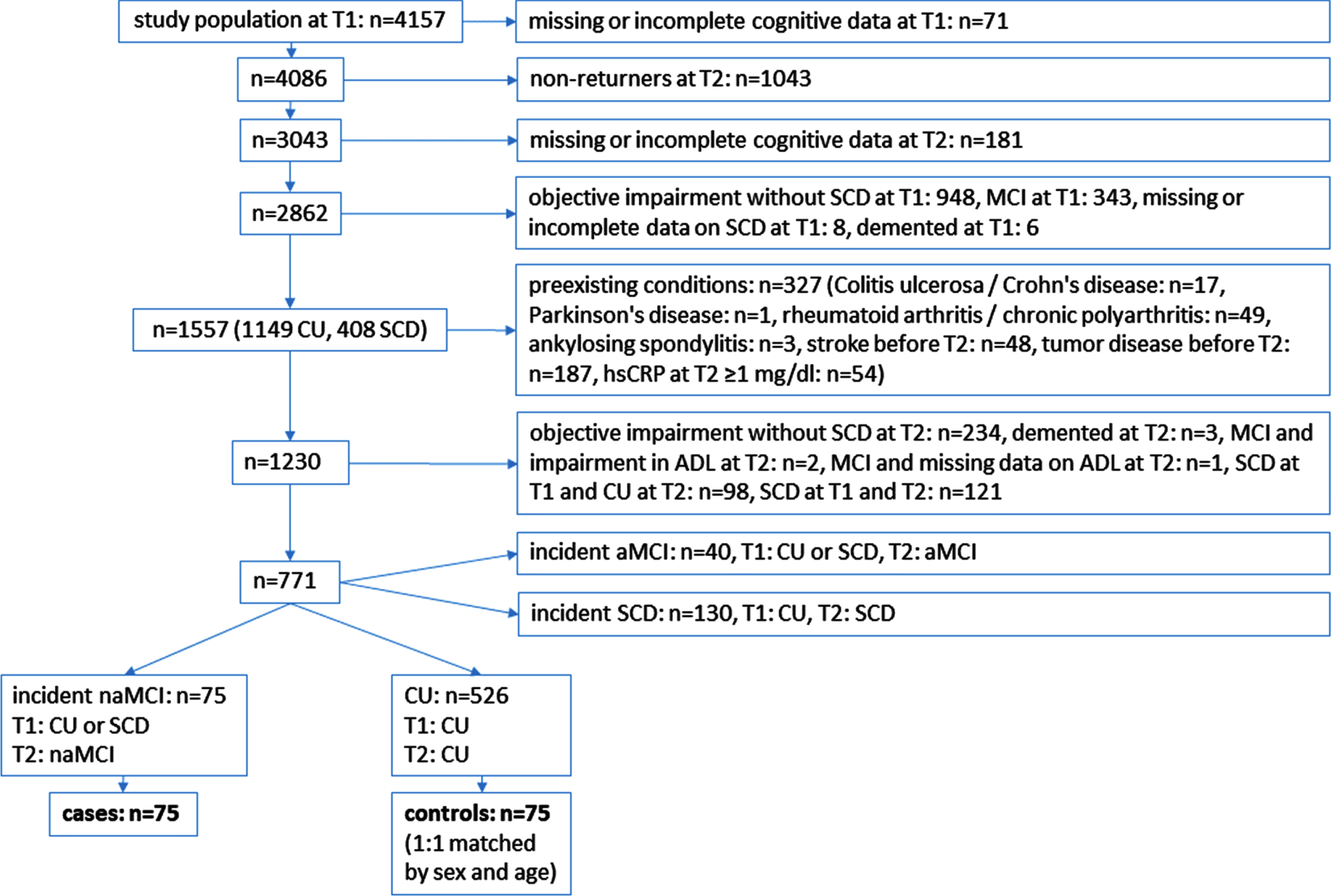
The cases of our nested case-control study were participants who were cognitively unimpaired (CU) or reported subjective cognitive decline (SCD) at T1 and had a naMCI diagnosis at T2 (see below for the exact definition of naMCI). Controls had to be CU at T1 and T2. The cases and controls were free of the secondary diseases mentioned below at least to T2. Each naMCI case was assigned a control partner matched for age (±3 years) and sex. The flowchart of the study population is shown in Fig. 1.
Fig. 1
Flow chart of the study population. ADL, activities of daily living; aMCI, amnestic mild cognitive impairment; CU, cognitively unimpaired; hsCRP, high-sensitivity C-reactive protein; naMCI, non-amnestic mild cognitive impairment; SCD, subjective cognitive decline; T1, first follow-up visit; T2, second follow-up visit.
The study was approved by the Institutional Review Board of the University of Duisburg-Essen and followed established guidelines of good epidemiological practice. Written informed consent was obtained from all participants.
Survey of secondary diseases
As in Schramm et al. [19] the following secondary conditions were recorded: Standardized computer-assisted interviews were performed and participants were asked of having ankylosing spondylitis, rheumatoid arthritis, chronic polyarthritis, Crohn’s disease, ulcerative colitis, stroke, Parkinson’s disease, or tumor disease before T2. In addition, participants were asked about stroke or tumor disease in annual post-follow-up questionnaires. Participants were included into our study if they did not report those diseases until T2. As Parkinson’s disease, stroke, and tumor disease, or their specific treatments have negative influence on cognition, we decided to exclude those participants. Because of evidence of an association between blood KLK8 and inflammation [40], participants with laboratory evidence of inflammation (high-sensitivity C-reactive protein (hsCRP)≥1 mg/dl (reference value: 0.3 mg/dl) at T2) and participants with the above inflammatory conditions were also excluded from the analyses.
Measurement of KLK8
Details our blood KLK8 measurements and technical specifications of the utilized KLK8 enzyme-linked immunosorbent assay (ELISA) kit have been described previously [14]. In March 2021, KLK8 levels were measured in duplicate in T2 blood serum in the central reference laboratory at the Institute of Neuropathology, University of Duisburg-Essen, Germany, according to the manufacturer’s instructions, using a commercially available ELISA kit (#EK0819, Boster Biological Technology, Pleasanton, CA, USA). The samples were previously frozen between 6.9 and 9.9 years at –80°C. Kits from one batch were used and blood serum samples were diluted 1:2 in sample buffer. Two experienced technicians who were blinded to the participants’ diagnoses performed the measurements. The high agreement between the two measurements has been published previously [19].
Survey of cognitive performance
At T1 and T2 standardized cognitive performance assessments were performed, the details were published previously [41, 42] and further details on the validity of the assessment in identifying MCI cases can be found in Wege et al. [43]. Five subtests were performed at T1: measures of immediate and delayed verbal memory (eight word list [44]), speed of processing/problem solving (Labyrinth test [44]), verbal fluency (semantic category “animals” [45]), and visuo-spatial ability (clock-drawing test [46]). At T2 the following cognitive tests were added: Trail Making Test A (TMT A), Trail Making Test B (TMT B) [47], and a short version of the Stroop task named Color-word test [44] (card 1: word reading; card 2: color naming; card 3: color-word interference condition; card 3 minus card 2: interference performance [48]).
The administered tests at T2 were grouped into four domains: 1) attention –TMT A, Color-word test card 1 and card 2; 2) executive function –TMT B, Labyrinth test, Color-word test interference performance, verbal fluency; 3) verbal memory –eight word list immediate and delayed recall; 4) visuoconstruction –clock-drawing test [42]. To assess whether cognitive impairment was present, a z-transformation of the raw data was performed, as described in detail in [42, 49]. Cognitive impairment at T2 was defined as a performance of more than one SD below the mean in at least one total domain score of the domains attention, executive function, verbal memory, or as a score of≥3 in visuoconstruction [42, 46].
Dementia diagnosis was defined as follows: There was a previous medical diagnosis of dementia; dementia diagnosis criteria were met according to the Diagnostic and statistical manual of mental disorders 4th Edition (DSM-IV) [50], participants reported intake of cholinesterase inhibitors (code: N06DA according to the anatomic-therapeutic-chemical classification issued by the World Health Organization (WHO) [51]), or the intake of other anti-dementia drugs (code: N06DX). Six participants had dementia at T1 and three at T2, so they were excluded (see Fig. 1).
Definition of cases and controls
Cases were participants with a naMCI diagnosis based on meeting all of the following published Winblad et al. naMCI criteria [33]: 1) cognitive impairment in a non-memory domain (attention, executive function, visuoconstruction); 2) subjective cognitive decline; 3) normal functional abilities of daily activities; 4) no dementia diagnosis (definition see above). To examine only incident naMCI at T2, participants with MCI, dementia, or participants with objective impairment without SCD at T1 were excluded.
The cognitive performance of controls was within the age- and education-adjusted range in all domains, and they did not report SCD at T1 or T2. Each case is assigned a control matched for age (±3 years) and sex.
Survey of Apolipoprotein E ɛ4 status
Cardio-MetaboChip BeadArrays were used for genotyping of two single nucleotide polymorphisms (rs7412 and rs429358) to distinguish between the APOE ɛ2, ɛ3, and ɛ4 alleles. Participants who had at least one allele 4 (2/4, 3/4, 4/4) were defined as APOE ɛ4 positive, and all other participants were defined as APOE ɛ4 negative [42].
Survey of covariates
As reported in Schramm et al. [19] covariates were defined as follows: Height and weight were measured in a standardized manner by trained study nurses and body mass index (BMI) in kg/m2 was calculated. Education until T2 was classified as total years of formal education, combining school and vocational training according to the International Standard Classification of Education (ISCED-97) [52]. Participants were categorized as “current smoking” if they answered yes to the smoking questions at T2. ‘Past smoking’ was defined as quitted smoking, otherwise the participants were categorized into ‘never smoking’. When participants practiced one or more sports in the last 4 weeks prior to the interview sports at T2 was defined as ‘yes’, otherwise ‘no’. According to the Joint National Committee (JNC) 7 guidelines [53], we defined blood pressure categories: ‘Hypertension yes’ was defined if stage 1 or stage 2 hypertension was present, otherwise ‘no’. If participants reported diabetes mellitus disease or were taking antidiabetic medication or had an elevated fasting blood glucose level of≥200 mg/dl or a nonfasting blood glucose level of≥125 mg/dl at time T2, we defined “diabetes mellitus” as present. Depression at T2 was defined as present at cut-off point for “elevated depressive symptoms” of≥18 [54], using the German 15-item short form of the Center for Epidemiologic Studies Depression Scale (CES-D). Blood freezing duration in years was defined as the time between blood collection at T2 and KLK8 measurement in 2021.
Statistical analysis
To perform our nested case-control study each naMCI case is assigned a CU control matched for age (±3 years) and sex. The number of subjects with incident naMCI from T1 to T2 in this population-based study with n = 75 is predetermined. However, minimal detectable effect size estimates can be calculated for the association between exposure and outcome. With a sample size of 75 naMCI cases, a case-control ratio of 1:1, a standard deviation for the predictor blood KLK8 of 630 pg/ml, a statistical power of 80% and an alpha level of 0.05 with a two-sided test, the minimal detectable odds ratio obtained by using a logistic regression model is 1.58 per standard deviation of blood KLK8. Thus, sample size of the proposed study is sufficient to detect even moderate associations between blood KLK8 and naMCI.
Descriptive statistics were performed. p-values were estimated with Wilcoxon two-sample test, 2-sided, (continuous variables, not normally distributed) or chi-square test (nominal variables) to compare cases and controls. A box and scatter plot was created to show the distribution of blood KLK8 according to cognitive status. The association between blood KLK8 and naMCI compared to CU was determined using conditional multiple logistic regression to estimate odds ratios (OR) and 95% confidence intervals (95% CI). Because of the small scale of blood KLK8 values, we divided the values by 500 to determine an OR per 500 pg/ml increase. We adjusted for freezing duration in years because it has been previously reported that the freezing time might affect KLK8 levels [14], and inter-experimenter variability which should be understood as a proxy for the inter-assay variability and hereinafter referred to as ‘inter-assay variability’. Analyses were performed using the SAS 9.4 program (Statistical Analysis System Corp., Cary, NC, USA).
RESULTS
We measured blood KLK8 at T2 in 75 cases with incident naMCI and free of the above-mentioned diseases. At T1 and T2, 526 participants were CU and free of the above-mentioned diseases. N = 75 controls with frozen blood samples at T2 were matched for age±3 years and sex (Fig. 1). Two naMCI cases had no frozen blood, in 17 cases blood KLK8 was below and in one case blood KLK8 was beyond the detection threshold (resulting in 55 cases), in nine controls blood KLK8 was below the detection threshold (resulting in 66 controls).
In Table 1, the characteristics of the study population according to the cognitive status are presented. Out of 121 participants, the mean age was 70.5±7.1 years, with an age range of 56 to 85 years, and 54.5% were women. The 55 participants with incident naMCI had a slightly higher blood KLK8 mean value compared to the 66 CU participants (922±797 pg/ml versus 884±782 pg/ml). Participants with naMCI had lower z scores in all four domains. According to the cut-off (CES-D≥18) 7.3% of the participants with naMCI had depression, CU were free of depression. At T1, five participants with incident naMCI and one CU participant had depression (data not shown). There were no major differences between naMCI cases and controls regarding the other variables (age, BMI, education, smoking status, sports, APOE ɛ4 status, hypertension, diabetes mellitus, and freezing duration).
Table 1
Characteristics of the study population at T2, n (%), mean±SD
| naMCI | CU | Total | p** | |
| n = 55 | n = 66 | n = 121 | ||
| KLK8, pg/ml | 922±797 | 884±782 | 901±786 | 0.66 |
| Sex | 0.71 | |||
| Men | 24 (43.6) | 31 (47.0) | 55 (45.5) | |
| Women | 31 (56.4) | 35 (53.0) | 66 (54.5) | |
| Age, y | 70.5±7.3 | 70.5±6.9 | 70.5±7.1 | 0.98 |
| BMI, kg/m2 | 27.4±5.6 | 27.7±4.4 | 27.6±5.0 | 0.48 |
| Missing | 1 | 1 | ||
| Years of education | 14.7±2.3 | 14.5±2.4 | 14.5±2.4 | 0.65 |
| Smoking status | 0.94 | |||
| Never | 25 (45.5) | 32 (48.5) | 57 (47.1) | |
| Past | 25 (45.5) | 28 (42.4) | 53 (43.8) | |
| Current | 5 (9.1) | 6 (9.1) | 11 (9.1) | |
| Sports | 0.23 | |||
| No | 26 (47.3) | 24 (36.4) | 50 (41.3) | |
| Yes | 29 (52.7) | 42 (63.6) | 71 (58.7) | |
| APOE ɛ4 positive | 0.97 | |||
| No | 40 (72.7) | 50 (75.8) | 90 (74.4) | |
| Yes | 13 (23.6) | 16 (24.2) | 29 (24.0) | |
| Missing | 2 (3.6) | 2 (1.7) | ||
| Hypertension | 0.88 | |||
| No | 41 (74.5) | 50 (75.8) | 91 (75.2) | |
| Yes | 14 (25.5) | 16 (24.2) | 30 (24.8) | |
| Diabetes mellitus | 0.10 | |||
| No | 38 (69.1) | 54 (81.8) | 92 (76.0) | |
| Yes | 17 (30.9) | 12 (18.2) | 29 (24.0) | |
| Z score attention | 8.9±3.0 | 11.5±1.9 | 10.3±2.8 | <0.01 |
| Z score executive function | 9.1±3.1 | 11.5±1.9 | 10.4±2.8 | <0.01 |
| Z score verbal memory | 10.5±2.2 | 11.4±2.0 | 11.0±2.1 | 0.01 |
| Z score visuoconstruction | 8.2±3.1 | 11.6±1.8 | 10.0±3.0 | <0.01 |
| Depression* | 0.03 | |||
| No | 50 (90.9) | 64 (97.0) | 114 (94.2) | |
| Yes | 4 (7.3) | 0 | 4 (3.3) | |
| Missing | 1 (1.8) | 2 (3.0) | 3 (2.5) | |
| Freezing duration, y | 8.6±0.8 | 8.5±0.6 | 8.5±0.7 | 0.49 |
*Elevated depressive symptoms (CES-D≥18). **p-values were estimated with Wilcoxon two sample test or chi-square test. APOE ɛ4, Apolipoprotein E ɛ4; BMI, body mass index; CU, cognitively unimpaired; KLK8, kallikrein-8; naMCI, non-amnestic mild cognitive impairment; SD, standard deviation; T2, 10-year follow-up.
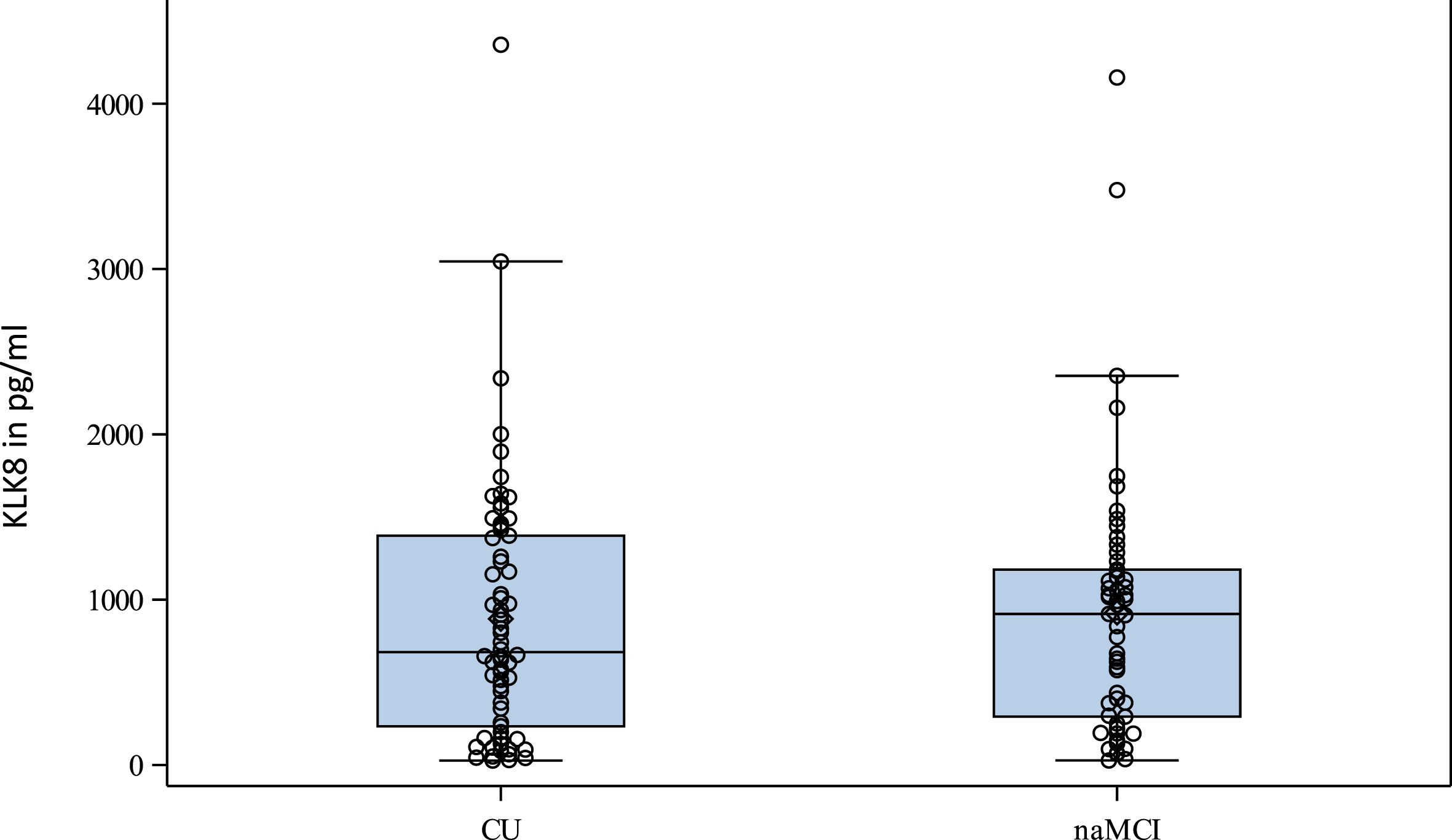
The distribution of blood KLK8 values according to the cognitive status are also shown in the box and scatter plot in Fig. 2. The distribution of blood KLK8 values is similar in participants with naMCI and CU.
Fig. 2
Distribution of KLK8 in pg/ml at T2 according to cognitive status. CU, cognitively unimpaired; KLK8, kallikrein-8; naMCI, non-amnestic mild cognitive impairment; T2, second follow-up visit.
In Table 2, the results of the conditional logistic regression analyses to estimate the association between blood KLK8 and cognitive status are presented. Crude and fully adjusted, blood KLK8 (per 500 pg/ml increase) was not associated with a higher chance of having naMCI compared to being CU (crude: OR: 1.01 [95% CI: 0.79–1.29], adjusted: 1.03 [0.80–1.32]. Even considering the upper confidence point of 1.3, the OR would be moderately elevated at best. When excluding participants with depression (CES-D≥18) at T2 from our analyses, our results on the association of blood KLK8 and naMCI did not change (1.01 [0.79–1.30]). We have reported previously that freezing duration affects KLK8 levels [14]. Therefore, we consider the freezing duration in our model. However, in contrast to our previous study, the freezing duration in the current study is very homogeneous between 6.9 and 9.9 years and showed no influence on the results (fully adjusted OR for freezing duration:1.01 [95% CI: 0.96; 1.06]).
Table 2
Association between KLK8 and cognitive status (naMCI versus CU) at T2
| OR [95% CI] | Model 1 | Model 2 |
| Per 500 pg/ml KLK8 | 1.01 [0.79–1.29] | 1.03 [0.80–1.32] |
| Experimenter 1 versus 2* | 0.59 [0.26–1.34] | |
| Freezing duration, y | 1.01 [0.96–1.06] |
Model 1: crude. Model 2: adjusted for freezing duration and experimenter. *We adjusted for experimenter as a proxy for the inter-assay variability. CU, cognitively unimpaired; naMCI, non-amnestic mild cognitive impairment; OR, odds ratio; 95% CI, 95% confidence interval; KLK8, kallikrein-8.
DISCUSSION
Previous studies of our laboratory have shown that blood KLK8 may be suitable as a biomarker for the diagnosis of aMCI and early AD [14, 19]. This is the first population-based study to show that blood KLK8 is not elevated in persons with naMCI, a precursor of non-AD type dementia, compared to CU. A blood KLK8 increase of 500 pg/ml was not associated with a higher chance of having naMCI compared to being CU. This highlights the blood KLK8 specificity for AD. Our finding that blood KLK8 is not elevated in individuals with naMCI compared with CU is another building block of evidence that blood KLK8 may be an early AD specific biomarker. The absence of an association between blood KLK8 and naMCI, which is considered a prodromal stage of non-AD type dementias [33, 35–37], is a necessary prerequisite for the suitability of blood KLK8 as an AD-specific biomarker. As the field of KLK8 and dementia research is still emerging, there is only one study available regarding blood KLK8 and non-AD type dementias. Li et al. [32] showed blood KLK8 levels to be higher in patients with vascular dementia and to be inversely correlated with the cognitive score of vascular dementia patients. If we assume that naMCI has a high probability of progression to vascular dementia, the results of Li et al. appear contrary to our findings. However, AD and vascular dementia often co-occur. While we excluded individuals with vascular dementia from the AD collective in our previous analyses [14, 18], Li et al. did not exclude patients with AD in their vascular dementia collective. Therefore, it is quite possible that the increase in KLK8 in their collective was due to co-occurring AD. Further research is needed regarding the role of blood KLK8 and other non-AD causes of dementia to support the specificity of blood KLK8for AD.
Strengths and limitations
Strengths and limitations of our study are as follows: Strengths are that the cases and controls are from a large population-based study. The high quality of data collection and processing in this study was confirmed by external certification. All cases and controls were free of severe diseases (like stroke and cancer) and had normal inflammatory parameters. Blood collection was performed in a central reference laboratory, and the storage period of the samples was similar. Furthermore, cases and controls were matched for sex and age. The major limitation is that our naMCI diagnosis was based on cognitive testing only, with no amyloid, tau, p-tau, or neurodegeneration biomarkers available. Therefore, we cannot rule out whether our naMCI participants are truly not on the ‘Alzheimer’s continuum’. Another limitation is the size of the population analyzed. Our study was primarily designed as a cardiovascular health study of myocardial infarction with long follow-up periods between examinations (every 5 years) and a high proportion of young participants (45–75 years at baseline). Therefore, the number of subjects with naMCI at study time T2, ten years after baseline, is not particularly high, and it is not yet clear how cognition has or will develop in these participants over time. Annual postal follow-up of these participants after T2 will continue to take place. Currently, data are being compiled from follow-up questionnaires and medical reports, and our dementia endpoint committee is identifying subjects that developed dementia in the further course of the study. Another limitation is that the measurement of blood KLK8 is not yet established for routine testing, and there are no established normal values of blood KLK8. Studies of blood KLK8 in large population-based collectives do not exist. In our study, however, the aim is not to establish or compare possible cut-offs, but first to clarify whether there is an effect at all or whether there is no effect. Last, it should also be noted that because of the possible different sources of KLK8 as described in the introduction, the major source of the KLK8 we measured in blood is not known.
Conclusion
Our exploratory study is the first population-based case-control study to show that blood KLK8 is not likely to be a biomarker for naMCI, which is considered to be a precursor for non-AD type dementia. In our study, blood KLK8 is not elevated in persons with naMCI compared with CU. As blood KLK8 is supposed to be an early biomarker for AD, this highlights its specificity for AD. However, further studies on this comparing more groups like CU, aMCI, naMCI, AD, vascular dementia, other dementia, and other neurodegenerative diseases are urgently needed.
ACKNOWLEDGMENTS
The authors express their gratitude to all study participants of the HNR study, the personnel of the HNR study center and the EBT-scanner facilities, the investigative group and all present and former employees of the HNR study. The authors also thank the Advisory Board of the HNR Study: T. Meinertz, Hamburg, Germany (Chair); C. Bode, Freiburg, Germany; P.J. de Feyter, Rotterdam, Netherlands; B. Güntert, Hall i.T., Austria; F. Gutzwiller, Bern, Switzerland; H. Heinen, Bonn, Germany; O. Hess, Bern, Switzerland; B. Klein, Essen, Germany; H. Löwel, Neuherberg, Germany; M. Reiser, Munich, Germany; G. Schmidt, Essen, Germany; M. Schwaiger, Munich, Germany; C. Steinmüller, Bonn, Germany; T. Theorell, Stockholm, Sweden; and S.N Willich, Berlin, Germany. The authors also thank U. Slomiany and Y. Hanel for technical support.
FUNDING
This subproject of the HNR study was not funded. The HNR study was supported by the Heinz Nixdorf Foundation [Chairman: Martin Nixdorf; Past Chairman: Dr jur. Gerhard Schmidt]. Parts of the HNR study were also supported by the German Research Council (DFG) [DFG project: EI 969/2-3, ER 155/6-1;6-2, HO 3314/2-1;2-2;2-3;4-3, INST 58219/32-1, JO 170/8-1, KN 885/3-1, PE 2309/2-1, SI 236/8-1;9-1;10-1,], the German Ministry of Education and Science [BMBF project: 01EG0401, 01GI0856, 01GI0860, 01GS0820_WB2-C, 01ER1001D, 01GI0205], the Ministry of Innovation, Science, Research and Technology, North Rhine-Westphalia (MIWFT-NRW), the Else Kröner-Fresenius-Stiftung [project: 2015_A119] and the German Social Accident Insurance [DGUV project: FF-FP295]. Furthermore the HNR study was supported by the Competence Network for HIV/AIDS, the deanship of the University Hospital and IFORES of the University Duisburg-Essen, the European Union, the German Competence Network Heart Failure, Kulturstiftung Essen, the Protein Research Unit within Europe (PURE), the Dr. Werner-Jackstädt Stiftung and the following companies: Celgene GmbH München, Imatron/GE-Imatron, Janssen, Merck KG, Philips, ResMed Foundation, Roche Diagnostics, Sarstedt AG&Co, Siemens HealthCare Diagnostics, Volkswagen Foundation.
CONFLICT OF INTEREST
SS, NK, UR, KHJ, and MJ have no competing interests.
AH and KK are inventors on the patent “Agents inhibiting Kallikrein-8 for use in the prevention or treatment of Alzheimer’s Disease”; patent no. 3353214, issued by The European Patent Office on 22/07/2021.
DATA AVAILABILITY
Data from the HNR study cannot be shared as a publicly accessible file for data security reasons. However, data requests can be sent to . The corresponding author has full access to all study data and has final responsibility for submission of the article for publication.
REFERENCES
[1] | ((2020) ) 2020 Alzheimer’s disease facts and figures, Alzheimers Dement 16: , 391–460. |
[2] | Jack CR Jr , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J , Liu E , Molinuevo JL , Montine T , Phelps C , Rankin KP , Rowe CC , Scheltens P , Siemers E , Snyder HM , Sperling R , Contributors ((2018) ) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease, Alzheimers Dement 14: , 535–562. |
[3] | Ritchie C , Smailagic N , Noel-Storr AH , Takwoingi Y , Flicker L , Mason SE , McShane R ((2014) ) Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI).CD, Cochrane Database Syst Rev 2014: , 008782. |
[4] | Ritchie C , Smailagic N , Noel-Storr AH , Ukoumunne O , Ladds EC , Martin S ((2017) ) CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI).CD, Cochrane Database Syst Rev 2017: , 010803. |
[5] | Nokkari A , Abou-El-Hassan H , Mechref Y , Mondello S , Kindy MS , Jaffa AA , Kobeissy F ((2018) ) Implication of the Kallikrein-Kinin system in neurological disorders: Quest for potential biomarkers and mechanisms, Prog Neurobiol 165: , 26–50. |
[6] | Yamamoto-Imoto H , Zamolodchikov D , Chen ZL , Bourne SL , Rizvi S , Singh P , Norris EH , Weis-Garcia F , Strickland S ((2018) ) A novel detection method of cleaved plasma high-molecular-weight kininogen reveals its correlation with Alzheimer’s pathology and cognitive impairment, Alzheimers Dement (Amst) 10: , 480–489. |
[7] | Singh PK , Chen ZL , Ghosh D , Strickland S , Norris EH ((2020) ) Increased plasma bradykinin level is associated with cognitive impairment in Alzheimer’s patients, Neurobiol Dis 139: , 104833. |
[8] | Lacoste B , Tong XK , Lahjouji K , Couture R , Hamel E ((2013) ) Cognitive and cerebrovascular improvements following kinin B1 receptor blockade in Alzheimer’s disease mice, J Neuroinflammation 10: , 57. |
[9] | Yousef GM , Diamandis EP ((2001) ) The new human tissue kallikrein gene family: Structure, function, and association to disease, Endocr Rev 22: , 184–204. |
[10] | Mella C , Figueroa CD , Otth C , Ehrenfeld P ((2020) ) Involvement of Kallikrein-related peptidases in nervous system disorders, Front Cell Neurosci 14: , 166. |
[11] | Kishi T , Cloutier SM , Kundig C , Deperthes D , Diamandis EP ((2006) ) Activation and enzymatic characterization of recombinant human kallikrein 8, Biol Chem 387: , 723–731. |
[12] | Shiosaka S ((2022) ) Kallikrein 8: A key sheddase to strengthen and stabilize neural plasticity, Neurosci Biobehav Rev 140: , 104774. |
[13] | Kishi T , Grass L , Soosaipillai A , Shimizu-Okabe C , Diamandis EP ((2003) ) Human kallikrein 8: Immunoassay development and identification in tissue extracts and biological fluids, Clin Chem 49: , 87–96. |
[14] | Teuber-Hanselmann S , Rekowski J , Vogelgsang J , von Arnim C , Reetz K , Stang A , Jockel KH , Wiltfang J , Esselmann H , Otto M , Tumani H , Herring A , Keyvani K ((2020) ) CSF and blood Kallikrein-8: A promising early biomarker for Alzheimer’s disease, J Neurol Neurosurg Psychiatry 91: , 40–48. |
[15] | Keyvani K , Munster Y , Kurapati NK , Rubach S , Schonborn A , Kocakavuk E , Karout M , Hammesfahr P , Wang YC , Hermann DM , Teuber-Hanselmann S , Herring A ((2018) ) Higher levels of kallikrein-8 in female brain may increase the risk for Alzheimer’s disease, Brain Pathol 28: , 947–964. |
[16] | Munster Y , Keyvani K , Herring A ((2020) ) Inhibition of excessivekallikrein-8 improves neuroplasticity in Alzheimer’s disease mousemodel, Exp Neurol 324: , 113115. |
[17] | Herring A , Kurapati NK , Krebs S , Grammon N , Scholz LM , Voss G , Miah MR , Budny V , Mairinger F , Haase K , Teuber-Hanselmann S , Dobersalske C , Schramm S , Jockel KH , Munster Y , Keyvani K ((2021) ) Genetic knockdown of Klk8 has sex-specific multi-targeted therapeutic effects on Alzheimer’s pathology in mice, Neuropathol Appl Neurobiol 47: , 611–624. |
[18] | Herring A , Munster Y , Akkaya T , Moghaddam S , Deinsberger K , Meyer J , Zahel J , Sanchez-Mendoza E , Wang Y , Hermann DM , Arzberger T , Teuber-Hanselmann S , Keyvani K ((2016) ) Kallikrein-8 inhibition attenuates Alzheimer’s disease pathology in mice, Alzheimers Dement 12: , 1273–1287. |
[19] | Schramm S , Jokisch M , Jockel KH , Herring A , Keyvani K ((2021) ) Is kallikrein-8 a blood biomarker for detecting amnestic mild cognitive impairment? Results of the population-based Heinz Nixdorf Recall study, Alzheimers Res Ther 13: , 202. |
[20] | Tamura H , Ishikawa Y , Hino N , Maeda M , Yoshida S , Kaku S , Shiosaka S ((2006) ) Neuropsin is essential for early processes of memory acquisition and Schaffer collateral long-term potentiation in adult mouse hippocampus}, J Physiol 570: , 541–551. |
[21] | Ishikawa Y , Horii Y , Tamura H , Shiosaka S ((2008) ) Neuropsin (KLK8)-dependent and -independent synaptic tagging in the Schaffer-collateral pathway of mouse hippocampus, J Neurosci 28: , 843–849. |
[22] | Kayser MS , Nolt MJ , Dalva MB ((2008) ) EphB receptors couple dendritic filopodia motility to synapse formation, Neuron 59: , 56–69. |
[23] | Konar A , Thakur MK ((2015) ) Neuropsin expression correlates with dendritic marker MAP2c level in different brain regions of aging mice, Mol Neurobiol 51: , 1130–1138. |
[24] | Srivastava N , Robichaux MA , Chenaux G , Henkemeyer M , Cowan CW ((2013) ) EphB2 receptor forward signaling controls cortical growth cone collapse via Nck and Pak, Mol Cell Neurosci 52: , 106–116. |
[25] | Robichaux MA , Chenaux G , Ho HY , Soskis MJ , Dravis C , Kwan KY , Sestan N , Greenberg ME , Henkemeyer M , Cowan CW ((2014) ) EphB receptor forward signaling regulates area-specific reciprocal thalamic and cortical axon pathfinding, Proc Natl Acad Sci U S A 111: , 2188–2193. |
[26] | Mao YT , Zhu JX , Hanamura K , Iurilli G , Datta SR , Dalva MB ((2018) ) Filopodia conduct target selection in cortical neurons using differences in signal kinetics of a single kinase, Neuron 98: , 767–782.e8. |
[27] | Tamura H , Kawata M , Hamaguchi S , Ishikawa Y , Shiosaka S ((2012) ) Processing of neuregulin-1 by neuropsin regulates GABAergic neuron to control neural plasticity of the mouse hippocampus, J Neurosci 32: , 12657–12672. |
[28] | Matsumoto-Miyai K , Ninomiya A , Yamasaki H , Tamura H , Nakamura Y , Shiosaka S ((2003) ) NMDA-dependent proteolysis of presynaptic adhesion molecule L1 in the hippocampus by neuropsin, J Neurosci 23: , 7727–7736. |
[29] | Shimizu C , Yoshida S , Shibata M , Kato K , Momota Y , Matsumoto K , Shiosaka T , Midorikawa R , Kamachi T , Kawabe A , Shiosaka S ((1998) ) Characterization of recombinant and brain neuropsin, a plasticity-related serine protease, J Biol Chem 273: , 11189–11196. |
[30] | Attwood BK , Bourgognon JM , Patel S , Mucha M , Schiavon E , Skrzypiec AE , Young KW , Shiosaka S , Korostynski M , Piechota M , Przewlocki R , Pawlak R ((2011) ) Neuropsin cleaves EphB2 in the amygdala to control anxiety, Nature 473: , 372–375. |
[31] | Shimizu-Okabe C , Yousef GM , Diamandis EP , Yoshida S , Shiosaka S , Fahnestock M ((2001) ) Expression of the kallikrein gene family in normal and Alzheimer’s disease brain, Neuroreport 12: , 2747–2751. |
[32] | Li J , Li SL , Song YH , Li ZP , Wang N , Zhang GH , Zhu CL ((2021) ) The association of serum Kallikrein-8 with cognitive function in vascular dementia, Eur Rev Med Pharmacol Sci 25: , 1997–2002. |
[33] | Winblad B , Palmer K , Kivipelto M , Jelic V , Fratiglioni L , Wahlund LO , Nordberg A , Backman L , Albert M , Almkvist O , Arai H , Basun H , Blennow K , de Leon M , DeCarli C , Erkinjuntti T , Giacobini E , Graff C , Hardy J , Jack C , Jorm A , Ritchie K , van Duijn C , Visser P , Petersen RC ((2004) ) Mild cognitive impairment–beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment, J Intern Med 256: , 240–246. |
[34] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease, Alzheimers Dement 7: , 270–279. |
[35] | Busse A , Hensel A , Guhne U , Angermeyer MC , Riedel-Heller SG ((2006) ) Mild cognitive impairment: Long-term course of four clinical subtypes, Neurology 67: , 2176–2185. |
[36] | Petersen RC , Doody R , Kurz A , Mohs RC , Morris JC , Rabins PV , Ritchie K , Rossor M , Thal L , Winblad B ((2001) ) Current concepts in mild cognitive impairment, Arch Neurol 58: , 1985–1992. |
[37] | Petersen RC ((2004) ) Mild cognitive impairment as a diagnostic entity, J Intern Med 256: , 183–194. |
[38] | Schmermund A , Möhlenkamp S , Stang A , Grönemeyer D , Seibel R , Hirche H , Mann K , Siffert W , Lauterbach K , Siegrist J , Jöckel KH , Erbel R ((2002) ) Assessment of clinically silent atherosclerotic disease and established and novel risk factors for predicting myocardial infarction and cardiac death in healthy middle-aged subjects: Rationale and design of the Heinz Nixdorf RECALL Study. Risk factors, evaluation of coronary calcium and lifestyle, Am Heart J 144: , 212–218. |
[39] | Stang A , Moebus S , Dragano N , Beck EM , Möhlenkamp S , Schmermund A , Siegrist J , Erbel R , Jöckel KH; Heinz Nixdorf Recall Study Investigation Group ((2005) ) Baseline recruitment and analyses of nonresponse of the Heinz Nixdorf Recall Study: Identifiability of phone numbers as the major determinant of response, Eur J Epidemiol 20: , 489–496. |
[40] | Stief TW ((2008) ) Kallikrein activates prothrombin, Clin Appl Thromb Hemost 14: , 97–98. |
[41] | Winkler A , Dlugaj M , Weimar C , Jockel KH , Erbel R , Dragano N , Moebus S ((2014) ) Association of diabetes mellitus and mild cognitive impairment in middle-aged men and women, J Alzheimers Dis 42: , 1269–1277. |
[42] | Muller-Gerards D , Weimar C , Abramowski J , Tebrugge S , Jokisch M , Dragano N , Erbel R , Jockel KH , Moebus S , Winkler A , Heinz Nixdorf Recall Study Investigative Group ((2019) ) Subjective cognitive decline, APOE epsilon4, and incident mild cognitive impairment in men and women, Alzheimers Dement (Amst) 11: , 221–230. |
[43] | Wege N , Dlugaj M , Siegrist J , Dragano N , Erbel R , Jockel KH , Moebus S , Weimar C , Heinz Nixdorf Recall Study Investigative Group ((2011) ) Population-based distribution and psychometric properties of a short cognitive performance measure in the population-based Heinz Nixdorf Recall Study, Neuroepidemiology 37: , 13–20. |
[44] | Oswald WD , Fleischmann UM (1994) Nürnberger Alters-Inventar (NAI). Hogrefe Verlag für Psychologie, Göttingen. |
[45] | Aschenbrenner S , Tucha O , Lange KW (2000) Regens-burger Wortflüssigkeits-Test (RWT). Hogrefe Verlag für Psychologie, Göttingen. |
[46] | Shulman KI ((2000) ) Clock-drawing: Is it the ideal cognitive screening test? Int J Geriatr Psychiatry 15: , 548–561. |
[47] | Tombaugh TN ((2004) ) Trail making test A and B: Normative data stratified by age and education, Arch Clin Neuropsychol 19: , 203–214. |
[48] | Stroop JR ((1935) ) Studies of interference in serial verbal reactions, J Exp Psychol 18: , 643–662. |
[49] | Ivnik RJ (1992)WAIS-R, WMS-R, and AVLT Norms for Ages 56 Through 97. Swets, The Netherlands. |
[50] | American Psychiatric Association ((1994) ) Diagnostic and statistical manual of mental disorders (DSM-IV), 4th Edition. American Psychiatric Association,Washington, DC. |
[51] | World Health Organization (2011) The 798 Selection and Use of Essential Medicines, Report of the WHO Expert Committee. |
[52] | UNESCO (1997) International standard classification ofeducation (ISCED). |
[53] | Joint National Committee (JNC) 7 guidelines, https://www.nhlbi.nih.gov/files/docs/guidelines/express.pdf,2003, accessed December 1, 2021. |
[54] | Hautzinger M , Bailer M , Hofmeister D , Keller F (1993) Allgemeine Depressions-Skala: ADS; Manual. Beltz-Test-GmbH, Göttingen. |


