
Updated Outlook of Cerebral Amyloid Angiopathy and Inflammatory Subtypes: Pathophysiology, Clinical Manifestations, Diagnosis and Management
Abstract
Cerebral amyloid angiopathy (CAA) is a common untreatable cause of lobar hemorrhages and cognitive decline in the older population. Subset of patients present with its inflammatory subtype with rapid decline in cognitive functions and neurological deficits. Most commonly the underlying pathophysiology of this disease is deposition of insoluble amyloid protein into blood vessel walls which results in vessel fragility leading to local neurotoxicity which may eventually leads to lobar hemorrhages and cognitive decline. The term “Amyloid Spell” encompasses transient focal neurological deficits which is commonly misdiagnosed as seizures or transient ischemic attack in the emergency department. Radiologic findings in these patients may reveal microbleeds, cortical superficial siderosis, white matter hyperintensities, and cerebral edema which support the clinical diagnosis which could be otherwise challenging. CAA diagnostic criteria require CT (Edinburgh Criteria) or MRI imaging, or neuropathology. The diagnosis can be suspected without imaging or neuropathology but cannot be confirmed. This review article provides a critical outlook on different types of presentations, updated diagnostic criteria and management of CAA patients illustrating underlying mechanisms associated with neuronal injury secondary to amyloid deposition.
INTRODUCTION
Amyloidosis is defined as abnormal folding of soluble proteins into insoluble fibrillar protein forms associating as ʲ-pleated sheets, a unique molecular conformation which is resistant to proteolytic degradation. The irreversible insoluble protein state and are responsible for binding affinity to Congo red stain. The main composition of amyloid protein responsible for insoluble properties are serum amyloid-ʲ protein precursor (AʲPP) and apolipoprotein E. The extracellular deposition of insoluble amyloid protein can be localized or systemic, which can be toxic to local cells. Amyloid-ʲ protein (Aʲ) is derived from AʲPP and is localized to the central nervous system (CNS), its wild type is acquired, and its variant is hereditary [1]. Local deposition of Aʲ into small to medium vessels of CNS and leptomeninges can lead to cerebral amyloid angiopathy (CAA), which is an untreatable disease mostly of the older population known for lobar cerebral hemorrhages and cognitive decline [2]. CAA is the second leading cause of spontaneous intracerebral hemorrhage (ICH) after hypertension [3]. Another long-term serious manifestation of this disease is disabling cognitive decline. Cerebral amyloid deposition is considered the overlapping cause of Alzheimer’s disease (AD) and cerebrovascular lesions [4]. AD primarily has amyloid deposition in the brain parenchyma in comparison to CAA, where amyloid deposition particularly involves vessel walls. The deposition of amyloid protein in aging brain vessels makes them fragile which contributes to microhemorrhages. CAA can also have acute on chronic presentation in a subset of patients with cerebral amyloid angiopathy-related inflammation (CAARI), or they may present with amyloid spells mimicking transient ischemic attack. Management of each patient depends on the severity and type of symptoms. The purpose of writing this updated review is to discuss cerebral amyloid angiopathy and its various types of presentation in the emergency department.
EPIDEMIOLOGICAL FEATURES
Incidence
Amyloid protein deposition is a common finding in the aging brain, and its incidence increases with advancing age. Block et al. reported the prevalence of CAA around 30% in the sixth decade, which increases to 50% by the seventh decade [5]. Due to the chronic course of the disease, patients may be asymptomatic until their first presentation with lobar hemorrhage or amyloid spell. CAA-related ICH is seen in 3.8% to 20% of non-traumatic hemorrhages, which makes CAA the second leading cause of spontaneous ICH [4, 6]. Some studies reported amyloid deposition in 50% of lobar hemorrhage [7]. In general, ICH is more common in men, but CAA-related cerebral hemorrhages are more prevalent in women [4, 8, 9]. When compared to AD, CAA is less prevalent in patients without AD [10, 11].
Risk factors
Apolipoprotein E is a major cholesterol carrier and helps in lipid transportation. APOE and its polymorphic alleles are the major determinants of AD. Genetic predilection of APOE ɛ4 is a significant risk factor of CAA [10, 12]. APOE ɛ4 carriers present with more severe symptoms in comparison to non-ɛ4 carriers [10]. However, the presence of APOE ɛ2 is associated with increased risk of ICH [13].
Various other mutated proteins such as presenilin 1, α1-antichymotrypsin, neprilysin, low-density lipoprotein-receptor related protein, and angiotensin-converting enzyme genes are associated with CAA [10, 14–16]. These mutated proteins may interact with AʲPP to increase its enzymatic cleavage to form insoluble toxic ʲ-pleated amyloid fibrils thus unbalance between the production and clearance of these proteins lead to disease progression. CAA can be present in patients without a history of hypertension, hyperlipidemia, or diabetes mellitus, which probably indicates that it has almost no relation to classic vascular risk factors [10]. However, the presence of hypertension markedly amplifies the risk of hemorrhage, and strict control of arterial blood pressure in patients with probable CAA may reduce the risk of hemorrhage by 77% [5].
Table 1
Classification of cerebral amyloid proteins
| Type of amyloid protein | Clinical characteristic |
| CAA due to Aʲ peptide deposition | Sporadic: associated with advanced age; APOE ɛ4 Hereditary: missense mutations in APP gene; Italian (e693 k), arctic (e693 g), Iowa (d694 n), and piedmont (l705 v) variants; associated with Down syndrome [2]. |
| CAA due to mutated cystatin c in hereditary cerebral hemorrhage with amyloidosis of Icelandic-type (HCHWA-I) | Autosomal dominant: point mutation at codon 68 of the cystatin c gene located on chromosome 20, associated with fatal strokes in Icelandic patients with familial cerebral hemorrhage secondary to a form of autosomal dominant amyloidosis [3]. |
| CAA due to variant transthyretins | Mutations of the TTR gene, located on chromosome 18; most common neurological phenotype is familial amyloid sensorimotor polyneuropathy with or without associated autonomic neuropathy; associated with meningocerebrovascular amyloidosis, producing dementia, ataxia, and spasticity in Hungarian kindred [4, 5]. |
| CAA in human prion diseases | Mutations of stop codon 145 in PRNP gene; y145stop or the y163stop variants leads to loss of glycosylphosphatidylinositol (GPI) anchor. In normal cells, GPI interferes with the ability of PRP to form amyloid fibrils in the cerebrovascular system [2, 6]. |
| Gelsolin related familial amyloidosis of the Finnish type/Meretoja disease | G654a and the G654t mutations of the gelsolin gene located on chromosome 9; ophthalmological (lattice corneal dystrophy), dermatological and neurological symptoms and signs (mild generalized polyneuropathy as well as the involvement of vessels) [7]. |
| Hereditary CAA in familial British dementia (FBD) and familial Danish dementia (FDD)-bri2 gene related dementias | A point mutation (T to A) of the normal stop codon of the bri2 gene; mutated proteins deposit in the cerebrovascular system [8]. |
CLASSIFICATION OF CEREBRAL AMYLOID ANGIOPATHY
There are seven different types of amyloid proteins reported to be associated with CAA. The most widely known amyloid protein associated with CAA is Aʲ. Its wild type is seen in sporadic cases, and its variant form is considered hereditary. A missense mutation in the APP gene produces AʲPP variants. Different types of variants have been reported, such as Italian, Iowa, and Piedmont variants. However, there are various other protein depositions that have been associated with CAA, such as mutated Cystatin C which is seen in Icelandic patients with familial autosomal dominant hemorrhagic strokes. In Hungarian kindreds, mutated transthyretin protein is reported with familial meningo-cerebrovascular amyloidosis. Mutated prion protein produces mutated glycophosphatidylinositol anchor which forms an insoluble amyloid fibril in patients with prion disease.
In 1969, Finnish ophthalmologist Jouko Meretoja described a systemic disease known as Meretoja disease or familial gelsolin amyloidosis due to mutations of the gelsolin gene [17]. Gelsolin is a principal actin-modulating protein involved in axonal transport, myelination, and neuroprotection. Its mutated variant presents as CAA with corneal lattice dystrophy, cutis laxa, polyneuropathy, and facial palsy [18]. CAA is also linked to a mutation of the BRI-2 gene, which has been associated with hereditary conditions such as Familial British Dementia and Familial Danish Dementia, which presents with widespread amyloid deposition in the brain parenchyma and vessels [19]. In a Chinese prospective study, high serum levels of neurofilament light chain (NfL) were seen in patients with recurrent intraparenchymal hemorrhage secondary to CAA, and higher serum NfL levels were associated with severity and prognosis of CAA-related ICH. NfL are important for structural stability of axons and for achieving conduction of electrical impulses along the axons and might be released into cerebrospinal fluid (CSF) or blood during ongoing axonal damage. Higher than normal levels have been reported in many other conditions such as frontotemporal dementia, AD, and vascular dementia therefore elevated NfL are not used for diagnosis of any of these disorders (Table 1) [20].
PATHOPHYSIOLOGY
CAA involves deposition of insoluble Aʲ fibrils in the capillaries, arterioles, small to medium-sized cortical vessels and leptomeninges, and less commonly veins resulting in degenerative vascular changes [21, 22]. The imbalance between excessive production or impaired clearance of amyloid protein plays an important role in disease pathogenesis [22]. The exact origin of Aʲ in vessels is still not clear, but its origin from neural tissues and drainage into periarterial interstitial fluid has been proposed. Deposition of amyloid fibrils into periarterial spaces are associated with the disease process [23]. The process of amyloidogenic protein buildup within the brain parenchyma and vessel walls is an area of ongoing investigation.
The highly characteristic microscopic appearance of CAA shows acellular thickening of vessels by an amorphous, intensely eosinophilic material which gives a “smudged” appearance on the light microscope (Fig. 1) [24]. Amyloid deposition is commonly studied using Congo Red stain and viewing histologic sections under polarized light which gives apple-green birefringence [25]. Deposition of amyloid protein into cerebral vessels is a multistep process that includes infiltration of amyloid proteins into media and adventitia of vessels leading to effacement of the affected blood vessels with loss of smooth muscle cells [24].
Fig. 1
Amyloid special stain showing vascular amyloid deposition in a cortical leptomeningeal vessel with preservation of some vascular smooth muscle cells, corresponding to CAA grade 1. Note the vascular microaneurysm.
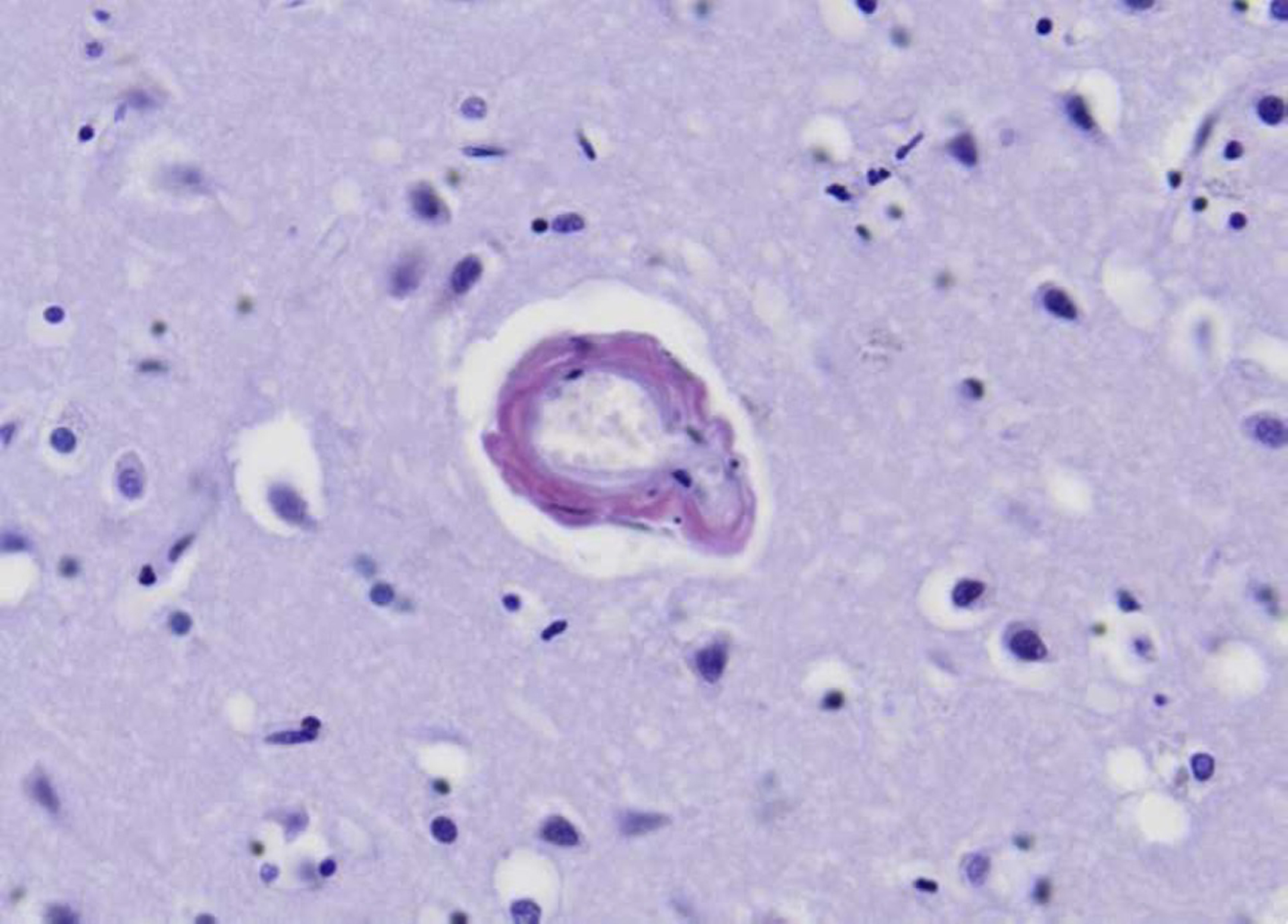
Deposition of insoluble proteins into the affected blood vessels makes them fragile and is responsible for vasculopathies which include fibrinoid necrosis with onion skin appearance of vessel wall, development of microaneurysms, hyaline degeneration, and perivascular infiltration of lymphocytes [26].
CAA can be graded into mild to severe forms based on histopathological features. 1) Mild: amyloid protein is restricted to congophilic rim around smooth muscle fibers. 2) Moderate: tunica media is thicker than normal and is replaced by amyloid protein with no microscopic evidence of leakage of blood. 3) Severe: extensive deposition of amyloid protein with focal wall fragmentation [27]. Fibrinoid necrosis is seen more commonly in moderate to severe forms of CAA and has a strong correlation with cerebral hemorrhage (Figs. 1–3) [27, 28].
Fig. 2
Leptomeningeal vascular biopsy in patient with suspected CAA showing complete replacement of vascular wall by homogenous eosinophilic amyloid categorized as CAA Grade 2. Confirmed by amyloid-ʲ immunohistochemistry (not shown). Hematoxylin and Eosin, 400x.
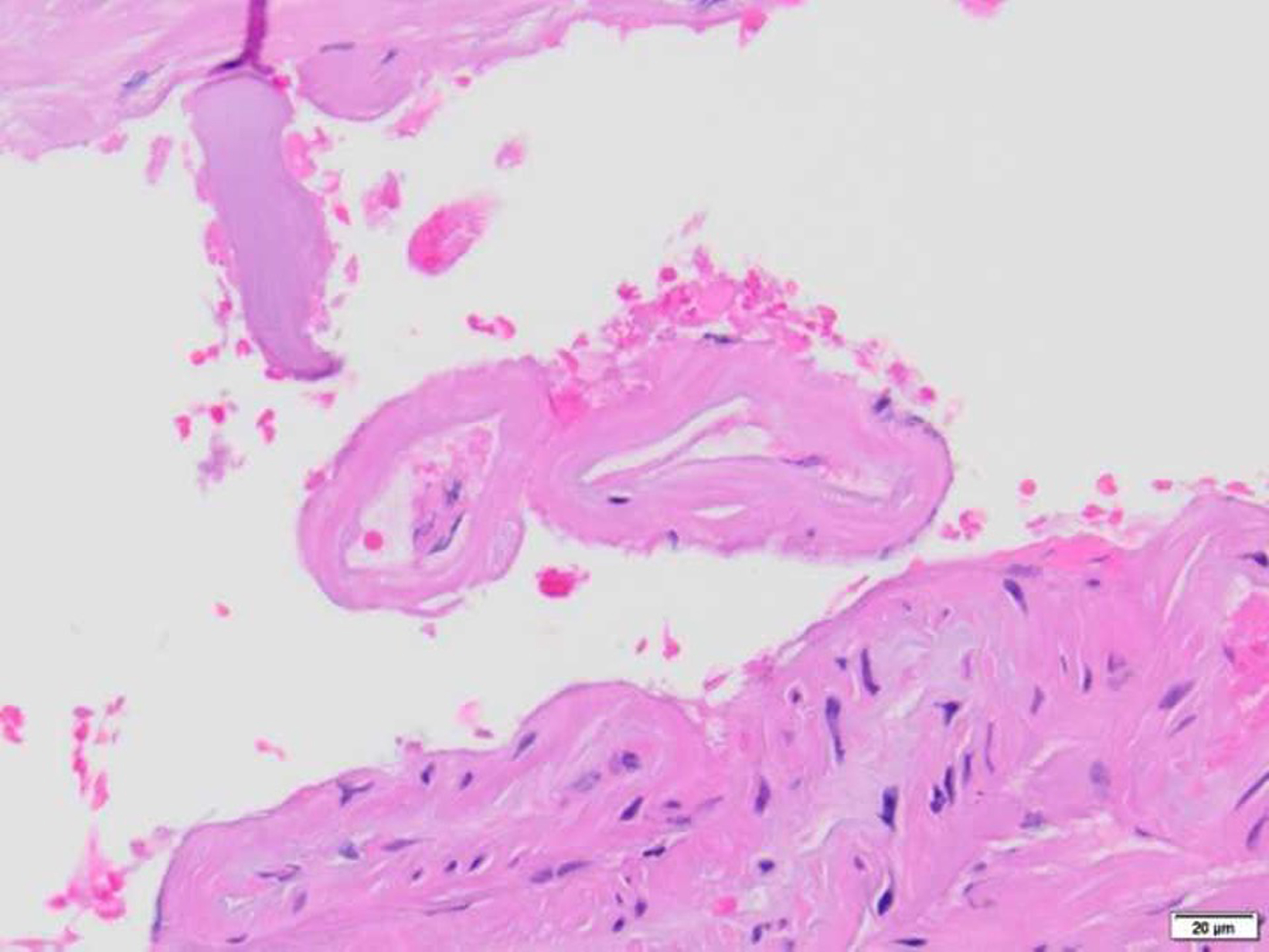
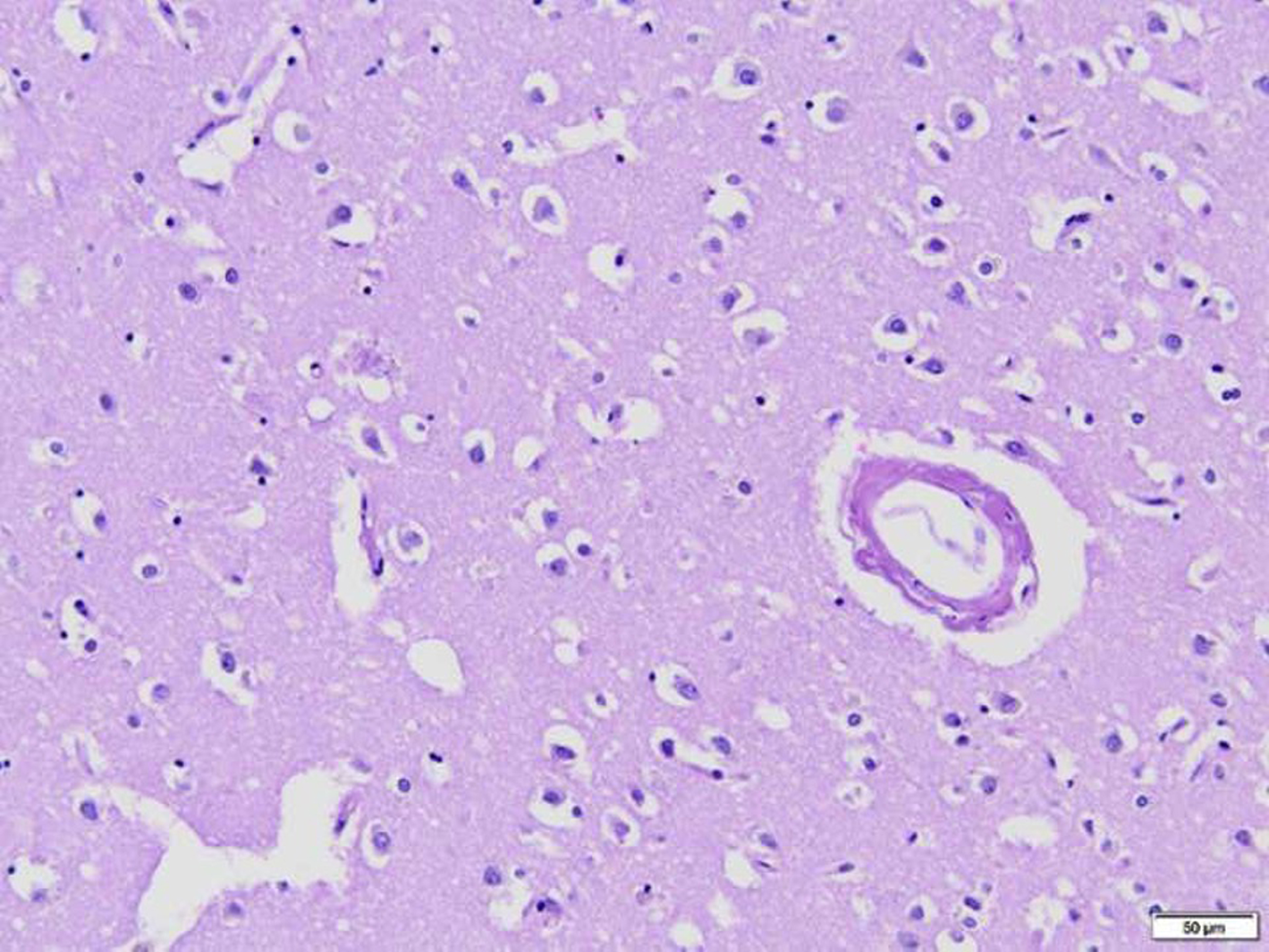
Fig. 3
Plaque and blood vessel with amyloid deposition. Hematoxylin and Eosin, 200x.
A subset of patients develops an inflammatory reaction to the amyloid protein known as CAA-associated inflammatory form (CAARI). The presence of perivascular inflammation in these cases may present in various clinical non-specific syndromes such as subacute cognitive decline, seizures, and leukoencephalopathy. Patients without CAARI more commonly presents with lobar hemorrhages [29].
4.1Cerebral amyloid angiopathy and Alzheimer’s disease
The deposition of Aʲ into parenchymal cells and vascular tissues occurs independently or may overlap (Fig. 4). Aʲ can aggregate into toxic oligomers. Aʲ42 has more tendency to aggregate into plaques, while Aʲ40 is predominantly seen as amyloid deposition in the walls of bleed vessels. AD is the most common type of dementia seen in elderly people. Underlying pathology starts decades before the first clinical manifestation of AD. People at risk in the preclinical phase of the disease can have an early presence of CSF biomarkers which include low Aʲ42 levels and high tau protein level. The risk of progression to clinical AD increases with age and in APOE ɛ4 carriers. Various modifiable risk factors such as hypertension, diabetes mellitus, smoking, and physical inactivity have been associated with AD.
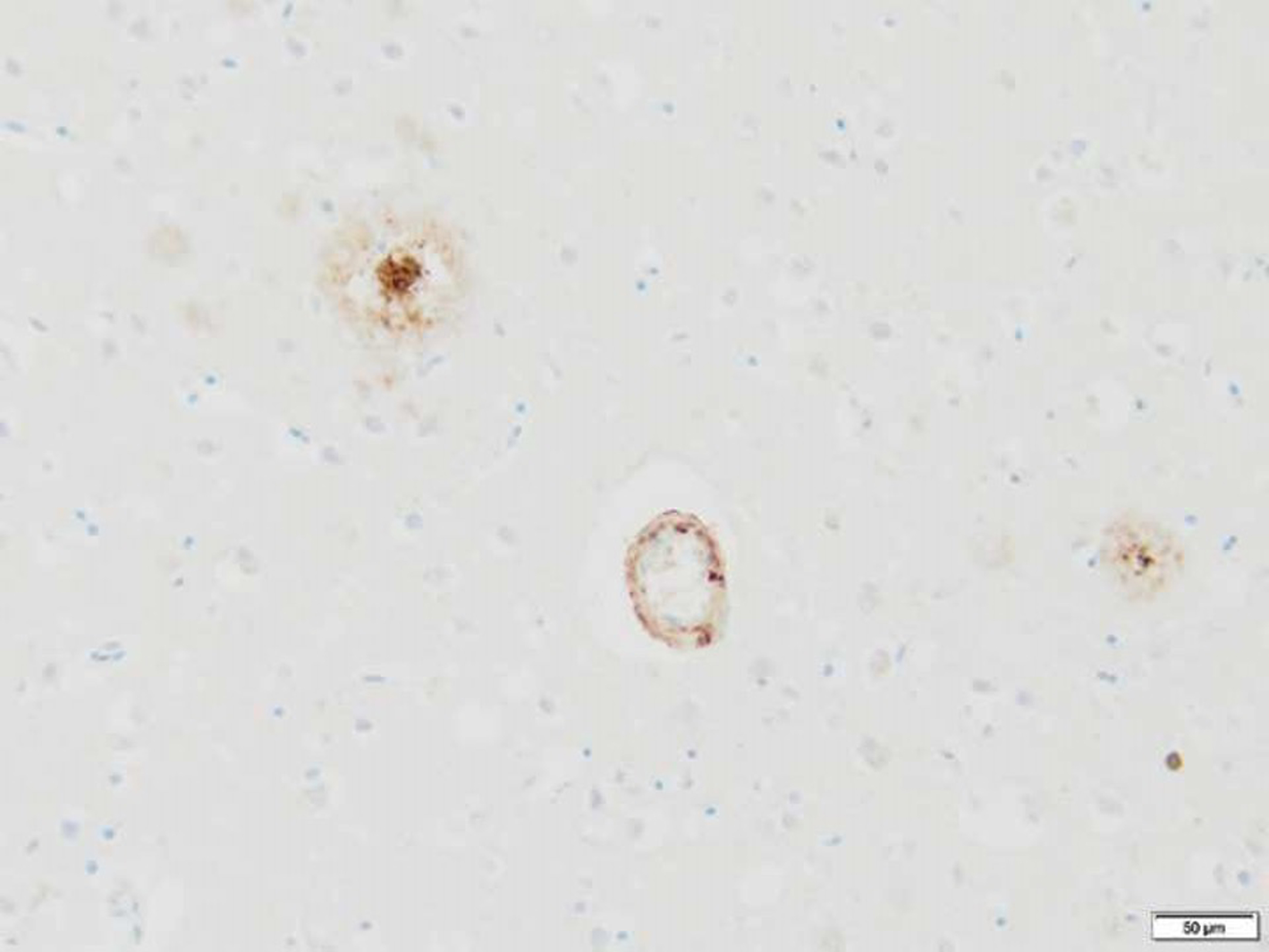
Fig. 4
Immunohistochemical stain highlighting cortically localized amyloid-ʲ plaques and vessel wall amyloid-ʲ deposition in Alzheimer’s disease (amyloid-ʲ ICH, 200x).
CLINICAL FEATURES
Intracerebral hemorrhage
Spontaneous ICH is the most common presentation of rupture of affected vessels in CAA [30]. Aʲ deposition has a predilection for cortical and leptomeningeal vessels and rarely for deep structures such as basal ganglia, thalamus, and pons; therefore CAA-related hemorrhage occurs more commonly in peripheral cortical and subcortical lobar locations [11, 30]. Localization of cerebral hemorrhages radiologically may help in the differentiation of CAA-related hemorrhages from hypertensive hemorrhages. Clinical and radiological analysis in SMASH-U classification (Structural lesion, Medication, Amyloid angiopathy, Systemic/other disease, Hypertension, Undetermined) showed that cortical hemorrhages are strongly associated with amyloid deposition compared to hypertension. Deep supratentorial involvement is rare in CAA (5%) as compared to hypertension (83%). In-hospital mortality of CAA-related hemorrhages is reduced (13%) when compared to hypertensive hemorrhages (23%). Study also showed that initial presentation with CAA is less severe with average NIH stoke scale (NIHSS) scoring 06 as compared to hypertension hemorrhages averaging NIHSS scores of 13. CAA-related hemorrhage is the second leading cause of spontaneous cerebral hemorrhage following chronic hypertension [31]. The risk of clinical signs of CAA increases with age, and it has shown female predominance, particularly in the range of 65 to 74 years of age [11]. The most common manifestations are motor paresis, change in mental status, abnormalities in higher brain functions, visual loss, seizures, sensory symptoms, speech disturbance, or ataxia [32]. Furthermore, an ICH may extend to the subarachnoid space leading to headache and meningeal signs [33]. CAA-associated hemorrhages tend to have less mortality however the recurrence rate is quite common and carries a higher risk of mortality [34].
Secondary effects of cortical microhemorrhages
Cortical surface microhemorrhages are commonly seen in the aging brain, and their incidence is associated with an increased risk of neurodegenerative conditions. The distribution of blood oozing from affected capillaries and small vessels is restricted to the immediate area surrounding the leaking vessel. The spatial limitation of blood is attributed to rapid clotting of the ruptured vessel. Anticoagulation increases the risk of extension of intracerebral hematoma by limiting the clotting in ruptured vessels; therefore, anticoagulants of warfarin, direct factor Xa, and direct thrombin inhibitors potentially may increase the severity of the disease [35]. Tissue compression by microhemorrhages is too low to cause ischemia; however, additional injury can be caused by sustained inflammatory response due to exposure of brain parenchyma to blood plasma components which may cause long-term neuronal cell dysregulation or death that underlies the cognitive decline in patients with CAA [35, 36].
Cortical superficial siderosis
Cortical superficial siderosis (cSS) is defined as the deposition of blood breakdown products on cortical sulci and subarachnoid spaces. Its clinical presentation is transient focal neurological deficits, often known as amyloid spells [37]. cSS has been reported as an independent risk factor for increased risk of recurrent ICH [38]. Advanced leptomeningeal amyloid disease is associated with cSS, likely due to the rupture of fragile vessels into the subarachnoid space with blood breakdown products eventually entering onto superficial cortical surfaces [39]. On magnetic resonance imaging (MRI), cSS is characterized by low signal intensity over cortical surfaces having a characteristic bi-linear track-like appearance [40].
Amyloid spells or transient focal neurological episodes (TFNE)
TFNE are defined as brief transient stereotypical neurological symptoms that mimic transient ischemic attacks (TIA) or may present as seizure-like activity [41]. TFNEs may occur from seconds to several minutes, often related to cortical superficial siderosis. The exact underlying mechanisms which may initiate these events are not yet clear. The proposed mechanisms include cortical spreading depression or possibly focal seizures resulting from cortical irritation of blood breakdown products which overly the cortical hemispheres [42]. For some patients, a TFNE will be the first clinical presentation of underlying CAA. It is not uncommon to have an initial diagnosis of unspecified seizure or TIA then, after undergoing a thorough clinical exam, assessment of cognitive function, and obtaining advanced radiographic imaging, be advised that the underlying concern is for a diagnosis of possible or probable CAA.
Cerebral amyloid angiopathy-related inflammation
CAARI is a rare form of the disease characterized by an inflammatory response to amyloid deposition in the vascular system. CAARI predominantly affects females with a mean age of 69 years [42]. Neuropathological findings include perivascular inflammatory changes with or without granulomatous formation. The most common clinical presentation in these patients is the subacute onset of cognitive impairment, acute encephalopathy, followed by headache, seizures, hemiparesis, aphasia, and visual symptoms [43]. CSF analysis predominantly shows elevated proteins, nucleated cells greater than 5, normal glucose levels, although pleocytosis is not seen in every patient [42]. However, CSF analysis is neither sensitive nor specific in diagnoses [42]. MRI findings may show symmetric or asymmetric cortical vasogenic edema and subcortical hyperintense T2 lesions [44]. CAARI can be diagnosed from the initial clinical presentation with complimentary MRI findings which provide good sensitivity as well as specificity. Definitive diagnosis requires brain biopsy but that should not delay the treatment in patients suspected to have CAARI. The subset of patients who fail to respond to immunosuppressant therapy may require brain biopsy within 3 weeks [45].
Amyloid beta-related angiitis (ABRA)
ABRA is another rare form of CAA which shares clinical features with CNS vasculitis and is distinct from the inflammatory CAA subtype. This variant presents earlier in life compared to typical CAA, but at a later age in comparison to patients with CNS vasculitis. ABRA is characterized as a perivascular and transmural inflammatory response to amyloid protein, often with granuloma formation and CAARI is characterized by inflammatory reaction around the blood vessels without angiodestruction [46, 47]. Unlike CAA patients, ABRA presents more often with a rapid decline in mental status followed by cognitive impairment, focal neurological deficits, seizures, and behavioral symptoms [48]. CSF commonly shows elevated protein and pleocytosis, findings which are not always present and are not very sensitive. Imaging findings in ABRA may vary from asymmetric white matter changes to leukoencephalopathy [49]. ABRA is an angio-destructive condition which may present with microinfarcts in addition to microhemorrhages and cortical superficial siderosis. Like CAARI, ABRA can also be diagnosed from clinical and MRI findings, obviating the need for brain biopsy; however, brain biopsy is the gold standard that shows granulomatous inflammatory findings [50]. ABRA is noted to have a clear response to immunosuppression (Table 2) [48].
Table 2
Various clinical manifestations of CAA
| Pathophysiology | Clinical manifestations |
| Peripheral cortical and subcortical hemorrhages | Most common manifestation is motor paresis, disturbance of consciousness, abnormalities in higher brain functions, visual loss, seizures, sensory symptoms, speech disturbances, or ataxia. |
| Sulcal subarachnoid hemorrhages leading to superficial cortical siderosis | Presents as transient focal neurological episodes (TFNE), aka amyloid spells. |
| Cerebral amyloid angiopathy-related inflammation | Presents as subacute onset of cognitive impairment followed by headache, seizures, hemiparesis, aphasia, and visual symptoms. |
| Amyloid beta-related angiitis (ABRA) | Presents as a rapid decline in mental status followed by focal neurological deficits, seizures, and behavioral symptoms. |
DIAGNOSIS
Clinical presentation and imaging findings together have good sensitivity and excellent specificity in the initial diagnosis of CAA and related subtypes; however, definitive diagnosis requires a brain biopsy. On initial computed tomography (CT) head, location and size of the hematoma should be noted. Sensitive MR imaging is more helpful than CT head to detect cerebral microhemorrhages. [51] Susceptibility weighted images (SWI) are superior to conventional gradient-echo techniques in diagnosing cerebral microhemorrhages and allow more precise analysis of the natural course of the disease. Brain imaging with SWI requires only additional few minutes and should be included in routine neuroimages (Fig. 5). Formal diagnostic subtracted angiography, or MR angiography, while potentially useful to exclude alternative causes of lobar hemorrhages such as vascular malformations, are not used to diagnose CAA because they do not visualize the small arteries and arterioles that are affected by it [52]. Definite CAA is diagnosed by postmortem autopsy. The modified Boston criteria version 2.0 is now the clinical standard to divide CAA into possible or probable disease by incorporating emerging MRI findings. The addition of multifocality of cSS is one of the main updates of the Boston Criteria Version 2.0. This modification increases sensitivity without lowering specificity (Table 3) [53]. Patients who are very sick in acute settings or in certain contraindications such as non-MRI compatible implants and in low income or middle-income countries where MRI brain might be unavailable, the Edinberg CT and genetic rule in and rule out diagnostic criteria can help the clinicians to make therapeutic decisions and inform prognosis by using three predictors: APOE ɛ4 possession, subarachnoid hemorrhage, and finger-like projections (Table 4) [54]. Additionally, CAA-associated inflammatory and ABRA need to be recognized, and their clinical presentations with associated radiological findings can help in those diagnoses. The presence of anti-Aʲ antibodies in CSF has been reported in CAARI, which further supports the CSF analysis, but routine CSF analysis is not a specific test [55].
Fig. 5
Radiographic findings in 65-year-old-woman presented with TFNE secondary to CAARI with microhemorrhages. Notes: Axial non-contrast CT scan of the head (a), axial MRI T2-FLAIR (b), and SWI sequence (c) show a pattern of inflammatory CAA with diffuse vasogenic edema involving predominantly the frontal, parietal, and temporal lobes (white arrows) resulting in effacement of adjacent sulci; there are associated multiple foci of petechial microhemorrhages (double arrows) and acute subarachnoid hemorrhage in the left central sulcus (dotted arrow).
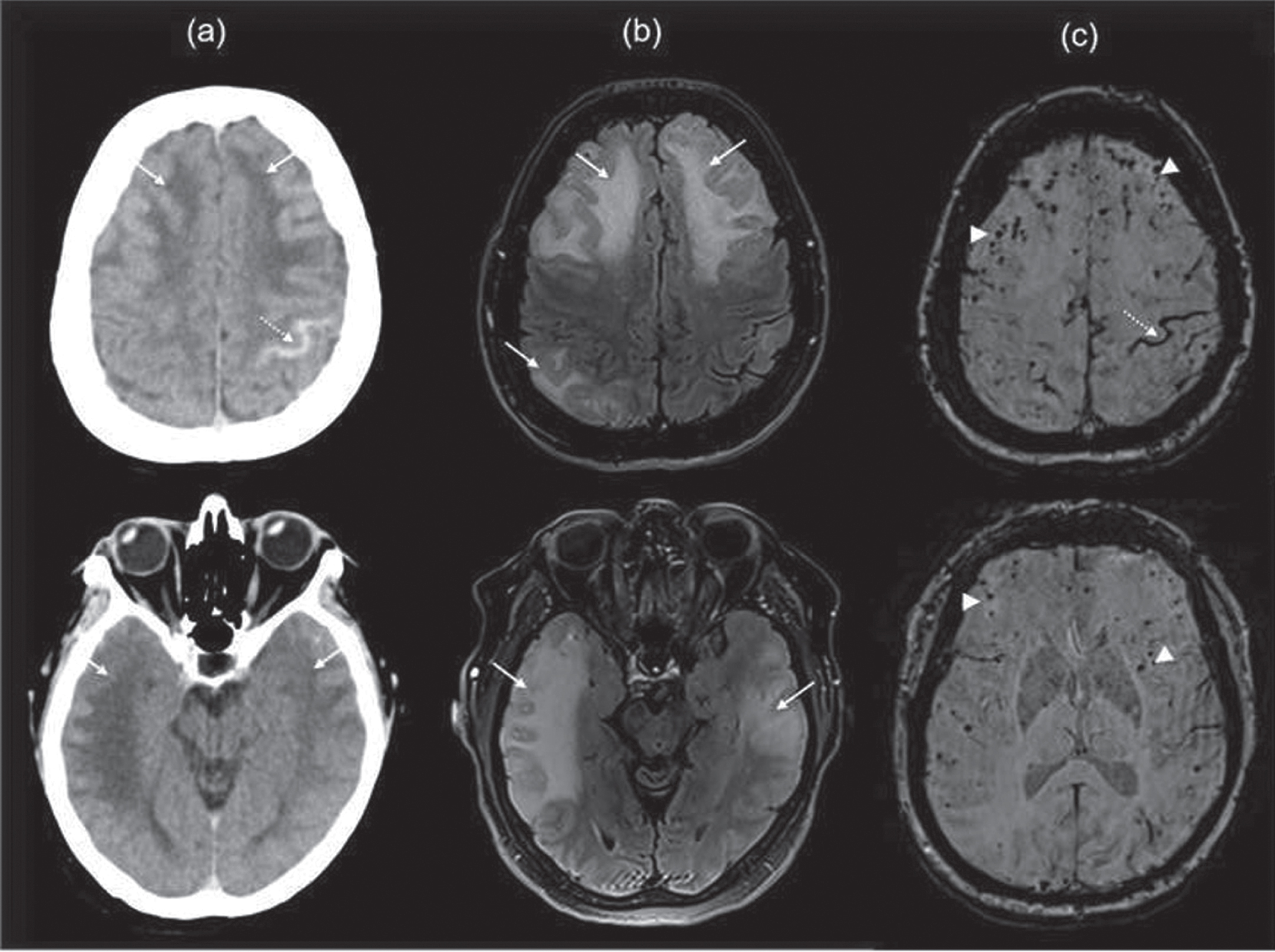
Table 3
Modified Boston criteria Version 2.0 for diagnosis of CAA
| Boston criteria Version 2.0 for diagnosis of CAA | |
| Definite CAA | Full postmortem examination demonstrating: |
| •Lobar, cortical, or cortico-subcortical hemorrhage | |
| •Severe CAA with vasculopathy | |
| •Absence of another diagnostic lesion | |
| Probable CAA with supporting pathology | Clinical data and pathological tissue (evacuated hematoma or cortical biopsy) demonstrating: |
| •Lobar, cortical, or cortico-subcortical hemorrhage | |
| •Some degree of CAA in the specimen | |
| •Absence of another diagnostic lesion | |
| Probable CAA | Clinical data and MRI or CT demonstrating: |
| •Age≥55 years | |
| •Presentation of spontaneous intracerebral hemorrhage. | |
| •Presence of at least 2 of the strictly lobar hemorrhagic lesions on T2 weighted MRI, in any combination: intracerebral hemorrhage, cerebral microbleeds, or foci of cortical superficial siderosis or convexity subarachnoid hemorrhage | |
| OR | |
| •One lobar hemorrhage with one of the white matter lesions (severe perivascular spaces in the central semiovale or white matter hyperintensities in a multisport pattern) | |
| •Absence of deep hemorrhagic lesions | |
| •Absence of other causes of hemorrhage or cSS | |
| Possible CAA | Clinical data and MRI or CT demonstrating: |
| •Presentation with spontaneous intracerebral hemorrhage | |
| •Presence of one strictly lobar hemorrhage on T2 weighted MRI, cerebral microhemorrhages, foci of cSS or convexity subarachnoid hemorrhage. | |
| •Age≥55 years | |
| •Absence of other causes of hemorrhage or cSS | |
| OR | |
| •One white matter lesion (severe perivascular spaces in the central semiovale or white matter hyperintensities in a multisport pattern) | |
| •Absence of deep hemorrhagic lesions | |
| •Absence of other causes of hemorrhages |
Table 4
The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral hemorrhage associated with cerebral amyloid angiopathy
| Probability of moderate to severe CAA | Low | Medium | Severe | |||
| Subarachnoid hemorrhage | – | + | – | + | + | + |
| APOE4 possession | – | – | + | + | – | + |
| Finger like projections on CT head | – | – | – | – | + | + |
| Diagnostic test accuracy | Rule out sensitivity 100% (CI 95%) | Rule in sensitivity 96% (CI 95%) | ||||
MANAGEMENT
Evaluation and management in the emergency department
Patients with CAA may present acutely to the emergency department with stroke-like symptoms involving deficits of motor, sensory, cranial nerve, language, speech, coordination, or gait dysfunction. They may present with first-ever or recurrent seizures, acute to subacute cognitive impairment even encephalopathy or headache. When stroke-like deficits are noted emergency personnel must rapidly screen for possible acute stroke interventions. Initial evaluation with CT head imaging is required to evaluate for spontaneous ICH which, if found, may indicate CAA as CAA-related ICH commonly have lobar location. These unique hemorrhages may also involve cortical surfaces allowing hemorrhaging blood to extend into the subarachnoid spaces. A thoughtful bedside history will include questions regarding anticoagulant use which raises the risk for rebleeding, and increases the severity of spontaneous hemorrhage, morbidity, and mortality and requires rapid measures to reverse anticoagulation effect. Sudden neurological deterioration in the emergency department mandates immediate re-imaging to assess for hematoma expansion, with considerations for intubation, transfer to intensive care unit, and neurosurgical consultation. Another challenge faced by emergency physicians is identification of underlying disease in patients presenting with transient neurological deficits which resolves within minutes. When TIA is suspected we recommend CAA be included in the working differential, thus highlighting the importance of admission for a complete stroke workup prior to discharge.
Intensive monitoring for intracranial spontaneous hemorrhage
The risk of neurological deterioration from rebleeding is highest within the first 24 hours of ICH. Following initial stabilization, patients should be observed in the intensive care unit with hourly neurological assessments. Blood pressure management is deemed critical to prevent hematoma expansion and blood pressure monitoring with systemic arterial pressure should be considered. American Heart Association recommends lowering systolic blood pressures in the setting of acute ICH to 140 is safe while maintaining mean arterial pressures greater than 65 [56]. Furthermore, critical care management in the emergency department should include monitoring for any change in the neurological examination. This warrants immediate CT imaging of the head to rule out hematoma expansion, worsening cerebral edema with cerebral compression, brain herniation, and development of acute hydrocephalus which may require neurosurgical consultation.
Management of long-term blood pressure
Chronic CAA leads to cerebral blood vessel fragility. In the presence of chronic hypertension, there is increased risk for hemorrhage and hemorrhagic recurrence. Long-term management of blood pressure plays an important role in reducing the risk of CAA-related ICH [57]. In general, effective blood pressure control also reduces the risk of all types of ICH. Increases of blood pressure variability significantly above the average baseline has an association with the increased size of cerebral microhemorrhages, particularly in the deep and infratentorial regions [58]. Poor control of blood pressure is associated with increased progression of CAA disease and subsequent mortality.
Antiplatelet use in patients with CAA
Current ACC/AHA guidelines recommend against the use of antiplatelets for primary prevention of cardiovascular events in CAA patients due to potential risk for hemorrhage related to CAA disease [59]. However, The REstart or STop Antithrombotics Randomised Trial (RESTART) study has showed no significant difference in ICH incidence between the two groups, those taking antiplatelet therapy for secondary prevention of all major occlusive vascular events such as ischemic stroke, myocardial infarction, and peripheral artery occlusion and those not on antiplatelet therapy. Hence RESTART authors conclude the benefits of using antiplatelets for secondary prevention is greater than the risk of hemorrhage in patients with CAA. Interestingly, all-cause mortality was less in patients on antiplatelet therapy for secondary prevention [60].
Anticoagulation use in patients with CAA
Non-valvular atrial fibrillation
The use of anticoagulants increases the risk of hematoma expansion and mortality associated with ICH, and there have been no specific randomized controlled trials examining patients with CAA. This places the burden of the decision to use anticoagulants onto the treating physician and family when faced with conditions requiring anticoagulants. Because patients with CAA are at increased risk of recurrent hemorrhages, potential alternatives to anticoagulation should be discussed when anticoagulation is required such as for risk reduction of ischemic stroke secondary to atrial fibrillation. In general, anticoagulants should be avoided in CAA patients if possible; however, emerging FDA-approved alternatives to anticoagulants such as left atrial appendage closure have expanded the non-pharmacological strategies of atrial fibrillation in patients with increased risk of ICH [61].
Valvular heart disease
Patients with mechanical valves require lifelong anticoagulation because of the thrombogenicity of a mechanical valve. The benefit of using anticoagulants to prevent vascular occlusive disease, including in patients with CAA, outweighs the risk of ICH [49]. Patients with a previous history of ICH can be a candidate for bioprosthetic valves; however, the decision of starting anticoagulants in these patients (even short-term anticoagulation) should be a multidisciplinary consideration.
Antiepileptic use in patients with CAA
The CAA patient may present with amyloid spells/TFNE with transient neurological symptoms, or seizures. One of the widely accepted theories for amyloid spells/TFNE is cortical depolarization spreading secondary to hemosiderin following microhemorrhages. Cortical depolarization spreading is also associated with migraine pathophysiology, and various antiepileptics have proven to be beneficial in migraine patients but there is no randomized study examining the effect of antiepileptic medications for the management of TFNE.
Immunosuppression for patients with CAARI and ABRA
A subset of patients may present with an inflammatory form of CAA which is an extremely rare and potentially reversible condition. Treatment relies on high-dose intravenous (IV) pulse steroids followed by steroid tapering for 6 months. One retrospective study showed that the early course of disease and recurrence rate is reduced with early use of high dose immunosuppressant. [42] In this study the typical steroid dosage used was 1 gram IV daily for 3 to 5 days, followed by oral prednisone 60 mg daily for 6 months followed by subsequent tapering.
Cyclophosphamide, mycophenolate, and rituximab are acceptable alternatives, especially for patients with ABRA, as this is severe variant of CAA disease has strong similarities to CNS vasculitis. [62] Despite immunosuppressive therapies, relapse can happen in certain patients. This population may benefit from trials of alternative immunosuppressants.
CONCLUSION
This review article highlights the importance of understanding the different clinical manifestations of CAA in the emergency department, intensive care unit, and in patient clinic follow-up. Considering that TFNE can mimic TIA as well as seizures. Initial diagnosis of CAA can easily be missed, and the inflammatory subtype can be misdiagnosed as tumor if MRI brain with SWI sequencing is not performed early in evaluation. The characteristic MRI radiographic findings of CAA include evidence for prior multiple microhemorrhages, hemosiderosis, and even large lobar hemorrhages. Systolic blood pressure for active CAA-related hemorrhage is recommend by AHA to be 140 mmHg, and we recommend always maintaining mean arterial pressures above 65. Anti-platelet therapy for secondary stroke prevention appears to be safe whereas anticoagulation should be multidisciplinary and other alternatives should be considered if possible. A randomized trial is required to determine the use of anti-epileptics in treating TFNE presenting as seizures. Our current understanding of amyloid deposition into the walls of small to medium cortical vessels is not well-understood. New treatments need to be developed which prevent or decrease amyloid deposition in affected vessels. Studies of genetic factors associated with CAA deposition are ongoing. Factors leading to inflammatory CAA subtype also need further study.
ACKNOWLEDGMENTS
Special thanks to my family for their love and support.
FUNDING
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Hazenberg BP ((2013) ) Amyloidosis: A clinical overview. Rheum Dis Clin North Am 39: , 323–345. |
[2] | Smith EE , Greenberg SM ((2009) ) Beta-amyloid, blood vessels, and brain function. Stroke 40: , 2601–2606. |
[3] | Qureshi AI , Tuhrim S , Broderick JP , Batjer HH , Hondo H , Hanley DF ((2001) ) Spontaneous intracerebral hemorrhage. N Engl J Med 344: , 1450–1460. |
[4] | Jellinger KA ((2002) ) Alzheimer disease and cerebrovascular pathology: An update. J Neural Transm (Vienna) 109: , 813–836. |
[5] | Block F , Dafotakis M ((2017) ) Cerebral amyloid angiopathy in stroke medicine. Dtsch Arztebl Int 114: , 37–42. |
[6] | Jellinger K ((2000) ) Inverse relation between Braak stage and cerebrovascular pathology in Alzheimer predominant dementia. J Neurol Neurosurg Psychiatry 68: , 799–800. |
[7] | Ikram MA , Wieberdink RG , Koudstaal PJ ((2012) ) International epidemiology of intracerebral hemorrhage. Curr Atheroscler Rep 14: , 300–306. |
[8] | Giroud M , Gras P , Chadan N , Beuriat P , Milan C , Arveux P , Dumas R ((1991) ) Cerebral haemorrhage in a French prospective population study. J Neurol Neurosurg Psychiatry 54: , 595–598. |
[9] | Bateman BT , Claassen J , Willey JZ , Hirsch LJ , Mayer SA , Sacco RL , Schumacher HC ((2007) ) Convulsive status epilepticus after ischemic stroke and intracerebral hemorrhage: Frequency, predictors, and impact on outcome in a large administrative dataset. Neurocrit Care 7: , 187–193. |
[10] | Yamada M ((2002) ) Risk factors for cerebral amyloid angiopathy in the elderly. Ann N Y Acad Sci 977: , 37–44. |
[11] | Yamada M ((2015) ) Cerebral amyloid angiopathy: Emerging concepts. J Stroke 17: , 17–30. |
[12] | Huynh TV , Davis AA , Ulrich JD , Holtzman DM ((2017) ) Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-beta and other amyloidogenic proteins. J Lipid Res 58: , 824–836. |
[13] | McCarron MO , Nicoll JA , Stewart J , Ironside JW , Mann DM , Love S , Graham DI , Dewar D ((1999) ) The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J Neuropathol Exp Neurol 58: , 711–718. |
[14] | Yamada M , Sodeyama N , Itoh Y , Suematsu N , Otomo E , Matsushita M , Mizusawa H ((1997) ) Association of presenilin-1 polymorphism with cerebral amyloid angiopathy in the elderly. Stroke 28: , 2219–2221. |
[15] | Yamada M ((2004) ) Cerebral amyloid angiopathy and gene polymorphisms. J Neurol Sci 226: , 41–44. |
[16] | Yamada M , Sodeyama N , Itoh Y , Suematsu N , Otomo E , Matsushita M , Mizusawa H ((1998) ) Association of alpha1-antichymotrypsin polymorphism with cerebral amyloid angiopathy. Ann Neurol 44: , 129–131. |
[17] | Kiuru-Enari S , Haltia M ((2010) ) Hereditary gelsolin amyloidosis–40 years of Meretoja disease. Duodecim 126: , 1162–1171. |
[18] | Kiuru-Enari S , Haltia M ((2013) ) Hereditary gelsolin amyloidosis. Handb Clin Neurol 115: , 659–681. |
[19] | Garringer HJ , Murrell J , D’Adamio L , Ghetti B , Vidal R ((2010) ) Modeling familial British and Danish dementia. Brain Struct Funct 214: , 235–244. |
[20] | Cheng X , Su Y , Wang Q , Gao F , Ye X , Wang Y , Xia Y , Fu J , Shen Y , Al-Shahi Salman R , Dong Q ((2020) ) Neurofilament light chain predicts risk of recurrence in cerebral amyloid angiopathy-related intracerebral hemorrhage. Aging (Albany NY) 12: , 23727–23738. |
[21] | Revesz T , Holton JL , Lashley T , Plant G , Frangione B , Rostagno A , Ghiso J ((2009) ) Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 118: , 115–130. |
[22] | Gatti L , Tinelli F , Scelzo E , Arioli F , Di Fede G , Obici L , Pantoni L , Giaccone G , Caroppo P , Parati EA , Bersano A ((2020) ) Understanding the pathophysiology of cerebral amyloid angiopathy. Int J Mol Sci 21: , 3435. |
[23] | Weller RO , Massey A , Newman TA , Hutchings M , Kuo YM , Roher AE ((1998) ) Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153: , 725–733. |
[24] | Vinters HV ((1987) ) Cerebral amyloid angiopathy. A critical review. Stroke 18: , 311–324. |
[25] | Howie AJ ((2015) ) “green(or apple-) birefringence” of Congo red-stained amyloid. Amyloid 22: , 205–206. |
[26] | Xu HQ , Wang YS ((1991) ) Pathological study on cerebral amyloid angiopathy. Chin Med J (Engl) 104: , 842–845. |
[27] | Vonsattel JP , Myers RH , Hedley-Whyte ET , Ropper AH , Bird ED , Richardson EP Jr. ((1991) ) Cerebral amyloid angiopathy without andwith cerebral hemorrhages: A comparative histological study. Ann Neurol 30: , 637–649. |
[28] | Keable A , Fenna K , Yuen HM , Johnston DA , Smyth NR , Smith C , Al-Shahi Salman R , Samarasekera N , Nicoll JA , Attems J , Kalaria RN , Weller RO , Carare RO ((2016) ) Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta 1862: , 1037–1046. |
[29] | Eng JA , Frosch MP , Choi K , Rebeck GW , Greenberg SM ((2004) ) Clinical manifestations of cerebral amyloid angiopathy-related inflammation. Ann Neurol 55: , 250–256. |
[30] | Knudsen KA , Rosand J , Karluk D , Greenberg SM ((2001) ) Clinical diagnosis of cerebral amyloid angiopathy: Validation of the Boston criteria. Neurology 56: , 537–539. |
[31] | Meretoja A , Strbian D , Putaala J , Curtze S , Haapaniemi E , Mustanoja S , Sairanen T , Satopaa J , Silvennoinen H , Niemela M , Kaste M , Tatlisumak T ((2012) ) SMASH-U: A proposal for etiologic classification of intracerebral hemorrhage. Stroke 43: , 2592–2597. |
[32] | Hirohata M , Yoshita M , Ishida C , Ikeda SI , Tamaoka A , Kuzuhara S , Shoji M , Ando Y , Tokuda T , Yamada M ((2010) ) Clinical features of non-hypertensive lobar intracerebral hemorrhage related to cerebral amyloid angiopathy. Eur J Neurol 17: , 823–829. |
[33] | Maia LF , Mackenzie IR , Feldman HH ((2007) ) Clinical phenotypes of cerebral amyloid angiopathy. J Neurol Sci 257: , 23–30. |
[34] | Greenberg SM ((1998) ) Cerebral amyloid angiopathy: Prospects for clinical diagnosis and treatment. Neurology 51: , 690–694. |
[35] | Rosidi NL , Zhou J , Pattanaik S , Wang P , Jin W , Brophy M , Olbricht WL , Nishimura N , Schaffer CB ((2011) ) Cortical microhemorrhages cause local inflammation but do not trigger widespread dendrite degeneration. PLoS One 6: , e26612. |
[36] | Lipowsky HH ((2005) ) Microvascular rheology and hemodynamics. Microcirculation 12: , 5–15. |
[37] | Charidimou A , Linn J , Vernooij MW , Opherk C , Akoudad S , Baron JC , Greenberg SM , Jager HR , Werring DJ ((2015) ) Cortical superficial siderosis: Detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 138: , 2126–2139. |
[38] | Charidimou A , Boulouis G , Greenberg SM , Viswanathan A ((2019) ) Cortical superficial siderosis and bleeding risk in cerebral amyloid angiopathy: A meta-analysis. Neurology 93: , e2192–e2202. |
[39] | Charidimou A , Perosa V , Frosch MP , Scherlek AA , Greenberg SM , van Veluw SJ ((2020) ) Neuropathological correlates of cortical superficial siderosis in cerebral amyloid angiopathy. Brain 143: , 3343–3351. |
[40] | Lioutas VA , Gattringer T ((2020) ) Cortical superficial siderosis: Fine dark lines, harbingers of doom. Neurology 94: , 729–730. |
[41] | Cooperman SS , Shah AK , Rajamani K ((2019) ) Amyloid spells. Neurol Clin Pract 9: , e17–e18. |
[42] | Regenhardt RW , Thon JM , Das AS , Thon OR , Charidimou A , Viswanathan A , Gurol ME , Chwalisz BK , Frosch MP , Cho TA , Greenberg SM ((2020) ) Association between immunosuppressive treatment and outcomes of cerebral amyloid angiopathy-related inflammation. JAMA Neurol 77: , 1261–1269. |
[43] | Castro Caldas A , Silva C , Albuquerque L , Pimentel J , Silva V , Ferro JM ((2015) ) Cerebral amyloid angiopathy associated with inflammation: Report of 3 cases and systematic review. J Stroke Cerebrovasc Dis 24: , 2039–2048. |
[44] | Kirshner HS , Bradshaw M ((2015) ) The inflammatory form of cerebral amyloid angiopathy or “cerebral amyloid angiopathy-related inflammation” (CAARI). Curr Neurol Neurosci Rep 15: , 54. |
[45] | Auriel E , Charidimou A , Gurol ME , Ni J , Van Etten ES , Martinez-Ramirez S , Boulouis G , Piazza F , DiFrancesco JC , Frosch MP , Pontes-Neto OV , Shoamanesh A , Reijmer Y , Vashkevich A , Ayres AM , Schwab KM , Viswanathan A , Greenberg SM ((2016) ) Validation of clinicoradiological criteria for the diagnosis of cerebral amyloid angiopathy-related inflammation. JAMA Neurol 73: , 197–202. |
[46] | Ng DW , Magaki S , Terashima KH , Keener AM , Salamon N , Karnezis S , Macyszyn L , Vinters HV ((2017) ) Amyloid-beta-related angiitis: A report of 2 cases with unusual presentations. Hum Pathol 64: , 191–197. |
[47] | Salvarani C , Morris JM , Giannini C , Brown RD , Jr. , Christianson T , Hunder GG ((2016) ) imaging findings of cerebral amyloid angiopathy, Abeta-related angiitis (ABRA), and cerebral amyloid angiopathy-related inflammation: A single-institution 25-year experience. Medicine (Baltimore) 95: , e3613. |
[48] | Scolding NJ , Joseph F , Kirby PA , Mazanti I , Gray F , Mikol J , Ellison D , Hilton DA , Williams TL , MacKenzie JM , Xuereb JH , Love S ((2005) ) Abeta-related angiitis: Primary angiitis of the central nervous system associated with cerebral amyloid angiopathy. Brain 128: , 500–515. |
[49] | Kozberg MG , Perosa V , Gurol ME , van Veluw SJ ((2021) ) A practical approach to the management of cerebral amyloid angiopathy. Int J Stroke 16: , 356–369. |
[50] | Danve A , Grafe M , Deodhar A ((2014) ) Amyloid beta-related angiitis–a case report and comprehensive review of literature of 94 cases. Semin Arthritis Rheum 44: , 86–92. |
[51] | Nandigam RN , Viswanathan A , Delgado P , Skehan ME , Smith EE , Rosand J , Greenberg SM , Dickerson BC ((2009) ) MR imaging detection of cerebral microbleeds: Effect of susceptibility-weighted imaging, section thickness, and field strength. AJNR Am J Neuroradiol 30: , 338–343. |
[52] | Haacke EM , DelProposto ZS , Chaturvedi S , Sehgal V , Tenzer M , Neelavalli J , Kido D ((2007) ) Imaging cerebral amyloid angiopathy with susceptibility-weighted imaging. AJNR Am J Neuroradiol 28: , 316–317. |
[53] | Charidimou A , Boulouis G , Frosch MP , Baron JC , Pasi M , Albucher JF , Banerjee G , Barbato C , Bonneville F , Brandner S , Calviere L , Caparros F , Casolla B , Cordonnier C , Delisle MB , Deramecourt V , Dichgans M , Gokcal E , Herms J , Hernandez-Guillamon M , Jager HR , Jaunmuktane Z , Linn J , Martinez-Ramirez S , Martinez-Saez E , Mawrin C , Montaner J , Moulin S , Olivot JM , Piazza F , Puy L , Raposo N , Rodrigues MA , Roeber S , Romero JR , Samarasekera N , Schneider JA , Schreiber S , Schreiber F , Schwall C , Smith C , Szalardy L , Varlet P , Viguier A , Wardlaw JM , Warren A , Wollenweber FA , Zedde M , van Buchem MA , Gurol ME , Viswanathan A , Al-Shahi Salman R , Smith EE , Werring DJ , Greenberg SM ((2022) ) The Boston criteria version 2.0for cerebral amyloid angiopathy: A multicentre, retrospective,MRI-neuropathology diagnostic accuracy study.. Lancet Neurol 21: , 714–725. |
[54] | Rodrigues MA , Samarasekera N , Lerpiniere C , Humphreys C , McCarron MO , White PM , Nicoll JAR , Sudlow CLM , Cordonnier C , Wardlaw JM , Smith C , Al-Shahi Salman R ((2018) ) The Edinburgh CT and genetic diagnostic criteria for lobar intracerebral haemorrhage associated with cerebral amyloid angiopathy: Model development and diagnostic test accuracy study. Lancet Neurol 17: , 232–240. |
[55] | DiFrancesco JC , Brioschi M , Brighina L , Ruffmann C , Saracchi E , Costantino G , Galimberti G , Conti E , Curto NA , Marzorati L , Remida P , Tagliavini F , Savoiardo M , Ferrarese C ((2011) ) Anti-Abeta autoantibodies in the CSF of a patient with CAA-related inflammation: A case report. Neurology 76: , 842–844. |
[56] | Zhang S , Wang Z , Zheng A , Yuan R , Shu Y , Zhang S , Lei P , Wu B , Liu M ((2020) ) Blood pressure and outcomes in patients with different etiologies of intracerebral hemorrhage: A multicenter cohort study. J Am Heart Assoc 9: , e016766. |
[57] | Arima H , Tzourio C , Anderson C , Woodward M , Bousser MG , MacMahon S , Neal B , Chalmers J , Group PC ((2010) ) Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: The PROGRESS trial. Stroke 41: , 394–396. |
[58] | Liu W , Liu R , Sun W , Peng Q , Zhang W , Xu E , Cheng Y , Ding M , Li Y , Hong Z , Wu J , Zeng J , Yao C , Huang Y , CASISP Study Group ((2012) ) Different impacts of blood pressure variability on the progression of cerebral microbleeds and white matter lesions. Stroke 43: , 2916–2922. |
[59] | Arnett DK , Blumenthal RS , Albert MA , Buroker AB , Goldberger ZD , Hahn EJ , Himmelfarb CD , Khera A , Lloyd-Jones D , McEvoy JW , Michos ED , Miedema MD , Munoz D , Smith SC Jr. , Virani SS , Williams KA Sr. , Yeboah J , Ziaeian B ((2019) ) 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 74: , e177–e232. |
[60] | Collaboration R ((2019) ) Effects of antiplatelet therapy after stroke due to intracerebral haemorrhage (RESTART): A randomised, open-label trial. Lancet 393: , 2613–2623. |
[61] | Gurol ME ((2018) ) Nonpharmacological management of atrial fibrillation in patients at high intracranial hemorrhage risk. Stroke 49: , 247–254. |
[62] | Corovic A , Kelly S , Markus HS ((2018) ) Cerebral amyloid angiopathy associated with inflammation: A systematic review of clinical and imaging features and outcome. Int J Stroke 13: , 257–267. |


