
Genetic Screening in Korean Patients with Frontotemporal Dementia Syndrome
Abstract
Background:
Frontotemporal dementia (FTD) syndrome is a genetically heterogeneous group of diseases. Pathogenic variants in the chromosome 9 open reading frame 72 (C9orf72), microtubule-associated protein tau (MAPT), and progranulin (GRN) genes are mainly associated with genetic FTD in Caucasian populations.
Objective:
To understand the genetic background of Korean patients with FTD syndrome.
Methods:
We searched for pathogenic variants of 52 genes related to FTD, amyotrophic lateral sclerosis, familial Alzheimer’s disease, and other dementias, and hexanucleotide repeats of the C9orf72 gene in 72 Korean patients with FTD using whole exome sequencing and the repeat-primed polymerase chain reaction, respectively.
Results:
One likely pathogenic variant, p.G706R of MAPT, in a patient with behavioral variant FTD (bvFTD) and 13 variants of uncertain significance (VUSs) in nine patients with FTD were identified. Of these VUSs, M232R of the PRNP gene, whose role in pathogenicity is controversial, was also found in two patients with bvFTD.
Conclusions:
These results indicate that known pathogenic variants of the three main FTD genes (MAPT, GRN, and C9orf72) in Western countries are rare in Korean FTD patients.
INTRODUCTION
Frontotemporal dementia (FTD) is the second most common type of early onset dementia and is characterized by progressive deterioration of behavior or language associated with frontal or temporal degeneration. It comprises of three clinical phenotypes: behavioral variant FTD (bvFTD), semantic variant primary progressive aphasia (svPPA), and non-fluent/agrammatic variant PPA (nfvPPA), which occasionally overlaps with motor neuron disease or atypical parkinsonian syndromes, such as progressive supranuclear palsy syndrome and corticobasal syndrome [1]. Although FTD is highly heritable in Western countries [2–4], genetic FTD is rare in Asian populations [5, 6]. We previously identified a single known pathogenic variant in the progranulin (GRN) gene and two novel variants in the colony stimulating factor 1 receptor (CSF1R) and alanyl-tRNA synthetase 2 (AARS2) genes in 107 Korean patients with sporadic FTD using next-generation sequencing (NGS) [6]. To elucidate the genetic characteristics of FTD in Asian population, large-scale, unbiased, and in-depth genetic screening using NGS technologies is continuously required.
In this study, we performed whole exome sequencing (WES) of 72 Korean patients with clinical FTD syndrome, without regarding familial status, to identify the presence of pathogenic variants of FTD, amyotrophic lateral sclerosis (ALS), or other dementia-related genes.
MATERIALS AND METHODS
Patients
Patients were prospectively recruited from ten neurology clinics across Korea between June 2016 and January 2018. All patients that were enrolled in this study met the FTD criteria proposed by Knopman et al. [7] or the new consensus diagnostic criteria for bvFTD [8], svPPA, and nfvPPA [9]. Patients with clinical and electrophysiological evidence of ALS were enrolled as having FTD-ALS, regardless of the clinical subtype of FTD. This study was conducted as part of the Clinical Research Center for Dementia of South Korea–FTD (CREDOS-FTD) registry study, which was carried out between 2010 and 2018 [10]. All participants were registered in the CREDOS-FTD registry and evaluated using the CREDOS-FTD protocol. The protocol is composed of a clinical evaluation form (clinical history, neurological examination, Korean version of Mini-Mental State Examination, Hachinski ischemic scale, Global Deterioration Scale, Unified Parkinson’s Disease Rating Scale, and Frontotemporal-Clinical Dementia Rating sum of boxes score), caregiver questionnaire form (Korean dementia screening questionnaire, Barthel Activities of Daily Living, Seoul Instrumental Activities of Daily Living, and Caregiver-Administered Neuropsychiatric Inventory and Frontal Behavioral Inventory), and cognitive test battery for FTD, consisting of subdomains assessing attention, language, visuospatial, memory, and frontal/executive functions [10]. All patients underwent brain magnetic resonance imaging (MRI) to exclude any structural lesions. Patients with current or past neurological or psychiatric illnesses were excluded from this study.
The institutional review boards (IRB) at all participating centers approved the study (IRB No. 2012-12), and informed consent was obtained from each patient and caregiver. As informed consent did not include open access to the data used in the study, our data is not publicly available.
Genetic analysis
Genomic DNA was extracted from peripheral blood leukocytes using a standard procedure. WES was performed using an Agilent SureSelect All Exon kit 50 Mb (Agilent Technologies, Santa Clara, CA, USA) on a NextSeq 500 platform (Illumina Inc., San Diego, CA, USA). Alignment of sequence reads, indexing of the reference genome (GRCh37/hg19), and variant calling were performed using a pipeline based on GATK Best Practices. Variants with allele frequencies >0.001 were filtered out based on public databases, including the Genome Aggregation Database (http://gnomad.broadinstitute.org/) and 1,722 ethnically matched controls from the Korean Reference Genome Database (KRGDB) [11]. Thereafter, 32 genes related to ALS-FTD and 20 genes related to familial Alzheimer’s disease and other dementias that have been explored by the authors from OMIM (https://www.omim.org/), GeneReview (http://www.ncbi.nlm.nih.gov/books/NBK1116/), and high-quality published literature [1, 4] and introduced as causative genes of FTD-ALS spectrum disorders or dementia, were screened for pathogenic variants (Table 1). Missense, frameshift, indel, nonsense, synonymous, and intronic variants within the exon-flaking regions were also evaluated. Splice site and conservation analyses were performed for all synonymous variants using Human Splice Finder and GERP scores. All amino acid variants were confirmed using Sanger sequencing or DeepVariant (https://github.com/google/deepvariant, version 1.2.0). The variants were classified according to the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines [12]. The BP7 rule of the ACMG/AMP guidelines was only applied in cases that were being predicted as ‘no impact and not highly conserved’. Hexanucleotide repeat expansion of chromosome 9 open reading frame 72 (C9orf72) was tested for all patients using the triplet repeat-primed polymerase chain reaction, as previously described [13].
Table 1
List of frontotemporal dementia (FTD), amyotrophic lateral sclerosis (ALS), familial Alzheimer’s disease and other dementia-related genes
| Gene symbol | RefSeq | Gene description | Chromosomal location | Inheritance |
| FTD and ALS-related genes | ||||
| ALS2 | NM_020919.3 | Amyotrophic lateral sclerosis 2 | 2q33.1 | AD |
| ANG | NM_001145.4 | Angiogenin, ribonuclease, RNase A family, 5 | 14q11.1-q11.2 | AD |
| CCNF | NM_001761.3 | Cyclin F | 16p13.3 | AD |
| CHCHD10 | NM_213720.3 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 10 | 22q11.23 | AD |
| CHMP2B | NM_014043.3 | Chromatin modifying protein 2B | 3p11.2 | AD |
| CHRNA4 | NM_000744.6 | Acetylcholine receptor, neuronal nicotinic, alpha-4 subunit | 20q13.2-q13.3 | AD |
| CYLD | NM_015247.3 | CYLD lysine 63 deubiquitinase | 16q12.1 | AD |
| DAO | NM_001917.4 | D-amino-acid oxidase | 12q24 | AD |
| DCTN1 | NM_004082.4 | Dynactin 1 | 2p13 | AD |
| FIG4 | NM_014845.5 | FIG4 phosphoinositide 5-phosphatase | 6q21 | AR |
| FUS | NM_004960.3 | FUS RNA binding protein | 16p11.2 | AD |
| GRN | NM_002087.2 | Granulin | 17q21.32 | AD |
| HNRNPA1 | NM_031157.2 | Heterogeneous nuclear ribonucleoprotein A1 | 12q13.1 | AD |
| HNRNPA2B1 | NM_031243.2 | Heterogeneous nuclear ribonucleoprotein A2/B1 | 7p15 | AD |
| MAPT | NM_005910.5 | Microtubule-associated protein tau | 17q21.1 | AD |
| MATR3 | NM_199189.2 | Matrin3 | 5q31.2 | AD |
| OPTN | NM_021980.4 | Optineurin | 10p13 | AD |
| PRNP | NM_000311.3 | Prion protein | 20p13 | AD |
| SETX | NM_015046.5 | Senataxin | 9q34.13 | AD/AR |
| SIGMAR1 | NM_005866.2 | Sigma non-opioid intracellular receptor 1 | 9p13.3 | AD/AR |
| SOD1 | NM_000454.4 | Superoxide dismutase 1 | 21q22.11 | AD |
| SPG11 | NM_025137.3 | SPG11, spatacsin vesicle trafficking associated | 15q14 | AR |
| SQSTM1 | NM_003900.4 | Sequestosome 1 | 5q35 | AD |
| TAF15 | NM_139215.2 | TATA-box binding protein associated factor 15 | 17q11.1-q11.2 | AD |
| TARDBP | NM_007375.3 | TAR DNA binding protein | 1p36.22 | AD |
| TBK1 | NM_013254.3 | Tank-binding kinase 1 | 12q14.2 | AD |
| TIA1 | NM_022173.4 | TIA1 cytotoxic granule-associated RNA-binding protein | 2p13.3 | AD |
| TREM2 | NM_018965.2 | Triggering receptor expressed on myeloid cells 2 | 6p21.1 | AR |
| TUBA4A | NM_006000.3 | Tubulin alpha 4a | 2q35 | AD |
| UBQLN2 | NM_013444.3 | Ubiquilin 2 | Xp11.21 | XL |
| VAPB | NM_004738.4 | VAMP (vesicle-associated membrane protein)-associated protein B and C | 20q13.33 | AD |
| VCP | NM_007126.3 | Valosin-containing protein | 9p13.3 | AD |
| Genes of familial Alzheimer’s disease | ||||
| APP | NM_000484.3 | Amyloid beta precursor protein | 21q21.2 | AD |
| PSEN1 | NM_000021.3 | Presenilin-1 (alzheimer disease 3) | 14q24.3 | AD |
| PSEN2 | NM_000447.2 | Presenilin-2 (alzheimer disease 4) | 1q31-q42 | AD |
| Other dementia-related genes | ||||
| AARS2 | NM_020745.3 | Alanyl-tRNA synthetase 2, mitochondrial | 6p21.1 | AR |
| ABCD1 | NM_000033.3 | ATP binding cassette subfamily D member 1 | Xq28 | XL |
| ARSA | NM_000487.5 | Arylsulfatase A | 22q13.33 | AR |
| CSF1R | NM_005211.3 | Colony-stimulating factor 1 receptor | 5q32 | AD |
| DARS2 | NM_018122.4 | Aspartyl-tRNA synthetase 2, mitochondrial | 1q25.1 | AR |
| EIF2B1 | NM_001414.3 | Eukaryotic translation initiation factor 2B subunit alpha | 12q24.31 | AR |
| EIF2B2 | NM_014239.3 | Eukaryotic translation initiation factor 2B subunit beta | 14q24.3 | AR |
| EIF2B3 | NM_020365.4 | Eukaryotic translation initiation factor 2B subunit gamma | 1p34.1 | AR |
| EIF2B4 | NM_015636.3 | Eukaryotic translation initiation factor 2B subunit delta | 2p23.3 | AR |
| EIF2B5 | NM_003907.2 | Eukaryotic translation initiation factor 2B subunit epsilon | 3q27.1 | AR |
| GALC | NM_000153.3 | Galactosylceramidase | 14q31.3 | AR |
| GBA | NM_001005741.2 | Glucosidase, beta acid | 1q21 | AD, susceptibility |
| GLA | NM_000169.2 | Galactosidase alpha | Xq22.1 | XL |
| ITM2B | NM_021999.5 | Integral membrane protein 2B | 13q14.2 | AD |
| NOTCH3 | NM_000435.2 | Notch 3 | 19q13.12 | AD |
| SNCB | NM_001001502.1 | Synuclein, beta | 5q35 | AD |
| TYROBP | NM_003332.3 | TYRO protein tyrosine kinase binding protein | 19q13.12 | AR |
AD, autosomal dominant; AR, autosomal recessive; XL, X-linked.
RESULTS
Demographic and clinical findings
A total of 72 patients (35 males and 37 females) with FTD were enrolled, including 38 with bvFTD, 26 with svPPA, six with nfvPPA, and two with FTD-ALS. One patient with FTD-ALS presented with abnormal behavior and the other presented with dysarthria followed by behavioral changes. The mean age was 65.8±10.3 years, and the mean age at disease onset was 63.9±11.5 years. The mean interval between disease onset and enrollment into the study was 3.6±2.3 years. Seventeen patients (23.6%) had a history of dementia or neuropsychiatric disease in a first-degree relative. The detailed demographic data is summarized in Table 2. During the preparation of this manuscript, a parallel study on telomere length in FTD syndrome was published by our group [14]. Of the 72 participants, 40 patients with sufficient DNA available for the Southern blotting analysis for telomere length after WES, were included in the parallel study.
Table 2
Demographics of patients
| Total | bvFTD | svPPA | nfvPPA | FTD-ALS | |
| Number | 72 | 38 | 26 | 6 | 2 |
| Age (y) | 65.8±10.3 | 64.3±11.5 | 67.3±7.1 | 69.8±11.9 | 61.0±21.2 |
| Onset Age (y) | 61.9±10.6 | 60.5±12.0 | 63.3±7.3 | 66.2±13.4 | 60.5±21.9 |
| Sex (M:F) | 35:37 | 20:18 | 12:14 | 2:4 | 1:1 |
| Interval between onset and enrollment (y) | 3.6±2.3 | 3.6±2.0 | 4.0±2.8 | 2.7±2.0 | 0.6±0.6 |
| FTD-CDR (SB) | 7.5±5.2 | 8.0±5.5 | 7.5±5.2 | 6.0±3.9 | 4.3±3.2 |
| MMSE | 17.2±9.1 | 18.5±8.0 | 15.0±10.7 | 16.2±8.9 | 25.5±4.9 |
| Family history* | 23.6% (17/72) | 23.7% (9/38) | 23.1% (6/26) | 33.3% (2/6) | 0% (0/2) |
ALS, amyotrophic lateral sclerosis; bvFTD, behavioral variant frontotemporal dementia; CDR, Clinical Dementia Rating; F, female; MMSE, Mini-Mental State Examination; M, male; nfvPPA, non-fluent/agrammatic variant primary progressive aphasia; SB, sum of boxes; svPPA, semantic variant primary progressive aphasia. *Family history of dementia or neuropsychiatric disease in first-degree relatives.
Genetic findings
WES yielded an average read depth of 121.81x and the average for 10× coverage was 99.33%. After variant analysis, one likely pathogenic variant in the MAPT gene, c.2116G>A (p.G706R), was identified in a patient with bvFTD (FTD-18), which has been reported in familial or sporadic FTD [15]. FTD-18 had H1/H1 haplotypes of the MAPT gene and had been enrolled in an FTD modeling study using induced pluripotent stem cell (iPSC) technology and had been reported elsewhere [16]. The four variants of uncertain significance (VUSs) from ALS-FTD-related genes were also detected in four patients with FTD: PRNP gene, c.695T>G (p.M232R) in two patients with bvFTD (FTD-13, FTD-39), VCP gene, c.278G>T (p.R93L) in a patient with svPPA, and UBQLN2 gene, c.20G>A (p.S7N) in a patient with bvFTD (Table 3). Of these, p.M232R of the PRNP gene has been reported previously in Creutzfeldt–Jacob disease (CJD) [17], but its pathogenicity has been controversial. p.R93L of the VCP gene and p.S7N of the UBQLN2 gene were novel variants. None of the patients had abnormal repeat expansions of C9orf72. Regarding genes related to familial Alzheimer’s disease and other dementias, nine VUSs were identified from seven patients and one of them, p.L136S of the APP gene, was novel (Table 3).
Table 3
Variants of uncertain significance in ALS-FTD, familial Alzheimer’s disease, and other dementia related genes
| Patient ID | Gene | Reference sequence | Nucleotide change | Amino acid change | ClinVar | rs number | Allele frequency | In-silico analysis | ||||
| gnomAD† | KRGDB‡ | Poly Phen-2 | SIFT | Mutation Taster | CADD§ | |||||||
| ALS-FTD related genes | ||||||||||||
| FTD-13 | PRNP | NM_000311.3 | c.695T>G | p.Met232Arg | VUS | rs74315409 | 0.0009 | 0.0040 | B | D | P | <20 |
| FTD-39 | PRNP | NM_000311.3 | c.695T>G | p.Met232Arg | VUS | rs74315409 | 0.0009 | 0.0040 | B | D | P | <20 |
| FTD-36 | VCP | NM_007126.3 | c.278G>T | p.Arg93Leu | N/A | N/A | 0 | 0 | D | D | DC | 29.90 |
| FTD-48 | UBQLN2 | NM_013444.3 | c.20G>A | p.Ser7Asn | N/A | N/A | 0 | 0 | B | T | P | <20 |
| Genes of familial Alzheimer’s disease | ||||||||||||
| FTD-48 | PSEN2 | NM_000447.3 | c.1262C>T | p.Thr421Met | VUS | rs756609078 | 0.0001 | 0 | PD | T | DC | 34.00 |
| FTD-61 | APP | NM_000484.4 | c.407T>C | p.Leu136Ser | N/A | N/A | 0 | 0 | PD | D | DC | 25.10 |
| Other dementia related genes | ||||||||||||
| FTD-41 | CSF1R | NM_005211.3 | c.110C>T | p.Thr37Met | B* | rs139635308 | 0.0004 | 0 | PD | T | P | <20 |
| FTD-19 | GBA | NM_000157.4 | c.902G>A | p.Arg301His | N/A | rs140955685 | 0.0003 | 0.0009 | B | T | P | <20 |
| FTD-39 | ITB2B | NM_021999.4 | c.454-3del | – | N/A | rs747826043 | <0.0001 | 0.0009 | N/A | N/A | N/A | N/A |
| FTD-71 | ITB2B | NM_021999.4 | c.454-3del | – | N/A | rs747826043 | <0.0001 | 0.0009 | N/A | N/A | N/A | N/A |
| FTD-32 | NOTCH3 | NM_000435.2 | c.5336G>T | p.Gly1779Val | N/A | rs771041592 | 0.0001 | 0.0009 | B | T | DC | 24.80 |
| FTD-39 | NOTCH3 | NM_000435.2 | c.6097C>A | p.Pro2033Thr | VUS | rs375213868 | <0.0001 | 0 | PD | D | DC | 23.80 |
| FTD-48 | GALC | NM_000153.4 | c.1912G>A | p.Gly638Ser | VUS | rs769851272 | 0.0009 | 0.0009 | PD | D | DC | 25.10 |
N/A, not applicable; VUS, variant of uncertain significance; PD, probably damaging; B, benign; D, deleterious; T, tolerable; DC, disease causing; P, polymorphism; CADD, Combined Annotation Dependent Depletion. *The variant was submitted by single institute without criteria (accessed on 28 July 2022). †gnomAD, gnome Aggregation Database (http://gnomad.broadinstitute.org/). ‡KRGDB, the Korean Reference Genome Database [11]. §The variants over the CADD score 20 are presented in bold. The variants were classified according to the guideline of ACMG [12].
Case carrying p.G706R in the MAPT gene
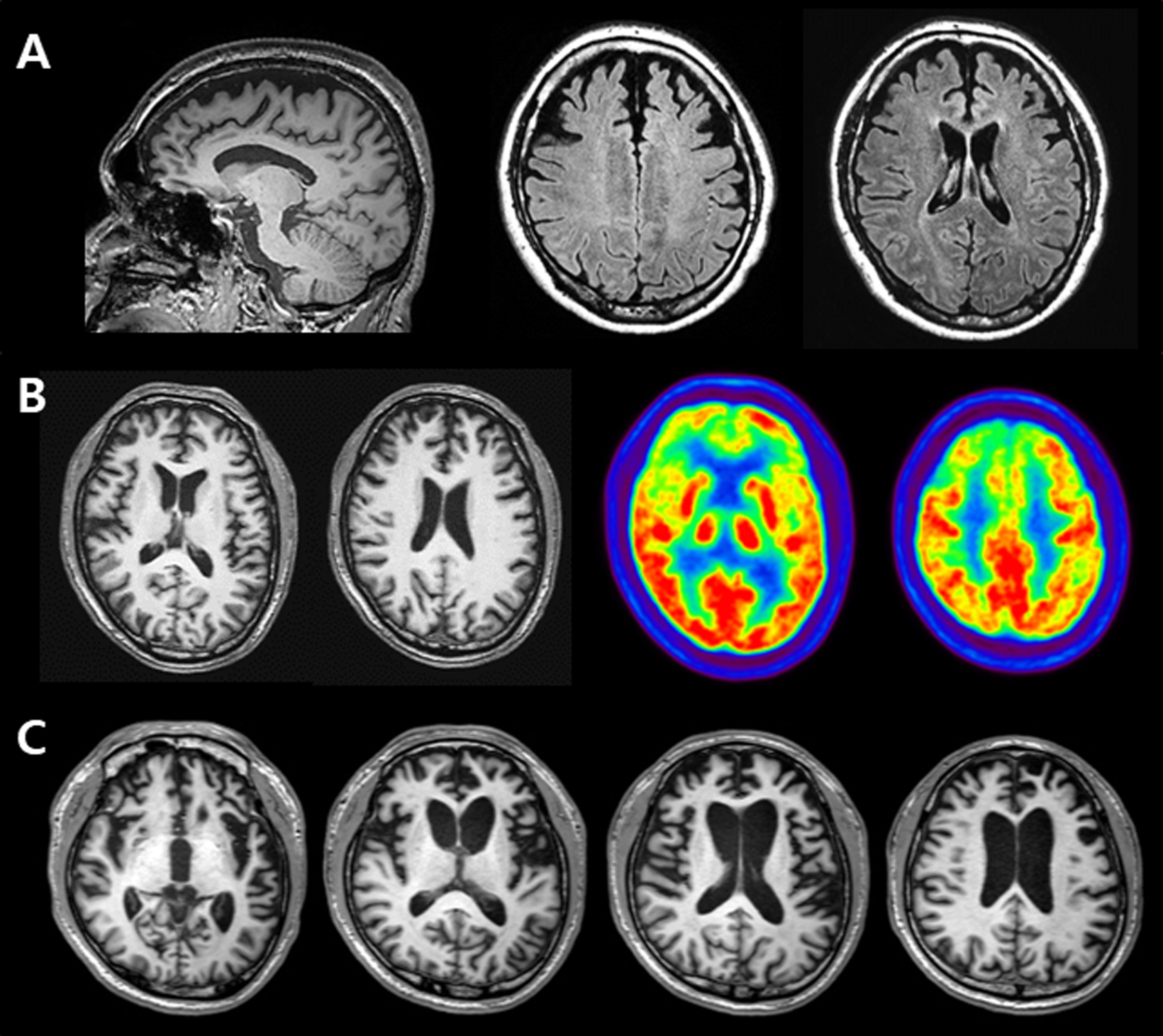
FTD-18 was a 32-year-old man with a three-year history of progressive behavioral changes and cognitive deficits. His symptoms included impatience, aggression, and emotional blunting. The patient became more inert and depressed with time. He also exhibited hypersexuality and an obsession with sweet foods and computer games. Episodic memory impairment, visuospatial dysfunction, language difficulties, and decline in personal hygiene and activities of daily living developed around the time of his first visit to the neurology clinic. His maternal grandfather had dementia at the age of 60. Neurological examinations were unremarkable. Neuropsychological evaluation revealed severely impaired memory, visuospatial, language, and executive functions. The MMSE score was 20 and the clinical dementia rating was 1. Brain MRI demonstrated bilateral and symmetric frontal atrophy (Fig. 1A). Florbetaben and flortaucipir PET revealed negative findings. The clinical syndromic diagnosis was bvFTD. His symptoms progressed rapidly, and two years after the initial evaluation, he was transferred to another hospital.
Fig. 1
A) Brain MRIs of FTD-18 who carried variant p.G706R of the MAPT gene, revealing bilateral and symmetric frontal atrophy. B) Brain MRIs and FDG-PET images of FTD-13 who carried variant M232R of the PRNP gene, showing cortical atrophy and severe glucose hypometabolism in the bifrontal areas. C) Brain MRIs of FTD-39 who carried variant M232R of the PRNP gene, demonstrating prominent bilateral frontotemporal atrophy, worse on the left.
Cases carrying p.M232R in the PRNP gene
FTD-13 was a 54-year-old man who worked as a construction manager. He was admitted to the psychiatric ward with rapidly progressive behavioral changes and memory impairment for six months. He repeatedly called friends to borrow money, although he was financially stable; he could not wait his turn at restaurants for food to be put on his plate; he obsessively called his son, until he convinced him to have lunch together; he woke his son up at 2 am to visit his parents’ grave and repeatedly asked the same questions. However, he was able to maintain normal daily living activities. Neurological examinations showed normal findings. The MMSE score was 26 and the global deterioration score (GDS) was 3. Neuropsychological test results showed impaired naming, memory, and frontal function. The patient had no family history of dementia. Initial brain MRI revealed prominent atrophy in bilateral frontal lobes. Diffusion-restricted areas were not detected on diffusion-weighted imaging (DWI). The follow-up MRIs, taken at one-month intervals, showed the same findings as the previous ones. Fluorodeoxyglucose-positron emission tomography (FDG-PET) demonstrated severe glucose hypometabolism in the bilateral frontal areas, which worsened on the right side (Fig. 1B). He was diagnosed with bvFTD and transferred to another hospital after six months of follow-ups.
FTD-39 was a 51-year-old man with a two-year history of behavioral changes. He was eating 15 meals per day due to increased appetite and gained 22 kg in a year; he went to the bathroom frequently, over 15 times a day; he provided short answers to questions and refused to take a bath; he showed poor spontaneity and sometimes talked to himself. Memory impairment, visuospatial dysfunction, language difficulties, and disorientation also developed in the patient. There was no family history of dementia. Neurological examinations did not reveal any significant changes. Initial MMSE score was 24, and the GDS was 4. However, brain MRI revealed bilateral frontotemporal atrophy, which worsened on the left side. Initially, he was diagnosed with schizophrenia in a psychiatric clinic and was referred to a neurology clinic where he was diagnosed with bvFTD. The 9-month follow-up MMSE score was 17, and the GDS was 5. Follow-up MRIs showed aggravated cortical atrophy in both frontal and temporal areas (Fig. 1C). After almost three years of follow-ups, the patient stopped visiting the hospital.
Cases carrying novel variants (p.R93L of the VCP gene, p.S7N of the UBQLN2 gene, and p.L136S of the APP gene)
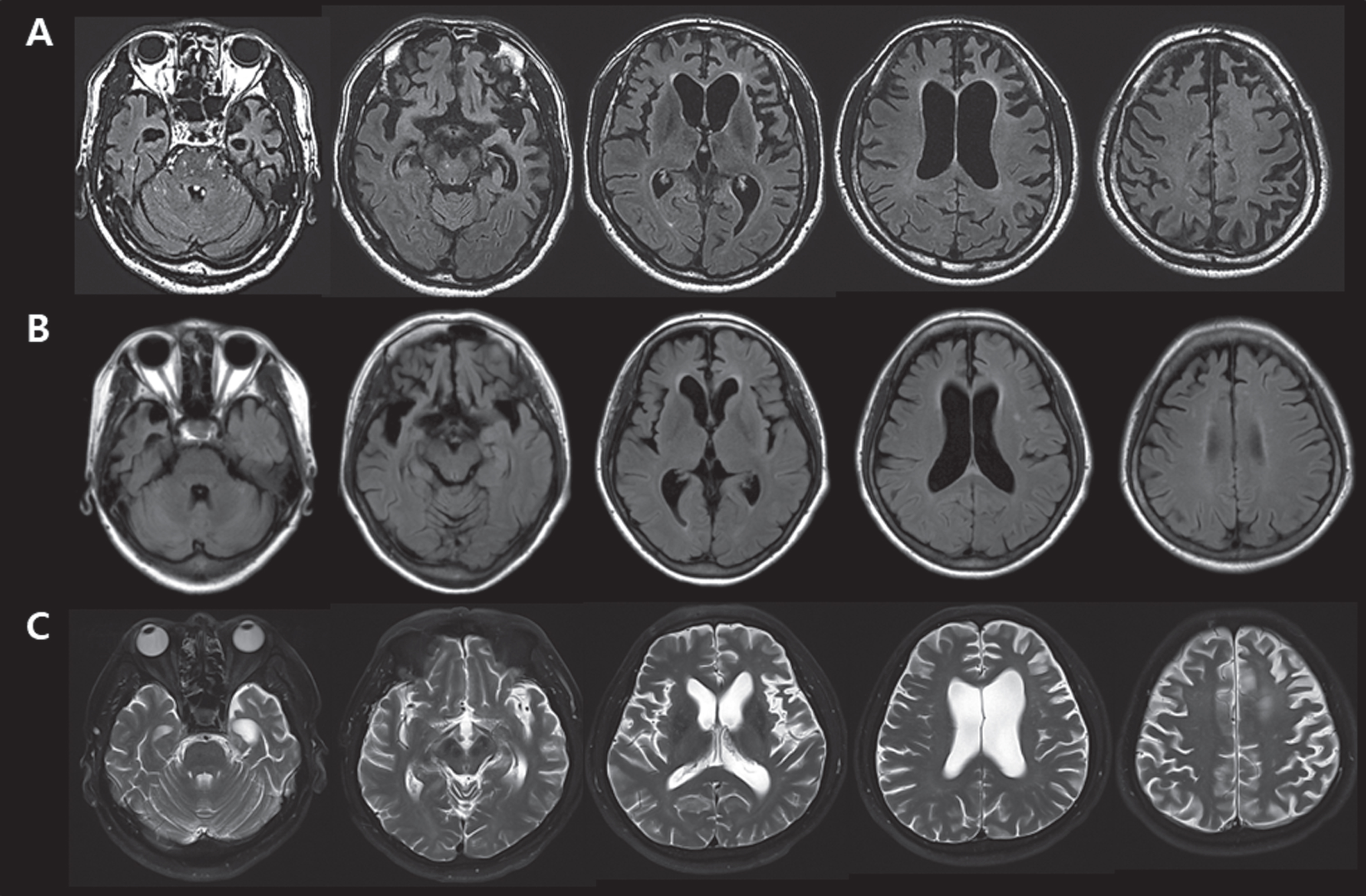
FTD-36 with p.R93L of the VCP gene was a 50-year-old man, whose illness began at the age of 45; he presented memory impairment, followed by fluent aphasia, disinhibition, and myoclonic seizures. Brain MRI showed severe bi-frontotemporal atrophy, worse on the left side, and left parietal atrophy (Fig. 2A). Electroencephalography revealed partial seizures arising in both frontal areas. A clinical syndromic diagnosis of svPPA was made. His mother had a stroke. FTD-48 with p.S7N of the UBQLN2 gene was a 59-year-old woman, whose personality changes including apathy, indifference, loss of empathy, obsession, and disinhibition started at the age of 57. In contrast to the behavioral changes, her memory, language, and visuospatial functions were relatively preserved and her APOE genotype was ɛ3/ɛ4. Brain MRI revealed right asymmetric frontotemporal atrophy (Fig. 2B). The clinical syndromic diagnosis was bvFTD. The patient’s family history was unremarkable. This patient carried two more VUS: p.T421M of the PSEN2 gene and p.G638S of the GALC gene. The p.T421M of PSEN2 has been reported in an early onset sporadic AD patient, carrying APOE ɛ4/ɛ4, from Japan [18]. FTD-61 with p.L136S of the APP gene was a 66-year-old man whose illness began at the age of 58; he presented with apathy, disinhibition, and hyperorality, followed by right-side rigidity and gait disturbances at the age of 60. A brain MRI demonstrated prominent atrophy in the bifrontal and left temporal areas (Fig. 2C). A clinical syndromic diagnosis of bvFTD was made. Notably, his father had Parkinson’s disease.
Fig. 2
A) Brain MRIs of FTD-36 who carried variant p.R93L of the VCP gene, showing bi-frontotemporal atrophy (worse on the left) and left parietal atrophy. B) Brain MRIs of FTD-48 who carried variant p.S7N of the UBQLN2 gene, variant p.T421M of the PSEN2 gene and variant p.G638S of the GALC gene, revealing right asymmetric frontotemporal atrophy. C) Brain MRIs of FTD-61 who carried variant p.L136S of the APP gene, demonstrating prominent atrophy in the bifrontal and left temporal area.
DISCUSSION
Pathogenic variants detected in common FTD genes, such as MAPT, GRN, and C9orf72, are generally rare in Asian populations. Our first study screening these in 75 Korean patients with sporadic FTD and the subsequent testing on multiple genes using NGS in 107 Korean patients with FTD confirmed the ethnic or geographical variability of the mutations in known FTD genes [5, 6]. In the present study, which is in accordance with previous studies, we identified only one patient with bvFTD harboring p.G706R in the MAPT gene.
The p.G706R variant, traditionally known as the G389R mutation in the MAPT gene, has been associated with rapidly progressive young-onset FTD, which was similar to those of our FTD-18 [15, 19–21]. This variant revealed the possibility of incomplete penetrance based on a lack of autosomal dominant inheritance patterns or unaffected mutation carriers, which was also observed in FTD-18, whose parents were clinically normal [19–22]. In vitro study, the p.G706R variant altered the affinity of tau to microtubules and decreased its ability to enhance microtubule assembly [15, 23]. MAPT p.P513A and p.L266V variants have recently been observed in two Korean patients with early onset Alzheimer’s disease and nfvPPA, respectively [24, 25].
Of the 13 VUSs we found in this study, the p.M232R variant of the PRNP gene is one of the five most frequent CJD mutations in Japan [26] and has mostly been reported in Asian populations [17, 27, 28]. The clinical characteristics of p.M232R are similar to those of sporadic CJD, including progressive dementia, 14-3-3 protein positivity, DWI hyperintensity, and no family history [26]. Previously, reported Korean cases of p.M232R were all suspected to be sporadic CJD based on their clinical symptoms at the time of diagnosis [29]. Notably, FTD-13 and FTD-39 in this study presented with frontal behavioral abnormalities associated with prominent bifrontal atrophy or hypometabolism, leading to the clinical diagnosis of bvFTD. Although the clinical course of FTD-13 and FTD-39 progressed somewhat rapidly, all DWIs repeatedly performed at one-month intervals were negative. Since the DWI negative p.M232R cases were presented recently [30], it is possible that both patients might have developed myoclonus, which is a typical feature of CJD, or demonstrated periodic sharp and wave complexes on EEG or positive DWIs later on.
As mentioned earlier, some researchers are concerned about the questionable pathogenicity of p.M232R since it has also been detected in healthy controls or non-CJD patients [31]. In addition to this, the allele frequency of the variant was 0.0008 in the East Asian cohort of gnomAD and 0.0040 in the ethnic-matched control of KRGDB, which is higher than the annual incidence rate of prion disease (0.85 per million population) [26]. Thus, pathological confirmation is required to resolve whether the p.M232R variant found in our patients with bvFTD is pathogenic or rare.
A recently published large international study of genetic FTD showed that approximately 25–30% of patients with FTD syndrome harbored pathogenic variants of 40% in C9orf72, 35% in GRN, 25% in MAPT, and only 1-2% in other genes [32]. Another study from the North American FTD cohort found 31 different pathogenic variants within the three main FTD genes in 223 of 302 sporadic and 390 familial participants (32.2%) [3]. However, our group has only identified two pathogenic variants in the three main FTD genes (one for MAPT and one for GRN) through CREDOS-FTD genetic studies (approximately 1%) [5, 6]. Given the extreme rarity of genetic FTD in Korea, genetic screening of sporadic and familial FTD through a longitudinal Korean cohort is needed to better understand the geographical or ethnic variability of genetic FTD. Furthermore, collaborating with worldwide genetic FTD cohorts would encourage the development of powerful biomarkers and construct trial-ready cohorts for new FTD syndrome therapies, eventually leading to its prevention.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This research was supported by a fund (2018-ER6203-02) by Research of Korea Disease Control and Prevention Agency and the “National Institute of Health” research project (project No. 2021-ER1004-00, 2021-ER1004-01).
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Bang J , Spina S , Miller BL ((2015) ) Frontotemporal dementia. Lancet 386: , 1672–1682. |
[2] | Rohrer JD , Guerreiro R , Vandrovcova J , Uphill J , Reiman D , Beck J , Isaacs AM , Authier A , Ferrari R , Fox NC , Mackenzie IR , Warren JD , de Silva R , Holton J , Revesz T , Hardy J , Mead S , Rossor MN ((2009) ) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73: , 1451–1456. |
[3] | Ramos EM , Dokuru DR , Van Berlo V , Wojta K , Wang Q , Huang AY , Miller ZA , Karydas AM , Bigio EH , Rogalski E , Weintraub S , Rader B , Miller BL , Gorno-Tempini ML , Mesulam MM , Coppola G ((2020) ) Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimers Dement 16: , 118–130. |
[4] | Greaves CV , Rohrer JD ((2019) ) An update on genetic frontotemporal dementia. J Neurol 266: , 2075–2086. |
[5] | Kim EJ , Kwon JC , Park KH , Park KW , Lee JH , Choi SH , Jeong JH , Kim BC , Yoon SJ , Yoon YC , Kim S , Park KC , Choi BO , Na DL , Ki CS , Kim SH ((2014) ) Clinical and genetic analysis of MAPT, GRN, and C9orf72 genes in Korean patients with frontotemporal dementia. Neurobiol Aging 35: , 1213.e13–17. |
[6] | Kim EJ , Kim YE , Jang JH , Cho EH , Na DL , Seo SW , Jung NY , Jeong JH , Kwon JC , Park KH , Park KW , Lee JH , Roh JH , Kim HJ , Yoon SJ , Choi SH , Jang JW , Ki CS , Kim SH ((2018) ) Analysis of frontotemporal dementia, amyotrophic lateral sclerosis, and other dementia-related genes in 107 Korean patients with frontotemporal dementia. Neurobiol Aging 72: , 186.e1–e7. |
[7] | Knopman DS , Kramer JH , Boeve BF , Caselli RJ , Graff-Radford NR , Mendez MF , Miller BL , Mercaldo N ((2008) ) Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 131: , 2957–2968. |
[8] | Rascovsky K , Hodges JR , Knopman D , Mendez MF , Kramer JH , Neuhaus J , van Swieten JC , Seelaar H , Dopper EG , Onyike CU , Hillis AE , Josephs KA , Boeve BF , Kertesz A , Seeley WW , Rankin KP , Johnson JK , Gorno-Tempini ML , Rosen H , Prioleau-Latham CE , Lee A , Kipps CM , Lillo P , Piguet O , Rohrer JD , Rossor MN , Warren JD , Fox NC , Galasko D , Salmon DP , Black SE , Mesulam M , Weintraub S , Dickerson BC , Diehl-Schmid J , Pasquier F , Deramecourt V , Lebert F , Pijnenburg Y , Chow TW , Manes F , Grafman J , Cappa SF , Freedman M , Grossman M , Miller BL ((2011) ) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134: , 2456–2477. |
[9] | Gorno-Tempini ML , Hillis AE , Weintraub S , Kertesz A , Mendez M , Cappa SF , Ogar JM , Rohrer JD , Black S , Boeve BF , Manes F , Dronkers NF , Vandenberghe R , Rascovsky K , Patterson K , Miller BL , Knopman DS , Hodges JR , Mesulam MM , Grossman M ((2011) ) Classification of primary progressive aphasia and its variants. Neurology 76: , 1006–1014. |
[10] | Kim EJ , Park KW , Lee JH , Choi S , Jeong JH , Yoon SJ , Kim BC , Kwon JC , Ku BD , Kim SH , Choi BO , Na DL ((2014) ) Clinical and neuropsychological characteristics of a nationwide hospital-based registry of frontotemporal dementia patients in Korea: A CREDOS-FTD Study. Dement Geriatr Cogn Dis Extra 4: , 242–251. |
[11] | Jung KS , Hong KW , Jo HY , Choi J , Ban HJ , Cho SB , Chung M ((2020) ) KRGDB: The large-scale variant database of 1722 Koreans based on whole genome sequencing. Database (Oxford) 2020: , baz146. |
[12] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL ; ACMG Laboratory Quality Assurance Committee ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[13] | Jang JH , Kwon MJ , Choi WJ , Oh KW , Koh SH , Ki CS , Kim SH ((2013) ) Analysis of the C9orf72 hexanucleotide repeat expansion in Korean patients with familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 34: , 1311.e7–e9. |
[14] | Kim EJ , Koh SH , Ha J , Na DL , Seo SW , Kim HJ , Park KW , Lee JH , Roh JH , Kwon JC , Yoon SJ , Jung NY , Jeong JH , Jang JW , Kim HJ , Park KH , Choi SH , Kim S , Park YH , Kim BC , Kim YE , Kwon HS , Park HH , Jin JH ((2021) ) Increased telomere length in patients with frontotemporal dementia syndrome. J Neurol Sci 428: , 117565. |
[15] | Murrell JR , Spillantini MG , Zolo P , Guazzelli M , Smith MJ , Hasegawa M , Redi F , Crowther RA , Pietrini P , Ghetti B , Goedert M ((1999) ) Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol 58: , 1207–1226. |
[16] | Kim M , Kim HJ , Koh W , Li L , Heo H , Cho H , Lyoo CH , Seo SW , Kim EJ , Nakanishi M , Na DL , Song J ((2020) ) Modeling of frontotemporal dementia using iPSC technology. Int J Mol Sci 21: , 5319. |
[17] | Kitamoto T , Ohta M , Doh-ura K , Hitoshi S , Terao Y , Tateishi J ((1993) ) Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun 191: , 709–714. |
[18] | Yagi R , Miyamoto R , Morino H , Izumi Y , Kuramochi M , Kurashige T , Maruyama H , Mizuno N , Kurihara H , Kawakami H ((2014) ) Detecting gene mutations in Japanese Alzheimer’s patients by semiconductor sequencing. Neurobiol Aging 35: , 1780.e1–e5. |
[19] | Bermingham N , Cowie TF , Paine M , Storey E , McLean C ((2008) ) Frontotemporal dementia and Parkinsonism linked to chromosome 17 in a young Australian patient with the G389R Tau mutation. Neuropathol Appl Neurobiol 34: , 366–370. |
[20] | Chaunu MP , Deramecourt V , Buée-Scherrer V , Le Ber I , Brice A , Ehrle N , El Hachimi K , Pluot M , Maurage CA , Bakchine S , Buée L ((2013) ) Juvenile frontotemporal dementia with parkinsonism associated with tau mutation G389R. J Alzheimers Dis 37: , 769–776. |
[21] | Sun L , Chen K , Li X , Xiao S ((2017) ) Rapidly progressive frontotemporal dementia associated with MAPT mutation G389R. J Alzheimers Dis 55: , 777–785. |
[22] | Rossi G , Marelli C , Farina L , Laurà M , Maria Basile A , Ciano C , Tagliavini F , Pareyson D ((2008) ) The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov Disord 23: , 892–895. |
[23] | Niewidok B , Igaev M , Sündermann F , Janning D , Bakota L , Brandt R ((2016) ) Presence of a carboxy-terminal pseudorepeat and disease-like pseudohyperphosphorylation critically influence tau’s interaction with microtubules in axon-like processes. Mol Biol Cell 27: , 3537–3549. |
[24] | Giau VV , Senanarong V , Bagyinszky E , An SSA , Kim S ((2019) ) Analysis of 50 neurodegenerative genes in clinically diagnosed early-onset Alzheimer’s disease. Int J Mol Sci 20: , 1514. |
[25] | Sung W , Kim YE , Kim SH ((2021) ) Hereditary frontotemporal dementia linked to the pathogenic p.L266V variant of the MAPT gene in Korea. J Clin Neurol 17: , 478–480. |
[26] | Nozaki I , Hamaguchi T , Sanjo N , Noguchi-Shinohara M , Sakai K , Nakamura Y , Sato T , Kitamoto T , Mizusawa H , Moriwaka F , Shiga Y , Kuroiwa Y , Nishizawa M , Kuzuhara S , Inuzuka T , Takeda M , Kuroda S , Abe K , Murai H , Murayama S , Tateishi J , Takumi I , Shirabe S , Harada M , Sadakane A , Yamada M ((2010) ) Prospective 10-year surveillance of human prion diseases in Japan. Brain 133: , 3043–3057. |
[27] | Takada LT , Kim MO , Cleveland RW , Wong K , Forner SA , Gala II , Fong JC , Geschwind MD ((2017) ) Genetic prion disease: Experience of a rapidly progressive dementia center in the United States and a review of the literature. Am J Med Genet B Neuropsychiatr Genet 174: , 36–69. |
[28] | Bagyinszky E , Giau VV , Youn YC , An SSA , Kim S ((2018) ) Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr Dis Treat 14: , 2067–2085. |
[29] | Choi BY , Kim SY , Seo SY , An SS , Kim S , Park SE , Lee SH , Choi YJ , Kim SJ , Kim CK , Park JS , Ju YR ((2009) ) Mutations at codons 178, 200-129, and 232 contributed to the inherited prion diseases in Korean patients. BMC Infect Dis 9: , 132. |
[30] | Kang YJ , Kim KH , Jang SH , Lee GH , Lee YJ , Kim YS , Kim EJ ((2019) ) Diffusion-weighted imaging negative M232R familial Creutzfeldt-Jakob disease. J Clin Neurosci 64: , 47–49. |
[31] | Beck J , Collinge J , Mead S ((2012) ) Prion protein gene M232R variation is probably an uncommon polymorphism rather than a pathogenic mutation. Brain 135: , e209. |
[32] | Moore KM , Nicholas J , Grossman M , McMillan CT , Irwin DJ , Massimo L , Van Deerlin VM , Warren JD , Fox NC , Rossor MN , Mead S , Bocchetta M , Boeve BF , Knopman DS , Graff-Radford NR , Forsberg LK , Rademakers R , Wszolek ZK , van Swieten JC , Jiskoot LC , Meeter LH , Dopper EG , Papma JM , Snowden JS , Saxon J , Jones M , Pickering-Brown S , Le Ber I , Camuzat A , Brice A , Caroppo P , Ghidoni R , Pievani M , Benussi L , Binetti G , Dickerson BC , Lucente D , Krivensky S , Graff C , Öijerstedt L , Fallström M , Thonberg H , Ghoshal N , Morris JC , Borroni B , Benussi A , Padovani A , Galimberti D , Scarpini E , Fumagalli GG , Mackenzie IR , Hsiung GR , Sengdy P , Boxer AL , Rosen H , Taylor JB , Synofzik M , Wilke C , Sulzer P , Hodges JR , Halliday G , Kwok J , Sanchez-Valle R , Lladó A , Borrego-Ecija S , Santana I , Almeida MR , Tábuas-Pereira M , Moreno F , Barandiaran M , Indakoetxea B , Levin J , Danek A , Rowe JB , Cope TE , Otto M , Anderl-Straub S , de Mendonça A , Maruta C , Masellis M , Black SE , Couratier P , Lautrette G , Huey ED , Sorbi S , Nacmias B , Laforce R Jr , Tremblay ML , Vandenberghe R , Damme PV , Rogalski EJ , Weintraub S , Gerhard A , Onyike CU , Ducharme S , Papageorgiou SG , Ng ASL , Brodtmann A , Finger E , Guerreiro R , Bras J , Rohrer JD ; FTD Prevention Initiative ((2020) ) Age at symptom onset and death and disease duration in genetic frontotemporal dementia: An international retrospective cohort study. Lancet Neurol 19: , 145–156. |


