
Lithium Benzoate Exerts Neuroprotective Effect by Improving Mitochondrial Function, Attenuating Reactive Oxygen Species, and Protecting Cognition and Memory in an Animal Model of Alzheimer’s Disease
Abstract
Background:
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disease affecting many cellular pathways, including protein aggregation, mitochondrial dysfunction, oxidative stress (OS), and neuroinflammation. Currently, no effective treatment for AD exists.
Objective:
We aim to determine the effect of lithium benzoate (LiBen) in protecting neurons from amyloid-β (Aβ) or other neurotoxin insults.
Methods:
Primary rat cortical neurons co-treated with neurotoxins and LiBen were used to examine its effect in cell viability, reactive oxygen species (ROS) clearance, and mitochondrial functions by MTT, CellRox fluorescence staining, and seahorse assay. Then, Barnes maze and prepulse inhibition test were performed in APP/PS1 mice that received chronic LiBen treatment to assess its effect on cognitive protection. Oral bioavailability of LiBen was also assessed by pharmacokinetic study in rat plasma.
Results:
In this study, we discovered that LiBen can attenuate cellular ROS level, improve mitochondrial function, increase cell viability against multiple different insults of mitochondrial dysfunction, Aβ accumulation, and neuroinflammation, and promote neurogenesis. We demonstrated that LiBen has advantages over lithium or sodium benzoate alone as LiBen displays superior neuroprotective efficacy and oral bioavailability than the other two agents when being applied either alone or in combination. Furthermore, chronic administration of LiBen showed protection for cognition as well as spatial memory and reduced the senile plaque deposition in brains of AD animal models.
Conclusion:
LiBen stands as a promising therapeutic agent for improving cognition and delaying the progression of AD.
INTRODUCTION
Alzheimer’s disease (AD), the most common type of dementia, is a neurodegenerative disease associated with progressive cognitive decline, without effective course-alteration treatment to impede the disease progression. AD is characterized by extracellular amyloid-β (Aβ) plaques composed of insoluble Aβ fibrils, and by intraneuronal deposits of neurofibrillary tangles (NFTs) constituted by hyperphosphorylated tau protein. Over the past decades, the therapeutic strategy for AD has been focused mostly on Aβ aggregation. Unfortunately, no signs of promising improvement have been shown. Therefore, the need to develop therapeutic to prevent and/or slow the progression of AD beyond the original Aβ hypothesis is urgent.
AD is a multifactorial disease caused by a combination of genetic, lifestyle, and environmental factors which affects brain over time. Most AD cases (90-95%) occur sporadically, while the remainder are familial. Although the etiology of sporadic AD is poorly understood, accumulative evidence suggests an important role of mitochondrial function and oxidative stress (OS) in the pathogenesis of the sporadic AD [1–3]. OS and Aβ are interactive since Aβ aggregation induces OS, and vice versa [4–9]. OS increases with age due to the imbalance in the generation of reactive oxygen species (ROS) and the dysfunction of the antioxidant system [10]. The brain is particularly vulnerable to OS-induced damage because of its high energy and oxygen consumption. Thus, the disturbance of the energy metabolism in the brain can lead to central nervous system diseases [1, 11]. The accumulation of ROS will result in oxidative modification and dysfunction of the major cell components including nucleic acids, lipids, and proteins. Indeed, the oxidative modified products of nucleic acid (e.g., 8-hydroxydeoxyguanosine and 8-hydroxyguanosine), proteins (e.g., 3-nitrotyrosine and protein carbonyls), and products of lipid peroxidation (e.g., 4-hydroxynonenal and malondialdehyde) are elevated in AD brains [12, 13]. Furthermore, mitochondria are considered as cellular power plants that generate most of adenosine triphosphate (ATP) which is a source of chemical energy for brain cells. Dysfunction of mitochondria is known to generate ROS and reduce mitochondrial ATP production, which is associated with neurodegeneration [14]. Therefore, targeting mitochondria to attenuate the ROS level has potential therapeutic benefits for AD.
To alleviate ROS stress, lithium, a well-known mood stabilizer, is a potential therapeutic option with neuroprotective and neurogenic properties [15]. Preclinical studies demonstrated that lithium can prevent neurons from oxidative damage across species, including hypoxia-induced Aβ-expression in flies [16], cerebral ischemia and 3-nitropropionic acid (3-NP)-induced neurotoxicity in rats [17, 18], and kainate and neuropeptide S-induced behavioral or neurological abnormalities in mice [19, 20]. Moreover, growing evidence suggests that lithium can reduce Aβ accumulation in brains through multiple mechanisms, including inhibiting Aβ protein synthesis [21], preventing Aβ maturation [22], and enhancing Aβ efflux across the blood-brain barrier [23]. In clinical trials, although short-term lithium treatment (10 weeks) did not show beneficial effect in AD patients [24], prolonged treatment, at least for one year, revealed that lithium has multi-target functions, including modulating the cerebrospinal fluid level of Aβ, tau, and GSK-3, as well as stabilizing cognition in AD patients [25, 26]. However, despite the attractive benefits for AD, the lithium formulations, such as lithium carbonate and lithium citrate, have a narrow therapeutic range while the dose required for the treatment of AD is much higher beyond the non-toxic range, which impedes its therapeutic application in AD. Therefore, finding an optimal salt form of lithium, which itself is therapeutic, is critical to develop long-term lithium therapy in AD.
Sodium benzoate (SB) is a widely used additive and preservative of food [27]. US FDA and JECFA of WHO placed SB in the Generally-Recognized-as-Safe (GRAS) category. They established the acceptable daily intake level at 5 mg/kg body weight [27–29]. SB is a well-known competitive inhibitor of D-amino acid oxidase (DAAO) [30]. Low levels of D-serine are associated with the initial phases of neuronal death. Inhibition of DAAO in the brain through binding benzoate leads to reducing degradation of D-serine, thereby enhancing NMDAR function [31]. Some studies suggested that SB has neuropharmacological property [32, 33]. Age-related reduction of D-serine level has been found to be associated with AD [34]. More recently, SB has been reported to attenuate mitochondria-mediated OS and inhibit neuronal apoptosis in cell levels and animal models [35–37]. Treatment of SB is associated with reduction of Aβ burden in the hippocampus and protection of memory and learning in the transgenic mice model of AD [35]. In addition, the result of a double-blind, placebo-controlled clinical study showed that SB can correct the cognitive impairment in the early phase of AD [38].
Based on the preclinical and clinical data, we hypothesize that benzoate is a favorable salt form of lithium. Furthermore, lithium and benzoate together in lithium benzoate (LiBen) can provide synergistic mechanisms of action for the treatment of AD. In the present study, we first evaluate the protective effects of LiBen against 3-NP, Aβ, or hypoxia-induced neurotoxicity in primary cortical neurons, as well as assess whether LiBen possesses synergy of the dual mechanism of lithium and benzoate. We further analyze and compare the bioavailability of LiBen versus lithium carbonate and SB. Lastly, we investigate whether chronic administration of LiBen to APP/PS-1 double transgenic mice can elicit a reduction of amyloid deposition and a greater effect than lithium carbonate alone in delaying disease progression and improving cognition and memory.
MATERIALS AND METHODS
Chemicals
3-nitropropionic acid (3-NP; Sigma-Aldrich) was dissolved in phosphate-buffered saline (PBS) as a stock solution of 1 M and adjusted to pH 7.4 with 10 M sodium hydroxide. The 3-NP stock solution was then dispensed into 1 ml aliquots, protected from light, and stored at -20°C until use. LiBen (Chem-Impex Int’l. Inc.), lithium chloride (LiCl; Sigma-Aldrich), and SB (Merck) were all reconstituted in sterile ddH2O to make stock solutions of 50μM before stored at 4°C until use.
Aβ25-35 was prepared as the published article [39]. Aβ25-35 (Cat. No. A4559, Sigma-Aldrich) was dissolved in autoclaved ddH2O to make a stock solution of 2 mM, dispensed into aliquots, and immediately stored at -80°C until use. One day prior to experimentation, aliquots of Aβs were incubated at 37°C for 24 h to allow aggregation.
Primary fetal rat cortical cultures
Primary neuronal cultures were prepared from cortices of fetal rat brains at embryonic day 18 (E18) as previously described [40, 41]. Briefly, E18 fetal Sprague-Dawley rat brains were dissected to separate cortices. The brain tissues were mechanically triturated and then filtrated through a 70-μm filter (BD Falcon) for removing the tissue debris. The flow-through containing cortical cells was then centrifuged at 100×g for 10 min and the pellets were subsequently suspended in minimum essential medium with 10% fetal bovine serum (Hyclone) and transferred into appropriate culture vessels previously coated with poly-D-lysine (Sigma-Aldrich). After 4 h of incubation, the culture medium was replaced with neurobasal medium supplemented with B27 (GIBCO/Invitrogen Corporation) and maintained at 37°C in a humidified incubator with 5% CO2 for at least 4 days. Cortical cells were used for experiments between days-in-vitro 7–9. The cortical culture used in the present study was a neuron-enriched co-culture system as evidenced by more cells immunostained with antibodies recognizing microtubule-associated protein-2 (approximately 85%) than with glial fibrillary acidic protein (approximately 5–15%), the respective cellular marker for neurons and astrocytes [40].
Hoechst staining and MTT assay for cell survival analysis
Hoechst staining is used to assess the extent of the cell survival. Cells were fixed in 4% paraformaldehyde in PBS, incubated with the Hoechst 33342 diluted in PBS (1 : 1000) for 5-15 min. The “cell survival (%)” shown in figures was defined as the mean numbers of surviving cells in experimental groups divided by those of control. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) reduction assay was performed as previously described [42]. Counting of Hoechst-stained nuclei with normal nuclear morphology to quantitatively assess the extent of cell survival was described detailly in [43–45]. The “Cell Survival (%)” in figures was defined as the mean numbers of surviving cells in experimental groups divided by those of concurrent control cultures in the same experiment and multiplied by 100%.
Detection of cellular reactive oxygen species
ROS are detected on primary cortical neuronal culture by CellROX® Oxidative Reagents (Invitrogen™, USA). CellROX® Green Reagent is a DNA dye, and upon oxidation, it binds to DNA; its signal is localized in the nucleus and mitochondria. To study their therapeutic effects, the cultured cells were treated with LiBen (0, 0.5, 1, or 3 mM), LiCl, or SB (0 or 0.5 mM) for 24 h. The cells were then exposed to 2.5 mM 3-NP for additional 24 h. Subsequently, 2 mM CellROX® Reagent was added, and incubated for 10 h. The cells were then washed with PBS and ROS levels were quantified by fluorescence intensity (Excitation: 485 nm; Emission: 520 nm). The samples were observed under a laser scanning confocal microscope Zeiss LSM700 equipped with filter sets to detect their fluorescent signal.
Mitochondrial respiration
The mitochondrial activity of the cells was determined by Seahorse XF Cell Mito Stress Test kit (Agilent Technologies, USA). The rat primary cortical neurons seeded on XF 96 well cell culture microplates were pre-treated with LiBen (3 mM), SB (3 mM), or LiCI (3 mM) for 24 h prior to the assay. On the day of assay, culture media were changed to XF Assay medium (Seahorse Biosciences, USA) and incubated at 37°C in an incubator without CO2 supplementation for 1 h. The oxygen consumption rate was measured in the Seahorse XF96 Flux Analyzer (Agilent Technologies). The XF Cell Mito Stress Test modulates respiration and targets the components of the electron transport chain in the mitochondria to reveal key parameters of metabolic function. The modulators (oligomycin, FCCP, and a mix of rotenone and antimycin A) are serially injected to measure the ATP production, maximal respiration, and non-mitochondrial respiration. Proton leak and spare respiratory capacity are then calculated by these parameters. In this study, oncostatin M served as a comparable group, which might protect mitochondrial dysfunction.
BRDU labeling and immunocytochemistry
Primary cortical cultures prepared from El8 fetal Sprague-Dawley rat brains were cultured on coverslips in 24-well plates. After 2 days of Aβ25-35 incubation, the cell culture medium was removed and replaced with BrdU labeling solution and incubated at 37°C for 4 days along with LiBen (0.5 mM), LiCl (0.5 mM), or SB (0.5 mM) treatments. Subsequently, cells were fixed in 4% paraformaldehyde solution and washed with PBS, blocked with 2% bovine serum albumin, permeabilized with 0.03% Triton X-100 in PBS, and acid-washed with 2 N HCl and phosphate/citric acid buffer (pH 7.4). Next, anti-BrdU primary antibody (1 : 250, Thermo Fisher Scientific) or anti-NeuN primary antibody (1 : 300, Abcam) were added. The goat anti-rat IgG Alexa Fluor® 488-conjugated secondary antibodies (1 : 200; Cat. No. A11006; Thermo Fisher Scientific); anti-mouse IgG Alexa Fluor® 546-conjugated secondary antibodies (1 : 500; Cat. No. A11003; Thermo Fisher Scientific) were applied to bind the primary antibodies of the BrdU and NeuN respectively. The coverslips were observed under a digital imaging fluorescence microscope (Olympus BX61, Japan) equipped with filter sets to detect the corresponding fluorescence signals.
Oxygen glucose deprivation (OGD) in rat primary cortical neuronal culture
The Neurobasal medium (Cat. No. 21130349, Gibco, USA) supplemented with B27 (Cat. No. 17504044, Gibco) was removed from the cultures before inducing OGD. To induce OGD, cultures were rinsed with Neurobasal-A Medium (Cat. No. 10888-022, Gibco) or Neurobasal-A Medium without D-glucose and sodium pyruvate (Cat. No. A24775-01, Gibco). The cultured cells were maintained in a humidified incubator at 37°C with 5% CO2 for 1 h to consume the glucose and pyruvate. The cultures were then subjected to an anaerobic environment of 95% N2 - 5% CO2 for 4 h. Oxygen concentration was monitored by an oxygen analyzer Proox-110 (Biospherix, USA), to be maintained at approximately 1.0% throughout the experiment.
In vivo pharmacokinetic study
Male Sprague-Dawley rats weighed between 240-260 grams were randomly assigned to two groups (n = 6 per group). Rats in Group 1 were orally administered with a single dose of SB (287.9 mg/kg) and lithium carbonate (Li2CO3, 74.2 mg/kg), whereas rats in Group 2 were orally administered with LiBen (255.3 mg/kg). These two regimens would provide the same molarity of lithium and benzoate. They were dissolved in PBS, and then administrated by oral gavage. All rats were fasted overnight with free access to water before experiments. After single dose administration of the drugs, the blood samples were then collected from rat jugular vein at the time point 0 (pre-dose), 5, 15, 30, 60, and 90 min, 2, 4, 6, 12, and 24 h post-dose in precoated heparinized polypropylene tubes. Plasma was harvested by centrifugation at 2500×g for 15 min at 4°C and stored at -80°C until analysis. The study protocol was approved by the Institutional Animal Care and Use Committee.
The plasma concentration of benzoic acid was determined by LC/MS/MS. The chromatographic separation was carried out on a ODS 5-μm column (4.6 × 150 mm, Biosil). The mobile phase consisted of a mixture of 1% mobile phase A (ACN)/H2O/1M H4HCO3 = 25/75/0.5 (v/v)) and 99% mobile phase B (H2O/1M H4HCO3 = 100/0.5 (v/v)). The eluent had a flow rate of l ml/min. The injection volume was 50μl and the column temperature was 23°C. All solvents were HPLC grade quality.
For lithium quantification, plasma samples were mixed with 2-5% nitric acid and incubated at room temperature for overnight, followed by filtration with a 0.22μm filter. Plasma lithium concentration was detected by ICP-MS. Plots of plasma concentration of benzoate and lithium versus time are constructed. The pharmacokinetic parameters were derived by non-compartmental analysis (NCA) using WinNonlin.
Animals and drug treatment
Male B6.Cg-Tg (APPswe, PSEN1dE9) 85Dbo/Mmjax transgenic mice (Stock No. 005864) were purchased from the Jackson Laboratory and were bred with female C57BL/6 mice to yield mutant APP/PS1 (APPswe/PS1-dE9) and wildtype (WT) offsprings. APP/PS1 mice develop Aβ deposits in the brain by 6 to 7 months of age. All mice were characterized by PCR genotyping for the presence of the transgenes in accordance with Jackson Laboratory protocols. Mice were group housed (3-5 same-gender mice per cage) and maintained on a 12/12-h light/dark cycle in a temperature 22±2°C and 60% humidity-controlled environment.
LiBen obtained from Chem-Impex Int’l. Inc. was dissolved in 0.9% normal saline and injected intraperitoneally to 12 APP/PS1 transgenic mice (AD/LiBen). LiBen injection once daily beginning at 5 months of age (the day after the assessment of baseline performance) for 8 months. 16 additional APP/PS1 mice received 0.9% normal saline as a vehicle control (AD/Veh). In addition, 50 WT littermates were either received LiBen (WT/LiBen; n = 27) or 0.9% normal saline (WT/Veh; n = 23). LiBen at 256 mg/kg/day (Li: 14 mg/kg/day) were applied initially for 5 months and the dose was lower to 200 mg/kg/day (Li: 10.9 mg/kg/day) to prevent toxicity.
Behavioral tests were conducted on mice at 5 months (prior to dose), 7 months, 10 months, and 13 months of age to assess the cognitive, and spatial learning and memory. The study protocol was approved by the Institutional Animal Care and Use Committee.
Behavioral analysis
Mice were tested in the Barnes maze to examine their spatial learning and memory as described previously [46]. The testing apparatus was an elevated (50 cm above the floor) circular PLEXIGLAS® plate (100 cm in diameter) with 20 holes (7 cm in diameter, 7 cm between holes) evenly spaced around the perimeter. The mice were trained on the plate to identify an escape box (25×8×6 cm) hidden behind the target hole. The location of the target hole for each mouse was randomized and remained the same throughout the test. Mice were initially placed at the center of the plate covered by an opaque cylinder, and the cylinder was removed 10 s after the beginning of the trial with both an aversive tone (440 Hz, 85 dB) and the strong light (100 lux) switched on. The mice were trained to locate the target hole according to surrounding visual cues and escape from the aversion for three training trials per day over 3 consecutive days. The spatial memory was measured by the “probe test.” All training trials and the probe trials were videotaped for 3 min. The escape latency for the training trials and the percentage of time spent in different quadrants (target, left, right, and opposite) during the probe test were analyzed.
The deficits in sensorimotor gating have been reported associating with cognitive fragmentation and psychotic symptoms [47]. Pre-pulse inhibition, using SR-LAB startle apparatus (San Diego Instruments, USA), was used to determine sensorimotor gating function in mice. Under 65 dB background noise, each session was composed of a 5-min accumulation period followed by 64 trials in four blocks. The pulse alone (PA) trial was a 40 ms, 120 dB white noise burst. In the prepulse (pp)+pulse trials, a 20 ms white noise prepulse stimuli of 71 dB (pp6), 75 dB (pp10), and 83 dB (pp18) were presented 100 ms before a 40 ms 120 dB pulse. The non-stimulus (NS) trials presented the background noise only. The initial and the last blocks were both composed of six PA trials. Two middle blocks consisted of PA, pp+pulse, and NS trials. These trials were presented pseudo-randomly and separated by intertrial intervals of 15 s on average (varying between 10 to 20 s). The percentage of prepulse inhibition was evaluated by the following formula: % PPI = 100×[(PA score) - (pp+P score)] / (PA score), where the PA score was the average of the PA value in the middle blocks.
The histopathological analysis of Congo red staining
Mice were euthanized by isoflurane at 16 months of age and perfused transcardially with cold PBS followed by 4% paraformaldehyde. Brains were isolated immediately and fixed in 4% paraformaldehyde for 24 h. Brains were dehydrated in ascending grades of alcohol before being embedded in paraffin, sectioned at 5μm thickness and placed on the glass slides for Congo red staining. Congo red staining was performed as followed. Brain sections were deparaffinized and stained in 0.5% Congo red solution in 50% alcohol for 30 min and rinsed with distilled H2O. The stain was differentiated by dipping the slides 5–10 times in alkaline alcohol solution, rinsed with water, dehydrated through graded alcohol, and fixed in xylene and coverslipped by Consul-Mount (Thermo Shandon Limited, UK). Numbers of amyloid plaque in cortex and hippocampus were quantified as Congo red positive staining plaques.
Statistical analysis
The data were presented as mean ± SD for in vitro study and as mean ± SEM for animal behavioral studies. For in vitro cell culture studies, multiple groups were first analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. Statistical analysis of the memory testing session of Barnes maze test and pre-pulse inhibition (PPI) test were performed by two-way ANOVA followed by post hoc analysis with Dunnett’s test. For histological analysis, Student’s t-test was used for comparing the numbers of Aβ plaques between groups. Statistical analysis was performed using GraphPad Prism 7.0 software. p-value of less than 0.05 was considered statistically significant.
RESULTS
Lithium benzoate protects neurons from ROS-induced neurotoxicity by the inhibition of ROS production in rodent primary cortical neurons
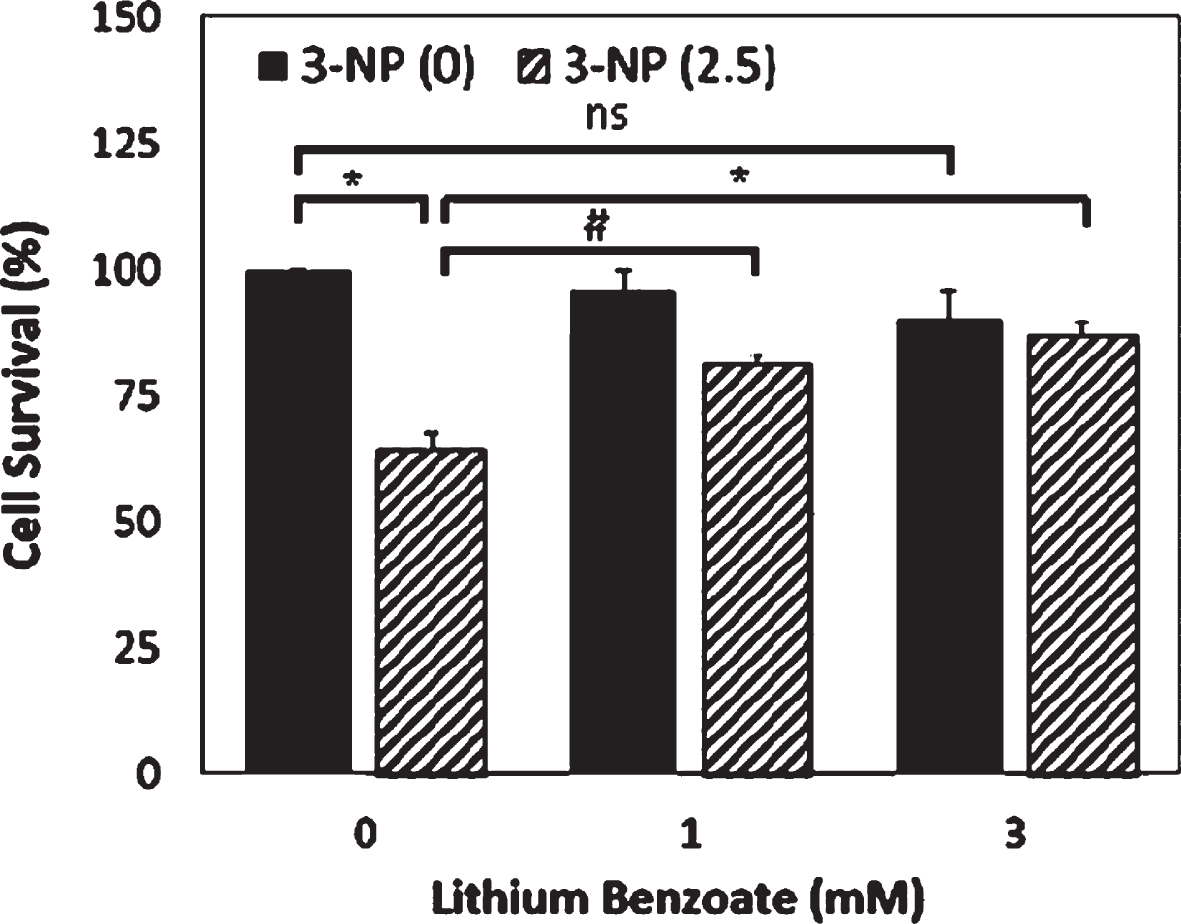
3-nitropropionic acid (3-NP), an irreversible mitochondrial complex II inhibitor of the electron transport chain, can cause OS and apoptosis by the overproduction of ROS. To examine whether LiBen is able to protect neuron death induced by ROS accumulation, rat primary cortical neurons were pre-treated with LiBen for 24 hrs and then exposed to 2.5 mM 3-NP for additional 24 hrs. As shown in Fig. 1, LiBen (1 or 3 mM) was shown to significantly increase (control vs 1 mM p < 0.005; control vs 3 mM p < 0.001) the viability of cultured cortical neurons exposed to 3-NP while the difference was not revealed in the absence of 3-NP analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test.
Fig. 1
Neuroprotective effect of lithium benzoate against 3-NP-induced neurotoxicity. Primary rat cortical neuron cultures were treated with lithium benzoate (LiBen) at indicated concentration for 24 h prior to exposure to 2.5 mM of 3-nitropropionic acid (3-NP) for additional 24 h. Cell survival was assessed by counting of Hoechst-stained nuclei of surviving cells. Data represent mean ± SD from N = 4 for each experimental condition and analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. # and * represents p < 0.005 and < 0.001, respectively, as compared to the cells challenged with 3-NP alone without LiBen treatment.
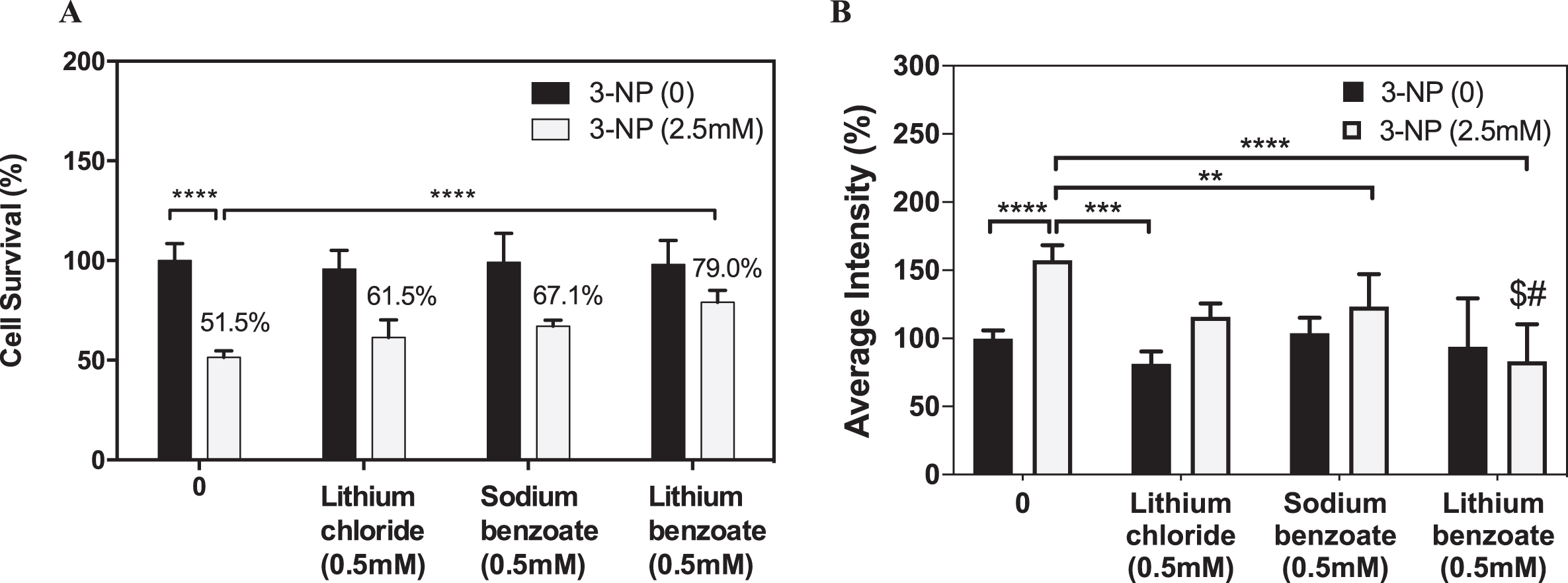
In order to assess whether LiBen exhibits better neuroprotective effect than monotreatment with either lithium or benzoate, rat cortical neurons were treated with LiCl, SB or LiBen at 0.5 mM for 24 h prior to 3-NP exposure, and the cell viability was measured by Hoechst staining. The results revealed that 3-NP caused nearly 50% neuronal cell death while LiCl, SB and LiBen significantly increased (p < 0.05) the cell survival rate, respectively, to 61.5%, 67.1% and 79.0% (Fig. 2A). Further, ROS production levels elicited by 3-NP exposure in rat cortical neurons were quantified with CellROX® fluorescence reagent. Compared to control group, the ROS level represented as fluorescent intensity was remarkably increased with 3-NP treatment by 57.5% (p < 0.05). LiCl and SB treatments showed significant decreases in ROS levels, compared with 3-NP-vehicle-treated group (vehicle versus LiCl p < 0.001; vehicle vs SB p < 0.01), analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test (Fig. 2B). Importantly, the effect of ROS reduction is much more significant by LiBen than the SB or LiCl treatment (vehicle versus LiBen p < 0.0001).
Fig. 2
Effects of lithium chloride, sodium benzoate, and lithium benzoate on neuron protection and ROS reduction in 3-NP-treated neuronal cells. Primary rat cortical neuron cultures were treated with lithium chloride (LiCl), sodium benzoate (SB), or lithium benzoate (LiBen) at 0.5 mM for 24 h prior to 3-nitropropionic acid (3-NP) at 2.5 mM exposure for additional 24 h. A) Cell survival was assessed by counting of Hoechst-stained nuclei of surviving cells from each group. B) The 3-NP-induced reactive oxygen species (ROS) production in the primary rat neuronal culture was quantified by using CellRox fluorescence staining. Data represent mean ± SD from N = 3 for each experiment condition and analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. **p<0.01; ***p<0.001; ****p<0.0001 as compared to the cells challenged with 3-NP alone without preconditioning. $ represents p < 0.05 as LiBen compared to LiCl with 3-NP treatment. # represents p < 0.01 as LiBen compared to SB with 3-NP treatment.
Lithium benzoate enhances spare respiratory capacity for the mitochondrial function
Mitochondria are the powerhouses and the major sources of free radicals in almost all mammalian cells. Mitochondrial dysfunction is implicated in numerous neuropathological diseases, including AD. We used a Seahorse cellular bioenergetic analyzer to examine the effects of LiBen, LiCl, and SB on mitochondrial respiration in rat primary cortical neurons. As shown in Fig. 3A, no differences were found in the maximal respiration among all groups, which showed that the general function of mitochondria to achieve the maximum rate of respiratory are the same. Under different states adjusted by modulators (oligomycin, carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone (FCCP), and a mix of rotenone (Rot.) and antimycin A (Antm.), the basal respiration, spare respiratory capacity, proton leak, and ATP production were determined respectively and shown in Fig. 3B. Pre-treatment of LiBen (3 mM) had reduced more basal oxygen consumption rate than the other treatment groups, which showed lower energetic demand of cells under basal conditions. However, this reduction did not reach the significant level due to relative small sample size. The increasing tendency of spare respiratory capacity in LiBen-treated (3 mM) group was observed and demonstrated better adaptability of cells, also indicating better capacity to respond to energy demands. Although SB (3 mM) and LiCl (3 mM) had the same tendency to increase spare respiratory capacity of cells, they were less effective than LiBen (3 mM).
Fig. 3
Analysis of the impact of lithium chloride, sodium benzoate and lithium benzoate on mitochondrial functions. Primary rat cortical neuron cultures were treated with lithium chloride (LiCl), sodium benzoate (SB), or lithium benzoate (LiBen) at 3 mM for 24 h prior to analysis of mitochondrial function. The Seahorse XF96 analyzer was employed to determine the mitochondrial oxygen consumption rate and mitochondrial ATP production. A) The mitochondrial oxygen consumption profile of mitochondrial stress test for neuronal culture. B) Bar graph represents the key parameters of metabolic function including basal respiration, ATP production, proton leak, and spare respiratory capacity of the treated neuronal cells. Data represent mean ± SD from N = 3 for each experiment condition. CTRL, Control; FCCP, Carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone; OCR, Oxygen consumption rate; ROT., Rotenone; Antm., Antimycin A.
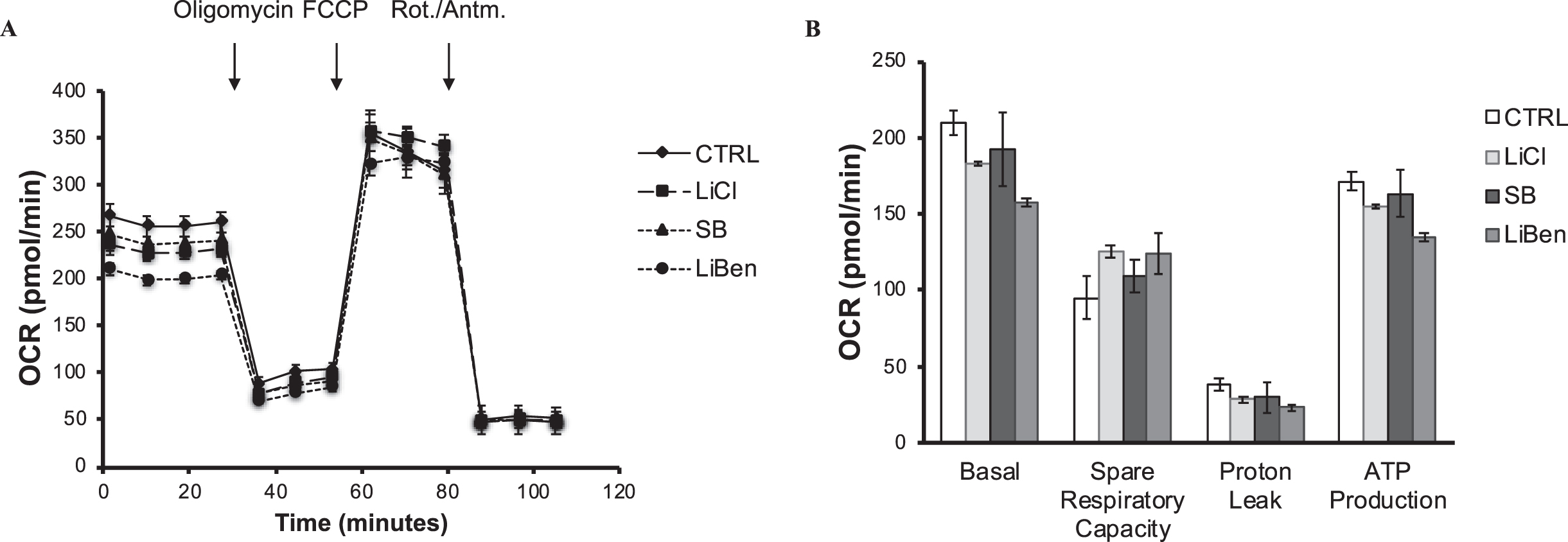
Moreover, Proton leak reflects mitochondrial damage. LiBen exhibited more reduction of proton leak and ATP production than the SB or LiCl treatment (Fig. 3B). Thus, the results indicate that LiBen may protect cells from mitochondria damage. The ATP produced by mitochondria is to meet the energy demand of cells. The reduction of ATP in the LiBen-treated group indicates the lower energetic demand of the basal respiration. In summary, LiBen reduced the basal energy demand of neurons, protected them from proton leak, and improved the adaptability on demands of the mitochondria activity by increasing the spare respiratory capacity.
Lithium benzoate protects rodent primary cortical neurons against Aβ25-35-induced cytotoxicity
Since Aβ has neurotoxic effects both in vitro and in vivo [48], and the accumulation of Aβ in brain is the key factor contributing to neuronal mitochondrial dysfunction and OS. The amyloid β protein fragment Aβ25–35 (GSNKGAIIGLM), the smallest fragment formed by brain proteases, retains the toxicity of the parent peptide as well as a capacity to form aggregates in vivo. As such, Aβ25–35 promotes the development of AD pathology and the resultant clinical symptoms [49, 50]. Deposition of Aβ25–35 in brain triggers tau protein phosphorylation and formation of intracellular NFT, subsequently leading to mitochondrial dysfunction and membrane rupture, prior to necrosis or apoptosis [51].
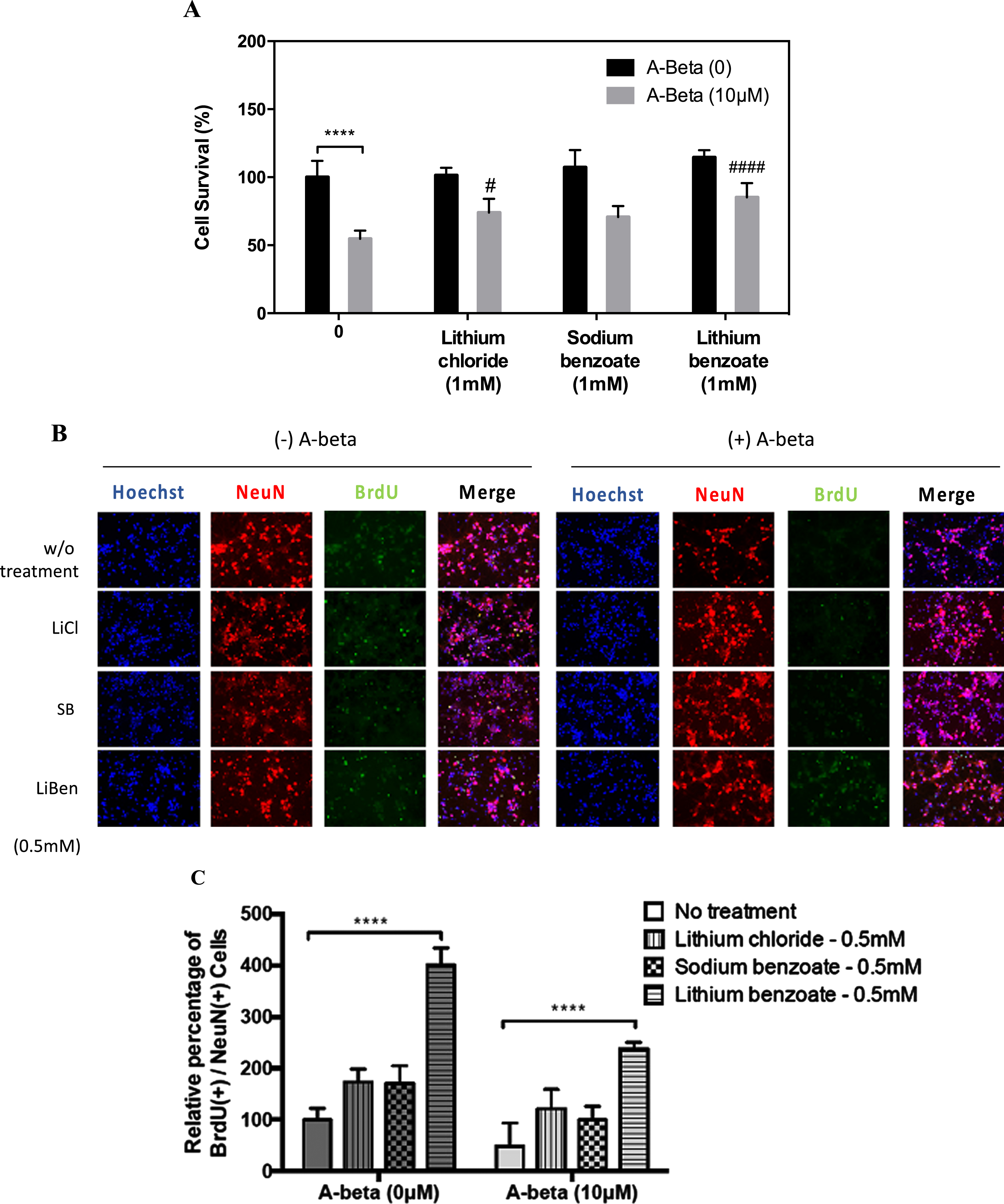
To assess whether LiBen possesses potential cytoprotective actions again Aβ-induced apoptosis, rat primary cortical neurons were exposed to 10μM Aβ25-35 for 2 days, and then followed by treating with LiBen (1 mM) for 4 days. The cell viability was assessed by Hoechst staining. In addition, the effects of LiCl (1 mM) and SB (1 mM) in the same condition were also examined. As shown in Fig. 4A, compared to control group, Aβ25-35 peptide treatment induced neurotoxicity and greatly reduced cell survival rate to 54.6% (p < 0.0001). Compared to Aβ25-35-vehicle-treated group, although all three treatments displayed improvement for cortical neuron survival, LiBen significantly improved the cell survival rate (p < 0.0001) compared with Aβ25-35-vehicle-treated group and showed much more favorable effects on protecting neurons from Aβ25-35 peptide-induced cell death as the cell survival rate was greater than the other two treatments (85.1% for LiBen vs 74.0% for LiCl and 70.7% for SB).
Fig. 4
Neuroprotective and neurogenesis effects of lithium chloride, sodium benzoate, and lithium benzoate on Aβ25–35-induced neurotoxicity. Rat cortical neurons were pre-treated with 10μM Aβ25-35 for 2 days, and then followed by treatment of lithium chloride (LiCl), sodium benzoate (SB), or lithium benzoate (LiBen) at 1 mM for 4 days. A) Cell viability was determined by MTT assay and expressed as percentage of control cells. B) Immunofluorescence micrographs of Hoechst (blue)/NeuN (red)/BrdU (green) triple staining for assessing neurogenesis. C) The bar graph represents the percentage of Hoechst/NeuN/BrdU triple positive cells to the control cells without any treatment. Data represent mean ± SD from N = 3 for each experiment condition and analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. ****p<0.0001 as compared to the corresponding control cells. #p<0.05 and # # # #p<0.0001 as compared to the cells challenged with Aβ25-35 alone. BrdU, Bromodeoxyuridine; NeuN, Neuronal nuclear protein.
Lithium benzoate exhibits superior effects on promoting neurogenesis
The impairment of neurogenesis in the adult hippocampus is involved in the pathogenesis of AD [52]. In order to assess whether LiBen exerts the neuroprotective effects by promoting neurogenesis as well, the rat primary cortical neurons exposed to Aβ25-35 peptide were post-conditioned with LiBen, LiCl or SB at 0.5 mM for 4 days and the proliferative changes of neuronal cells were detected using Bromo-deoxyuridine (BrdU) incorporation and NeuN (the neuronal cell marker) immunostaining. We found that the mean percentage of BrdU+/NeuN+ cells in cultured cortical neuronal cells exposed to 10μM of Aβ25-35 peptide decreased to about 50% compared to the group without Aβ25-35 peptide treatment. Interestingly, while all three compounds could alleviate the Aβ25-35-induced neurogenesis impairment, LiBen significantly improve neurogenesis, with or without Aβ25-35 peptide exposure, compared with corresponding control group (No Aβ25-35 exposure, p < 0.0001 and Aβ25-35 exposure, p < 0.0001) and displays enhanced neurogenesis much greater than the other two compounds (Fig. 4B and C).
The neuroprotective effect of lithium benzoate is through NMDA receptor-independent pathway
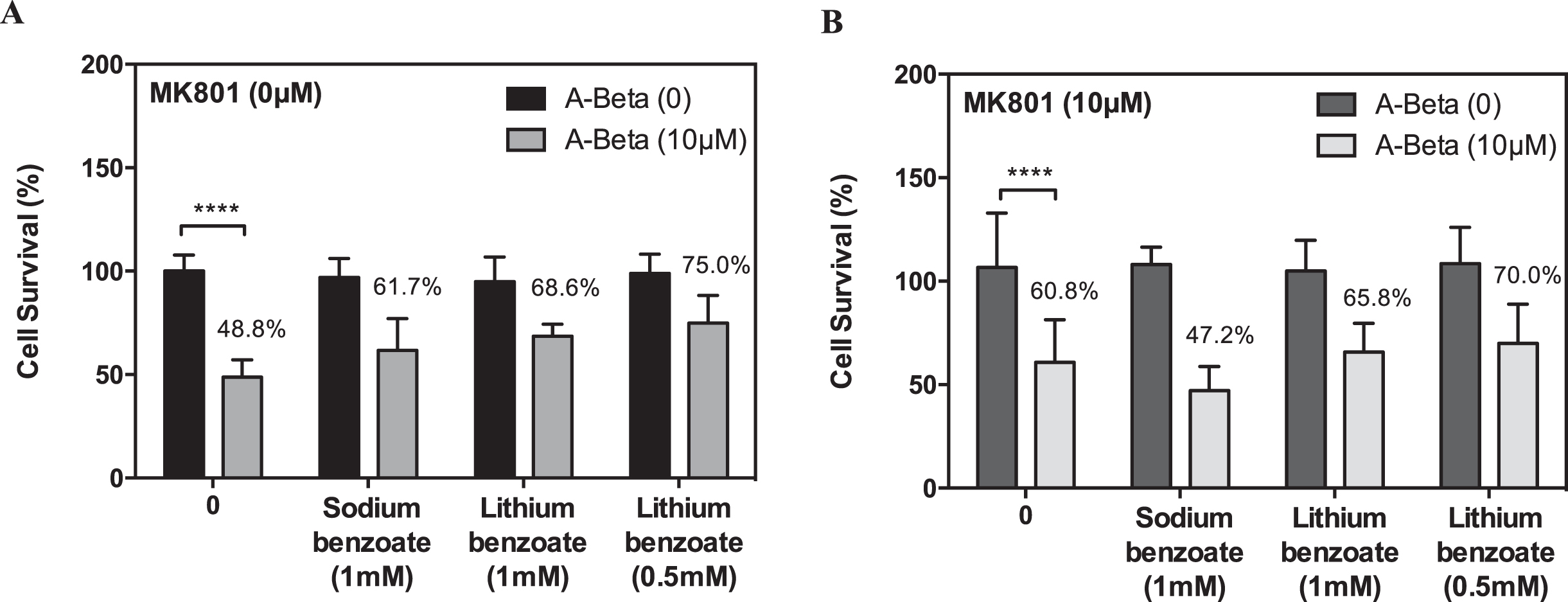
N-methyl-D-aspartate receptor (NMDA receptor) is an ionotrophic glutamate receptor mediating calcium permeability in neuronal cells. The excitotoxicity caused by excessive calcium influx is considered a major mechanism of neuronal death associated with many neurodegenerative diseases including AD. Moreover, recent studies indicate that NMDA receptor-mediated glutamate neurotoxicity may trigger Aβ-induced cell death [53, 54]. To determine whether LiBen treatment improves the viability of cortical neuronal cells exposed to Aβ25-35 peptide through NMDA receptor, we compared the survival rate of rat cortical neurons in the presence or absence of MK-801 (10μM), an uncompetitive antagonist of NMDA receptor. As shown in Fig. 5A, LiBen and SB effectively promote cell survival in the cortical cells with Aβ25-35 exposure at 0.5 mM and 1 mM, respectively, whereas this protective effect of SB was blocked by MK-801 treatment (Fig. 5B). However, no differences were observed in the LiBen group in the presence or absence of MK-801, which indicated that the neuroprotective effect of LiBen was not mediated through the NMDA receptor pathway.
Fig. 5
Effect of MK-801 on sodium benzoate and lithium benzoate-elicited protection against Aβ25–35-induced neurotoxicity. Rat cortical neurons were pre-treated with 10μM Aβ25-35 (Amyloid-β peptide 25-35) for 2 days, and then followed by treatment of sodium benzoate (SB; 1 mM) or lithium benzoate (LiBen; 0.5 and 1 mM) for 4 days in the (A) absence or (B) presence of MK-801 (10μM). Cell viability was determined by MTT assay and expressed as percentage of the corresponding control cells. Data represent mean ± SD from N = 3 for each experiment condition and analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. ****p<0.0001 as compared to the cells challenged with Aβ25-35 alone without postconditioning.
Lithium benzoate protects the primary cortical neurons from hypoxia-induced apoptosis
Appropriate blood circulation and tissue perfusion, for sufficient nutrient and oxygen delivery or the clearance of metabolic waste products, are essential for the brain development and function [55]. Microvascular dysfunction, resulting in hypoperfusion, is considered as an integral part of AD pathogenesis. Hypoxia is a direct consequence of hypoperfusion, a common vascular component among the risk factors of AD [56]. To assess whether LiBen can protect hypoxic neuronal cells from apoptosis, LiBen was applied to the in vitro oxygen-glucose deprivation (OGD) cellular model and the cell viability was evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Two treatment conditions were used in this study: (1) LiBen was present in the primary cortical neuronal culture for 53 hrs of the whole culture period (Fig. 6A); (2) LiBen was applied to the primary cortical neuronal culture only for 48 hrs post-OGD (Fig. 6B). The results show that, compared to the normoxia condition, the cell survival was reduced about 50% under oxygen-glucose deprivation. In the presence of 3 mM LiBen for 53 hrs of the culture period, the OGD-induced cell death was significantly recovered and comparable to the normoxia group (Fig. 6C). However, the neuroprotective property of LiBen vanished at higher concentration (5 mM). In this concentration, the cell survival was also reduced in normoxia group. When LiBen was added to the neuronal culture after deprivation, LiBen significantly alleviates OGD-induced cell death at the dose levels from 0.3 to 3 mM, compared with the 0 mM group (Fig. 6D). We speculated that 5 mM of LiBen might be too high to produce protective effect, in other words, too high LiBen may cause adverse effect instead.
Fig. 6
Protective effect of lithium benzoate against hypoxia-induced neurotoxicity. A) Diagram of the experimental design in which rat cortical neuronal cultures were treated with lithium benzoate (LiBen) for 53 h of the whole culture period. B) Diagram of the experimental design in which neuronal cultures were treated with LiBen for 48 h after 4 h of hypoxia treatment. C) Cell viability of rat primary cortical neuronal culture treated with LiBen (0, 0.3, 1, 3, and 5 mM) for 53 h of the whole culture period. D) Cell viability of rat primary cortical neuronal culture treated with LiBen (0, 0.3, 0.5, 1, 2, and 3 mM) for 48 h after 4 h of hypoxia treatment. Cell viability was determined by MTT assay and expressed as percentage of the control cells cultured under normoxia condition without LiBen treatment. Data presented as mean ± SD from N = 3 for each experiment condition and analyzed by one-way analysis of variance (ANOVA) followed by a post hoc Student-Newman-Keuls test. **p<0.01, ***p<0.001, and ****p<0.0001 as compared to cells treated with hypoxia condition alone.

Lithium benzoate exhibits higher oral bioavailability than lithium chloride and sodium benzoate
Peak plasma concentration (Cmax) and time to Cmax (Tmax) of benzoic acid were determined by HPLC analysis. The plasma concentration-time profile of benzoic acid after oral administration of either the mixture of SB and Li2CO3 (SB+Li2CO3) or LiBen were illustrated in Fig. 7A. The half-life (T1/2), area under curve (AUC) and other pharmacokinetic parameters were calculated and presented in Table 1. The mean Cmax of benzoic acid following the single administration of the mixture of SB and Li2CO3 was 71.38 μg/ml at 0.14 hrs, and the administration of LiBen was 109.32 μg/ml at 0.19 hr (Table 1). It is clear on the plasma concentration-time profile that benzoic acid in both groups quickly reached the peak in plasma around 10 min and was rapidly removed from plasma with a half-life of 1.45 hrs in the group of SB+Li2CO3 mixture, and 2.52 hrs in LiBen group. Benzoic acid concentrations in both groups were undetectable in 12th hr, that might indicate the elimination in rats. Interestingly, we found that the Cmax, AUC and T1/2 values of benzoic acid in LiBen group were much higher than the SB+Li2CO3 mixture group, suggesting its superior bioavailability, though the difference did not reach significant level due to relatively small sample size.
Fig. 7
Plasma concentration-time profiles of benzoic acid and lithium. A) The mean plasma benzoic acid and B) lithium concentration changes over time after a single oral administration of lithium benzoate (LiBen; 255.3 mg/kg) or the mixture of sodium benzoate (SB; 287.9 mg/kg) and lithium carbonate (Li2CO3; 74.2 mg/kg). Data were presented as mean±SD from N = 6 for each experiment condition.
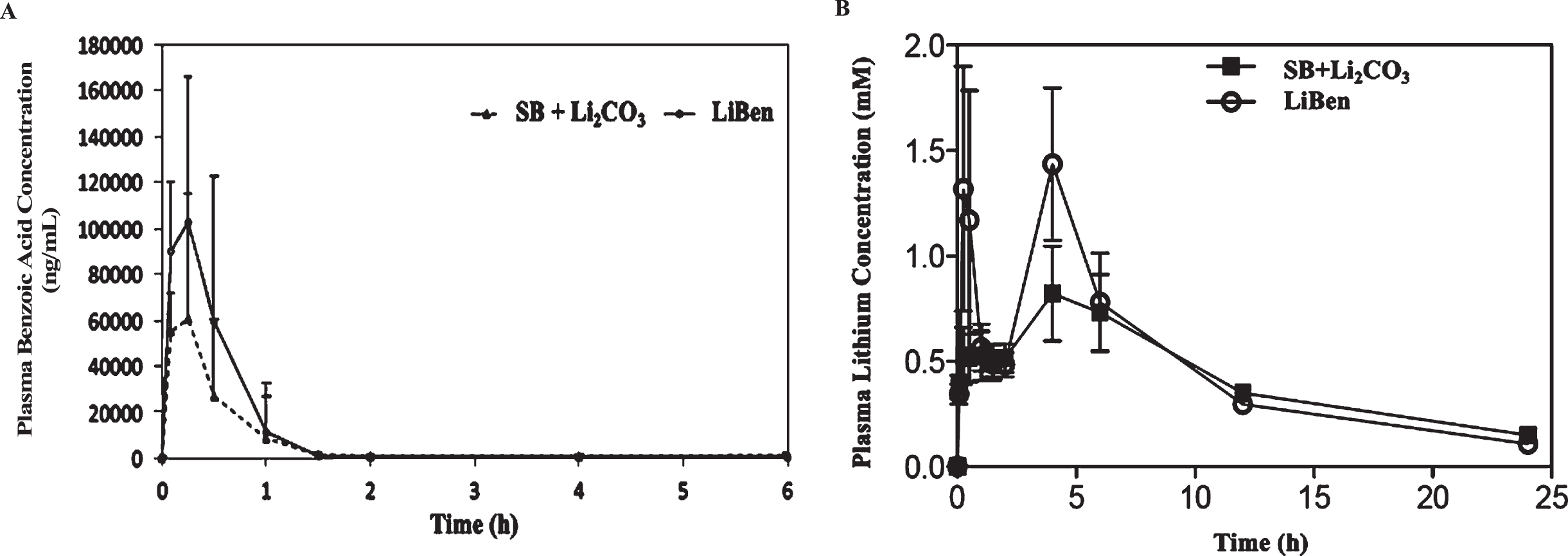
Table 1
Pharmacokinetics parameters of benzoic acid in rat plasma
| Parameters | Tmax (h) | Cmax (μg/mL) | T½ (h) | AUC0-t (hr*μg/mL) | AUC0-Inf (hr*μg/mL) |
| LiBen (255.3 mg/kg) | 0.19 | 109.3 | 2.52 | 62,059 | 62,874 |
| SB (287.9 mg/kg)+Li2CO3 (74.2 mg/kg) | 0.14 | 71.4 | 1.46 | 39,201 | 41,577 |
LiBen, lithium benzoate; SB, sodium benzoate; Li2CO3, Lithium carbonate.
Additionally, plasma lithium concentrations in both groups were analyzed by ICP-MS. Consist with benzoic acid results, the Cmax of lithium for LiBen group was significantly higher than the group of SB + Li2CO3 mixture. Moreover, the AUC was also higher in the LiBen group than that in the group of SB + Li2CO3 mixture. Overall, LiBen has a better pharmacokinetic profile than the combination of SB and Li2CO3 (Fig. 7B and Table 2). The above results demonstrated that, under the same molar concentration, LiBen has better bioavailability than the combination of SB and Li2CO3.
Table 2
Pharmacokinetics parameters of lithium in rat plasma
| Parameters | Tmax (h) | Cmax (mM) | AUC0-t |
| LiBen (255.3 mg/kg) | 2.2 | 1.172 | 620.59 |
| SB (287.9 mg/kg)+Li2CO3 (74.2 mg/kg) | 3.7 | 0.906 | 605.87 |
LiBen, lithium benzoate; SB, sodium benzoate; Li2CO3, Lithium carbonate.
Administration of lithium benzoate mitigates the impairments of cognition and spatial memory in APP/PS1 mutant mice
To verify whether the neuroprotective effects of LiBen observed in vitro is able to present in vivo, LiBen or control vehicle were intraperitoneally (i.p.) injected into APP/PS1 mice or wildtype (WT) littermates starting at 5-month of age for a period of 8 months and conducted behavior tests every 2 to 3 months. Since the major symptom in AD is the progressive loss of memory and cognition, Barnes circular maze and prepulse inhibition tests were performed. Barnes circular maze test is a hippocampus-dependent cognitive task which requires spatial reference memory. Spatial reference memory was assessed by the ratio of time mice spent in the target quadrant to total exploring time in Barnes maze. There was no difference in the baseline performances of Barnes maze between genotypes, indicating normal spatial memory formation of APP/PS1 mice at 5-month of age. Five months later, vehicle-treated APP/PS1 mice exhibit significant lower exploring time in the target quadrant compared with baseline performance of WT mice. This cognitive decline, however, was not observed in the LiBen-treated APP/PS1 mice (AD/LiBen) at the same age (Fig. 8A), which tended to preferentially spend more time in the target quadrant (p = 0.074 for AD/LiBen vs AD/Veh), suggesting a protective effect of LiBen in the rodent model of AD.
Fig. 8
Effect of lithium benzoate on the cognition and memory impairment in AD transgenic mice model. AD transgenic mice (AD, APPswe/PS1-dE9 line) received intraperitoneal injection of vehicle (AD/Veh, N = 16) or lithium benzoate (AD/LiBen, N = 12), and wild-type (WT) littermates received intraperitoneal injection of vehicle (WT/Veh, N = 27) or lithium benzoate (WT/LiBen, N = 23) for 8 months beginning at 5 months of age. A) Cognition and memory abilities of AD mice and their wildtype littermates were assessed using the Barnes maze test. The bar graph represents the ratio of time spending in the target quadrant divided by total exploring time during the probe trial of the test at the indicated age. LiBen-treated APP/PS1 mice (AD/LiBen) at 10 months of age tended to spend more time in target quadrant (p = 0.074), compared with vehicle-treated APP/PS1 mice (AD/Veh). B) Bar graphs represents the prepulse inhibition (PPI%) of AD mice and WT littermates at the indicated age. Some of the AD mice showed negative PPI suggesting of hearing impairment. Data presented as mean ± S.E.M. Two-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test was used. * Represents p < 0.05 as compared to WT mice at 5 months of age for Barnes maze test and to the concurrent AD/Veh mice for PPI test. AD, Alzheimer’s disease transgenic mice; WT, Wildtype littermates; Veh, Vehicle; LiBen, Lithium benzoate.
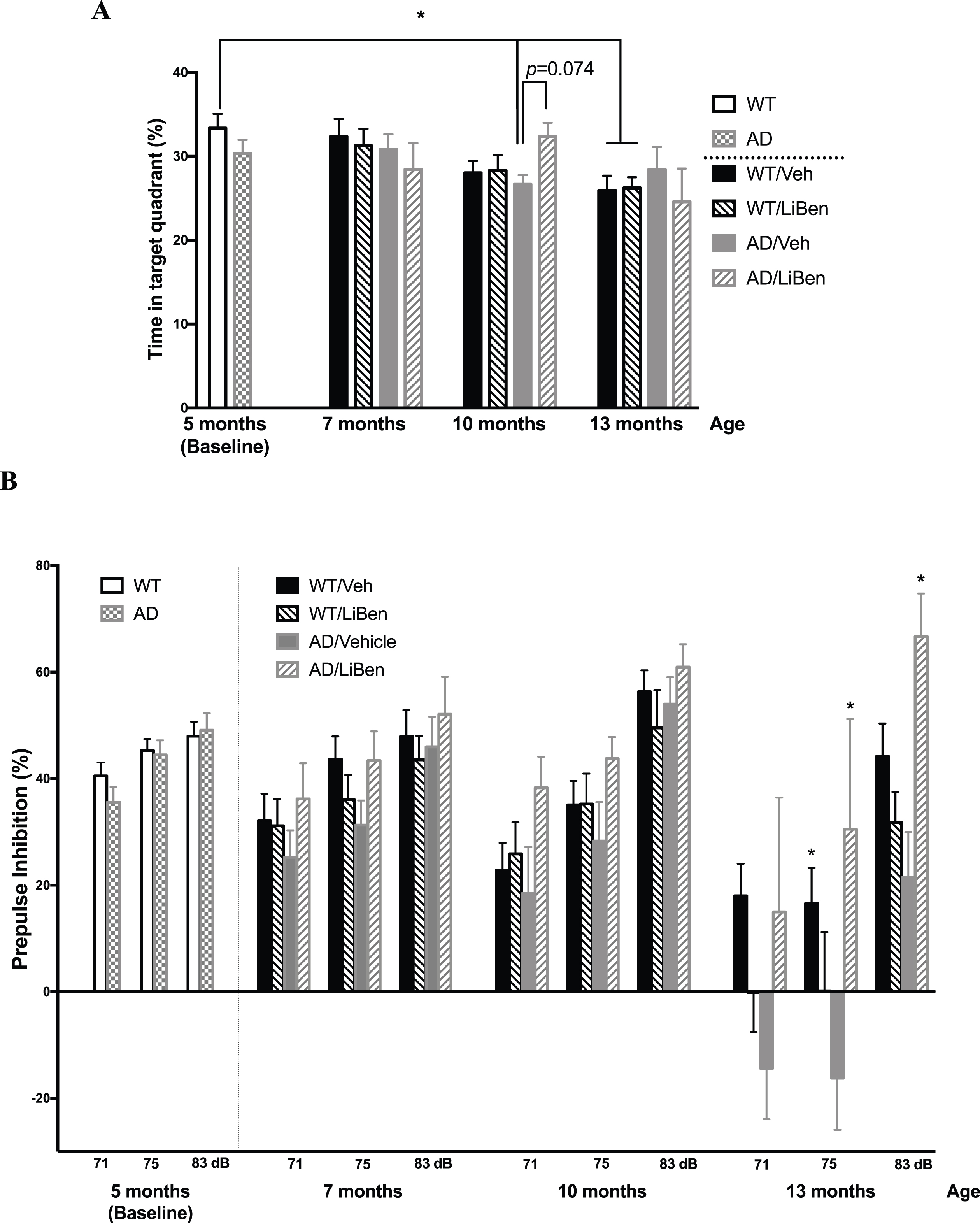
AD patients in the mild to moderate stages of the disease frequently fail to direct attention to novel or interesting aspects of their environment [57]. The lack of inhibition of competing and conflicting responses, known as sensorimotor gating, has been shown to be the most affected aspect of selective attention in early AD [58]. Sensorimotor gating assessed by PPI assay revealed PPI deficit in AD mice and patients. We then tested the sensorimotor gating function in the same cohort of mice by a PPI assay. The results showed that no genotype effect was observed for startle response at 5 months of age (baseline). Compared to WT mice, vehicle-treated APP/PS1 mice (AD/Veh) displayed gradual declines in PPI along with age, while LiBen-treated APP/PS1 mice (AD/LiBen) exhibited normal PPI at all time points (Fig. 8B). Overall, these results reveal the prevention of impairment of sensorimotor gating in APP/PS1 mice by the 6- to 8-month treatment regimen of LiBen.
Lithium benzoate alleviates the brain deposition of Aβ plaques in APP/PS1 mutant mice
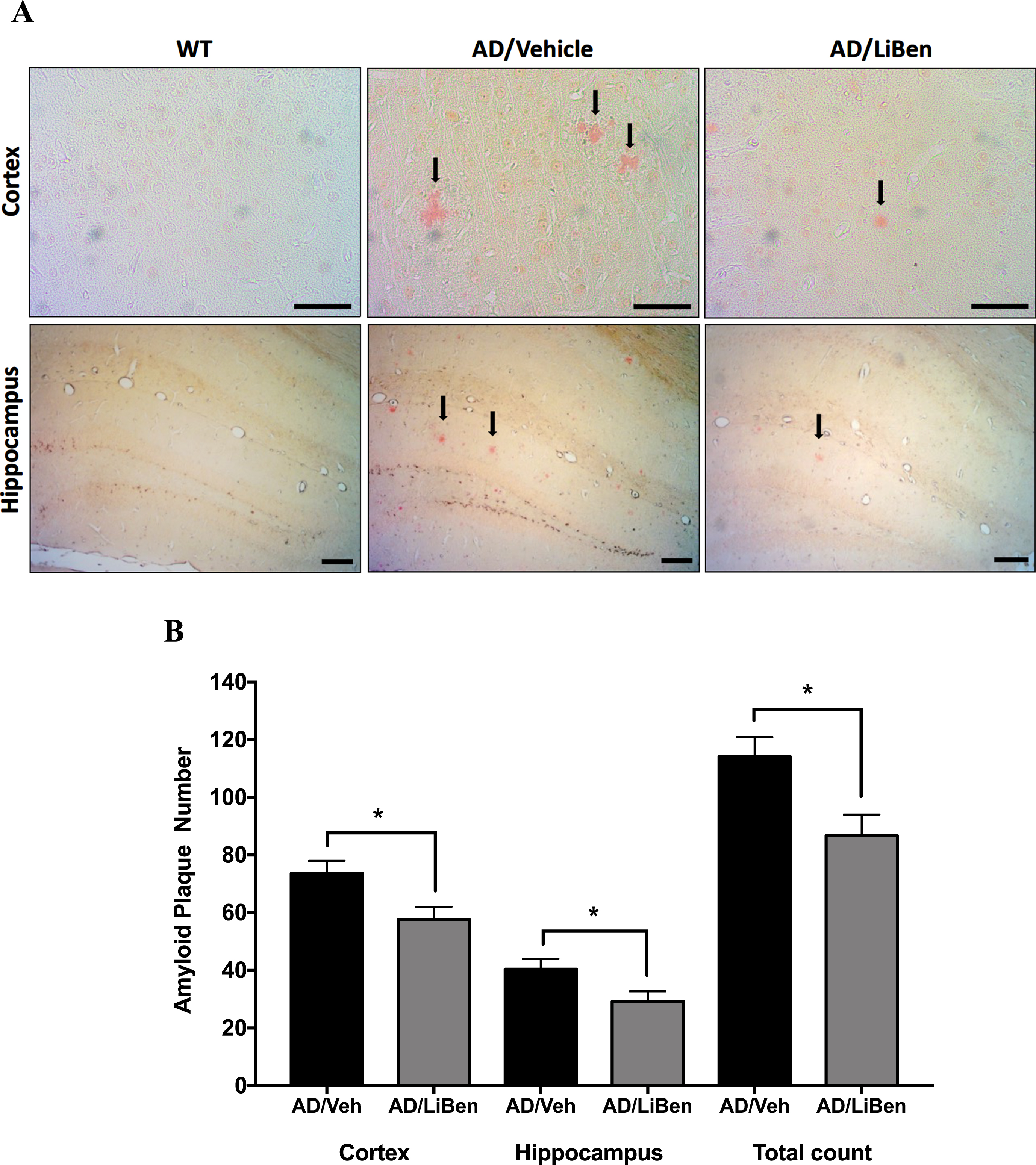
The accumulation of extracellular Aβ plaques composed of insoluble Aβ fibrils is one of the typical pathological changes in AD patients and animal models. To determine whether LiBen treatment prevents or reduces Aβ plaque deposit in brain, Congo red was used to quantify the amyloid plaque deposit on brain sections of wild-type and AD mice at 16 months of age (Fig. 9A). After 11 months of LiBen treatment, mice showed significantly decreased number of senile plaque in both cortical and hippocampal areas, compared with the control animals (Fig. 9B; p < 0.05 for AD/LiBen versus AD/Veh; Student’s t-test). The number of plaques in LiBen-treated APP/PS1 mice (AD/LiBen) was decreased by 27.8% in hippocampus and by 21.8% in cortex. These results indicate the substantial beneficial effects of LiBen in reducing Aβ plaque deposition in brains.
Fig. 9
Deposition of amyloid-β plaque in the brains of mice with or without LiBen. A) Congo red staining of cortex and hippocampus from the APP/PS1 (AD) and wild type (WT) mice. Close-up of brain sections showed staining of amyloid plaques with red color (Cortex at 400 magnification, Scale bar = 50μm; hippocampus at 100 magnification, Scale bar = 100μm). B) Quantification of amyloid plaque numbers in the cortex and hippocampus. Compared to the AD/Veh group, the number of plaques in both cortex and hippocampus showed significant decrease in AD/LiBen groups. Data were presented as mean±SEM and significant differences between AD/Veh and AD/LiBen at *p<0.05 were analyzed by Student’s t-test.
DISCUSSION
OS and mitochondrial dysfunction both are involved in the AD pathogenesis. The source of OS in AD includes altered mitochondrial function, hypoperfusion, Aβ peptides, and the presence of unbound transition metals [59]. Our present study of both in vitro and in vivo models evaluates the neuroprotective properties of LiBen. 3-NP is a mitochondrial respiratory complex II inhibitor. The inhibition of complex II mediates ATP level decrease, subsequently leads to a rise in ROS, and eventually results in mitochondria fission and neuronal cell death [60]. We have assessed the cell survival and ROS reduction rate of LiBen in 3-NP-treated rat cortical neuronal culture and found that LiBen increases cell survival rate more effectively than LiCl and SB. These results indicate that LiBen might elicit its neuroprotective effect through other mechanism apart from ROS reduction. In addition, the cell mitochondria stress test shows that LiBen treatment profoundly decline the basal energy demand of neurons, protected them from proton leak, and improved the adaptability on the demands of the mitochondria activity.
In addition to aging, brain hypoxia caused by brain hypoperfusion also plays an important role in Aβ accumulation. As brain injuries may cause hypoperfusion, a history of stroke and traumatic brain injury can increase AD prevalence [61]. It has been reported that hypoxia can alter amyloid-β protein precursor (AβPP) processing and increase Aβ deposition by increasing the activity of β- and γ-secretases [62]. Hypoxia inducible factor-1α (HIF-1α), a molecular upregulated by hypoxic condition, has been revealed in binding to beta-site AβPP-cleaving enzyme 1 (BACE1) promoter, increasing the expression and the enzymatic activity of β-secretase, and resulting in overproduction of Aβ [63]. In addition, hypoxia reduces aerobic oxidative respiration and decreases electron-transport rate in the mitochondria leading to mitochondrial membrane potential (Δψm) reduction, therefore, increases ROS generation, enhances nitric oxide (NO) synthase, and eventually induces apoptosis. In the OGD test, we find that LiBen exerts its best performance by the post-hypoxia treatment, although significant neuroprotective effect was observed in both pre- and post-hypoxia treatment.
Aβ aggregation, formation of extracellular senile plaques, and intraneuronal deposits of NFTs are key steps of the pathological process of AD. Growing evidence suggest that Aβ is not only the product of AD pathogenesis, but also a contributor of mitochondrial abnormalities and ROS elevation. Reddy and Beal [59] reported that Aβ could enter the mitochondria where it would increase the generation of ROS and induce OS. Moreover, studies of postmortem brain of AD and the AD transgenic-mice showed that Aβ and AβPP can be found in mitochondrial membranes where they block transport of protein and disrupt the electron transport chain with final, irreversible cell damage [59]. In the present study, we find that LiBen significantly reduces Aβ-induced cell death, and the effect is superior to LiCl and SB. Interestingly, we also find that LiBen can not only protect cells from the insults of Aβ, but also promote neurogenesis even under Aβ treatment. Although lithium is known as an important role in neuroprotection by preventing oxidative and neuroinflammatory damage, maintaining protein homeostasis, and enhancing neurogenesis and synaptic maintenance [64], LiBen shows more profound effect on neurogenesis than LiCl in our study.
The mechanism of action of LiBen attributes to the unique dual molecular functions of lithium and benzoate. Evidences have demonstrated that the benefits of SB, a metabolite of cinnamon and a widely used food flavor, would increase the levels of neurotrophic factors such as brain-derived neurotrophic factor and neurotrophin-3 in neurons [38, 65, 66]. Our observation is also supported by in vivo studies in the animal model of AD, which demonstrate that SB protects learning and memory by decreasing ROS generation and increasing glutathione level in the hippocampus of 5xFAD transgenic mice [35]. Furthermore, It is reported that SB improves the neurocognitive function in patients with mild AD [38].
Consistent with the in vitro cell culture findings, we demonstrate that chronic administration of subtoxic level of LiBen protects cognition and memory as well as prevents PPI deficits in APP/PS-1 transgenic mice. The deficiency of prepulse inhibition was observed in AD patients and suggested as a potential biomarker of AD progression [67, 68]. In addition to behavioral changes, we also investigated the effect of LiBen in deposition of Aβ plaques, the most notorious feature of AD, in APP/PS1 mice. Inconsistent results of lithium’s effects were reported in Aβ plaque formation of this double-transgenic model. Pan et al. reported that oral administration of LiCl (Li dose: 49.4 mg/kg) for 26 days did not affect the number of amyloid plaque in 8 month old APP/PS1 mice [23]. However, in another study, three-month administration of LiCl (Li dose: 41.7 mg/kg) by intraperitoneal route can significantly reduce amyloid plaques in this AD model at 13 months of age [69]. Our study demonstrate that even in very late stage of the AD (16 months of age), LiBen can substantially reduce the Aβ deposition in cortical as well as hippocampal regions at a much lower oral-dose. In different behavioral tests, the protective effect of LiBen might be varied depending on different stages of AD progression. PPI deficit of APP/PS-1 transgenic mice was improved in all three measured time points and the improvement reached significant level at 13 months of age. Nevertheless, in the Barnes maze test, the protective effect of LiBen was only observed in one measured time point (10 months of age) and seemed not long-lasting in the AD animal model. Although LiBen’s effect in different stages of AD might require further examination, the two improvements from PPI and histological assays observed at late stage of AD progression still suggest the sustained beneficial effect of LiBen. The combination of lithium and SB in LiBen is an innovative approach to achieve the additive functions of these two compounds. In addition, we find that oral bioavailability of LiBen is approximate 1.6 times the combination of SB plus Li2CO3 in the PK study, indicating less amount of LiBen is required to achieve equal therapeutic effect of mixing SB and Li2CO3, and with lower risk of lithium-related toxicity.
As a novel medication for AD, the potential toxicity of LiBen is also concerned. The prolonged use of lithium could affect the nervous system and induce neurological symptoms, including tremor, decreasing verbal learning, memory, creativity, and motor function in human subjects [70, 71]. Intriguingly, growing evidence also indicates the neuroprotective effect of lithium against multiple neurotoxins including ethanol [72], Aβ [69], 1-methyl-4-phenylpyridinium ions [73], and rotenone [74]. However, no lithium-related toxic events were observed in our study. After 8-month administration of lithium benzoate, WT mice showed similar cognitive performance as control vehicle-treated group in Barnes maze test. Moreover, these mice could survive for at least 16 months (the termination time point of this experiment) without significant body weight change during the experimental period. Overall, the chronic use of LiBen within the tested dose range did not display any toxicity, hence its safety assumed. We consider LiBen as a viable option for lithium therapy for AD when lithium is not administered in the toxic range.
Up to date, there is no effective therapy that can cure or arrest the progression of AD. Six drugs are approved for the treatment of AD including four cholinesterase inhibitors (tacrine, donepezil, rivastigmine, and galantamine), one NMDA receptor antagonist (memantine) [75, 76] and recently, the controversial aducanumab. Tacrine was the first approved acetylcholinesterase inhibitor for the treatment of mild-to moderate AD in 1993. However, it is no longer prescribed because of its poor tolerability and risk of hepatotoxicity. Many attempts in AD drug development had failed due to failing to show efficacy (drug/placebo difference) or having unacceptable adverse effects. A summarization of a review article of the AD drug development pipeline shows that most agents undergoing clinical studies have mechanism of action directedly at disease modification (63%), 23% are cognitive-enhancing agents, and 12% are drugs directedly at controlling neuropsychiatric symptoms. Since AD is a multifactorial neurodegenerative disease, combination of agents to target multiple mechanisms of action would be a favorable therapeutic approach. For instance, combination of donepezil and memantine is more effective than single treatment for delaying the degradation of cognitive function in patients with moderate or severe disease level [76, 77]. The mechanism of action of LiBen is complex, including disease modification by the multiple mechanisms we reported in this study to improve amyloid deposit, cognition, and sensorimotor gating.
In summary, LiBen can provide a safe alternative therapeutic approach over the existing anti-AD therapies. We have demonstrated that LiBen is superior to LiCl and SB in reducing cellular ROS level, improving mitochondrial function, increasing cell viability against multiple insults (mitochondrial dysfunction, Aβ accumulation, and OGD), and promoting neurogenesis. A proof of concept in preclinical study demonstrate that chronic administration of LiBen can protect cognition and memory and reduce the amyloid deposit in APP/PS-1 double transgenic mice. Furthermore, single oral administration of LiBen in rodent displayed better bioavailability than the combination of Li2CO3 and SB.
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
The patent portfolio of lithium benzoate was assigned to SyneuRx International (Taiwan) Corp.
REFERENCES
[1] | Praticò D ((2008) ) Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol Sci 29: , 609–615. |
[2] | Guglielmotto M , Tamagno E , Danni O ((2009) ) Oxidative stress and hypoxia contribute to Alzheimer’s disease pathogenesis: Two sides of the same coin. ScientificWorldJournal 9: , 781–791. |
[3] | Perez Ortiz JM , Swerdlow RH ((2019) ) Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol 176: , 3489–3507. |
[4] | Harkany T , Abrahám I , Timmerman W , Laskay G , Tóth B , Sasvári M , Kónya C , Sebens JB , Korf J , Nyakas C , Zarándi M , Soós K , Penke B , Luiten PG ((2000) ) beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci 12: , 2735–2745. |
[5] | Mattson MP ((2004) ) Pathways towards and away from Alzheimer’s disease. Nature 430: , 631–639. |
[6] | Paola D , Domenicotti C , Nitti M , Vitali A , Borghi R , Cottalasso D , Zaccheo D , Odetti P , Strocchi P , Marinari UM , Tabaton M , Pronzato MA ((2000) ) Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells. Biochem Biophys Res Commun 268: , 642–646. |
[7] | Misonou H , Morishima-Kawashima M , Ihara Y ((2000) ) Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 39: , 6951–6959. |
[8] | Atwood CS , Bowen RL , Smith MA , Perry G ((2003) ) Cerebrovascular requirement for sealant, anti-coagulant and remodeling molecules that allow for the maintenance of vascular integrity and blood supply. Brain Res Brain Res Rev 43: , 164–178. |
[9] | Tong Y , Zhou W , Fung V , Christensen MA , Qing H , Sun X , Song W ((2005) ) Oxidative stress potentiates BACE1 gene expression and Abeta generation. J Neural Transm (Vienna) 112: , 455–469. |
[10] | Barja G ((2004) ) Aging in vertebrates, and the effect of caloric restriction: A mitochondrial free radical production-DNA damage mechanism? Biol Rev Camb Philos Soc 79: , 235–251. |
[11] | Terman A , Kurz T , Navratil M , Arriaga EA , Brunk UT ((2010) ) Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12: , 503–535. |
[12] | Beal MF ((2002) ) Oxidatively modified proteins in aging and disease. Free Radic Biol Med 32: , 797–803. |
[13] | Nunomura A , Perry G , Pappolla MA , Wade R , Hirai K , Chiba S , Smith MA ((1999) ) RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J Neurosci 19: , 1959–1964. |
[14] | Trushina E , McMurray CT ((2007) ) Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 145: , 1233–1248. |
[15] | Chuang D-M , Chen R-W , Chalecka-Franaszek E , Ren M , Hashimoto R , Senatorov V , Kanai H , Hough C , Hiroi T , Leeds P ((2002) ) Neuroprotective effects of lithium in cultured cells and animal models of diseases. Bipolar Disord 4: , 129–136. |
[16] | Kerr F , Sofola-Adesakin O , Ivanov DK , Gatliff J , Gomez Perez-Nievas B , Bertrand HC , Martinez P , Callard R , Snoeren I , Cochemé HM , Adcott J , Khericha M , Castillo-Quan JI , Wells G , Noble W , Thornton J , Partridge L ((2017) ) Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet 13: , e1006593. |
[17] | Khan A , Jamwal S , Bijjem KRV , Prakash A , Kumar P ((2015) ) Neuroprotective effect of hemeoxygenase-1/glycogen synthase kinase-3β modulators in 3-nitropropionic acid-induced neurotoxicity in rats. Neuroscience 287: , 66–77. |
[18] | Chen X , Liu Y , Zhu J , Lei S , Dong Y , Li L , Jiang B , Tan L , Wu J , Yu S , Zhao Y ((2016) ) GSK-3β downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion. Sci Rep 6: , 20196. |
[19] | Rojo AI , Rada P , Egea J , Rosa AO , López MG , Cuadrado A ((2008) ) Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol Cell Neurosci 39: , 125–132. |
[20] | Castro AA , Casagrande TS , Moretti M , Constantino L , Petronilho F , Guerra GCB , Calo’ G , Guerrini R , Dal-Pizzol F , Quevedo J , Gavioli EC ((2009) ) Lithium attenuates behavioral and biochemical effects of neuropeptide S in mice. Peptides 30: , 1914–1920. |
[21] | Sofola-Adesakin O , Castillo-Quan JI , Rallis C , Tain LS , Bjedov I , Rogers I , Li L , Martinez P , Khericha M , Cabecinha M ((2014) ) Lithium suppresses Aβ pathology by inhibiting translation in an adult Drosophila model of Alzheimer’s disease. Front Aging Neurosci 6: , 190. |
[22] | Rockenstein E , Torrance M , Adame A , Mante M , Bar-On P , Rose JB , Crews L , Masliah E ((2007) ) Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci 27: , 1981–1991. |
[23] | Pan Y , Short JL , Newman SA , Choy KH , Tiwari D , Yap C , Senyschyn D , Banks WA , Nicolazzo JA ((2018) ) Cognitive benefits of lithium chloride in APP/PS1 mice are associated with enhanced brain clearance of β-amyloid. Brain Behav Immunity 70: , 36–47. |
[24] | Hampel H , Ewers M , Bürger K , Annas P , Mörtberg A , Bogstedt A , Frölich L , Schröder J , Schönknecht P , Riepe MW ((2009) ) Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 70: , 10490. |
[25] | Forlenza OV , Diniz BS , Radanovic M , Santos FS , Talib LL , Gattaz WF ((2011) ) Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: Randomised controlled trial. Br J Psychiatry 198: , 351–356. |
[26] | Nunes MA , Viel TA , Buck HS ((2013) ) Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res 10: , 104–107. |
[27] | World Health Organization & International Programme on Chemical Safety ((2000) ) Benzoic acid and sodium benzoate. World Health Organization. https://apps.who.int/iris/handle/10665/42310 |
[28] | Nair B ((2001) ) Final report on the safety assessment of benzyl alcohol, benzoic acid, and sodium benzoate. Int J Toxicol 20 Suppl 3: , 23–50. |
[29] | Reddy TL , Krishnarao PS , Rao GK , Bhimireddy E , Venkateswarlu P , Mohapatra DK , Yadav JS , Bhadra U , Bhadra MP ((2015) ) Para amino benzoic acid-derived self-assembled biocompatible nanoparticles for efficient delivery of siRNA. Int J Nanomed 10: , 6411–6423. |
[30] | Mattevi A , Vanoni MA , Todone F , Rizzi M , Teplyakov A , Coda A , Bolognesi M , Curti B ((1996) ) Crystal structure of D-amino acid oxidase: A case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci U S A 93: , 7496–7501. |
[31] | Coyle JT , Tsai G ((2004) ) NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. Int Rev Neurobiol 59: , 491–515. |
[32] | Lane H-Y , Lin C-H , Green MF , Hellemann G , Huang C-C , Chen P-W , Tun R , Chang Y-C , Tsai GE ((2013) ) Add-on treatment of benzoate for schizophrenia: A randomized, double-blind, placebo-controlled trial of d-amino acid oxidase inhibitor. JAMA Psychiatry 70: , 1267–1275. |
[33] | Lin C-H , Lin C-H , Chang Y-C , Huang Y-J , Chen P-W , Yang H-T , Lane H-Y ((2018) ) Sodium benzoate, a D-amino acid oxidase inhibitor, added to clozapine for the treatment of schizophrenia: A randomized, double-blind, placebo-controlled trial. Biol Psychiatry 84: , 422–432. |
[34] | Billard J-M ((2015) ) D-Serine in the aging hippocampus. J Pharm Biomed Anal 116: , 18–24. |
[35] | Modi KK , Roy A , Brahmachari S , Rangasamy SB , Pahan K ((2015) ) Cinnamon and its metabolite sodium benzoate attenuate the activation of p21rac and protect memory and learning in an animal model of Alzheimer’s disease. PLoS One 10: , e0130398. |
[36] | Arabsolghar R , Saberzadeh J , Khodaei F , Borojeni RA , Khorsand M , Rashedinia M ((2017) ) The protective effect of sodium benzoate on aluminum toxicity in PC12 cell line. Res Pharm Sci 12: , 391–400. |
[37] | Xu W , Li T , Gao L , Lenahan C , Zheng J , Yan J , Shao A , Zhang J ((2019) ) Sodium benzoate attenuates secondary brain injury by inhibiting neuronal apoptosis and reducing mitochondria-mediated oxidative stress in a rat model of intracerebral hemorrhage: Possible involvement of DJ-1/Akt/IKK/NFκB pathway. Front Mol Neurosci 12: , 105. |
[38] | Lin C-H , Chen P-K , Chang Y-C , Chuo L-J , Chen Y-S , Tsai GE , Lane H-Y ((2014) ) Benzoate, a D-amino acid oxidase inhibitor, for the treatment of early-phase Alzheimer disease: A randomized, double-blind, placebo-controlled trial. Biol Psychiatry 75: , 678–685. |
[39] | Chen S-D , Wu C-L , Lin T-K , Chuang Y-C , Yang D-I ((2012) ) Renin inhibitor aliskiren exerts neuroprotection against amyloid beta-peptide toxicity in rat cortical neurons. Neurochem Int 61: , 369–377. |
[40] | Ju T-C , Yang Y-T , Yang D-I ((2004) ) Protective effects of S-nitrosoglutathione against neurotoxicity of 3-nitropropionic acid in rat. Neurosci Lett 362: , 226–231. |
[41] | Wu C-L , Yin J-H , Hwang C-S , Chen S-D , Yang D-Y , Yang D-I ((2012) ) c-Jun-dependent sulfiredoxin induction mediates BDNF protection against mitochondrial inhibition in rat cortical neurons. Neurobiol Dis 46: , 450–462. |
[42] | Ju T-C , Chen S-D , Liu C-C , Yang D-I ((2005) ) Protective effects of S-nitrosoglutathione against amyloid beta-peptide neurotoxicity. Free Radic Biol Med 38: , 938–949. |
[43] | Wu Y , Connors D , Barber L , Jayachandra S , Hanumegowda UM , Adams SP ((2009) ) Multiplexed assay panel of cytotoxicity in HK-2 cells for detection of renal proximal tubule injury potential of compounds. Toxicol In Vitro 23: , 1170–1178. |
[44] | Chen H-M , Hsu J-H , Liou S-F , Chen T-J , Chen L-Y , Chiu C-C , Yeh J-L ((2014) ) Baicalein, an active component of Scutellaria baicalensis Georgi, prevents lysophosphatidylcholine-induced cardiac injury by reducing reactive oxygen species production, calcium overload and apoptosis via MAPK pathways. BMC Complement Altern Med 14: , 233. |
[45] | Chang G , Chen Y , Li Y , Li S , Huang F , Shen Y , Xie A ((2015) ) Self-healable hydrogel on tumor cell as drug delivery system for localized and effective therapy. Carbohydr Polym 122: , 336–342. |
[46] | Barnes CA ((1979) ) Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93: , 74–104. |
[47] | Kapur S ((2003) ) Psychosis as a state of aberrant salience: A framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 160: , 13–23. |
[48] | Cheignon C , Tomas M , Bonnefont-Rousselot D , Faller P , Hureau C , Collin F ((2018) ) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14: , 450–464. |
[49] | Hellström-Lindahl E , Viitanen M , Marutle A ((2009) ) Comparison of Abeta levels in the brain of familial and sporadic Alzheimer’s disease. Neurochem Int 55: , 243–252. |
[50] | Basha MR , Wei W , Bakheet SA , Benitez N , Siddiqi HK , Ge Y-W , Lahiri DK , Zawia NH ((2005) ) The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25: , 823–829. |
[51] | Tabaton M , Piccini A ((2005) ) Role of water-soluble amyloid-β in the pathogenesis of Alzheimer’s disease. Int J Exp Pathol 86: , 139–145. |
[52] | Mu Y , Gage FH ((2011) ) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6: , 85. |
[53] | Arias C , Arrieta I , Tapia R ((1995) ) beta-Amyloid peptide fragment 25-35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. J Neurosci Res 41: , 561–566. |
[54] | Kessels HW , Nabavi S , Malinow R ((2013) ) Metabotropic NMDA receptor function is required for β-amyloid-induced synaptic depression. Proc Natl Acad Sci U S A 110: , 4033–4038. |
[55] | Zlokovic BV ((2011) ) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12: , 723–738. |
[56] | Koistinaho M , Koistinaho J ((2005) ) Interactions between Alzheimer’s disease and cerebral ischemia–focus on inflammation. Brain Res Brain Res Rev 48: , 240–250. |
[57] | Craig AH , Cummings JL , Fairbanks L , Itti L , Miller BL , Li J , Mena I ((1996) ) Cerebral blood flow correlates of apathy in Alzheimer disease. Arch Neurol 53: , 1116–1120. |
[58] | Levinoff EJ , Li KZH , Murtha S , Chertkow H ((2004) ) Selective attention impairments in Alzheimer’s disease: Evidence for dissociable components. Neuropsychology 18: , 580–588. |
[59] | Reddy PH , Beal MF ((2008) ) Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14: , 45–53. |
[60] | Liot G , Bossy B , Lubitz S , Kushnareva Y , Sejbuk N , Bossy-Wetzel E ((2009) ) Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Differ 16: , 899–909. |
[61] | Li Y , Li Y , Li X , Zhang S , Zhao J , Zhu X , Tian G ((2017) ) Head injury as a risk factor for dementia and Alzheimer’s disease: A systematic review and meta-analysis of 32 observational studies. PLoS One 12: , e0169650. |
[62] | Sun X , He G , Qing H , Zhou W , Dobie F , Cai F , Staufenbiel M , Huang LE , Song W ((2006) ) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A 103: , 18727–18732. |
[63] | Zhang X , Zhou K , Wang R , Cui J , Lipton SA , Liao F-F , Xu H , Zhang Y ((2007) ) Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282: , 10873–10880. |
[64] | Kerr F , Bjedov I , Sofola-Adesakin O ((2018) ) Molecular mechanisms of lithium action: Switching the light on multiple targets for dementia using animal models. Front Mol Neurosci 11: , 297. |
[65] | Jana A , Modi KK , Roy A , Anderson JA , van Breemen RB , Pahan K ((2013) ) Up-regulation of neurotrophic factors by cinnamon and its metabolite sodium benzoate: Therapeutic implications for neurodegenerative disorders. J Neuroimmune Pharmacol 8: , 739–755. |
[66] | Khasnavis S , Pahan K ((2014) ) Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9: , 569–581. |
[67] | Ueki A , Goto K , Sato N , Iso H , Morita Y ((2006) ) Prepulse inhibition of acoustic startle response in mild cognitive impairment and mild dementia of Alzheimer type. Psychiatry Clin Neurosci 60: , 55–62. |
[68] | Aziz VM ((2019) ) Deficit in sensorimotor gating in Alzheimer’s disease (AD): Measuring pre pulse inhibition (PPI) as a measure of liability to AD. Ann Psychiatr Clin Neurosci 2: , 1008. |
[69] | Zhang X , Heng X , Li T , Li L , Yang D , Zhang X , Du Y , Doody RS , Le W ((2011) ) Long-term treatment with lithium alleviates memory deficits and reduces amyloid-β production in an aged Alzheimer’s disease transgenic mouse model. J Alzheimers Dis 24: , 739–749. |
[70] | Gitlin M ((2016) ) Lithium side effects and toxicity: Prevalence and management strategies. Int J Bipolar Disord 4: , 27. |
[71] | Fountoulakis KN , Vieta E , Bouras C , Notaridis G , Giannakopoulos P , Kaprinis G , Akiskal H ((2008) ) A systematic review of existing data on long-term lithium therapy: Neuroprotective or neurotoxic? Int J Neuropsychopharmacol 11: , 269–287. |
[72] | Zhong J , Yang X , Yao W , Lee W ((2006) ) Lithium protects ethanol-induced neuronal apoptosis. Biochem Biophys Res Commun 350: , 905–910. |
[73] | Chi-Tso C , Chuang D-M ((2011) ) Neuroprotective action of lithium in disorders of the central nervous system. Zhong Nan Da Xue Xue Bao Yi Xue Ban 36: , 461. |
[74] | Hou L , Xiong N , Liu L , Huang J , Han C , Zhang G , Li J , Xu X , Lin Z , Wang T ((2015) ) Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci 16: , 82. |
[75] | Hyde C , Peters J , Bond M , Rogers G , Hoyle M , Anderson R , Jeffreys M , Davis S , Thokala P , Moxham T ((2013) ) Evolution of the evidence on the effectiveness and cost-effectiveness of acetylcholinesterase inhibitors and memantine for Alzheimer’s disease: Systematic review and economic model. Age Ageing 42: , 14–20. |
[76] | Howard R , McShane R , Lindesay J , Ritchie C , Baldwin A , Barber R , Burns A , Dening T , Findlay D , Holmes C , Hughes A , Jacoby R , Jones R , Jones R , McKeith I , Macharouthu A , O’Brien J , Passmore P , Sheehan B , Juszczak E , Katona C , Hills R , Knapp M , Ballard C , Brown R , Banerjee S , Onions C , Griffin M , Adams J , Gray R , Johnson T , Bentham P , Phillips P ((2012) ) Donepezil and memantine for moderate-to-severe Alzheimer’s disease. N Engl J Med 366: , 893–903. |
[77] | Campos C , Rocha NB , Vieira RT , Rocha SA , Telles-Correia D , Paes F , Yuan T , Nardi AE , Arias-Carrión O , Machado S , Caixeta L ((2016) ) Treatment of cognitive deficits in Alzheimer’s disease: A psychopharmacological review. Psychiatr Danub 28: , 2–12. |


