
Intracerebral Infection with E. coli Impairs Spatial Learning and Induces Necrosis of Hippocampal Neurons in the Tg2576 Mouse Model of Alzheimer’s Disease
Abstract
Background:
In patients with Alzheimer’s disease (AD), bacterial infections are often associated with a cognitive decline. Animal models of genuine acute infections with viable bacteria which induce deterioration of neurodegenerative diseases are missing.
Objective:
We assessed the effect of an intracerebral infection with E. coli in a mouse model of AD.
Methods:
13-month-old Tg2576 +/- mice and transgene negative littermates (Tg2576 -/-) received an intracerebral injection with E. coli K1 or saline followed by treatment with ceftriaxone starting 41 h post infection (p.i.) for 5 days. For 4 weeks, mice were monitored for clinical status, weight, motor functions, and neuropsychological status using the Morris water maze. ELISAs, stainings, and immunohistochemistry in brains were performed at the end of the experiment.
Results:
Mortality of the infection was approximately 20%. After 4 weeks, spatial learning of infected Tg2576 +/- mice was compromised compared to non-infected Tg2576 +/- mice (p < 0.05). E. coli infection did not influence spatial learning in Tg2576 -/- mice, or spatial memory in both Tg2576 +/- and -/- mice within 4 weeks p.i.. Necrosis of hippocampal neurons was induced in infected compared to non-infected Tg2576 +/- mice 4 weeks p.i., whereas brain concentrations of Aβ1–40, Aβ1–42, and phosphoTau as well as axonal damage and microglia density were not altered.
Conclusion:
Here, we proved in principle that a genuine acute bacterial infection can worsen cognitive functions of AD mice. Mouse models of subacute systemic infections are needed to develop new strategies for the treatment of bacterial infections in patients with AD in order to minimize their cognitive decline.
INTRODUCTION
Alzheimer’s disease (AD), the most common form of dementia, currently affects more than 30 million people worldwide and is one of the principal causes of disability in the elderly [1]. Concomitant with the increased life expectancy, the number of AD patients will further increase. The cause of AD is still not known, and a curative treatment is not available. Therefore, prevention and optimized handling of all conditions which can worsen neurological and neuropsychological symptoms of AD patients obtain highest relevance in the clinical routine.
The deterioration of neurological and neuro-psychological symptoms of patients suffering from neurodegenerative diseases during acute systemic infections is a common clinical problem [2, 3]. Resolution of neurological symptoms which developed during infection is often incomplete [4]. By now, there is strong evidence for an infection-related progression of neurodegeneration from studies in humans [for review: 2, 5, 6]. Common bacterial infections like sepsis, pneumonia, urinary tract infections, and cellulitis are associated with an increased risk and/or an accelerated development of dementia in elderly persons [7–10]. In AD patients, systemic infections were associated with an increased cognitive decline over a period of 2 and 6 month [11, 12]. Furthermore, in aged persons and patients with dementia, infections often cause delirium [13, 14] which can accelerate the loss of mnestic functions [15–17]. Systemic inflammatory responses, e.g., as consequence of respiratory or urinary tract infections, frequently occur in elderly people living in a nursing home [18] and their incidence is higher in AD patients than in age-matched controls [19]. Therefore, prevention and optimized treatment of acute bacterial infections are of high importance for minimizing infection-induced cognitive decline in AD patients.
Experimental models of acute systemic inflammation in animals with existing neurodegenerative pathology, e.g., in mouse models of prion disease or AD, have revealed important mechanisms of inflam-mation-driven neurodegeneration and cognitive decline [20]. In most experimental studies in this field, lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria with strong immuno-stimulatory properties, was used to induce inflammation [21, 22]. Although LPS-induced inflammation is a reliable and reproducible stimulus, it does not accurately imitate an infection with viable bacteria [23, 24], and caution is necessary, when translating results obtained with this artificial stimulus to real infections in humans [21, 25]. For the development of optimized treatment strategies, there is still need of an animal model in which a genuine acute bacterial infection with a common pathogen worsens clinical symptoms of a neurodegenerative disease [26].
The gram-negative bacterium Escherichia coli (E. coli) frequently causes urinary tract infections and is also a prevalent cause of bacterial meningitis in elderly and immunocompromised persons [27, 28]. Here, we analyzed the impact of an intracerebral infection with E. coli K1—posing a strong inflammatory stimulus in the central nervous system (CNS)—on the course of disease in the Tg2576 +/- mouse model of AD.
MATERIAL AND METHODS
Animals
All animal experiments were approved by the Animal Care Committee of the University Medical Center Göttingen (UMG) and by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), Braunschweig, Lower Saxony, Germany.
Transgenic Tg2576 mice heterozygous for the TgN(APP-Sw)2576 transgene express the human amyloid precursor protein (APP695) carrying the double-mutation Lys670-Asn and Met671-Leu found in a Swedish family with early onset of AD under the hamster prion protein promoter. These mice develop amyloid deposits in the brain at an age of 6 months and spatial memory deficits at an age of 9 months [29–31]. Despite some features of human AD are missing, e.g., severe neuronal loss and neurofibrillary tangles, the Tg2576 model is one of the most well characterized and most widely used mouse models of AD [32]. Mice were genotyped to identify transgene positive mice (Tg2576 +/-) and transgene negative control littermates (Tg2576 -/-). Infection was performed when mice were older than 12 months (Tg2576 +/- mice 13.0 + 1.8 months; Tg2576 -/- mice 13.1 + 1.4 months).
Infection
An E. coli K1 strain (serotype O18:K1:H7) originally isolated from an adult with meningitis (gift of Dr. G. Zysk, University of Düsseldorf, Germany) was used for inoculations [33]. E. coli K1 belongs to the group of MNEC (Meningitis/sepsis-associated E. coli), its key characteristic is its capsule preventing phagocytosis [34]. The inoculum was grown on blood agar plates, stored at –70°C, and diluted in 0.9% NaCl before infection. Bacterial concentrations in the inoculum were determined by plating of serial 1:10-dilutions on blood-agar-plates. Mice were anaesthetized by intraperitoneal injection of ketamine (100 mg/kg body weight) and xylazine (10 mg/kg body weight). Intracerebral infection was induced by injection of 10μl NaCl 0.9% containing 4.25 (3.5/5.75) ×103 CFU E. coli K1 (4 independent experiments) into the superficial right frontal cortex and subarachnoid space through the right frontolateral skull [33]. Non-infected control mice received 10μl NaCl 0.9% intracerebrally. Starting 41 h after infection, all mice received antibiotic treatment with ceftriaxone 100 mg/kg body weight (Rocephin; Hoffmann-LaRoche, Grenzach-Wyhlen, Germany) administered subcutaneously twice daily for five consecutive days. Mice were clinically monitored every 12 h and weighed daily during the acute phase of the infection. Mice which were unable to walk or lost more than 15% of their body weight were sacrificed.
Rotarod test
Motor performance was assessed regularly using a rotarod device (Neuroscience Inc., Tokyo, Japan). Mice were placed on a cylinder at a slow rotational speed that was gradually increased from 4 to 40 rpm within a period of 300 s. Time mice remained on the rotarod was recorded up to 300 s (minimum 0 s, maximum 300 s) [35].
Morris water maze
To assess spatial memory and learning deficits, the water maze task [36] was performed in a white-painted pool (diameter 104 cm, height 35 cm) containing water maintained at a temperature of 22°C. A transparent platform of 10 cm in diameter was located 1 cm beneath the water surface. A video camera fixed at the ceiling above the pool and linked to a computer (TSE Systems, Bad Homburg, Germany) recorded the swim tracks and the latency to find the platform. Because of the high intra-individual variation of each mouse with respect to its performance in the water maze, the median of 3 consecutive runs was used for further analyses.
3 days before the infection (day -3, -2, -1), all mice were trained to find the hidden under-water platform in less than 90 s (3×6 runs over 3 days). Mice which did not find the platform within 90 s were placed onto it for 30 s. Directly before infection (day 0) and beginning at 7 days after the infection, 3 water maze runs with the hidden platform located at the pre-infection site were repeated once a week for 4 weeks (days 7, 14, 21, 27) to assess spatial memory. To investigate deficits in spatial learning, 4 weeks after infection, the platform was moved into the center of the adjacent quadrant, and 3×6 runs spread over 3 days were performed (day 29, 30, 31).
Tissue preparation, processing, and quantification of bacterial concentrations
Mice were sacrificed by cervical dislocation. Brains were removed and sagittally divided into two parts. For staining and immunohistochemical analyses, the right hemisphere was fixated in 4% formaldehyde and embedded in paraffin. 1-2μm sections were produced. The left hemisphere was divided into three parts (anterior and posterior cerebrum, and cerebellum) which were immediately frozen in liquid nitrogen and stored frozen at –80°C.
Of mice that died from the infection, the left half of the cerebellum and one half of the spleen were homogenized in NaCl 0.9%. Bacterial concentrations in the homogenates were determined by plating of serial 1:10-dilutions on blood-agar-plates.
Quantification of Aβ and phosphorylated Tau protein (phosphoTau)
Aβ enzyme-linked immunosorbent assay (ELISA) and phosphoTau ELISA
Protein extraction from the posterior half of the left cerebral hemisphere was performed according to the manufacturer's instructions. Protein concentrations were determined using the Pierce™ BCA Protein Assay (Thermo Fisher Scientific). ELISAs for Aβ1–40 and Aβ1–42 (hAmyloid β40/ β42; The Genetics Company) and phosphoTau (Tau [pS199] mouse ELISA Kit; Thermo Fisher Scientific) were performed according to the manufacturer’s instructions.
Thioflavine S staining
After deparaffinization and washing in distilled water, sections were treated with 0.25% KMnO4 for 20 min and bleached in a solution consisting of 1% K2S2O5 and 1% oxalic acid for 2 min. After that, sections were treated with a solution containing 1 g NaOH and 3 ml of 30% H2O2 in 100 ml H2O for 20 min and dipped for 5 s in 0.25% acetic acid. Between all steps, sections were washed with H2O. Then sections were stained with 1% thioflavine S in 50% ethanol for 5 min. Finally, sections were rinsed twice with 50% ethanol and twice with distilled H2O and mounted in fluoromount. The area of thioflavin S-stained plaques was quantified using an automated computer-assisted analysis (Definiens Developer XD 1.0.0, Definiens AG, Munich, Germany).
Quantification of necrotic neurons, axonal damage, and microglial cells
Hematoxylin-eosin (HE) staining of brain sections was performed in mice dying from the infection in order to document meningitis as the cause of death as well as in mice sacrificed at the end of the experiment in order to quantify necrotic neurons in hippocampus (CA 1–4), dentate gyrus and neocortex using a semiquantitative score: 0 = no necrotic cells, 1 = single necrotic cells, 2 = groups of necrotic cells, 3 = large parts of hippocampus or dentate gyrus necrotic [37].
Axonal damage was detected by amyloid-β protein precursor (AβPP) immunohistochemistry and quantified by two blinded investigators using a semiquantitative score: 0 = no damage or just single axons, 1 = minimal damage (1-2 lesions), 2 = moderate damage (3-4 lesions), 3 = severe damage (> 4 lesions) [38].
Ionized calcium-binding adaptor molecule 1 (Iba-1) was used to identify microglial cells. In each animal, Iba-1-positive cells were quantified in the subiculum of the hippocampal formation and in three different neocortical regions and (x 20 objective) [39].
Statistics
GraphPad Prism 5.0 Software (GraphPad Software, San Diego, California, USA) was used to perform statistical analyses and graphical presentation. Parametric data are expressed as means±standard deviations (SD), nonparametric data are expressed as medians (25th percentile/75th percentile). Student’s t-test was performed to compare two groups of parametric data, Mann-Whitney U-test to compare two groups of nonparametric data. For comparison of the rotarod test performance-versus-time curves and the water maze test performance-versus-time curves areas under the curves (AUCs) were compared using Mann-Whitney U-test. The Bonferroni method was applied in case of multiple comparisons to correct for repeated testing. Log-rank test was performed for the comparison of survival curves. Repeated measures ANOVA followed by followed by Tukey's multiple comparisons test was performed for analysis of weight loss. p values < 0.05 were considered statistically significant.
RESULTS
Intracerebral injection of E. coli K1 induced acute signs of a strong infectious disease in Tg2576 +/+ and Tg2576 -/- mice
22 Tg2576 +/- mice and 20 Tg2576 -/- mice received an intracerebral injection of E. coli K1. Despite antibiotic treatment after 41 h, the overall mortality of the infection was 21.4%. Mortality did not differ between transgenic and non-transgenic mice: 18.2% of Tg2576 +/- mice (4 of 22) and 25% of Tg2576 -/- mice (5 of 20) died due to the infection after 2 to 5 days (log-rank test: p = 0.64; Fig. 1A).
Fig. 1
Acute signs of infection in Tg2576 +/- and Tg2576 -/- mice after intracerebral injection of E. coli. A) Mortality after intracerebral E. coli injection was 18.2% in Tg2576 +/- mice and 25.0% in Tg2576 -/- mice (Kaplan-Meier curve; log-rank test: p = 0.64). B) Representative HE-stainings of the right cerebral hemisphere of a deceased Tg2576 +/- mouse (left image) and a deceased Tg2576 -/- mouse (right image) showing meningeal inflammation. C) Concentrations of E. coli in cerebellum and spleen of deceased Tg2576 +/- and Tg2576 -/- mice [medians and single values; Mann-Whitney U-test: p = 0.11 (cerebellum) and p = 0.22 (spleen)].
![Acute signs of infection in Tg2576 +/- and Tg2576 -/- mice after intracerebral injection of E. coli. A) Mortality after intracerebral E. coli injection was 18.2% in Tg2576 +/- mice and 25.0% in Tg2576 -/- mice (Kaplan-Meier curve; log-rank test: p = 0.64). B) Representative HE-stainings of the right cerebral hemisphere of a deceased Tg2576 +/- mouse (left image) and a deceased Tg2576 -/- mouse (right image) showing meningeal inflammation. C) Concentrations of E. coli in cerebellum and spleen of deceased Tg2576 +/- and Tg2576 -/- mice [medians and single values; Mann-Whitney U-test: p = 0.11 (cerebellum) and p = 0.22 (spleen)].](https://content.iospress.com:443/media/adr/2022/6-1/adr-6-1-adr210049/adr-6-adr210049-g001.jpg)
HE-stainings of the right cerebral hemispheres of deceased Tg2576 +/- and Tg2576 -/- mice showed inflammation and/or disruption of the meninges (Fig. 1B).
In all deceased mice, bacterial concentrations in cerebellum and spleen were determined by quantitative plating. In mice which died before beginning of the antibiotic therapy, E. coli concentrations were > 106 CFU/ml in cerebellum and > 104 CFU/ml in spleen, demonstrating the growth of bacteria in the CNS and their systemic spreading. In both cerebellum and spleen, bacterial concentrations decreased after initiation of antibiotic treatment. As more Tg2576 +/- mice than Tg2576 -/- mice died before the first antibiotic administration, slightly higher bacterial titers were observed in deceased Tg2576 +/- mice compared to Tg2576 -/- mice. However, bacterial concentrations did not differ significantly between deceased Tg2576 +/- and Tg2576 -/- mice in cerebellum (2.25×106 (9.75×105/3×108) versus 4×105 (2.5×104/1.5×106) CFU/ml; p = 0.11) and spleen (6.5×104 (2.6×104/3×106) versus 1.7×103 (< 1×103/7.5×104) CFU/ml; p = 0.22; Fig. 1C).
Mice surviving the infection showed a significant weight loss during the acute disease phase as a clinical sign of infection (Tg2576 + /–: p < 0.05 compared to weight before infection (day 0) on days 2 to 5 p.i. / Tg2576 -/-: p < 0.05 compared to weight before infection on days 2 to 5 p.i.). Minimum weight in both groups was reached on day 3 p.i. (Tg2576 + /–: 26.9±3.1 g before infection (day 0) versus 24.8±3.7 g on day 3 p.i.; p < 0.001 / Tg2576 -/-: 30.9±4.8 g before infection (day 0) versus 28.8±5.8 g on day 3 p.i.; p < 0.001). This weight loss was transient, weight at the end of the experiment did not differ significantly from weight before infection (Tg2576 + /–: 25.9±3.6 g on day 31 p.i.; p > 0.05 / Tg2576 -/-: 29.6±5.2 g on day 31 p.i.; p > 0.05).
Non-infected mice that were intracerebrally injected with NaCl 0.9% showed neither significant weight loss during the period of antibiotic treatment nor at the end of the experiment (Tg2576 + /–: 26.5±3.5 g on day 0; 26.2±3.5 g on day 3, 25.9±3.8 g on day 31, p > 0.05, respectively / Tg2576 -/-: 32.9±4.5 g on day 0; 32.1±4.4 g on day 3; 31.9±4.0 g on day 31; p > 0.05, respectively). A slight reduction of weight from the start until the end of the experiment was seen in all groups, most probably due to the increased physical activity during the monitoring tests.
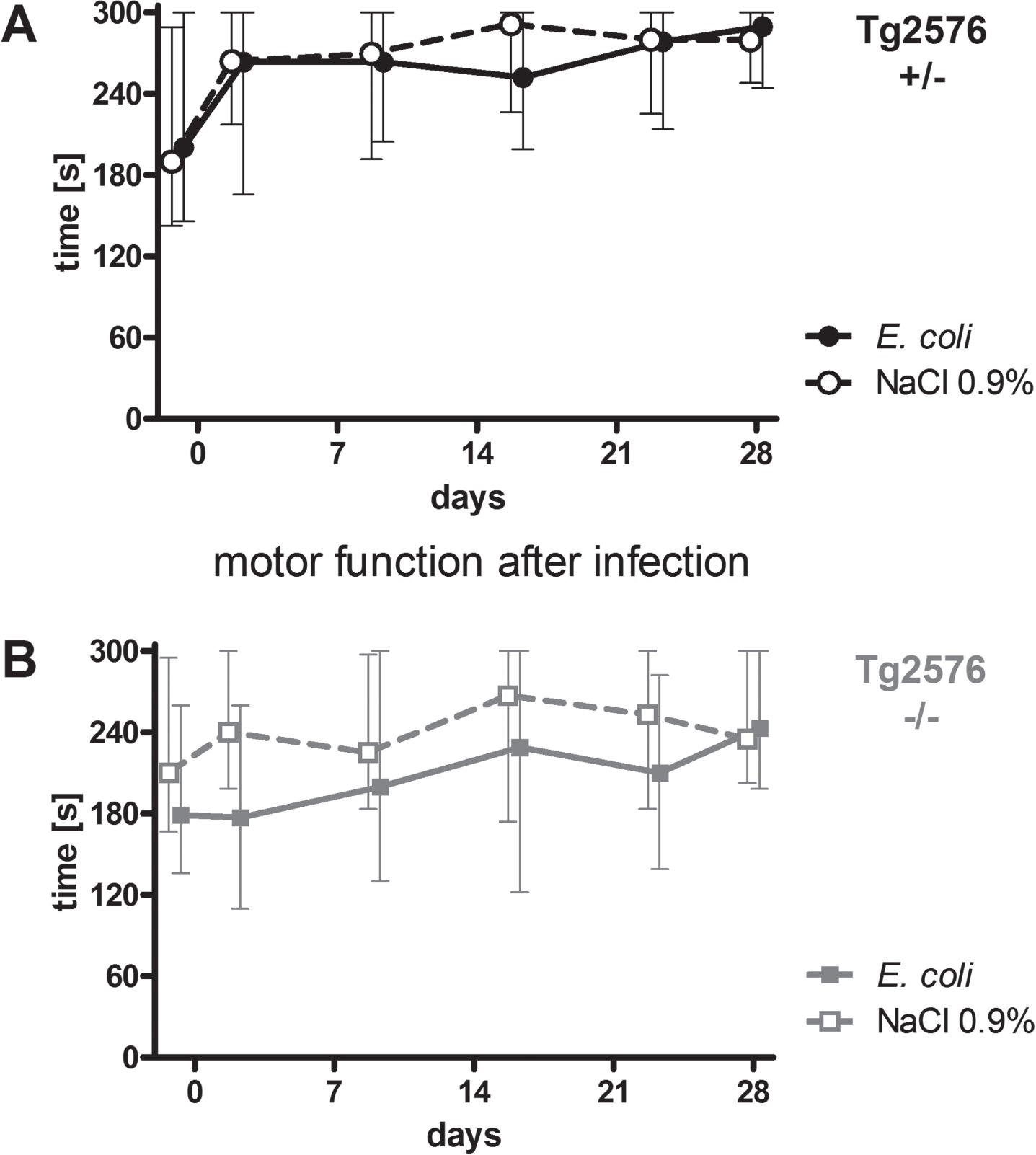
Intracerebral infection with E. coli did not lead to persistent deficits of motor functions of Tg2576 +/- and Tg2576 -/- mice
Motor functions of all mice were assessed regularly throughout the experiment (day -1, 2, 9, 16, 23, and 28) using the rotarod test. Intracerebral infection with E. coli did not affect performance of Tg2576 +/- mice (Fig. 2A) and Tg2676 -/- mice (Fig. 2B) in the rotarod test (AUC (seconds×days) infected versus non-infected Tg2576 +/- mice: 7574 (6169/7865) versus 7795 (6174/8684), p = 0.61; AUC (seconds x days) infected versus non-infected Tg2576 -/- mice: 5654 (4025/7745) versus 7340 (5546/8178), p = 0.19). As water maze results are dependent on the ability of mice to swim, a stable motor function after infection was essential for the reliable analysis of spatial memory and spatial learning based on the performance of mice in the Morris water maze.
Fig. 2
Motor function of Tg2576 +/- and Tg2576 -/- mice after surviving an intracerebral E. coli infection. A) Performance of surviving infected Tg2576 +/- mice (n = 18) in the rotarod test did not differ from that of uninfected Tg2576 +/- mice (n = 14) up to 4 weeks after intracerebral E. coli infection (comparison of AUCs, Mann-Whitney U-test, p = 0.61). B) Performance of surviving infected Tg2576 -/- mice (n = 15) in the rotarod test did not differ from that of uninfected Tg2576 -/- mice (n = 17) up to 4 weeks after intracerebral E. coli infection (comparison of AUCs, Mann-Whitney U-test, p = 0.19). Medians (25./75. percentiles) of times that mice remained on the rotarod are presented (minimum 0, maximum 300 s).
Intracerebral infection with E. coli impaired spatial learning in Tg2576 +/- mice, but did not influence spatial memory
36 Tg2576 +/- mice and 37 Tg2576 -/- mice learned the location of a hidden platform in three water maze sessions three, two and one day before infection (day -3, -2, and -1). There was no difference between Tg2576 +/- and Tg2576 -/- mice concerning their ability to learn the location of the platform prior to infection (AUC (seconds x days): 142.6 (87.0/172.3) versus 137.1 (79.1/165.1), p = 0.29).
After this training episode, mice were randomly allocated to the infection and non-infection groups considering a similar sex distribution, and intracerebral injections were performed. Mice that died from the infection were excluded from the following analysis shown in Fig. 3. Surviving Tg2576 +/- (n = 32) and Tg2576 -/- mice (n = 32) significantly improved their performance in the Morris water maze [latency (s)] prior to infection (Tg2576 + /–: 91 (64/91) on day -3 versus 69 (23/91) on day -1, p = 0.0066; Tg2576 -/-: 88 (58/91) on day -3 versus 36 (15/79) on day -1, p = 0.0006).
Fig. 3
Spatial memory and spatial learning of Tg2576 +/- and Tg2576 -/- mice after intracerebral E. coli infection. A-C) Water maze performances of surviving infected Tg2576 +/- mice (n = 18) and uninfected Tg2576 +/- mice (n = 14) assessed by the latencies to find the hidden platform. A) The ability to learn the location of the platform did not differ between Tg2576 +/- mice allocated to the infection-group and the non-infection-group. B) The ability to remember the location of the hidden platform, assessed once a week during four weeks after infection, did not differ significantly between infected and non-infected Tg2576 +/- mice. C) Four weeks after infection, the ability of infected Tg2576 +/- mice to learn the new location of the platform was impaired compared to non-infected Tg2576 +/-. D-F) Water maze performances of surviving infected Tg2576 -/- mice (n = 15) and uninfected Tg2576 -/- mice (n = 17) assessed by the latencies to find the hidden platform. There were no differences between Tg2576 -/- mice of the infection group and Tg2576 -/- mice of the non-infection group concerning their ability to learn the location of the platform before infection (D), to remember the location of the platform during four weeks after infection (E), and to learn the new location of the platform four weeks after infection (F). Medians (25./75. percentiles) of median times mice needed to find the platform in 3 to 6 water maze runs are presented (maximum 90 s).
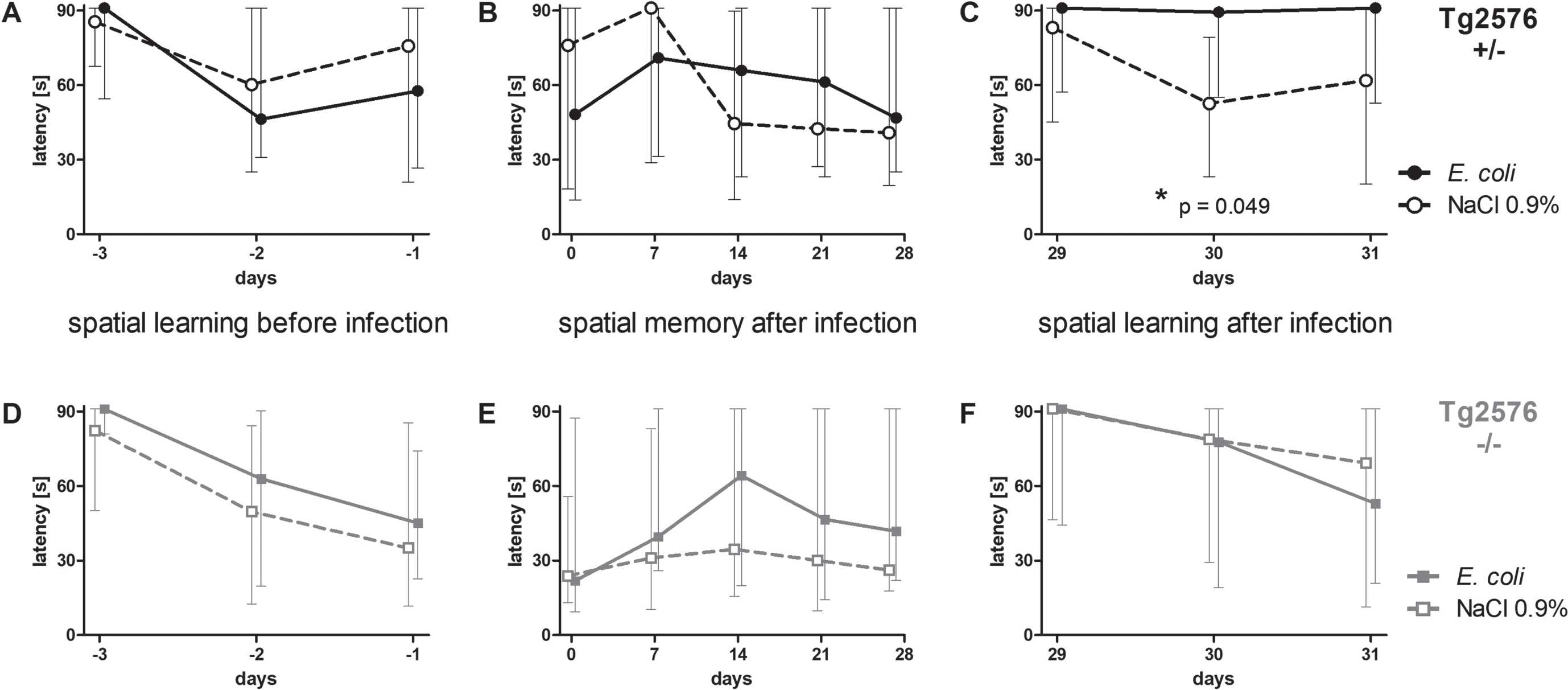
Surviving Tg2675 +/- mice allocated to the infection group (n = 18) did not differ from Tg2675 +/- mice allocated to the NaCl 0.9% -treated group (n = 14) concerning their ability to learn the location of the hidden platform before infection (AUC (seconds x days): 115.3 (77.7/158.4) versus 132.3 (80.57/174.7), p = 0.56; Fig. 3A). Also, surviving Tg2675 -/- mice allocated to the infection group (n = 15) did not differ from Tg2675 -/- mice allocated to the NaCl 0.9% -treated group (n = 17) concerning spatial learning before infection (AUC (seconds x days): 138.2 (79.2/170.7) versus 106.6 (58.2/145.5), p = 0.37; Fig. 3D). Sex ratio was almost equal in all groups (Tg2576 +/- E. coli: 6 male, 12 female (33.3% male); Tg2576 +/- NaCl 0.9% : 5 male, 9 female (35.7% male); Tg2576 -/- E. coli: 5 male, 10 female (33.3% male); Tg2576 -/- NaCl 0.9% : 6 male, 11 female (35.3% male)).
To assess spatial memory after intracerebral infection, water maze was performed once per week in the 4 weeks after infection; the location of the platform was not changed during this period. Spatial memory was not affected by intracerebral infection with E. coli in both Tg2576 +/- mice and Tg2576 -/- mice: latency to find the platform did not differ between infected and non-infected Tg2576 +/- mice (AUC (seconds x days): 1627 (1118/2028) versus 1555 (611.3/2141), p = 0.78; Fig. 3B) and infected and non-infected Tg2576 -/- mice (AUC (seconds x days): 1487 (539.8/2286) versus 845.6 (449.5/2029), p = 0.21; Fig. 3E).
Four weeks after infection, the platform was moved to the center of the adjacent quadrant, and the ability to learn the new location of the platform was assessed in the following three days—similar to the training sessions before infection. Here, infected Tg2576 +/- mice showed deficits in spatial learning and were not able to learn the new location of the platform, whereas non-infected Tg2576 +/- mice showed no deficits: latencies to find the platform were significantly higher in infected Tg2576 +/- than in non-infected Tg2576 +/- mice (AUC (seconds x days): 168.6 (109/182) versus 112.8 (88.91/142.2), p = 0.049; Fig. 3C). In Tg2576 -/- mice, spatial learning did not differ between the post-infection and the NaCl 0.9% -treated group four weeks after infection (AUC (seconds x days): 163 (62.31/182) versus 139.1 (61.04/182), p = 0.91; Fig. 3F).
Aβ load and phosphoTau levels in brains of Tg2576 +/- mice were not altered 4 weeks after intracerebral infection with E. coli
At the end of the experiment, 31 days after infection, all mice were sacrificed. In Tg2576 +/- mice, concentrations of soluble and insoluble Aβ1–40 and Aβ1–42 were determined by ELISA. Concentrations of the different Aβ subforms did not differ between brains of infected and non-infected Tg2576 +/- mice (Table 1). Amyloid plaques in brains of Tg2576 +/- mice were stained with thioflavine, and the cumulative plaque area was assessed software-assisted (Fig. 4A). Cumulative plaque area did not differ significantly between infected and non-infected Tg2576 +/- mice (794 (324.5/1612) μm2 versus 900 (292.5/1756) μm2, p = 0.92; Fig. 4B).
Table 1
Concentrations of Aβ1 - 40 and Aβ1 - 42 in brains of Tg2576 +/- mice after intracerebral infection with E. coli
| Soluble Aβ | |||
| E. coli | NaCl 0.9% | p | |
| Aβ1–40 (pg/mg brain) | 8.35±2.64 | 7.23±3.10 | 0.38 |
| Aβ1–42 (pg/mg brain) | 5.22±1.08 | 5.33±2.15 | 0.88 |
| Aβ1–42/Aβ1–40 | 0.66±0.17 | 0.94±0.72 | 0.23 |
| Insoluble Aβ | |||
| E. coli | NaCl 0.9% | p | |
| Aβ1–40 (pg/mg brain) | 1.78±0.28 | 1.72±0.12 | 0.51 |
| Aβ1–42 (pg/mg brain) | 6.67±1.91 | 6.29±0.40 | 0.49 |
| Aβ1–42/Aβ1–40 | 1.34±0.49 | 1.54±1.1 | 0.58 |
Fig. 4
Aβ load and phosphoTau concentrations in brains of Tg2576 +/- mice after intracerebral infection with E. coli. A) Example of a plaque stained with thioflavine (immunofluorescence, left image), and example of thioflavin stained cortical areas automatically detected using the Definiens software (immunofluorescence, right image). B) Comparison of the plaque area (median, 25th/75th percentile) in brains of infected and non-infected Tg2576 +/- mice (p = 0.92, Mann-Whitney U-test). C) Comparison of phosphoTau concentrations (means±SD) in brains of infected and non-infected Tg2576 +/- mice as measured by ELISA (p = 0.1, Student’s t-test).

PhosphoTau concentrations in brains of infected Tg2576 +/- mice (92.1±11.6 ng/ml) and non-infected Tg2576 + /–(82.2±20.9 ng/ml) were also not different (p = 0.1; Fig. 4C).
Density of necrotic neurons was increased in the hippocampus of Tg2576 +/- mice 4 weeks after intracerebral infection with E. coli
At the end of the experiment, 31 days p.i., necrotic neurons in HE-stainings were assessed by a semiquantitative score (Fig. 5A, B). In the majority of non-infected Tg2576 +/- mice, only few single necrotic neurons in dentate gyrus and hippocampus were detected. In the hippocampal areas CA1-4, the density of necrotic neurons was significantly higher in infected than in non-infected Tg2576 +/- mice (1 (1/2) versus 0 (0/1), p = 0.038; Fig. 5C), whereas the density of necrotic neurons did not differ in the dentate gyrus (1 (0/2) versus 1 (0/1), p = 0.76; Fig. 5D). In neocortex, no necrotic neurons were detected in non-infected and only few single necrotic neurons were detected in 2 of 18 infected Tg2576 +/- mice. Density of necrotic neurons in neocortex did not differ between infected and non-infected Tg2576 +/- mice (0 (0/0) versus 0 (0/0), p = 0.64).
Fig. 5
Necrotic neurons in brains of Tg2576 +/- mice after intracerebral infection with E. coli. A, B) Representative HE stainings with high numbers of necrotic neurons in the hippocampus regions CA1-4 (score 3; A), and low numbers of necrotic neurons in the hippocampus regions CA1-4 (score 0; B) [34]. C) Necrotic neurons in the hippocampus regions CA1-4 (median and single values) of infected and non-infected Tg2576 +/- mice (p = 0.038, Mann-Whitney U-test). D) Necrotic neurons in the dentate gyrus (median and single values) of infected and non-infected Tg2576 +/- mice (p = 0.76, Mann-Whitney U-test).
![Necrotic neurons in brains of Tg2576 +/- mice after intracerebral infection with E. coli. A, B) Representative HE stainings with high numbers of necrotic neurons in the hippocampus regions CA1-4 (score 3; A), and low numbers of necrotic neurons in the hippocampus regions CA1-4 (score 0; B) [34]. C) Necrotic neurons in the hippocampus regions CA1-4 (median and single values) of infected and non-infected Tg2576 +/- mice (p = 0.038, Mann-Whitney U-test). D) Necrotic neurons in the dentate gyrus (median and single values) of infected and non-infected Tg2576 +/- mice (p = 0.76, Mann-Whitney U-test).](https://content.iospress.com:443/media/adr/2022/6-1/adr-6-1-adr210049/adr-6-adr210049-g005.jpg)
Axonal damage and density of microglial cells were not altered in Tg2576 +/- mice 4 weeks after intracerebral infection with E. coli
Axonal damage was detected by AβPP immunohistochemistry (Fig. 6A) and quantified by a semi-quantitative score. The extent of axonal damage did not differ between brains of infected and non-infected Tg2576 +/- mice 31 days p.i. (1 (0.5/1.5) versus 0.5 (0.5/1.125); p = 0.074; Fig. 6D).
Fig. 6
Axonal damage and microglia density in brains of Tg2576 +/- mice after intracerebral infection with E. coli. A) Representative amyloid-β protein precursor (AβPP) immunohistochemistry showing small lesions of axonal damage (pink) [35]. B) Representative Iba-1 staining showing microglial cells in the hippocampus (brown). C) Representative Iba-1 staining showing microglial cells in the neocortex (brown). D) Axonal damage quantified by a semiquantitative score (0 = no axonal damage, 3 = severe axonal damage) in brains of infected and non-infected Tg2576 +/- (medians and single values; p = 0.074, Mann-Whitney U-test). E) Numbers of microglial cells (means and single values) in the hippocampus of infected and non-infected Tg2576 +/- mice (p = 0.84, Student’s t-test). F) Numbers of microglial cells (means and single values of means of 3 areas) in the neocortex of infected and non-infected Tg2576 +/- mice (p = 0.37, Student's t-test).
![Axonal damage and microglia density in brains of Tg2576 +/- mice after intracerebral infection with E. coli. A) Representative amyloid-β protein precursor (AβPP) immunohistochemistry showing small lesions of axonal damage (pink) [35]. B) Representative Iba-1 staining showing microglial cells in the hippocampus (brown). C) Representative Iba-1 staining showing microglial cells in the neocortex (brown). D) Axonal damage quantified by a semiquantitative score (0 = no axonal damage, 3 = severe axonal damage) in brains of infected and non-infected Tg2576 +/- (medians and single values; p = 0.074, Mann-Whitney U-test). E) Numbers of microglial cells (means and single values) in the hippocampus of infected and non-infected Tg2576 +/- mice (p = 0.84, Student’s t-test). F) Numbers of microglial cells (means and single values of means of 3 areas) in the neocortex of infected and non-infected Tg2576 +/- mice (p = 0.37, Student's t-test).](https://content.iospress.com:443/media/adr/2022/6-1/adr-6-1-adr210049/adr-6-adr210049-g006.jpg)
Microglial cells were identified by Iba-1 staining (Fig. 6B, C). Density of Iba-1 positive microglial cells did not differ between infected and non-infected Tg2576 +/- mice in the hippocampus (14.3±3.8 versus 14.6±4.1; p = 0.84; Fig. 6E) and in the neocortex (11.0±2.9 versus 11.8±2.0 (means of 3 areas); p = 0.37; Fig. 6F).
DISCUSSION
Chronic infections with microbes, e.g., spirochetal bacteria, Chlamydophila pneumoniae or Helicobacter pylori, have been linked to the pathogenesis of AD and are associated with cognitive decline (for reviews, see [5, 40–43]). Both clinical and experimental studies provide evidence for a progression of neurodegeneration related to an acute infection (for reviews, see [2, 5, 6]). Current knowledge about this phenomenon is mainly based on transgenic mouse models of neurodegenerative diseases in which LPS injections are used to mimic bacterial infections [21, 44]. For the development of optimized treatment strategies, there is still need of an animal model in which a real acute bacterial infection worsens clinical symptoms of a neurodegenerative disease.
However, it is difficult to induce a genuine systemic infection in mice with live bacteria which is potent enough and long-lasting enough to cause measurable changes in the neuropsychological tests and on the other hand does not kill the animals. In our previous study, repeated systemic infections with Streptococcus pneumoniae (S. pneumoniae) that were antibiotically treated already 12 h after infection to prevent death of the mice did not affect spatial memory and learning of Tg2576 +/- mice in the Morris water maze [35]. In the current study, we used the gram-negative bacterium E. coli which frequently induces urinary tract infections and sepsis and less often meningitis especially in elderly and immunocompromised persons. To achieve a strong and uniform infectious stimulus in the CNS, we chose an intracerebral way of infection with E. coli by directly injecting the bacteria into the right forebrain. The infection was left more than 40 h without antibiotic treatment, resulting in multiplication of bacteria in the CNS, systemic spreading of bacteria and an overall mortality of 20%. Without antibiotic treatment this infection would have been fatal in almost all animals. After performance of the same infection protocol, high levels of the proinflammatory cytokines IL-6 and KC (CXCL1) were observed in cerebellum and spleen of C57BL/6-N mice 24 h p.i. [33], and cerebellum levels of IL-1β, IL-6, KC, and MIP-2 (CXCL2) strongly correlated with bacterial titers in cerebellum [39]. Antibiotic treatment was performed with the bacteriolytic antibiotic ceftriaxone resulting in a rapid release of high amounts of bacterial products acting as strong stimuli of the innate immune system in the CNS and the systemic circulation [45]. Using this protocol, we demonstrate for the first time in an animal model, that a real acute infection exacerbates clinical symptoms in a neurodegenerative disease.
Infection was performed when mice were slightly older than 12 months. At this age, amyloid deposits are present in Tg2576 +/- mice and deficits of spatial memory have been detected [29]. With our water maze protocol, we were not able to detect differences between Tg2576 +/- and Tg2576 -/- mice concerning spatial learning at the beginning of the experiment before infection. This implies that we performed the intracerebral E. coli infection at a preclinical or mild state of AD.
We previously demonstrated that mortality after intracerebral E. coli infection was increased in aged mice (24 months) compared to young mice [33] reflecting the clinical situation in geriatric patients [28]. Clinical observations suggest that mortality of intracerebral infections also is higher in subjects with neurodegenerative diseases. However, in the present study, mortality after intracerebral infection with E. coli including antibiotic treatment with ceftriaxone at a preclinical or mild state of AD did not differ between Tg2576 +/- mice (18.2%) and Tg2576 -/- mice (25%; Fig. 1). An intracerebral infection at more advanced disease stages might have led to differences in mortality. Motor functions did not differ between transgenic and non-transgenic mice or infected and non-infected mice throughout the whole experiment which is a precondition for the reliable analysis of the water maze results.
During four weeks after intracerebral infection with E. coli, spatial memory was assessed once a week in the water maze. Two and three weeks after infection, infected Tg2576 +/- mice tended to need more time to find the hidden platform than non-infected Tg2576 +/- mice (difference not significant). Conversely, water maze performance four weeks after infection was nearly equal in both groups. Furthermore, comparison of the AUCs of the water maze performances over four weeks after infection showed no significant difference between infected and non-infected Tg2576 +/- mice suggesting that spatial memory of Tg2576 +/- mice was not affected by intracerebral infection with E. coli. Four weeks after infection, spatial learning was assessed by moving the platform in the water maze to the center of the adjacent quadrant. Mice were trained to learn the new position of the platform at day 29, 30, and 31 after infection. Here, infected Tg2576 +/- mice were not able to remember the new position of the platform in 3 x 6 runs, whereas non-infected Tg2576 +/- mice showed no deficits in the water maze task during these three days. Before infection at day -3, -2, and -1, Tg2576 +/- mice allocated to the infection group had learned to find the platform at a similar speed as mice allocated to the non-infection group. These results clearly show that intracerebral infection with E. coli caused spatial learning deficits in Tg2576 +/- mice four weeks p.i..
Several studies have already demonstrated a detrimental effect of systemic administration of the bacterial cell wall component LPS on cognition in animal models of neurodegenerative disease including mouse models of AD. Systemic inflammatory challenge with LPS in mice with prion disease led to enhanced brain inflammation, increased neurodegeneration, exacerbated cognitive and motor symptoms, and accelerated disease progression [44, 46, 47]. Marottoli et al. showed in E4FAD mice, which express human apolipoprotein E4 (APOE4) and 5 familial AD mutations (5xFAD), that nine intraperitoneal injections of LPS 0.5 mg/kg/week at a preclinical state of the disease (between 4 and 6 months of age) caused cognitive deficits: LPS treatment had no effect on spatial memory; however, it significantly impaired recognition memory [48]. In 3xTg-AD mice, triple-transgenic mice harboring three mutations of familial AD (PS1(M146V), APP(Swe), and tau(P301L)), at an age of 12 months, intraperitoneal injections of LPS 0.5 mg/kg twice per week for 6 weeks mimicking a chronic bacterial infection induced learning and memory impairments [49]. In the same mouse model (3xTg-AD) a single intraperitoneal injection of LPS 1 mg/kg at an age of 4 months induced a deficit in spatial memory 2 months later [50].
Our present study and these studies are difficult to compare, as different mouse models of AD, different tests for cognitive function, and different paradigms of administration of the infectious stimulus were used. However, they all demonstrate inflammation-induced cognitive decline in models of AD. To our knowledge, the present study is the first study demonstrating negative effects of a genuine infection with live bacteria on cognition in a mouse model of AD.
Studies in wild-type mice and rats also demonstrated negative effects of systemic LPS administration on performance in the water maze [51–53]. Intracerebral infection with S. pneumoniae in young C57BL/6-mice induced long-term deficits in spatial learning [36]. The single intracerebral infection with E. coli performed in our study neither caused deficits in spatial memory nor deficits in spatial learning in non-transgenic Tg2576 -/- mice. The exclusive occurrence of spatial learning deficits in AD mice supports the presumption of a higher vulnerability of AD patients for an infection-induced cognitive decline.
As in brains of patients with AD, in Tg2576 +/- mice Aβ accumulates in the brains in plaques and as cerebral amyloid angiopathy in the walls of arteries and arterioles [54, 55]. Aβ activates microglia, and microglial cells are able to phagocytose Aβ. The processing of Aβ plays a significant role in AD pathogenesis [56]. In brains of wild-type rats, Aβ accumulation and Tau phosphorylation were observed upon systemic inflammation induced by LPS or cecal ligation and perforation [57, 58]. After intraperitoneal or intracerebral administration of LPS to Tg2576 +/- mice, both an increase of Aβ brain concentrations [59, 60] and a reduction of cerebral Aβ levels or cortical plaque burden [61–63] have been observed. Marottoli et al. found higher extracellular levels of Aβ1–42 in the cortex of LPS-treated compared to control E4FAD mice [48]. In our model, we did not find differences concerning the Aβ load in brains of infected and non-infected Tg2576 +/- mice four weeks p.i.: plaques sizes and brain tissue levels of soluble and insoluble Aβ1–40 and Aβ1–42 did not differ. The E. coli infection performed in our model was of relatively short duration, and the Aβ load was assessed after the relatively long period of four weeks. Possibly we could have detected an alteration in Aβ load earlier after infection. In accordance with our results, Sy et al. also did not detect any differences concerning Aβ concentrations and plaques between LPS-treated and control 3xTg-AD mice in their study mentioned above [49]. They found elevated levels of phosphoTau as well as a shift of Tau from the soluble to the insoluble fraction [49], suggesting that the inflammation-mediated changes of Tau metabolism are more relevant for the impairment of cognition during infections than Aβ pathology. In the present study, however, levels of phosphoTau in brains of infected and uninfected Tg2576 +/- mice did not differ four weeks p.i. (Fig. 4).
Spatial learning and memory deficits have been linked to lesions in the hippocampal formation which are regularly observed in animal models of bacterial meningitis [36, 64–67]. In the present study, the number of necrotic neurons in the hippocampus regions CA1-4 was increased in Tg2576 +/- mice 4 weeks after intracerebral infection with E. coli (Fig. 5). However, we did not observe differences concerning axonal damage in brains of infected and non-infected Tg2576 +/- mice (Fig. 6). The infection-induced neuronal damage in the hippocampus most probably accounts for the spatial learning deficits in Tg2576 +/- mice 4 weeks p.i.
Glia-mediated inflammation is considered to play a crucial role for progression of neurodegeneration and clinical symptoms in AD during infections. The joint activation of microglia by bacterial products and endogenous proteins such as Aβ leading to an additive neuronal damage, which we previously demonstrated in vitro, may be an important link between systemic infections and neurodegenerative diseases. Further stimulation of already primed microglia by infections may drive disease progression in chronic inflammatory conditions [2, 71]. In a neurodegenerative mouse model of prion disease, systemic inflammation induced by LPS or polyinosinic:polycytidylic acid (Poly(I:C)) led to changes in microglial density and morphology; however, these changes did not last long [72, 73]. The only transient changes of microglial cells might explain that we did not detect differences in microglia density and morphology in brains of infected and non-infected Tg2576 +/- mice after the relatively long period of 4 weeks. Nonetheless, transiently activated microglial cells can induce longer-lasting inflammatory effects contributing to neurodegeneration. Thus, minimizing microglial activation by bacteria and bacterial products, e.g., by an antibiotic regimen aiming at a reduction of proinflammatory bacterial compounds [45, 74, 75], appears a promising approach for an optimized treatment of acute bacterial infections in patients with neurodegenerative diseases.
Bacterial CNS infections are more frequent in elderly than in young persons and show a worse outcome in aged subjects [28] which we could previously reproduce in our geriatric mouse model of E. coli meningitis [33]. However, due to their many times higher incidence, the impact of systemic infections on the disease progression of AD patients is of much higher clinical relevance. Despite leading to a fast systemic spreading of bacteria and a systemic inflammatory response (see Fig. 1C and [33]), the intracerebral way of E. coli injection used in our current mouse model does not accurately imitate systemic infection, particularly regarding bacterial concentrations in the CNS and the impact of the peripheral immune system. However, with the intracerebral infection paradigm of the current study, we proved in principle that a genuine acute bacterial infection can worsen cognitive functions of AD mice. Because of the greater clinical relevance of infections originating outside the CNS, the development of subacute mouse models of systemic infections in AD mice is urgent. One first approach could be the inoculation of bacteria into subcutaneously implanted tissue cages causing a chronic mild infection (e.g., [76, 77]). Models simulating common clinically relevant subacute infections (e.g., catheter-related infections, osteomyelitis or soft tissue infections) may be even more suitable for the assessment of anti-infective treatment options for bacterial infections in the expanding group of patients suffering from AD.
ACKNOWLEDGMENTS
We acknowledge support by the Open Access Publication Funds of the Göttingen University.
FUNDING
This work was supported by the Robert Bosch Foundation, Stuttgart, Germany – Forschungskolleg Geriatrie (SSch).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | GBD 2017 Disease and Injury Incidence and Prevalence Collaborators ((2018) ) Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 392: , 1789–1858. |
[2] | Perry VH , Cunningham C , Holmes C ((2007) ) Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 7: , 161–167. |
[3] | Brugger F , Erro R , Balint B , Kägi G , Barone P , Bhatia KP ((2015) ) Why is there motor deterioration in Parkinson’s disease during systemic infections-a hypothetical view. NPJ Parkinsons Dis 1: , 15014. |
[4] | Umemura A , Oeda T , Tomita S , Hayashi R , Kohsaka M , Park K , Sugiyama H , Sawada H ((2014) ) Delirium and high fever are associated with subacute motor deterioration in Parkinson disease: A nested case-control study. PLoS One 9: , e94944. |
[5] | McManus RM , Heneka MT ((2017) ) Role of neuroinflammation in neurodegeneration: New insights. Alzheimers Res Ther 9: , 14. |
[6] | Giridharan VV , Masud F , Petronilho F , Dal-Pizzol F , Barichello T ((2019) ) Infection-induced systemic inflammation is a potential driver of Alzheimer’s disease progression. Front Aging Neurosci 11: , 122. |
[7] | Muzambi R , Bhaskaran K , Brayne C , Davidson JA , Smeeth L , Warren-Gash C ((2020) ) Common bacterial infections and risk of dementia or cognitive decline: A systematic review. J Alzheimers Dis 76: , 1609–1626. |
[8] | Muzambi R , Bhaskaran K , Smeeth L , Brayne C , Chaturvedi N , Warren-Gash C ((2021) ) Assessment of common infections and incident dementia using UK primary and secon-dary care data: A historical cohort study. Lancet Healthy Longev 2: , 426–435. |
[9] | Dunn N , Mullee M , Perry VH , Holmes C ((2005) ) Association between dementia and infectious disease: Evidence from a case-control study. Alzheimer Dis Assoc Disord 19: , 91–94. |
[10] | Shah FA , Pike F , Alvarez K , Angus D , Newman AB , Lopez O , Tate J , Kapur V , Wilsdon A , Krishnan JA , Hansel N , Au D , Avdalovic M , Fan VS , Barr RG , Yende S ((2013) ) Bidirectional relationship between cognitive function and pneumonia. Am J Respir Crit Care Med 188: , 586–592. |
[11] | Holmes C , El-Okl M , Williams AL , Cunningham C , Wilcockson D , Perry VH ((2003) ) Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry 74: , 788–789. |
[12] | Holmes C , Cunningham C , Zotova E , Woolford J , Dean C , Kerr S , Culliford D , Perry VH ((2009) ) Systemic inflammation and disease progression in Alzheimer disease. Neurology 73: , 768–774. |
[13] | Pieralli F , Vannucchi V , Mancini A , Grazzini M , Paolacci G , Morettini A , Nozzoli C ((2014) ) Delirium is a predictor of in-hospital mortality in elderly patients with community acquired pneumonia. Intern Emerg Med 9: , 195–200. |
[14] | Chae JH , Miller BJ ((2015) ) Beyond urinary tract infections (UTIs) and delirium: A systematic review of UTIs and neuropsychiatric disorders. J Psychiatr Pract 21: , 402–411. |
[15] | Fong TG , Jones RN , Shi P , Marcantonio ER , Yap L , Rudolph JL , Yang FM , Kiely DK , Inouye SK ((2009) ) Delirium accelerates cognitive decline in Alzheimer disease. Neurology 72: , 1570–1575. |
[16] | Gross AL , Jones RN , Habtemariam DA , Fong TG , Tommet D , Quach L , Schmitt E , Yap L , Inouye SK ((2012) ) The long-term effect of delirium on the cognitive trajectory of persons with dementia. Arch Intern Med 172: , 1324–1331. |
[17] | Korgseth M , Watne LO , Juliebo V , Skovlund E , Engedal K , Frihagen F , Wyller TB ((2016) ) Delirium is a risk factor for further cognitive decline in cognitively impaired hip fracture patients. Arch Gerontol Geriatr 64: , 38–44. |
[18] | Engelhart ST , Hanses-Derendorf L , Exner M , Kramer MH ((2005) ) Prospective surveillance for healthcare-associated infections in German nursing home residents. J Hosp Infect 60: , 46–50. |
[19] | Natalwala A , Potluri R , Uppal H , Heun R ((2008) ) Reasons for hospital admissions in dementia patients in Birmingham, UK, during 2002-2007. Dement Geriatr Cogn Disord 26: , 499–505. |
[20] | Cunningham C , Hennessy E ((2015) ) Co-morbidity and systemic inflammation as drivers of cognitive decline: New experimental models adopting a broader paradigm in dementia research. Alzheimers Res Ther 7: , 33. |
[21] | Catorce MN , Gevorkian G ((2016) ) LPS-induced murine neuroinflammation model: Main features and suitability for pre-clinical assessment of nutraceuticals. Current Neuropharmacol 14: , 155–164. |
[22] | Schreuder L , Eggen BJ , Biber K , Schoemaker RG , Laman JD , de Rooij SE ((2017) ) Pathophysiological and behavioral effects of systemic inflammation in aged and diseased rodents with relevance to delirium: A systematic review. Brain Behav Immun 62: , 362–381. |
[23] | Remick DG , Ward PA ((2005) ) Evaluation of endotoxin models for the study of sepsis. Shock 24: , 7–11. |
[24] | Diesselberg C , Ribes S , Michel U , Redlich S , Brück W , Nau R , Schütze S ((2012) ) Follistatin does not influence the course of E. coli K1 sepsis in a mouse model. Shock 38: , 615–619. |
[25] | Barron M , Gartlon J , Dawson LA , Atkinson PJ , Pardon MC ((2017) ) A state of delirium: Deciphering the effect of inflammation on tau pathology in Alzheimer's disease. Exp Gerontol 94: , 103–107. |
[26] | Patrick KL , Bell SL , Weindel CG , Watson RO ((2019) ) Exploring the “multiple-hit hypothesis” of neurodegenerative disease: Bacterial infection comes up to bat. Front Cell Infect Microbiol 9: , 138. |
[27] | Choi C ((2001) ) Bacterial meningits in aging adults. Clin Infect Dis 33: , 1380–1385. |
[28] | Cabellos C , Verdaguer R , Olmo M , Fernández-Sabé N , Cisnal M , Ariza J , Gudiol F , Viladrich PF ((2009) ) Community-acquired bacterial meningitis in elderly patients: Experience over 30 years. Medicine (Balitmore) 88: , 115–119. |
[29] | Hsiao K , Chapman P , Nilsen S , Eckman C , Harigaya Y , Younkin S , Yang F , Cole G ((1996) ) Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science 274: , 99–102. |
[30] | Münch G , Apelt J , Kientsch-Engel R , Stahl P , Lüth HJ , Schliebs R ((2003) ) Advanced glycation endproducts and pro-inflammatory cytokines in transgenic Tg2576 mice with amyloid plaque pathology. J Neurochem 86: , 283–289. |
[31] | Apelt J , Bigl M , Wunderlich P , Schliebs R ((2004) ) Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity in beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int J Dev Neurosci 22: , 475–484. |
[32] | Jankowsky JL , Zheng H ((2017) ) Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol Neurodegener 12: , 89. |
[33] | Schütze S , Ribes S , Kaufmann A , Manig A , Scheffel J , Redlich S , Bunkowski S , Hanisch UK , Brück W , Nau R ((2014) ) Higher mortality and impaired elimination of bacteria in aged mice after intracerebral infection with E. coli are associated with an age-related decline of microglia and macrophage functions. Oncotarget 5: , 12573–12592. |
[34] | Kaper JB , Nataro JP , Mobley HL ((2004) ) Pathogenic Escherichia coli. Nat Rev Microbiol 2: , 123–140. |
[35] | Ebert S , Goos M , Rollwagen L , Baake D , Zech W-D , Esselmann H , Wiltfang J , Mollenhauer B , Schliebs R , Gerber J , Nau R ((2010) ) Recurrent systemic infections with Streptococcus pneumoniae do not aggravate the course of experimental neurodegenerative diseases. J Neurosci Res 88: , 1124–1136. |
[36] | Wellmer A , Noeske C , Gerber J , Munzel U , Nau R ((2000) ) Spatial memory and learning deficits after experimental pneumococcal meningitis in mice. Neurosci Lett 296: , 137–140. |
[37] | Gerber J , Raivich G , Wellmer A , Noeske C , Kunst T , Werner A , Brück W , Nau R. ((2001) ) A mouse model of Streptococcus pneumoniae meningitis mimicking several features of human disease. Acta Neuropathol 101: , 499–508. |
[38] | Tauber SC , Ribes S , Ebert S , Heinz T , Fingerle V , Bunkowski S , Kugelstadt D , Spreer A , Jahn O , Eiffert H , Nau R ((2011) ) Long-term intrathecal infusion of outer surface protein C from Borrelia burgdorferi causes axonal damage. J Neuropathol Exp Neurol 70: , 748–757. |
[39] | Ribes S , Regen T , Meister T , Tauber SC , Schütze S , Mildner A , Mack M , Hanisch UK , Nau R ((2013) ) Resistance of the brain to Escherichia coli K1 infection depends on MyD88 signaling and the contribution of neutrophils and monocytes. Infect Immun 81: , 1810–1819. |
[40] | Maheshwari P , Eslick GD ((2015) ) Bacterial infection and Alzheimer’s disease: A meta-analysis. J Alzheimers Dis 43: , 957–966. |
[41] | Itzhaki RF , Lathe R , Balin BJ , Ball MJ , Bearer EL , Braak H , Bullido MJ , Carter C , Clerici M , Cosby SL , Del Tredici K , Field H , Fülöp T , Grassi C , Griffin WS , Haas J , Hudson AP , Kamer AR , Kell DB , Licastro F , Letenneur L , Lövheim H , Mancuso R , Miklossy J , Otth C , Palamara AT , Perry G , Preston C , Pretorius E , Strandberg T , Tabet N , Taylor-Robinson SD , Whittum-Hudson JA ((2016) ) Microbes and Alzheimer’s disease. J Alzheimers Dis 51: , 979–984. |
[42] | Fülöp T , Witkowski JM , Bourgade K , Khalil A , Zerif E , Larbi A , Hirokawa K , Pawelec G , Bocti C , Lacombe G , Dupuis G , Frost EH ((2018) ) Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front Aging Neurosci 10: , 224. |
[43] | Schnaider L , Arnon ZA , Gazit E ((2020) ) Reevaluating the microbial infection link to Alzheimer’s disease. J Alzheimers Dis 73: , 59–62. |
[44] | Cunningham C , Wilcockson DC , Campion S , Lunnon K , Perry VH ((2005) ) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25: , 9275–9284. |
[45] | Nau R , Eiffert H ((2002) ) Modulation of release of proinflammatory bacterial compounds by antibacterials: Potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Micorbiol Rev 15: , 95–110. |
[46] | Combrinck MI , Perry VH , Cunningham C ((2002) ) Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience 112: , 7–11. |
[47] | Cunningham C , Campion S , Lunnon K , Murray CL , Woods JFC , Deacon RMJ , Rawlins JN , Perry VH ((2009) ) Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol Psychiatry 65: , 304–312. |
[48] | Marottoli FM , Katsumata Y , Koster KP , Thomas R , Fardo DW , Tai LM ((2017) ) Peripheral inflammation, apolipoprotein E4, and amyloid-β interact to induce cognitive and cerebrovascular dysfunction. ASN Neuro 9: , 1759091417719201. |
[49] | Sy M , Kitazawa M , Medeiros R , Whitman L , Cheng D , Lane TE , La Ferla FM ((2011) ) Inflammation induced by infection potentiates Tau pathological features in transgenic mice. Am J Pathol 178: , 2811–2822. |
[50] | Valero J , Mastrella G , Neiva I , Sanchez S , Malva JO ((2014) ) Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front Neurosci 8: , 83. |
[51] | Shaw KN , Commins S , O'Mara SM ((2001) ) Lipopolysaccharide causes deficits in spatial learning in the watermaze but not in BDNF expression in the rat dentate gyrus. Behav Brain Res 124: , 47–54. |
[52] | Sparkman NL , Martin LA , Calvert WS , Boehm GW ((2005) ) Effects of intraperitoneal lipopolysaccharide on Morris maze performance in year-old and 2-month-old female C57BL/6J mice. Behav Brain Res 159: , 145–151. |
[53] | Lee JW , Lee YK , Yuk DY , Choi DY , Ban SB , Oh KW , Hong JT ((2008) ) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflamm 5: , 37. |
[54] | Kuo YM , Crawford F , Mullan M , Kokjohn TA , Emmerling MR , Weller RO , Roher AE ((2000) ) Elevated A beta and apolipoprotein E in A betaPP transgenic mice and its relationship to amyloid accumulation in Alzheimer's disease. Mol Med 6: , 430–439. |
[55] | Kawarabayashi T , Younkin LH , Saido TC , Shoji M , Hsiao Ashe K , Younkin SG ((2001) ) Age-dependent changes in brain, CSF, and plasma amyloid beta protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: , 373–381. |
[56] | García-González L , Pilat D , Baranger K , Rivera S ((2019) ) Emerging alternative proteinases in APP metabolism and Alzheimer’s disease pathogenesis: A focus on MT1-MMP and MT5-MMP. Front Aging Neurosci 11: , 244. |
[57] | Gasparotto J , Girardi CS , Somensi N , Ribeiro CT , Moreira JCF , Michels M , Sonai B , Rocha M , Steckert AV , Barichello T , Quevedo J , Dal-Pizzol F , Gelain DP ((2018) ) Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J Biol Chem 293: , 226–244. |
[58] | Wang LM , Wu Q , Kirk RA , Horn KP , Ebada Salem AH , Hoffman JM , Yap JT , Sonnen JA , Towner RA , Bozza FA , Rodrigues RS , Morton KA ((2018) ) Lipopolysaccharide endotoxemia induces amyloid-beta and p-tau formation in the rat brain. Am J Nucl Med Mol Imaging 8: , 86–99. |
[59] | Sheng JG , Bora SH , Xu G , Borchelt DR , Price DL , Koliatsos VE ((2003) ) Lipopolysaccharide-induced-neuro-inflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis 14: , 133–145. |
[60] | Sly LM , Krzesicki RF , Brashler JR , Buhl AE , McKinley DD , Carter DB , Chin JE ((2001) ) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 mouse model of Alzheimer's disease. Brain Res Bull 56: , 581–588. |
[61] | DiCarlo G , Wilcock D , Henderson D , Gordon M , Morgan D ((2001) ) Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging 22: , 1007–1012. |
[62] | Quinn J , Montine T , Morrow J , Woodward WR , Kulhanek D , Eckenstein F ((2003) ) Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer's disease. J Neuroimmunol 137: , 32–41. |
[63] | Herber DL , Mercer M , Roth LM , Symmonds K , Maloney J , Wilson N , Freeman MJ , Morgan D , Gordon MN ((2007) ) Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmun Pharmacol 2: , 222–231. |
[64] | Leib SL , Kim YS , Chow LL , Sheldon RA , Täuber MG ((1996) ) Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest 98: , 2632–2639. |
[65] | Zysk G , Brück W , Gerber J , Brück Y , Prange HW , Nau R ((1996) ) Anti-inflammatory treatment influences neuronal apoptotic cell death in the dentate gyrus in experimental pneumococcal meningitis. J Neuropathol Exp Neurol 55: , 722–728. |
[66] | Braun JS , Sublett JE , Freyer D , Mitchell TJ , Cleveland JL , Tuomanen EI , Weber JR ((2002) ) Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J Clin Invest 109: , 19–27. |
[67] | Spreer A , Gerber J , Hanssen M , Schindler S , Hermann C , Lange P , Eiffert H , Nau R ((2006) ) Dexamethasone increases hippocampal neuronal apoptosis in a rabbit model of Escherichia coli meningitis. Pediatr Res 60: , 210–215. |
[68] | Ebert S , Gerber J , Bader S , Mühlhauser F , Brechtel K , Mitchell TJ , Nau R ((2005) ) Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J Neuroimmunol 159: , 87–96. |
[69] | Lotz M , Ebert S , Esselmann H , Iliev AI , Prinz M , Wiazewicz N , Wiltfang J , Gerber J , Nau R ((2005) ) Amyloid beta peptide 1-40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem 94: , 289–298. |
[70] | Schütze S , Loleit T , Zeretzke M , Bunkowski S , Brück W , Ribes S , Nau R ((2012) ) Additive microglia-mediated neuronal injury caused by amyloid-β and bacterial TLR agonists in murine neuron-microglia co-cultures quantified by an automated image analysis using cognition network technology. J Alzheimers Dis 31: , 651–657. |
[71] | Cunningham C ((2013) ) Microglia and neurodegeneration: The role of systemic inflammation. Glia 61: , 71–90. |
[72] | Field R , Campion S , Warren C , Murray C , Cunningham C ((2010) ) Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegeneration. Brain Behav Immun 24: , 996–1007. |
[73] | Murray C , Sanderson DJ , Barkus C , Deacon RM , Rawlins JN , Bannerman DM , Cunningham C ((2012) ) Systemic inflammation induces acute working memory deficits in the primed brain: Relevance for delirium. Neurobiol Aging 33: , 603–616. e3. |
[74] | Nau R , Djukic M , Spreer A , Ribes S , Eiffert H ((2015) ) Bacterial meningitis: An update of new treatment options. Expert Rev Anti Infect Ther 13: , 1401–1423. |
[75] | Muri L , Grandgirard D , Buri M , Perny M , Leib SL ((2018) ) Combined effect of non-bacteriolytic antibiotic and inhibition of matrix metalloproteinases prevents brain injury and preserves learning, memory and hearing function in experimental paediatric pneumococcal meningitis. J Neuroinflammation 15: , 233. |
[76] | Lucet JC , Herrmann M , Rohner P , Auckenthaler R , Waldvogel FA , Lew DP ((1990) ) Treatment of experimental foreign body infection caused by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 34: , 2312–2317. |
[77] | Kumar P , Kretzschmar B , Herold S , Nau R , Kreutzfeldt M , Schütze S , Bähr M , Hein K ((2015) ) Beneficial effect of chronic Staphylococcus aureus infection in a model of multiple sclerosis is mediated through the secretion of extracellular adherence protein. J Neuroinflammation 12: , 22. |


