
Neuronal Deposition of Amyloid-β Oligomers and Hyperphosphorylated Tau Is Closely Connected with Cognitive Dysfunction in Aged Dogs
Abstract
Background:
Canine cognitive dysfunction (CCD) is a progressive syndrome recognized in mature to aged dogs with a variety of neuropathological changes similar to human Alzheimer’s disease (AD), for which it is thought to be a good natural model. However, the presence of hyperphosphorylated tau protein (p-Tau) in dogs with CCD has only been demonstrated infrequently.
Objective:
The aim of the present study was to investigate the presence of p-Tau and amyloid-β oligomer (Aβo) in cerebral cortex and hippocampus of dogs with CCD, with focus on an epitope retrieval protocol to unmask p-Tau.
Methods:
Immunohistochemical and immunofluorescence analysis of the cortical and hippocampal regions of five CCD-affected and two nondemented aged dogs using 4G8 anti-Aβp, anti-Aβ1 - 42 nanobody (PrioAD13) and AT8 anti-p-Tau (Ser202, Thr205) antibody were used to demonstrate the presence of Aβ plaques (Aβp) and Aβ1 - 42 oligomers and p-Tau deposits, respectively.
Results:
The extracellular Aβ senile plaques were of the diffuse type which lack the dense core normally seen in human AD. While p-Tau deposits displayed a widespread pattern and closely resembled the typical human neuropathology, they did not co-localize with the Aβp. Of considerable interest, however, widespread intraneuronal deposition of Aβ1 - 42 oligomers were exhibited in the frontal cortex and hippocampal region that co-localized with p-Tau.
Conclusion:
Taken together, these findings reveal further shared neuropathologic features of AD and CCD, supporting the case that aged dogs afflicted with CCD offer a relevant model for investigating human AD.
INTRODUCTION
The human life expectancy is increasing with the advancement of medicine, nutrition, technology, health, and social care. According to the World Health Organization (WHO), the number of people aged over 60 years reached 1 billion in 2020 and is expected to increase to 2.1 billion by 2050 [1]. One of the most common neurological disorders in the aging population is Alzheimer’s disease (AD) and advancing age is the single most important risk factor. The worldwide prevalence of AD is currently at 50 million and will likely triple to 150 million people by 2050 [2]. AD is a progressive neurodegenerative disorder causing gradual decline in cognitive functions believed to begin at least two decades before any obvious cognitive symptoms [3, 4]. Thus far, the most commonly used animal model to investigate human AD employs rodents. The common strategy to generate these models is by overexpressing mutant human APP and/or PS1 genes [5, 6]. Although the importance of such transgenic models is undisputed, most cases of AD are sporadic, and these models do not fully recapitulate key clinical and pathological aspects. Sarasa and colleagues reported that human transgenes such as APP, PS1, or PS2 encode variable amyloid-β (Aβ) peptides which might be impeded by mouse endogenous AβPP and AβPP processing enzymes [6]. In addition, transgenic animal models currently used to investigate human AD, only replicate a particular aspect of the disease (amyloidogenic versus tauopathic) and often fail to reproduce all the associated pathological features. Significantly, favorable immunotherapeutic outcomes using AD transgenic animal models [7, 8] have failed to translate into effective human therapy, which raises serious doubts as to the validity of these amyloid-focused experimental models.
To understand the multifactorial nature of human AD pathogenesis, it has become vital to characterize a non-transgenic natural disease model. In this context, aged dogs have been proposed as a preclinical and clinical model for AD, because they naturally develop considerable human like AD neuropathology associated with cognitive deficits [9, 10].
Advancement of veterinary medical prophylaxis, interventions, and nutrition has helped increase the life expectancy of pet dogs, which in parallel with man has increased the age-related disease burden [11]. Human AD neuropathology is characterized by the presence of extracellular Aβ plaques (Aβp), intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau (p-Tau) protein and ubiquitin; cerebral amyloid angiopathy (CAA); and also severe synaptic loss and neuronal death [12]. Aged dogs with cognitive decline have already been shown to display human-like AD neuropathological features with the exception of intracellular p-Tau, the evidence for which has infrequently been found in this species [13–15]. To that end, we investigated the deposition and accumulation of p-Tau in seven aged dogs using an improved and refined protocol. Our study demonstrated intracellular p-Tau depositions in the hippocampus and frontal cortex of these seven dogs [16]. The overall aim of this study is to further characterize aged dogs as a translational model for human AD and validate p-Tau as a hallmark of canine cognitive dysfunction (CCD) or ‘Dog-zheimer’.
MATERIALS AND METHODS
Case presentation
Detailed information about the dogs used in this study is found in a previously published clinical report by Ozawa and colleagues [17]. Briefly, 37 brains were removed following routine necropsies performed at the Department of Veterinary Pathology, University of Tokyo. The brains of seven out of the 37 dogs were used in the current study following confirmation and scoring of CCD [18]. We included small and medium size breeds according to the American Kennel Club breed size division such as Papillon, Pomeranian, Lhasa Apso, and Shiba Inu, and their ages were 14 (n = 1), 15 (n = 1), 16 (n = 1), and 17 (n = 1) years respectively (Table 1). The size of the mongrels was determined according to their weight as < 15 kg is considered small, 15–25 kg medium, and > 25 kg large. In the current study, one small size Mongrel aged 16 years and two medium size Mongrels aged 14 and 17 years were used [17]. CCD scores were calculated from an established questionnaire [18] that was completed by the dogs’ owners and dogs that scored ≥50 were considered as suffering from CCD [17]. Five dogs were diagnosed with CCD (CCD score ≥50) and two dogs were considered nondemented (CCD score < 50), including a 16-year-old small size Lhasa Apso and 14-year-old small size Papillion with CCD scores of 38 and 44 respectively (Table 1).
Table 1
List of demographics, clinical,and pathological information of seven aged dogs affected with canine cognitive dysfunction
| Animal No | Age (y) | Breed | Size | Sex | CCD Score | Pathological Score | ||
| Hippocampus (H) and Frontal cortex (Fc) | ||||||||
| mTFS-Aβo | mTFS-p-Tau | PC-Aβp | ||||||
| 1 | 14 | Papillion | Small | Castrated male | 44 | 37.2 (H) | 36.4 (H) | 21(H) |
| 41.5 (Fc) | 36.3 (Fc) | 15 (Fc) | ||||||
| 2 | 14 | Mongrel | Medium | Castrated male | 58 | 53.6 (H) | 47.2 (H) | 20 (H) |
| 46.3 (Fc) | 45.1 (Fc) | 22 (Fc) | ||||||
| 3 | 15 | Pomeranian | Small | Male | 58 | 55.1 (H) | 64.2 (H) | 30 (H) |
| 63.5 (Fc) | 67.2 (Fc) | 32 (Fc) | ||||||
| 4 | 16 | Lhasa Apso | Small | Male | 38 | 52 (H) | 45.9 (H) | 16 (H) |
| 57 (Fc) | 66.5 (Fc) | 20 (Fc) | ||||||
| 5 | 16 | Mongrel | Small | Female | 50 | 11.9 (H) | 56 (H) | 15 (H) |
| 41.1 (Fc) | 15.3 (Fc) | 10 (Fc) | ||||||
| 6 | 17 | Shiba Inu | Medium | Spayed female | 64 | 44.8 (H) | 62.1 (H) | 5 (H) |
| 41.3 (Fc) | 25.9 (Fc) | 14 (Fc) | ||||||
| 7 | 17 | Mongrel | Medium | Spayed female | 60 | 60 (H) | 53.9 (H) | 23 (H) |
| 54.1 (Fc) | 68.6 (Fc) | 18 (Fc) | ||||||
Aβo indicates amyloid-β (Aβ1 - 42) oligomers, p-Tau indicates hyperphosphorylated tau and Aβp indicates amyloid beta plaques. TFS (Ten Field Score) refers to positively stained cells in 10 consecutive high-power fields (40 x) which resulted in a score. mTFS is the mean value of the 10 high-power fields. PC is plaque count. mTFS of amyloid-β oligomers (Aβo) in the hippocampus and frontal cortex: < 42 considered as low, 42–52 as moderate, > 52 as high. mTFS of p-Tau < 40 considered as low, 40–50 as moderate, > 50 as high in the hippocampus and frontal cortex. Plaque count (PC) of Aβ plaques: < 10 considered as low, 10–20 as moderate, and > 20 as high in the hippocampus and frontal cortex.
Dog brain samples
Following fixation in a 10%phosphate-buffered formalin solution and routine processing [17], congo red (CR) staining, immunohistochemistry (IHC), and immunofluorescence (IF) were performed using 4-μm-thick Paraffin-embedded tissue sections of brains of the seven aged dogs. Two regions of these brains, the hippocampus and frontal cortex, were used in this study.
Congo red staining
Initially, we performed CR staining to identify amyloid fibrils as described previously [19, 20]. Briefly, deparaffinized sections were placed in CR working solution for 20 min then rinsed in 5–8 changes of deionized water. This was followed by staining with Gill ll Haematoxylin (Leica bio systems, Wetzlar, Germany) for 1–3 min and rinsing in three changes of deionized water. Sections were then dehydrated in two changes of 95%alcohol followed by three changes of absolute alcohol for 1 min each. Finally, sections were cleared in two changes of xylene and mounted in a xylene miscible medium. Sections were then visualized under bright and polarized light microscopy (Olympus CX 43, Shinjuku, Tokyo, Japan).
Immunohistochemical staining of Aβ and p-Tau
Brain hippocampal and cortical sections were pre-treated for 1 h using the 2100 antigen retriever (Aptum biologics Ltd, Southampton, UK) followed by 90%formic acid then 0.1%triton-X treatment for 5 min and 1 min, respectively. Sections were then blocked with 0.3%H2O2 for 15 min to inactivate endogenous peroxidases and with Protein Block Serum-Free (Agilent, Santa Clara, CA, USA) for 15 min before adding the primary antibodies. Sections were then stained with the following primary antibodies: 4G8 anti-Aβp (1:500; Bio legend, San Diego, CA, USA) and AT8 anti-p-Tau (Ser202, Thr205) antibody (1:500; Thermo-fisher Scientific, MA, USA) respectively for 1 h at room temperature (RT). After washing three times with Tris buffered saline/0.05%Tween (TBST), sections were stained with secondary antibodies in TBS: HRP-conjugated anti-mouse IgG (1:500; Sigma-Aldrich) or anti-rabbit IgG (1:500; Sigma-Aldrich) respectively for 1 h at RT. Sections were again washed three times with TBST before addition of 3,3’-Diaminobenzidine (DAB) substrate chromogen system and incubated for 5–10 min. Slides were then counterstained with hematoxylin for 1 min. After mounting, slides were finally imaged using the Olympus CX 43 light microscope (Shinjuku, Tokyo, Japan) [20].
Immunofluorescence co-localization study
Double immunofluorescence labelling was performed to determine whether Aβ1 - 42 oligomers co-localized with Aβp in the cortex and hippocampus. Sections were co-stained with 4G8 antibody (1:500; Bio legend, San Diego, CA, USA) and anti-Aβ1 - 42 oligomer nanobody (PrioAD13; 1:500) [21]. Double immunofluorescence labelling was also performed to determine whether Aβ1 - 42 oligomers co-localized with p-Tau in the cortex and hippocampus using anti-Aβ1 - 42 oligomer nanobody (PrioAD13; 1:500) and AT8 anti-p-Tau (Ser202, Thr205) antibody (Thermo-fisher Scientific, MA, USA). Sections were incubated with primary antibodies overnight at 4°C. Sections were washed three times with TBST then incubated with secondary antibodies: goat anti-llama IgG conjugated to FITC (Bethyl Laboratories, Inc, TX, USA) and donkey-anti-mouse IgG conjugated to Texas red (Sigma-Aldrich, MO, USA) at a dilution of 1:500 for 2 h at RT. For IHC and IF studies, duplicate sections were also stained with secondary antibodies with omission of primary antibodies as negative control. After washing with TBST sections were cover slipped with paramount aqueous mounting medium (Dako, Agilent, Santa Clara, CA, USA), sealed and dried overnight. Finally, images were acquired using the LSM800 Confocal microscope with a standard FITC/Texas Red double band-pass filter set.
Neuropathological scoring of Aβ and p-Tau
Counting the number of positively stained cells in 10 consecutive high-power fields (40 x) resulted in a score (“Ten Field Score”–TFS) for Aβ1 - 42, Aβp, and p-Tau in the hippocampus and cortex. Our scoring system was calculated by quantifying neurons and plaques showing intracellular immunofluorescence stain of Aβ1 - 42 oligomers/p-Tau and extracellular deposits (plaques). For both Aβ1 - 42 oligomers and p-Tau scoring, we first counted the number of positively stained cells in 10 consecutive high-power fields (40 x) which resulted in a TFS. After calculating the mean TFS values (mTFS) of Aβ oligomers or p-Tau intensity count from the 10 consecutive high-power fields, they were categorized as low, moderate, or high mTFS. For Aβ oligomers, a mTFS < 42 was considered as low, 42–52 as moderate, and > 52 as high in the hippocampus and in the frontal cortex. For p-Tau, a mTFS < 40 was considered as low, 40–50 as moderate, and > 50 as high in the hippocampus and in the frontal cortex. Finally, for Aβp scoring, as the number of plaques was scarce, we instead counted the total number of plaques (PC) per section. A PC < 10 was considered as low, 10–20 as moderate, and > 20 as high in the hippocampus and in the frontal cortex. The extent to which both mTFS and PC scores were compared with CCD scores (Table 1) was determined [18].
RESULTS
Aged dogs with canine cognitive dysfunction display similar neuropathology to human AD
CAA is one of the histological hallmarks observed in the brains of AD patients and is characterized by the accumulation of Aβ in cerebral blood vessels [12, 16]. In this study, we used CR staining to identify amyloid fibrils and CAA deposits in the cortex and hippocampus of the aged dogs affected with CCD. Congophilic deposits were detected in CR-hematoxylin-stained sections and substantiated by cross-polarized light and appeared as apple-green birefringence in the brain hippocampal and cortical region confirming the presence of CAA (Fig. 1). However, as anticipated, CR positivity was not observed in the diffuse plaques in the hippocampal and cortical regions of the aged dogs (Supplementary Figure 1), indicating lack of the β pleated-sheet conformations [10]. Further, the hippocampus and cortex displayed widespread deposition of diffuse Aβp following IHC and IF staining with 4G8 antibody (Figs. 2 and 3 and Supplementary Figure 2).
Fig. 1
Photomicrographs of Congo red (CR) staining in cerebral amyloid angiopathy (CAA) in the brain hippocampal and cortical region of an aged dog. Distinctive red brick staining of cerebral blood vessels (largely the tunica media) (A) in the hippocampus and (C) in the frontal cortex (black arrows) of a 15-year-old small size Pomeranian following CR-hematoxylin staining. Vascular amyloid, confirming CAA, with the presence of apple-green birefringence (B) in the hippocampus and (D) in the frontal cortex (white arrows) of a 15-year-old small size Pomeranian under polarized light. Representative of all aged dogs. Scale bar = 20μm.
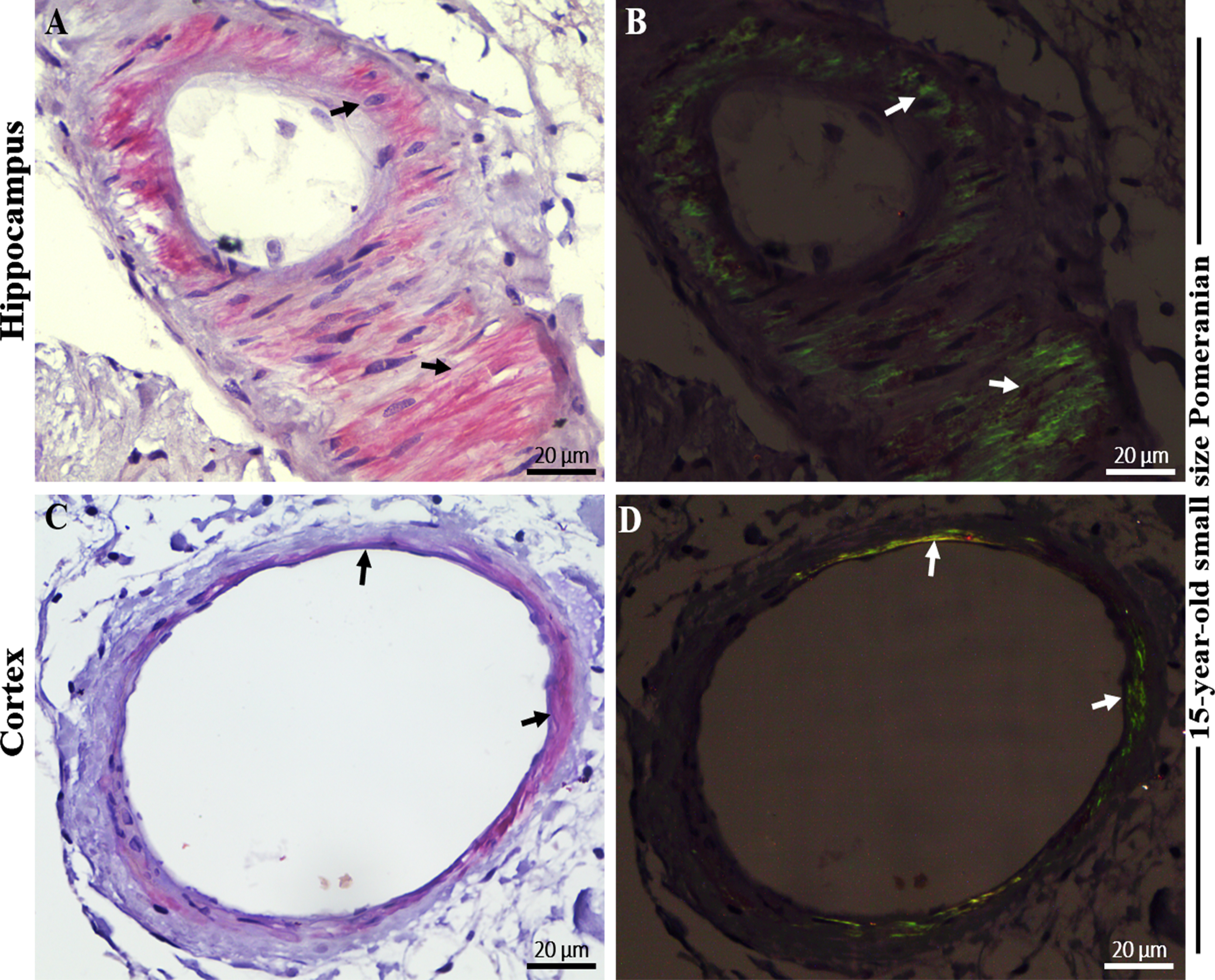
Fig. 2
Immunohistochemical staining with 4G8 anti-Aβ and AT8 anti-pTau antibody in the brain hippocampal and cortical region of an aged dog. Immunohistochemical staining showed extensive extracellular Aβ plaques in the A) hippocampus and B) frontal cortex (black arrows) of a 15-year-old small size Pomeranian. Staining of vascular amyloid (cerebral amyloid angiopathy) was prominent in the C) hippocampus and D) frontal cortex of a 15-year-old small size Pomeranian respectively (black arrows). Immunohistochemical staining showed extensive intracellular hyperphosphorylated tau (p-Tau) staining in the E) hippocampus and F) frontal cortex respectively (black arrows) of a 15-year-old small size Pomeranian. Representative of all aged dogs. Scale bar = 20μm and inserts scale bar = 5μm.
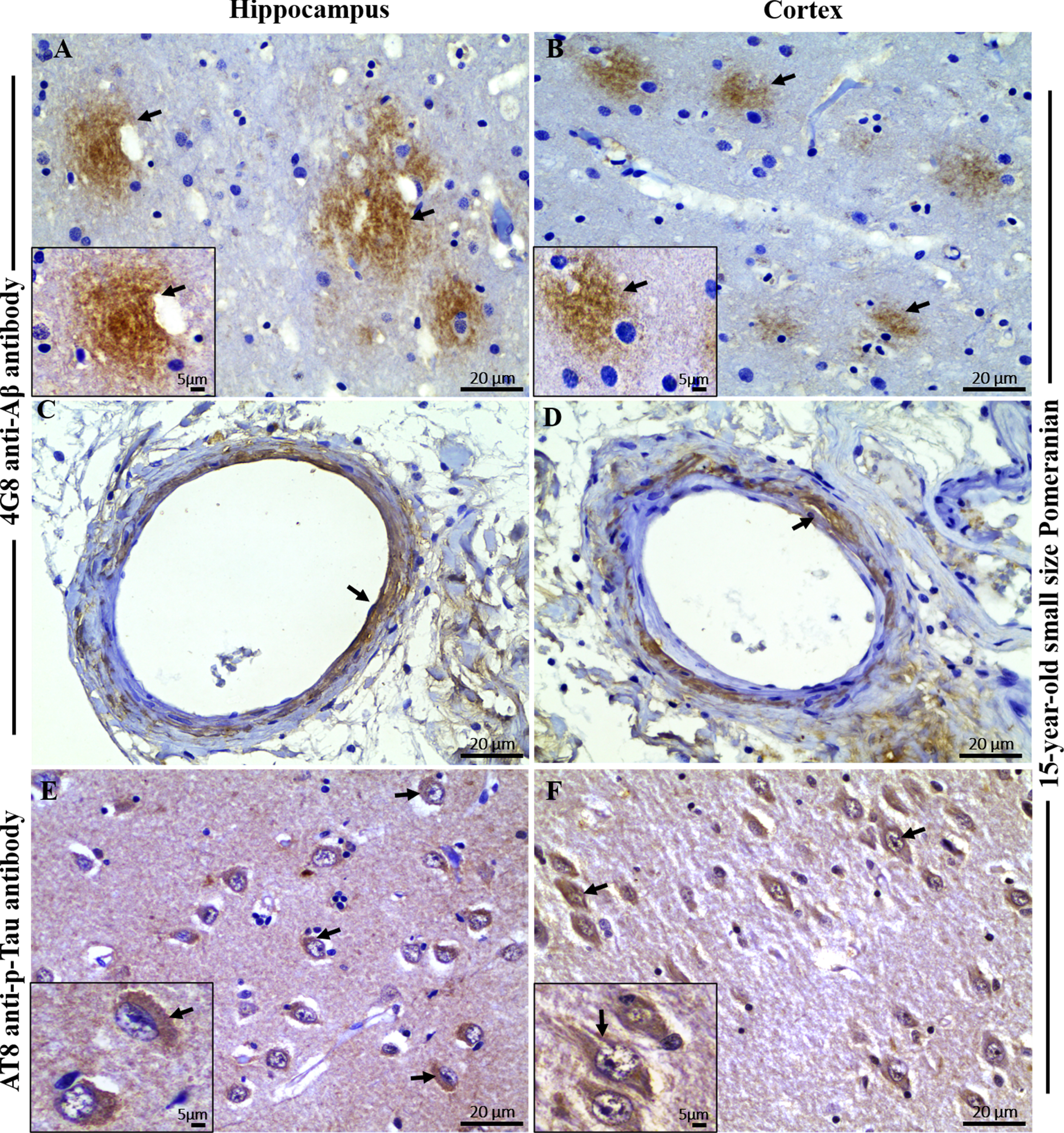
Fig. 3
Immunofluorescence staining with PrioAD13 anti-Aβ1 - 42 oligomer nanobody, 4G8 anti-Aβ, and AT8 anti phospho-Tau (Ser202, Thr205) antibody in brain hippocampal and cortical region of an aged dog. LSM800 confocal images were taken at 40x and 100x respectively for each tissue section with a standard FITC/Texas Red double band-pass filter set. Staining with anti-Aβ1 - 42 (PrioAD13) nanobody (GREEN) demonstrated extensive intracellular deposition of Aβ1 - 42 oligomers (white arrows) in the hippocampus at A) 40x magnification, scale bar = 20μm and B) higher magnification at 100x, scale bar = 5μm and also in the frontal cortex at C) 40x magnification, scale bar = 20μm and D) higher magnification at 100x, scale bar = 5μm of a 15-year-old small size Pomeranian; Staining with 4G8 anti-Aβ antibody (red) displayed extracellular diffuse plaque (white arrows) in the hippocampus both at E) 40x magnification, scale bar = 20μm and F) higher magnification at 100x, scale bar = 5μm and in the frontal cortex at G) 40x magnification, scale bar = 20μm and H) higher magnification at 100x, scale bar = 5μm of a 15-year-old small size Pomeranian; Staining with AT8 anti-p-Tau antibody (red) exhibited widespread intracellular p-Tau (white arrows) in the hippocampus both at I) 40x magnification, scale bar = 20μm and J) higher magnification at 100x, scale bar = 5μm and in the frontal cortex both at K) 40x magnification, scale bar = 20μm and L) higher magnification at 100x, scale bar = 5μm of a 15-year-old small size Pomeranian. Representative of all aged dogs.
We then compared the PC among the seven aged dogs including five affected with CCD (Fig. 5, Table 1). In general, dogs with the higher CCD scores also had among the higher PC-Aβp counts (dogs numbers 2, 3, and 7), but dog number 6 with the highest CCD score of 64 showed the lowest plaque count with a PC = 5 in the hippocampal region and a moderate PC = 14 in the cortex (Fig. 5, Table 1), indicating that the senile plaques may not be responsible for the cognitive deficits in these aged dogs as demonstrated for human AD [12, 22]. Aβ formation progresses from monomers to oligomers to fibrils and then to plaques. In order to identify the earlier assembly and most toxic isoform of Aβ, namely Aβ1 - 42 oligomers, and determine whether Aβ1 - 42 oligomers co-localized with Aβp, we co-stained the hippocampal and cortical regions with both 4G8 and PrioAD13 anti-Aβ1 - 42 nanobody [21]. Of importance, intraneuronal Aβ1 - 42 oligomers deposition was widespread but did not co-localize with Aβp (Fig. 3 and Supplementary Figure 2). Highest mTFS of Aβ1 - 42 oligomers (hippocampus = 55.1; cortex = 63.5) were observed in a 15-year-old small size male Pomeranian with a CCD score of 58 and a 17-year-old medium size spayed female Mongrel (hippocampus = 60; cortex = 54.1) with a CCD score 60 (Fig. 6, Table 1). Moderate to high Aβ1 - 42 oligomers mTFS were observed in a 16-year-old small size Lhasa Apso (hippocampus = 52; cortex = 57) with a CCD score of 38 and a 14-year-old medium size castrated male Mongrel (hippocampus = 53.6; cortex = 46.3) with a CCD score of 58 respectively (Fig. 6, Table 1). A 14-year-old castrated male Papillion with a CCD score of 44 displayed low mTFS (hippocampus = 37.2; cortex = 41.5) and finally a 16-year-old female Mongrel with a CCD score of 50 exhibited the lowest mTFS (hippocampus = 11.9; cortex = 41.1) (Fig. 6, Table 1).
Fig. 5
Comparing the CCD Scores with hippocampal and cortical Aβ plaque count (PC) in seven aged dogs. CCD score of ≥50 were considered as CCD or dog dementia. Aβ PC was calculated from whole tissue sections at high power fields (40x). PC of Aβ plaques: < 10 considered as low, 10–20 as moderate, and > 20 as high in the hippocampus and frontal cortex. A small size male Pomeranian with CCD score of 58 displayed the highest PC in the (A) hippocampus, PC = 30 and in the (B) frontal cortex, PC = 32. In contrast, a medium size spayed female Shiba Inu with highest CCD score of 64 exhibited the lowest PC in the (A) hippocampus, PC = 5 and a 16-year-old small size female Mongrel with CCD score of 50 showed the lowest PC in the (B) frontal cortex, PC = 10.
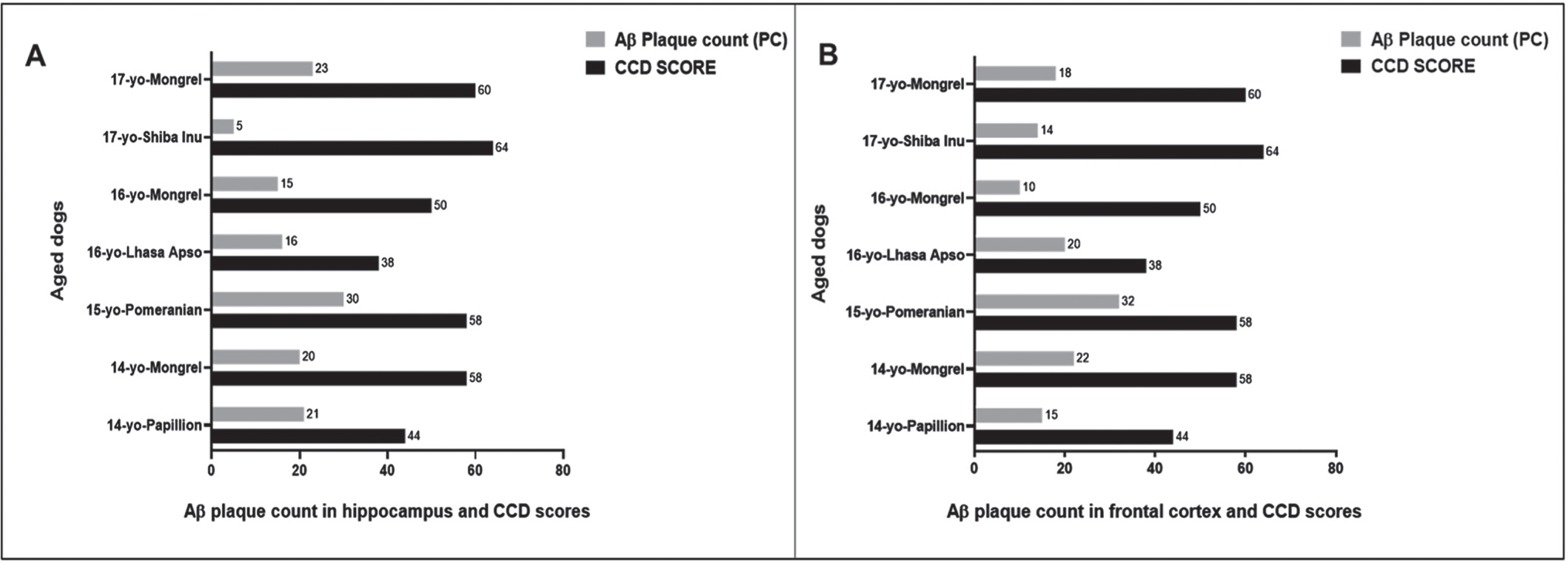
Fig. 6
Comparing the CCD Scores with hippocampal and cortical Aβ1 - 42 oligomers (Aβo), hyperphosphorylated tau (p-Tau) mean Ten Field Score (mTFS) in seven aged dogs. CCD score of ≥50 were considered as CCD or dog dementia. mTFS is the mean value calculated from 10 consecutive high-power fields (40x). mTFS of Aβo in the hippocampus and frontal cortex: < 42 considered as low, 42–52 as moderate, > 52 as high and mTFS of p-Tau < 40 considered as low, 40–50 as moderate, > 50 as high in the hippocampus and frontal cortex respectively. A small size male Pomeranian with CCD score of 58 has the highest Aβo mTFS = 55.1 and 63.5 and p-Tau mTFS = 64.2 and 67.2 in the (A) hippocampus and in the (B) frontal cortex respectively. In the (A) hippocampus a 16-year-old small size female Mongrel with CCD score of 50 exhibited the lowest Aβo mTFS = 11.9 and a 14-year-old small size castrated male Papillion with CCD score of 44 showed the lowest p-Tau mTFS = 36.4 whereas, in the (B) frontal cortex a 16-year-old small size female Mongrel with CCD score of 50 showed the lowest Aβo mTFS = 41.1 and p-Tau mTFS = 15.3 respectively.
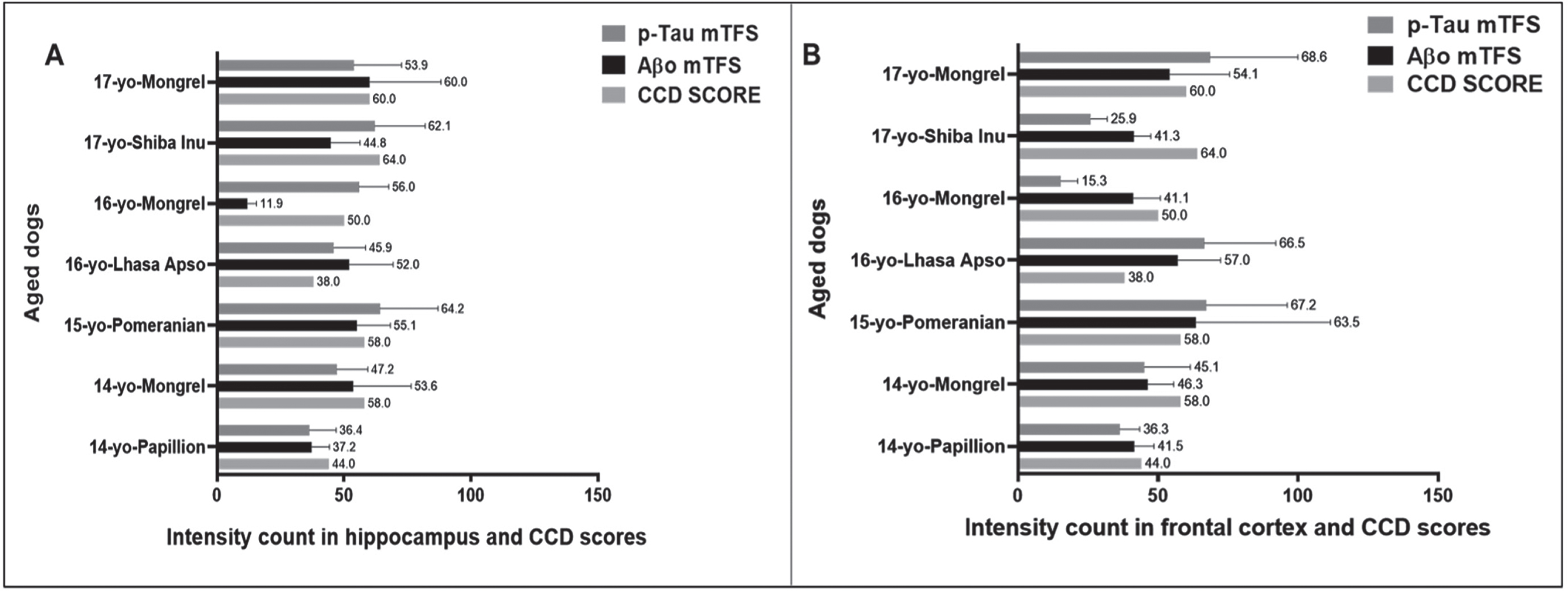
Finally, AT8 anti-p-Tau (Ser202, Thr205) monoclonal antibody strongly bound to p-Tau in the hippocampal and cortical regions following staining using IHC (Fig. 2) and IF (Fig. 3 and Supplementary Figure 2) and displayed intracellular structures [16, 23]. Highest mTFS of p-Tau were seen in a 15-year-old small size Pomeranian (hippocampus =64.2; cortex = 67.2) with a CCD score 58 and in a 17-year-old medium size Mongrel (hippocampus =53.9; cortex = 68.6) with a CCD score of 60 (Fig. 6, Table 1). Moderate to high mTFS of p-Tau were displayed in a 16-year-old small size Lhasa Apso (hippocampus = 45.9; cortex = 66.5) with a CCD score of 38 and a 14-year-old medium size Mongrel (hippocampus = 47.2; cortex = 45.1) with a CCD score of 58. Finally, lowest mTFS of p-Tau were seen in a 14-year-old small size Papillion (hippocampus = 36.4; cortex = 36.3) with a CCD score of 44. Interestingly a 17-year-old medium size Shiba Inu with a CCD score of 64 and a 16-year-old small size female Mongrel with a CCD score of 50 exhibited high mTFS for p-Tau in the hippocampus (62.1 and 56, respectively) and low mTFS for p-Tau in the cortex (25.9 and 15.3, respectively) (Fig. 6, Table 1).
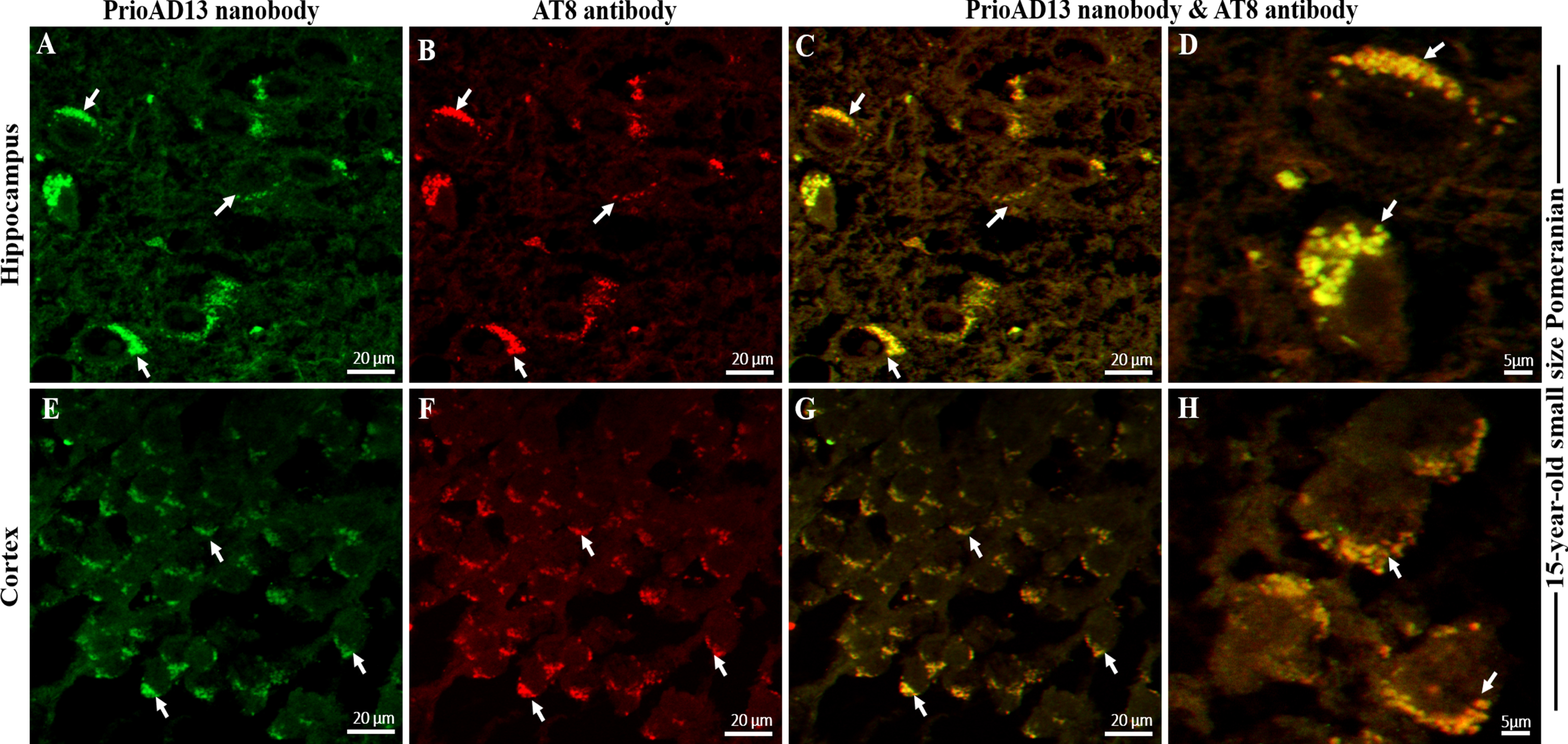
We then investigated whether the toxic Aβ1 - 42 oligomers and p-Tau co-localized to the same cellular compartments by double labelling of Aβ1 - 42 oligomers (PrioAD13) and p-Tau (AT8) in the hippocampal and cortical regions of the aged dog brains. We found widespread intracellular co-localization of oligomers with p-Tau deposits in the hippocampus and cortex (Fig. 4). High levels of Aβ1 - 42 oligomers and p-Tau which co-localized in a large number of neurons were found in a 15-year-old small size Pomeranian. Of note, this particular dog displayed the highest mTFS for both Aβ1 - 42 oligomer and p-Tau (Fig. 6, Table 1). This concurrent accumulation of both Aβ1 - 42 oligomer and p-Tau [24, 25], independent of Aβp co-localization, suggests that they may act synergistically to enhance synaptic dysfunction and neuronal death as shown in human AD [26, 27].
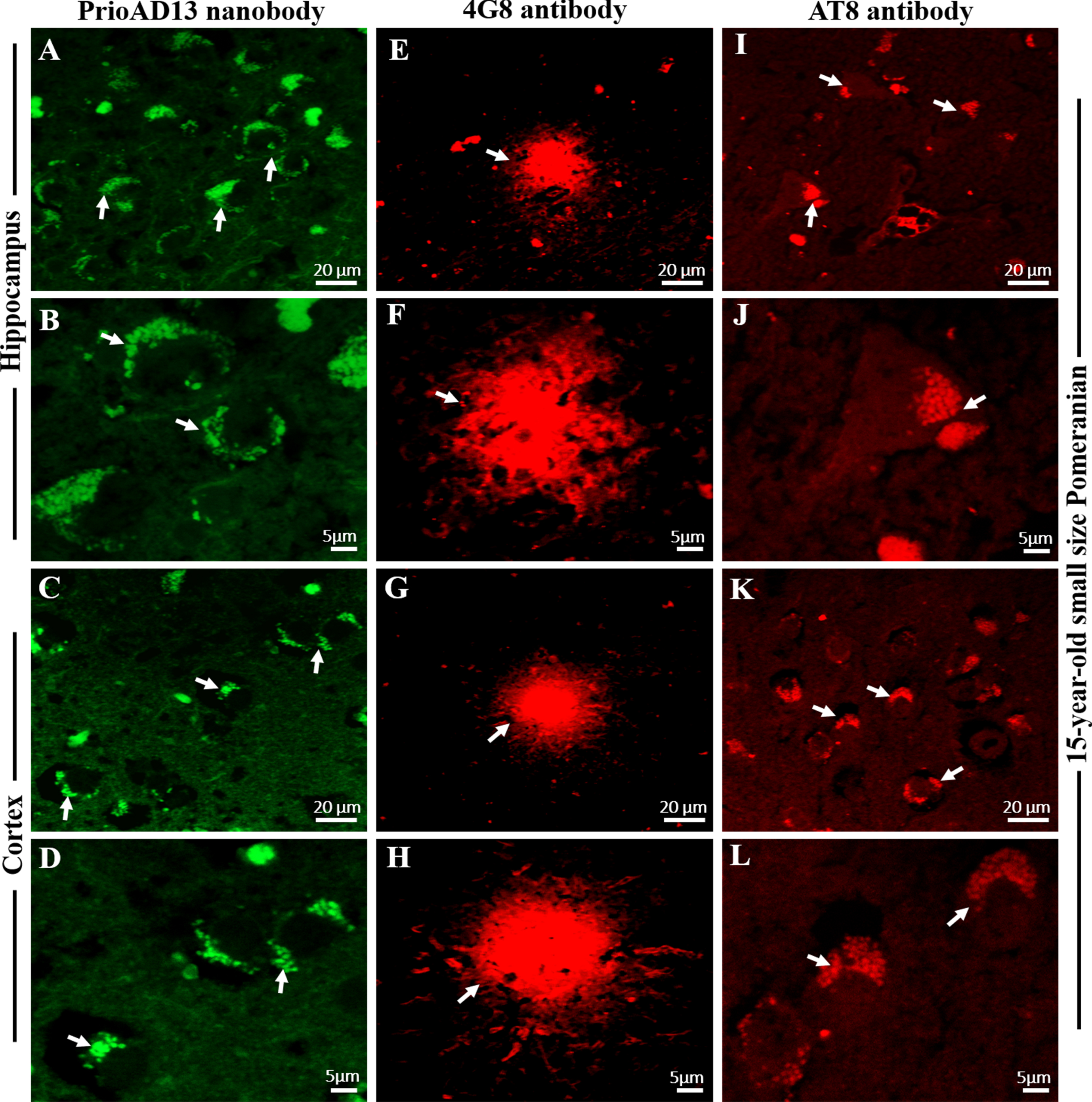
Fig. 4
Co-localization Aβ oligomers and phosphorylated Tau (p-Tau) in the hippocampus and frontal cortex of an aged dog. LSM800 confocal images were taken at 40x and 100x respectively with a standard FITC/Texas Red double band-pass filter set. Staining with anti-Aβ1 - 42 (PrioAD13) nanobody (GREEN) exhibited intracellular deposition of Aβ1 - 42 oligomers (white arrows) in the A) hippocampus and in the E) frontal cortex of a 15-year-old small Pomeranian, Scale bar = 20μm. Staining with AT8 anti-p-Tau antibody (red) displayed widespread intracellular p-Tau (white arrows) in the B) hippocampus and in the F) frontal cortex of a 15-year-old small Pomeranian respectively, Scale bar = 20μm. Co-localization, such as indicated by arrows, of anti-Aβ1 - 42 (PrioAD13) nanobody and AT8 anti-p-Tau antibody demonstrated the intracellular co-accumulation of Aβ1 - 42 oligomers and p-Tau in the hippocampus at C) 40x magnification, scale bar = 20μm and D) higher magnification at 100x, scale bar = 5μm and in the frontal cortex at G) 40x magnification, scale bar = 20μm and H) higher magnification at 100x, scale bar = 5μm, respectively. Representative of all aged dogs.
Clinical and demographic predictors of neuropathology in aged dogs with canine cognitive dysfunction
Details of the demographics of the aged dogs in this study reveal ages of 14 to 17 years with two castrated males, two males, one female, and two spayed females. There were four small size and three medium size dogs (Table 1). Previous studies showed that small size dogs usually have a longer life span than the larger dogs and are more prone to have age-related disorder [28, 29].
In our study, a 15-year-old small size male Pomeranian and a 17-year-old medium size spayed female Mongrel displayed the highest Aβp count (Fig. 5, Table 1) as well as mTFS for Aβ1 - 42 oligomer and p-Tau (Fig. 6, Table 1). However, while a 14-year-old Papillion displayed one of the lowest pathological scores we also found that a 16-year-old small size female Mongrel also had many low scores (Figs. 5 and 6, Table 1). In addition, a medium size breed Shiba Inu is one of the oldest dogs included in our study and showed the highest CCD score of 64 but exhibited the lowest number of plaques and moderate levels of Aβ1 - 42 oligomers and p-Tau (Figs. 5 and 6, Table 1), which suggests that CCD score might be triggered by the age and pathological scores including Aβ1 - 42 oligomers and p-Tau depositions but not with Aβp deposition [22].
DISCUSSION
In the present study we investigated the presence of neuropathological hallmarks normally associated with human AD in a few breeds of aged dogs affected with a syndrome called canine cognitive dysfunction (CCD) [9, 22]. Transgenic mice, used extensively to investigate human AD [5], have provided crucial information related to AD pathogenesis but their translational use has been inadequate. Therapeutic successes in treating AD in mice models have failed in numerous human AD clinical trials [30]. Dogs with CCD appear to replicate many neuropathological hallmarks of human AD, including accumulation of Aβ oligomers and plaques as well as CAA [9, 11, 31]. However, dogs with CCD are not universally accepted as a valid natural disease model of AD because the presence of a fundamental lesion, namely p-Tau, has been demonstrated in only a minority of cases and thus its association with this syndrome remains controversial [10, 11, 13–15]. To that end, we used a refined antigen retrieval protocol that enabled us to conclusively identify p-Tau in the hippocampal and cortical regions of aged dogs. The newly refined protocol for both immunohistochemical and immunofluorescence p-Tau staining relied on the use of the 2100 antigen retriever [32], which helped maintain optimal temperature, pressure and cycle time automatically controlled by an in-built sensor. The p-Tau are composed of microtubule-associated hyperphosphorylated tau protein. In human AD, p-Tau are deposited within the neurons and their morphology may vary according to the type of neurons [16, 23]. Braak and Braak proposed six distinct stages for the progression of p-Tau [33, 34] which include Braak and Braak stage III/IV where NFTs is present in the hippocampus [12, 33, 34]. In the current study, aged dogs exhibited extensive deposition of intracellular AT8 positive p-Tau in the hippocampal and frontal cortex region, therefore replicating morphological and regional distribution similar to human AD pathology [12]. Aged dogs also exhibited widespread distribution of both Aβ1 - 42 oligomers [35] and diffuse Aβp in the frontal cortex and hippocampal region [14, 17]. In human AD brain, regional distribution of Aβp is less predictable as compared to p-Tau distribution [12]. According to the Braak and Braak staging of amyloid accumulation, Aβp first deposits in the basal portions of the frontal, temporal, and occipital lobes (stage A) and then expand towards the hippocampal region (stage B) [34, 36]. In the canine brain, widespread Aβp deposition has been found involving prefrontal, parietal, occipital, and entorhinal cortices in dogs [37]. However, Aβo have not been well characterized histopathologically in dog brains affected with CCD. A previous study by Head and colleagues demonstrated accumulation of Aβo in the CSF of aged beagles which inversely correlated with brain total Aβ load [35]. Furthermore, Rusbridge and colleagues were able to immunodetect Aβo in blood and CSF of an aged dog affected with CCD using single domain camelid antibody fragments PrioAD12 (Aβ1–40), or PrioAD13 (Aβ1–42) and also reported the neurotoxic effect of dog Aβo in a human neuroblastoma cell line [38]. In the present study, we identified Aβ1–42 oligomers using single domain camelid antibody in our immunofluorescence assay. We showed extensive distribution of intracellular Aβ1–42 oligomers in the frontal cortex and hippocampus which failed to co-localize with diffuse Aβ plaques. In contrast, co-accumulation of p-Tau and Aβ1 - 42 oligomers deposits were conspicuous and closely linked. This strongly supports the hypothesis that it is the co-accumulation of both Aβ oligomers and p-Tau, and not Aβ plaques and p-Tau that are responsible for the neurotoxic effects and resultant cognitive deficits associated with AD [24] and CCD [38–40]. This might also indicate that Aβo may be responsible for initiating accumulation and spread of p-Tau as shown previously in human AD [41, 42]. Shin and colleagues reported a synergistic interaction between Aβo and tau aggregates [25]. The authors used different assemblies of Aβ in a tau cell-based model, including human neuroblastoma cells and primary hippocampal neurons and showed that tau seeding was facilitated by Aβo rather than Aβ fibrils or aggregates [25]. Interestingly, Chambers and colleagues showed for the first time that aged domestic cats develop hippocampal Aβ accumulation and NFT formation [43]. The authors demonstrated that Aβo accumulate in the cytosol of hippocampal pyramidal cells and suggested that these lead to p-Tau and NFT development [43, 44]. Moreover, we developed a new scheme that allowed us to count the number of positively stained cells in 10 consecutive high-power fields (40 x) resulted in a score, similar to the “mitotic index” widely used for assessing cancer pathology [45], to quantify Aβ1 - 42 oligomers and p-Tau (mTFS); we also quantified the total number of plaques (PC) in the hippocampal and cortical regions of the seven aged dogs. We then compared the quantified brain pathologies (mTFS versus PC) with cognitive performance (CCD scores). The cognitive scores were determined by a reference study by Salvin and colleagues who had developed a comprehensive questionnaire consisting of 13 questions to enable dog owners to rate cognitive function/dysfunction in aged dogs with 98.9%diagnostic accuracy [22]. This questionnaire was used to score CCD (CCD score) in the seven dogs investigated in this study [21]. Among the seven aged dogs, five dogs were diagnosed with CCD (score≥50) and two dogs were nondemented (CCD score < 50) which included a 16-year-old small size Lhasa Apso with a CCD score of 38 and a 14-year-old small size Papillion with a CCD scores of 44.
In AD, both neuritic and diffuse plaques are usually found in human brain where the former are believed to be associated with synaptic loss and glial cell reactivity and the latter related to ageing. The effects of neuritic and/or diffuse plaques was previously investigated in cognitively unimpaired individuals and dogs [11, 12, 17, 46]. Ahmadi and colleagues [47] assessed the link between cognitive performance and diffuse as well as neuritic plaque depositions in cognitively unimpaired individuals. The authors showed that cognitively unimpaired individuals with neuritic plaques exhibited low cognitive performance whereas diffuse plaques were shown to not influence cognition. Moreover, this study demonstrated that while diffuse plaques were not involved in cognitive deficits, increased neuritic plaques deposition paralleled a decrease in cognitive performance [47]. Similarly, Rofina and colleagues [22] performed a semiquantitative analysis of diffuse Aβ plaque deposition in a group of 30 dogs aged between 1 month to 19 years. The authors showed that diffuse plaques deposition was significantly higher in dogs older than 11 years. Furthermore, the authors demonstrated that this increased diffuse plaque burden did not influence the cognitive ability of the dogs. [22]. Similarly, our study also confirmed that aged non-demented dogs (14-year-old Papillion and 16-year-old Lhasa Apso) with low CCD scores (< 50) also displayed high PC scores reflecting a high burden of diffuse Aβp. Our study supports the hypothesis that diffuse plaques might be the part of normal ageing process, however, larger cohorts of dogs should be investigated in order to reach a conclusive outcome.
CONCLUSION
In conclusion, aged dogs affected with CCD display neuropathological features resembling those observed in human AD, such as intracellular Aβ oligomers, Aβp, CAA, and p-Tau as well as cognitive dysfunction [12]. To date no studies have conclusively demonstrated this extensive accumulation of p-Tau in dog brains; our study achieved this goal through the crucial use of an improved antigen retrieval methodology. Remarkably, early middle-aged dogs (6–10 years), equivalent to humans age 40–60 years [48], displayed cognitive decline and neurobehavioral changes which worsened with age, similar to human mild cognitive impairment [11, 49]. The ability to characterize aged dogs as a strong translational model for human AD offers a real possibility to efficiently investigate molecular determinants underlying AD pathogenesis, and to develop and test molecules for effective detection and treatment of AD. However, the use of imaging modalities, such as PET and MRI, currently used to aid in the diagnosis of human AD, is limited in aged dogs affected with CCD [50, 51]. There are substantial advantages of incorporating PET/MRI neuroimaging platforms to study/monitor AD in a large animal model such as dogs and will certainly inform on disease status, correlate blood and eye biomarkers with brain pathology, and predict treatment outcomes. Taken together, our results confirm the presence of the major neuropathological hallmarks of human AD associated with cognitive decline and argue for the use of this natural model to investigate AD.
ACKNOWLEDGMENTS
This study was funded by The Ainsworth Medical Research Innovation Fund. The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-210035.
REFERENCES
[1] | World Health Organization, Ageing,https://www.who.int/health-topics/ageing#tab=tab_1, |
[2] | Rosenberg A , Mangialasche F , Ngandu T , Solomon A , Kivipelto M ((2020) ) Multidomain interventions to prevent cognitive impairment, Alzheimer’s disease, and dementia: From FINGER to World-Wide FINGERS. J Prev Alzheimers Dis 7: , 29–36. |
[3] | Viola KL , Klein WL ((2015) ) Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129: , 183–206. |
[4] | Lesné SE , Sherman MA , Grant M , Kuskowski M , Schneider JA , Bennett DA , Ashe KH ((2013) ) Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 136: , 1383–1398. |
[5] | Kitazawa M , Medeiros R , Laferla FM ((2012) ) Transgenic mouse models of Alzheimer disease: Developing a better model as a tool for therapeutic interventions. Curr Pharm Des 18: , 1131–1147. |
[6] | Sarasa M , Pesini P ((2009) ) Natural non-trasgenic animal models for research in Alzheimer’s disease. Curr Alzheimer Res 6: , 171–178. |
[7] | Banik A , Brown RE , Bamburg J , Lahiri DK , Khurana D , Friedland RP , Chen W , Ding Y , Mudher A , Padjen AL , Mukaetova-Ladinska E , Ihara M , Srivastava S , Padma Srivastava MV , Masters CL , Kalaria RN , Anand A ((2015) ) Translation of pre-clinical studies into successful clinical trials for Alzheimer’s disease: What are the roadblocks and how can they be overcome? J Alzheimers Dis 47: , 815–843. |
[8] | Cummings JL , Morstorf T , Zhong K ((2014) ) Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res Ther 6: , 37. |
[9] | Adams B , Chan A , Callahan H , Milgram NW ((2000) ) The canine as a model of human cognitive aging: Recent developments. Prog Neuropsychopharmacol Biol Psychiatry 24: , 675–692. |
[10] | Cummings BJ , Head E , Ruehl W , Milgram NW , Cotman CW ((1996) ) The canine as an animal model of human aging and dementia. Neurobiol Aging 17: , 259–268. |
[11] | Youssef SA , Capucchio MT , Rofina JE , Chambers JK , Uchida K , Nakayama H , Head E ((2016) ) Pathology of the aging brain in domestic and laboratory animals, and animal models of human neurodegenerative diseases. Vet Pathol 53: , 327–348. |
[12] | Serrano-Pozo A , Frosch MP , Masliah E , Hyman BT ((2011) ) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: , a006189. |
[13] | Papaioannou N , Tooten PC , van Ederen AM , Bohl JR , Rofina J , Tsangaris T , Gruys E ((2001) ) Immunohistochemical investigation of the brain of aged dogs. I. Detection of neurofibrillary tangles and of 4-hydroxynonenal protein, an oxidative damage product, in senile plaques. Amyloid 8: , 11–21. |
[14] | Pugliese M , Mascort J , Mahy N , Ferrer I ((2006) ) Diffuse beta-amyloid plaques and hyperphosphorylated tau are unrelated processes in aged dogs with behavioral deficits. Acta Neuropathol 112: , 175–183. |
[15] | Schmidt F , Boltze J , Jäger C , Hofmann S , Willems N , Seeger J , Härtig W , Stolzing A ((2015) ) Detection and quantification of β-amyloid, pyroglutamyl Aβ, and tau in aged canines. J Neuropathol Exp Neurol 74: , 912–923. |
[16] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener 14: , 32. |
[17] | Ozawa M , Chambers JK , Uchida K , Nakayama H ((2016) ) The relation between canine cognitive dysfunction and age-related brain lesions. J Vet Med Sci 78: , 997–1006. |
[18] | Salvin HE , McGreevy PD , Sachdev PS , Valenzuela MJ ((2011) ) The canine cognitive dysfunction rating scale (CCDR): A data-driven and ecologically relevant assessment tool. Vet J 188: , 331–336. |
[19] | Habiba U , Merlin S , Lim JKH , Wong VHY , Nguyen CTO , Morley JW , Bui BV , Tayebi M ((2020) ) Age-specific retinal and cerebral immunodetection of amyloid-β plaques and oligomers in a rodent model of Alzheimer’s disease. J Alzheimers Dis 76: , 1135–1150. |
[20] | Habiba U , Descallar J , Kreilaus F , Adhikari UK , Kumar S , Morley JW , Bui BV , Hamaoui MK , Tayebi M ((2021) ) Detection of retinal and blood Aβ oligomers with nanobodies. Alzheimers Dement (Amst) 13: , e12193. |
[21] | David MA , Jones DR , Tayebi M ((2014) ) Potential candidate camelid antibodies for the treatment of protein-misfolding diseases. J Neuroimmunol 272: , 76–85. |
[22] | Rofina JE , van Ederen AM , Toussaint MJ , Secrève M , van der Spek A , van der Meer I , Van Eerdenburg FJ , Gruys E ((2006) ) Cognitive disturbances in old dogs suffering from the canine counterpart of Alzheimer’s disease. Brain Res 1069: , 216–226. |
[23] | Perl DP ((2010) ) Neuropathology of Alzheimer’s disease. Mt Sinai J Med 77: , 32–42. |
[24] | Manczak M , Reddy PH ((2013) ) Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: Implications to synaptic dysfunction and neuronal damage. J Alzheimers Dis 36: , 285–295. |
[25] | Shin WS , Di J , Cao Q , Li B , Seidler PM , Murray KA , Bitan G , Jiang L ((2019) ) Amyloid β-protein oligomers promote the uptake of tau fibril seeds potentiating intracellular tau aggregation. Alzheimers Res Ther 11: , 86. |
[26] | Kayed R , Head E , Thompson JL , McIntire TM , Milton SC , Cotman CW , Glabe CG ((2003) ) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: , 486–489. |
[27] | Haass C , Selkoe DJ ((2007) ) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8: , 101–112. |
[28] | Wallis LJ , Szabó D , Erdélyi-Belle B , Kubinyi E ((2018) ) Demographic change across the lifespan of pet dogs and their impact on health status. Front Vet Sci 5: , 200. |
[29] | Greer KA , Canterberry SC , Murphy KE ((2007) ) Statistical analysis regarding the effects of height and weight on life span of the domestic dog. Res Vet Sci 82: , 208–214. |
[30] | Drummond E , Wisniewski T ((2017) ) Alzheimer’s disease: Experimental models and reality. Acta Neuropathol 133: , 155–175. |
[31] | Head E ((2013) ) A canine model of human aging and Alzheimer’s disease. Biochim Biophys Acta 1832: , 1384–1389. |
[32] | Zhao Y , Flandin P , Long JE , Cuesta MD , Westphal H , Rubenstein JL ((2008) ) Distinct molecular pathways for development of telencephalic interneuron subtypes revealed through analysis of Lhx6 mutants. J Comp Neurol 510: , 79–99. |
[33] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[34] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[35] | Head E , Pop V , Sarsoza F , Kayed R , Beckett TL , Studzinski CM , Tomic JL , Glabe CG , Murphy MP ((2010) ) Amyloid-beta peptide and oligomers in the brain and cerebrospinal fluid of aged canines. J Alzheimers Dis 20: , 637–646. |
[36] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[37] | Head E , McCleary R , Hahn FF , Milgram NW , Cotman CW ((2000) ) Region-specific age at onset of beta-amyloid in dogs. Neurobiol Aging 21: , 89–96. |
[38] | Rusbridge C , Salguero FJ , David MA , Faller KME , Bras JT , Guerreiro RJ , Richard-Londt AC , Grainger D , Head E , Brandner SGP , Summers B , Hardy J , Tayebi M ((2018) ) An aged canid with behavioral deficits exhibits blood and cerebrospinal fluid amyloid beta oligomers. Front Aging Neurosci 10: , 7. |
[39] | Smolek T , Madari A , Farbakova J , Kandrac O , Jadhav S , Cente M , Brezovakova V , Novak M , Zilka N ((2016) ) Tau hyperphosphorylation in synaptosomes and neuroinflammation are associated with canine cognitive impairment. J Comp Neurol 524: , 874–895. |
[40] | Abey A , Davies D , Goldsbury C , Buckland M , Valenzuela M , Duncan T ((2021) ) Distribution of tau hyperphosphorylation in canine dementia resembles early Alzheimer’s disease and other tauopathies. Brain Pathol 31: , 144–162. |
[41] | Bennett RE , DeVos SL , Dujardin S , Corjuc B , Gor R , Gonzalez J , Roe AD , Frosch MP , Pitstick R , Carlson GA , Hyman BT ((2017) ) Enhanced tau aggregation in the presence of amyloid β. Am J Pathol 187: , 1601–1612. |
[42] | Bilousova T , Miller CA , Poon WW , Vinters HV , Corrada M , Kawas C , Hayden EY , Teplow DB , Glabe C , Albay R , 3rd Cole GM , Teng E , Gylys KH ((2016) ) Synaptic amyloid-β oligomers precede p-Tau and differentiate high pathology control cases. Am J Pathol 186: , 185–198. |
[43] | Chambers JK , Tokuda T , Uchida K , Ishii R , Tatebe H , Takahashi E , Tomiyama T , Une Y , Nakayama H ((2015) ) The domestic cat as a natural animal model of Alzheimer’s disease. Acta Neuropathol Commun 3: , 78. |
[44] | Gołaszewska A , Bik W , Motyl T , Orzechowski A ((2019) ) Bridging the gap between Alzheimer’s disease and Alzheimer’s-like diseases in animals. Int J Mol Sci 20: , 1664. |
[45] | Meuten DJ , Moore FM , George JW ((2016) ) Mitotic count and the field of view area: Time to standardize. Vet Pathol 53: , 7–9. |
[46] | Cummings BJ , Head E , Afagh AJ , Milgram NW , Cotman CW ((1996) ) Beta-amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiol Learn Mem 66: , 11–23. |
[47] | Malek-Ahmadi M , Perez SE , Chen K , Mufson EJ ((2016) ) Neuritic and diffuse plaque associations with memory in non-cognitively impaired elderly. J Alzheimers Dis 53: , 1641–1652. |
[48] | American Kennel Club, Dog Years to Human Years, https://www.akcpetinsurance.com/blog/year-of-the-dog |
[49] | Studzinski CM , Christie LA , Araujo JA , Burnham WM , Head E , Cotman CW , Milgram NW ((2006) ) Visuospatial function in the beagle dog: An early marker of cognitive decline in a model of human aging and dementia. Neurobiol Learn Mem 86: , 197–204. |
[50] | Su MY , Tapp PD , Vu L , Chen YF , Chu Y , Muggenburg B , Chiou JY , Chen C , Wang J , Bracco C , Head E ((2005) ) A longitudinal study of brain morphometrics using serial magnetic resonance imaging analysis in a canine model of aging. Prog Neuropsychopharmacol Biol Psychiatry 29: , 389–397. |
[51] | Thompkins AM , Deshpande G , Waggoner P , Katz JS ((2016) ) Functional magnetic resonance imaging of the domestic dog: Research, methodology, and conceptual issues. Comp Cogn Behav Rev 11: , 63–82. |


