
Upregulation of IL-1 Receptor Antagonist by Aspirin in Glial Cells via Peroxisome Proliferator-Activated Receptor-Alpha1
Abstract
Background:
Neuroinflammation is a recognized aspect of Alzheimer’s disease (AD) and other neurological illnesses. Interleukin 1 receptor antagonist (IL-1Ra) is an anti-inflammatory molecule, which inhibits inflammatory molecules in different cells including brain cells. However, mechanisms for upregulating IL-1Ra in brain cells are poorly understood.
Objective:
Since aspirin is a widely available pain reliever that shows promise beyond its known pain-relieving capacity, we examined whether aspirin could upregulate the IL-1Ra in the brain.
Methods:
We employed PCR, real-time PCR, western blot, immunostaining, chromatin immunoprecipitation (ChIP), and lentiviral transduction in glial cells. 5xFAD mice, an animal model of AD, were treated with aspirin orally via gavage.
Results:
Aspirin increased the expression of IL-1Ra mRNA and protein in primary mouse astrocytes and mouse BV-2 microglial cells. While investigating the mechanism, we found that the IL-1Ra gene promoter harbors peroxisome proliferator response element (PPRE) and that aspirin upregulated IL-1Ra in astrocytes isolated from peroxisome proliferator-activated receptor-beta knockout (PPARβ–/–), but not PPARα–/–, mice. Moreover, we observed that aspirin bound to tyrosine 314 residue of PPARα to stimulate IL-1Ra and that aspirin treatment also increased the recruitment of PPARα to the IL-1Ra promoter. Accordingly, aspirin increased IL-1Ra in vivo in the brain of wild type and PPARβ–/–, but not in PPARα–/– mice. Similarly, aspirin treatment also increased astroglial and microglial IL-1Ra in the cortex of 5xFAD, but not 5xFAD/PPARα–/– mice.
Conclusion:
Aspirin may reduce the severity of different neurological conditions by upregulating IL-1Ra and reducing the inflammation.
INTRODUCTION
Inflammation is a recognized aspect of neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease [1]. Inflammation in the brain is mediated by two distinct glial cell types, astrocytes and microglia [2–4]. Astrocytes fulfill numerous roles in the central nervous system (CNS) and maintain neuronal homeostasis by providing nutrients and by the production of neurotrophic factors [5, 6]. On the other hand, microglia are CNS-derived macrophages that provide the immune support responding to pathogens and other insults [7, 8].
The interleukin-1 receptor antagonist (IL-1Ra), encoded by the IL1RN gene, is a member of the interleukin 1 cytokine family [9]. IL-1Ra is known to compete with interleukin 1-alpha (IL-1α) and interleukin 1-beta (IL-1β) while binding to the IL-1 receptor (IL-1R) thereby reducing inflammatory signaling and preventing IL-1α and IL-1β from inducing the expression of other pro-inflammatory molecules [10]. Additionally, these cytokines induce IL-1Ra expression that acts as a negative feedback regulator for limiting its effects. However, mechanisms by which IL-1Ra could be stimulated in brain cells are poorly understood.
Acetylsalicylic acid commonly known as aspirin is a member of the nonsteroidal, anti-inflammatory drugs (NSAIDs) [11]. NSAIDs are widely available and frequently used to treat fevers and pain. Aspirin is effective in the treatment of pain by inhibiting the pro-inflammatory enzyme cyclooxygenase. While aspirin has been used for millennia as a pain reduction agent, current research is increasingly finding new uses for it. There is emerging research indicating that aspirin has beneficial effects on various cancers, cardiovascular diseases, and atherosclerosis [12]. Aspirin also combats neurodegenerative disorders such as Parkinson’s disease and AD [13]. Recent research has found that aspirin improves hippocampal plasticity and increases lysosomal activity effectively, increasing the clearance of toxic protein aggregates from the brain and improving memory [14, 15]. Aspirin protects mice from experimental autoimmune encephalomyelitis via IL-11-mediated upregulation of regulatory T cells [16, 17]. We have also demonstrated that low-dose aspirin stimulates dopamine production from dopaminergic neurons [18]. Here, we examined the efficacy of aspirin in upregulating the expression of IL-1Ra. We demonstrate for the first time that aspirin is capable of elevating the expression of IL-1Ra in glial cells via the activation of peroxisome proliferator-activated receptor α (PPARα).
MATERIALS AND METHODS
Animals
We obtained B6SJL-Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas/J transgenic (5×FAD) mice from The Jackson Laboratory. These mice exhibit glial activation and widespread plaque pathology to serve as a mouse model of AD [19–21]. 5×FAD/PPARα–/– (5×FAD mice lacking PPARα) were generated in our laboratory as described [22]. Animals were maintained and experiments were conducted in accordance with National Institutes of Health guidelines and were approved by the Rush University Medical Center Institutional Animal Care and Use Committee. Six-month-old male and female 5xFAD mice were treated with low-dose (2 mg/kg body weight/d) aspirin (solubilized in 0.1%methyl cellulose) via gavage for 30 d.
Reagents
Cell culture materials (DMEM/F-12, L-glutamine, Hanks’ balanced salt solution, 0.05%trypsin, and antibiotic/antimycotic) were purchased from ThermoFisher. Fetal bovine serum (FBS) was obtained from Atlas Biologicals. Aspirin and all molecular biology-grade chemicals were obtained from Sigma-Aldrich Inc. Sources of primary antibodies used in this study are mentioned in Table 1. Alexa-fluor antibodies used in immunostaining were obtained from Jackson Immuno Research and IR dye-labeled reagents used for immunoblotting were from Li-Cor Biosciences.
Table 1
Antibodies, sources, applications, and dilutions used in this paper
| Target | Antibody (Clone) | Immunogen | Application/Dilution | Source; Catalog |
| β-Actin | Mouse monoclonal (AC-15) | a.a. 1–15 of Xenopus laevis β-actin | WB –1:5000 | Abcam; ab6276 |
| IL-1Ra | Rabbit monoclonal [EPR6483] to IL-1RA | Synthetic peptide within Human IL-1RA aa 150 to the C-terminus (C terminal). The exact sequence is proprietary. | WB –1:10000 IF –1:100 | Abcam; ab124962 |
| GFAP | Rabbit polyclonal | Synth peptide to cow GFAP | IHC –1:2000 | Dako; z0334 |
| Iba-1 | Goat polyclonal | A synthetic peptide corresponding to Human Iba1 aa 135–147 (C terminal) | IF –1:1500 | Abcam; ab5076 |
| PPARα | Mouse monoclonal (3B6) | Recombinant full-length protein | ChIP –1:200 | Abcam; ab2779 |
| PPARβ | Mouse monoclonal (F-10) X TransCruz antibody | Amino acids 2–75 of PPARβ of mouse origin | ChIP –1:50 | Santa Cruz Biotechnology Inc, sc-74517 |
| PPARγ | Rabbit monoclonal (81B8) | A synthetic peptide corresponding to residues surrounding His494 of human PPARγ | ChIP –1:100 | Cell signaling Technology, 2443S |
IL-1Ra, interleukin 1 receptor antagonist; GFAP, glial fibrillary acidic protein; Iba-1, ionized calcium-binding adaptor molecule 1; PPAR, peroxisome, proliferator-activated receptor; WB, western blot; IF, immunofluorescence; ChIP, chromatin immunoprecipitation.
Isolation of primary mouse astroglia
Astroglia was isolated from 2- to 3-day-old mouse pups as described by us in several studies [23, 24]. Briefly, on day 9, the mixed glial cultures were subjected to shaking at 240 rpm for 2 h at 37°C on a rotary shaker to remove microglia followed by another round of shaking on day 11 at 180 rpm for 18 h to remove oligodendroglia and residual microglia. The adherent cells were washed and seeded onto new plates for further studies.
BV-2 microglial cells
Mouse BV-2 microglial cells (kind gift from Virginia Bocchini of University of Perugia) were also maintained in DMEM/F-12 as described by us in many studies [25, 26].
Semi-quantitative RT-PCR analysis
To remove any contaminating genomic DNA, total RNA was digested with DNase. Semi-quantitative RT-PCR was carried out as described earlier using an RT-PCR kit from Zymo Research [27]. Two μg of total RNA was reverse-transcribed using oligo(dT) as a primer and Moloney murine leukemia virus reverse transcriptase (Invitrogen). The resulting cDNA was appropriately diluted and amplified [28, 29]. Amplified products were electrophoresed on 1.5%agarose gels and visualized by ethidium bromide staining. The primers used are:
IL-1Ra (mouse): Sense, 5′-GCAGCACAGGCTGGTGAATGAC-3′
Antisense, 5′-TGCCCCCGTGGATGCCCAAG-3′
IL-1β (mouse): Sense, 5′-ATGGCAACTGTTCCTGAACTCAACT-3′
Antisense, 5′-CAGGACAGGTATAGATTCTTTCCTTT-3′
IL-1R (mouse): Sense, 5′-GGTGCCTCTGCTGTCGCTGG-3′
Antisense, 5′-CGCTGTGGGAAGGTGGCCTG-3′
GAPDH (mouse): Sense 5′-GGTGAAGGTCGGTGTGAACG-3′
Antisense 5′-TTGGCTCCACCCTTCAAGTG-3′
Real-time PCR analysis
It was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems) as described earlier. The mRNA expressions of the respective genes were normalized to the level of GAPDH mRNA. Data were processed by the ABI Sequence Detection System 1.6 software and analyzed by analysis of variance [28, 29].
Western blotting
Western blotting was conducted as described before with modifications [28, 30]. Briefly, cells were scraped in 1X RIPA buffer, the protein was measured using Bradford reagent and sodium dodecyl sulfate (SDS) buffer was added and electrophoresed on NuPAGE® Novex® 4–12%Bis-Tris gels (Life Technologies, Grand Island, NY) and proteins transferred onto a nitrocellulose membrane (Bio-Rad) using the Thermo-Pierce Fast Semi-Dry Blotter. The membrane was then washed for 15 min in TBS plus Tween 20 (TBST) and blocked for 1 h in TBST containing BSA. Next, membranes were incubated overnight at 4°C under shaking conditions with primary antibodies. The next day, membranes were washed in TBST for 1 h, incubated in secondary antibodies against for 1 h at room temperature, washed for one more hour, and visualized under the Odyssey® Infrared Imaging System (Li-COR, Lincoln, NE).
Immunofluorescence analysis
The analysis was performed as described [31]. Briefly, coverslips containing 100–200 cells/mm2 were fixed with (100%) methanol followed by treatment with cold ethanol and two rinses in phosphate-buffered saline (PBS). Samples were blocked with 3%bovine serum albumin (BSA) in PBS/Tween 20 (PBST) for 30 min and incubated in PBST containing 1%BSA, anti-IL-1Ra, and anti-GFAP antibodies. For dilutions of primary antibodies, please see Table 1. After three washes in PBST (15 min each), slides were further incubated with Cy2 or Cy5 (Jackson ImmunoResearch). The samples were mounted and observed under an Olympus BX-41 fluorescence microscope.
Immunostaining of tissue was performed by fixing the brains in 4%paraformaldehyde followed by 30%sucrose [32]. Tissue was sealed into OCT and sectioned every 40 microns on a Leica Cryostat CM3050 S and kept in cryoprotectant. Frozen sections were treated with cold ethanol (–20°C), followed by two rinses in PBS, blocking with 2%BSA in PBST, and double-labeling with two primary antibodies (Table 1). After three washes in PBST, sections were further incubated with Cy2 and Cy5 (Jackson ImmunoResearcdh Laboratories). The samples were mounted and observed under an Olympus BX-41 fluorescence microscope. Counting analysis was performed using Olympus Microsuite V software with the help of a touch counting module.
Chromatin immunoprecipitation (ChIP)
Recruitment of PPRE to the IL-1Ra gene promoter was determined by using the ChIP assay as described [33]. Briefly, 1×106 astrocytes were treated with aspirin, and after 30 min of stimulation, cells were fixed, and cross-linked adducts were resuspended and sonicated. ChIP was performed on the cell lysate by overnight incubation at 4°C with 2μg of antibodies against PPARα, PPARβ, PPARγ, RNA Polymerase II and IgG followed by overnight incubation with protein G-agarose. The beads were washed and incubated with elution buffer. Reversing the cross-linkages and purifying the DNA, precipitates were incubated in a 65°C incubator overnight and digested with proteinase K. DNA samples were then purified and precipitated, and precipitates were washed with 75%ethanol, air-dried, and resuspended in Tris/EDTA buffer. The following primers were used to amplify the IL-1Ra gene promoter region spanning the PPRE:
Sense: 5′-CCACAGGCAGGACTAAGTCAGAAT-3′
Antisense: 5′-AGGCGTGACTCAGTCTAGCAGAAGC-3′
PCR products were electrophoresed on 2%agarose gels.
Statistical analysis
Statistical analyses were performed with Student’s t test (for two-group comparisons) and one-way ANOVA, followed by Tukey’s multiple-comparison tests, as appropriate (for multiple groups comparison), using Prism 7 (GraphPad Software). Data represented as mean±SD or mean±SEM as stated in figure legends. A level of p < 0.05 was considered statistically significant.
RESULTS
Aspirin upregulates IL-1Ra in primary mouse astrocytes
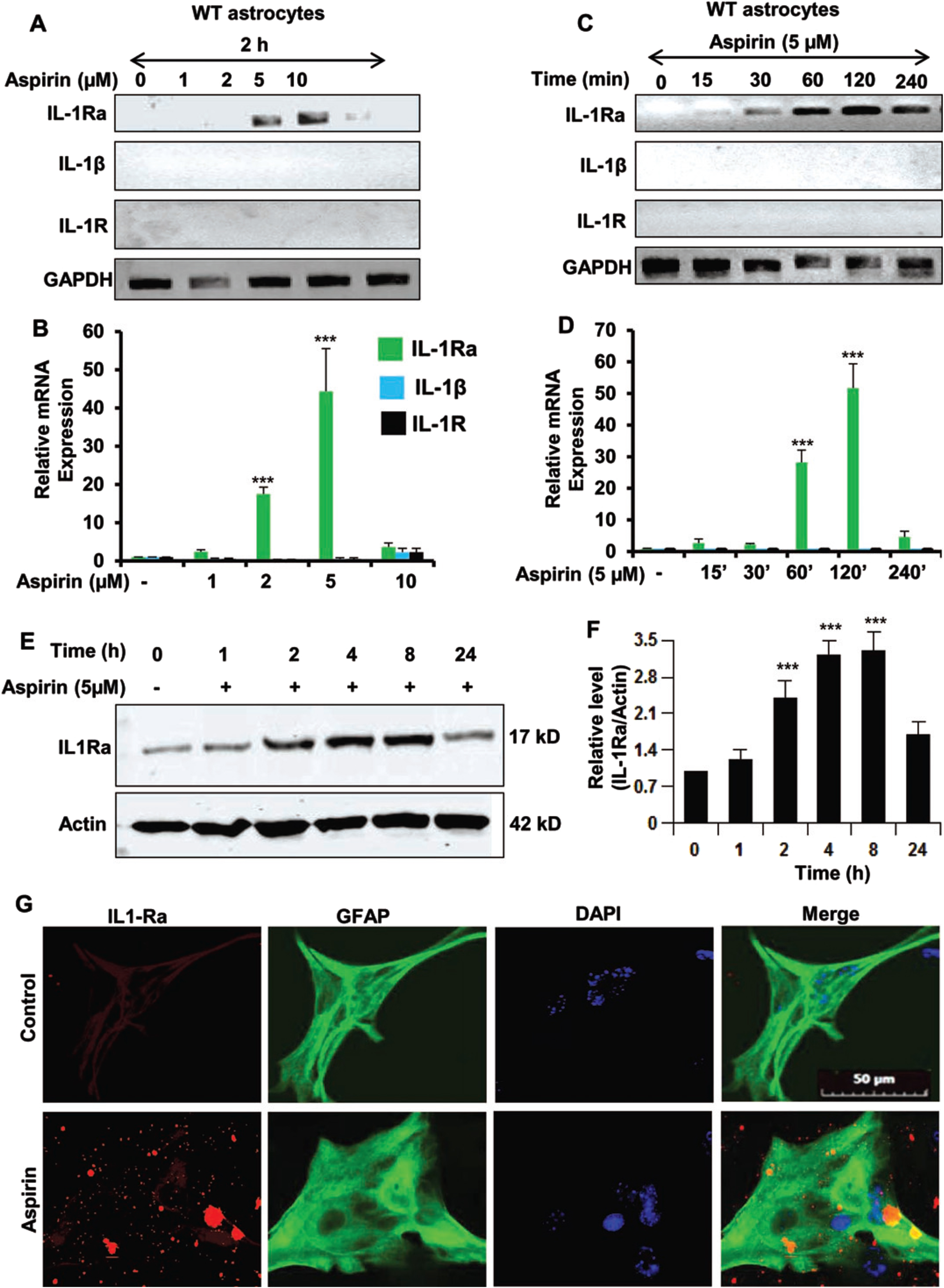
Our first step was to determine if IL-1Ra was upregulated by aspirin. Therefore, primary mouse astrocytes were cultured from C57BL/6 mice. The astrocytes were then treated with increasing dosages of aspirin (Fig. 1A, B). It was found that aspirin could upregulate IL-1Ra mRNA in a dose-dependent manner with maximum induction seen at 5μM. Next, a time course was performed using 5μM aspirin (Fig. 1C, D). The 5μM dose of aspirin induced IL-1Ra mRNA in a time-dependent manner peaking at 120 min (2 h) and then receding. The next step was to evaluate IL-1Ra protein expression by western blotting (Fig. 1E, F) and we found that IL-1Ra protein levels increased in a time-dependent manner after treatment with 5μM of aspirin. IL-1Ra protein levels increased beginning at two hours post-treatment with the highest point at 4 h and then the levels gradually decreased over time. Even at 24 h post-treatment there was still significantly increased IL-1Ra levels indicating that the beneficial effect of aspirin on IL-1Ra levels is more than a short-term effect. Immunostaining for IL-1Ra in astrocytes further confirmed that aspirin increased the level of IL-1Ra (Fig. 1G). This indicates that aspirin increases mRNA and protein levels IL-1Ra in astroglial cells.
Fig. 1
Aspirin upregulates IL-1Ra in mouse primary astrocytes. A, B) WT mouse astrocytes were treated with 1, 2, 5, and 10μM of aspirin for 2 h under serum-free condition. An increase in IL-1Ra mRNA was significant with p < 0.001. However, increase in either IL-1β or IL-1R was not significant. C, D) Time course of WT astrocytes beginning with 15 min to 240 min using 5μM of aspirin. All treatments after 120 min were significant at the p < 0.001 level by one-way ANOVA. E, F) Time course and related quantification of IL-1Ra protein immunoreactivity showing IL-1Ra with significance at the p < 0.001 level and the p < 0.01 level by one-way ANOVA. G) Representative images of immunostaining of astrocytes showing an increase in IL-1Ra immunoreactivity after aspirin treatment. Results are mean±SEM of three experiments. ***p < 0.001.
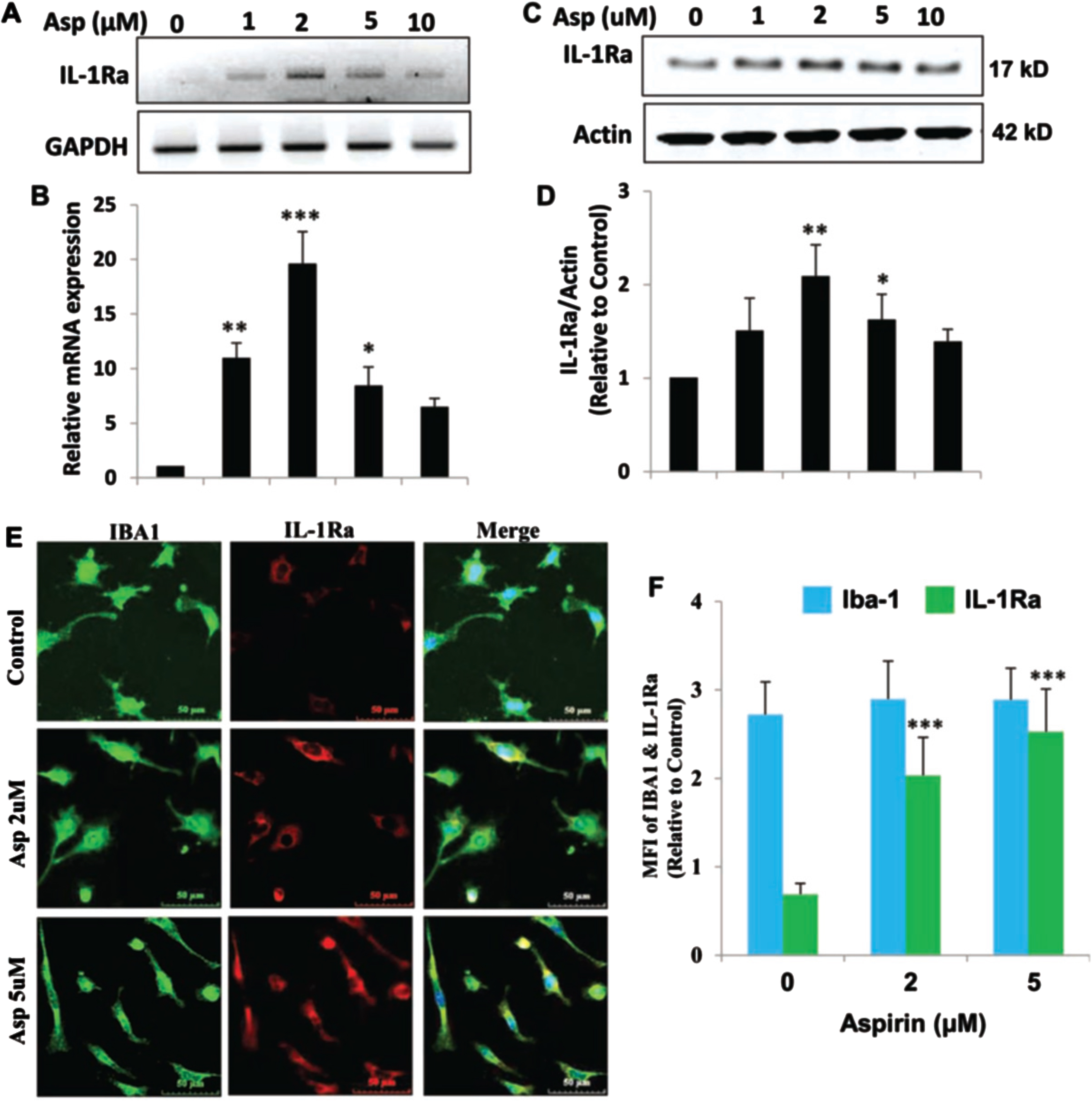
Aspirin upregulates IL-1Ra in mouse BV-2 microglial cells
Next, we determined if IL-1Ra was upregulated in microglial cells by aspirin. Therefore, BV-2 microglial cells were treated with increasing dosages of aspirin (Fig. 2A, B). It was found that aspirin upregulated IL-1Ra mRNA in a dose-dependent manner with 5μM of aspirin increasing IL-1Ra mRNA 2 h after treatment (Fig. 2). Western blotting analysis (Fig. 2C, D) also showed that IL-1Ra protein levels increased in response to aspirin in a dose-dependent manner. IL-1Ra protein levels increased at 2μM at the highest point at 4 h and then the levels gradually decreased in BV-2 cells. Immunostaining for IL-1Ra in microglia further confirmed that aspirin could increase the level of IL-1Ra (Fig. 2E). Therefore, similar to astroglia, aspirin could also increase the mRNA and protein level of IL-1Ra in microglial cells.
Fig. 2
Aspirin upregulates IL-1Ra in mouse BV-2 microglial cells. A, B) BV-2 cells were treated with 1, 2, 5, and 10μM of aspirin for 2 h under serum-free condition. An increase in IL-1Ra mRNA was significant with p < 0.001 by one-way ANOVA. C, D) An increase in IL-1Ra protein by aspirin was significant at 2μM with p < 0.01 by one-way ANOVA. E, F) Immunostaining and quantification of IL-1Ra showed significance at the p < 0.001 level with 2 and 5μM aspirin by one-way ANOVA. On the other hand, no change in Iba-1 was observed at these doses of aspirin. Results are mean±SEM of three experiments. *p < 0.05, **p < 0.01 and ***p < 0.001.
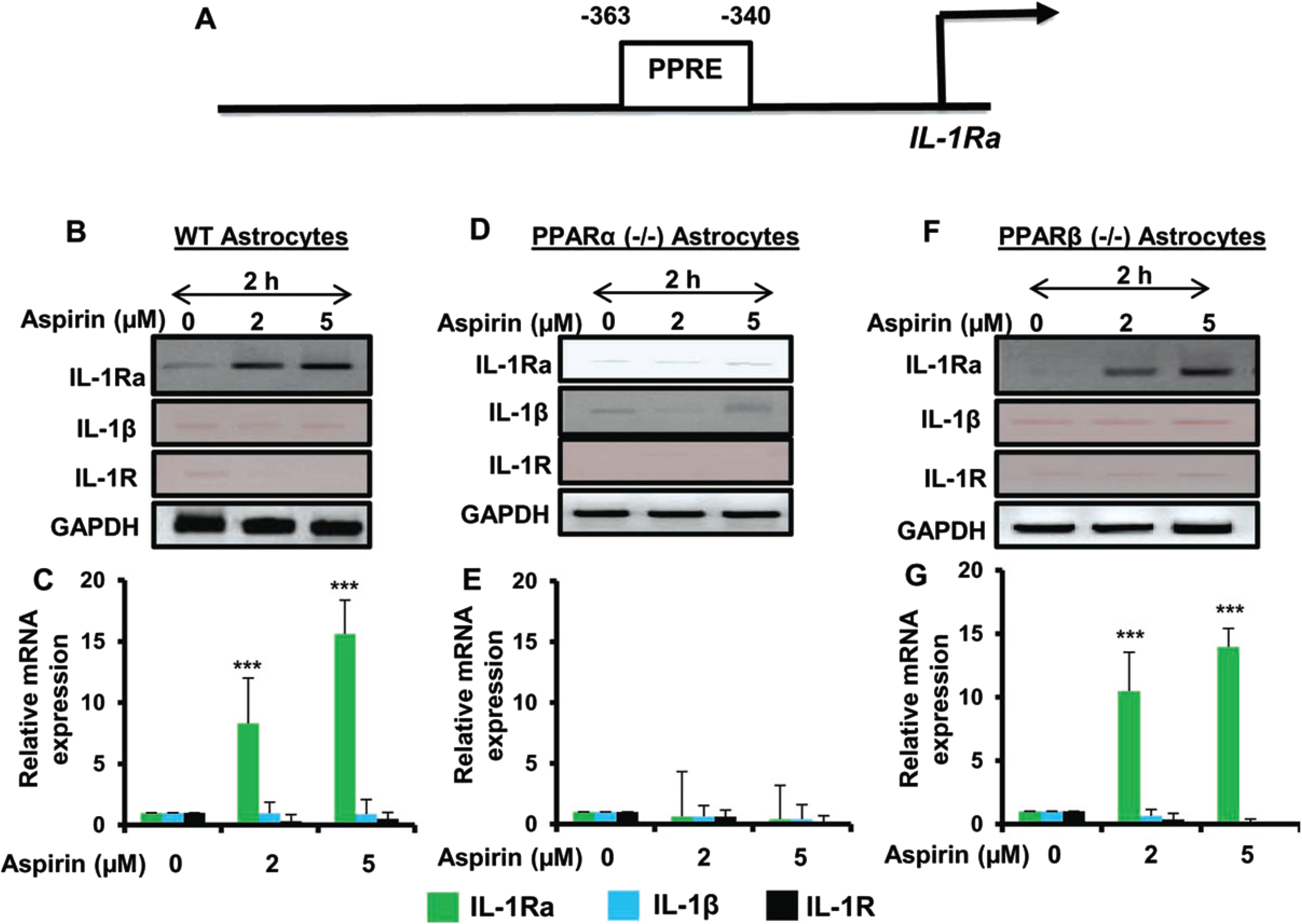
Aspirin upregulated IL-1Ra via PPARα, but not PPARβ
Our next step was to determine the mechanism by which IL-1Ra is upregulated by aspirin. Therefore, the genomic database was searched for known transcriptional activators that are located in front of the IL-1Ra gene. We found the presence of a consensus peroxisome proliferator response element (PPRE) in the promoter of IL-1Ra gene (Fig. 3A), indicating that PPAR could be involved in aspirin-mediated upregulation of IL-1Ra. Therefore, primary astrocytes isolated from C57BL/6, PPARα–/–, and PPARβ–/– mouse pups were treated with 2μM or 5μM of aspirin for 2 h. It was found by both RT-PCR and quantitative real-time PCR that aspirin upregulated IL-1Ra mRNA in wild type and PPARβ–/– astrocytes, but not PPARα–/– astrocytes (Fig. 3B-G). The relative expression of IL-1Ra was significant at the p < 0.001 level in wild type and PPARβ–/– astrocytes, but in PPARα–/– astrocytes there was no change IL-1Ra mRNA expression upon aspirin treatment. This indicates that aspirin upregulates IL-1Ra via PPARα and that functional PPARα is required for aspirin to activate IL-1Ra. Again, we did not find any expression of either IL-1β or IL-1R in WT, PPARβ–/–, and PPARα–/– astrocytes upon aspirin treatment (Fig. 3B-G).
Fig. 3
Aspirin-mediated increase in IL-1Ra mRNA is dependent on PPARα, but not PPARβ. Map of the IL-1Ra gene promoter harboring two PPREs at –363 to –340 bp (A). Cultured primary mouse astrocytes from wild type, PPARα–/– and PPARβ–/– treated with increasing doses of aspirin (2 and 5μM) show increasing induction of IL-1Ra mRNA in wild type and PPARβ–/–, but not PPARα–/–, astrocytes (B-G). Additionally, there were no significant changes in levels of IL-1β or IL-1R mRNA. Results are mean±SD of three experiments. *p < 0.05, **p < 0.01 and ***p < 0.001 by one-way ANOVA.
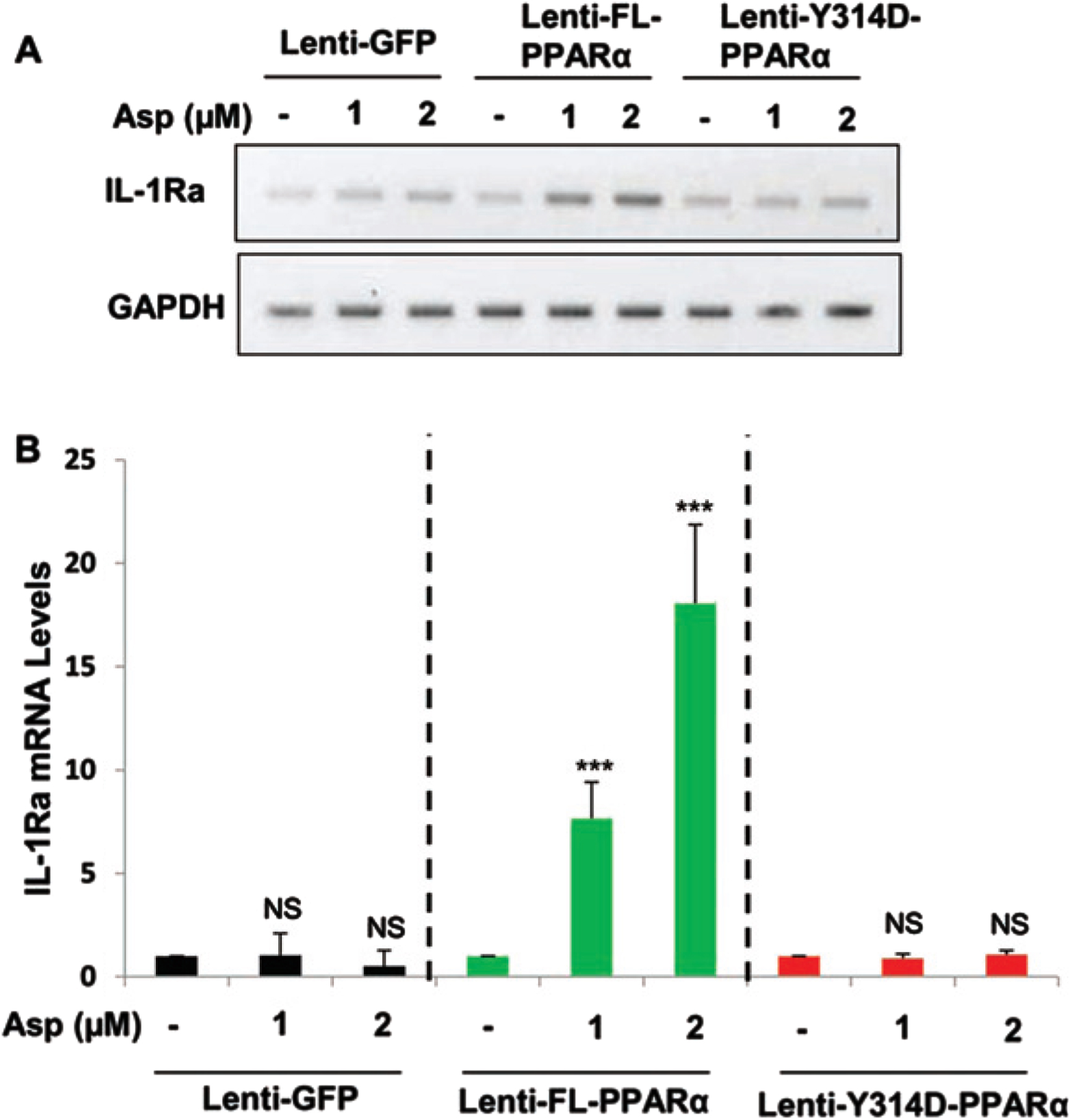
Aspirin induces IL-1Ra in PPARα–/– astrocytes that are transduced by full length PPARα, but not Y314D mutated PPARα
Recently, we have described that aspirin is a ligand of PPARα and that aspirin binds to tyrosine 314 of the ligand-binding domain of PPARα. Therefore, to confirm that aspirin binds to PPARα to stimulate IL-1Ra, mouse PPARα–/– astrocytes were transduced with lentivirus containing empty vector GFP, FL-PPARα, or Y314D-PPARα for 48 h followed by the treatment with 1 and 2μM aspirin for 2 h. Interestingly, treatment with aspirin (Fig. 4A, B) stimulated the expression of IL-1Ra when PPARα–/– astrocytes were transduced with lenti-FL-PPARα. In contrast, aspirin remained unable to stimulate IL-1Ra mRNA in PPARα–/– astrocytes transduced with either lenti-GFP or lenti-Y314D-PPARα (Fig. 4A, B). These results suggest that aspirin interacts with Y314 residue of PPARα for the upregulation of IL-1Ra in astrocytes.
Fig. 4
Aspirin induces IL-1Ra in PPARα null mouse astrocytes transduced by full length PPARα, but not Y314D mutated PPARα. PPARα-null mouse astrocytes were transduced with lentivirions containing GFP (vector), FL-PPARα, and Y314D- PPARα for 48 h followed by treatment with 1 and 2μM Aspirin for 2 h. Then check the mRNA expression in aspirin treated PPARα -null astroglial cell transduced with lentivirions containing FL-PPARα (FL-PPARα) and Y314D- PPARα. A, B) An increase in IL-1Ra, mRNA was significant in FL-PPARα transduced astrocytes with p < 0.001, but not lentivirions containing GFP or Y314D-PPARα. Significance of mean between control and aspirin-treated cells was analyzed by a two-tailed paired t-test.
Aspirin treatment induces the recruitment of PPARα to the IL-1Ra gene promoter
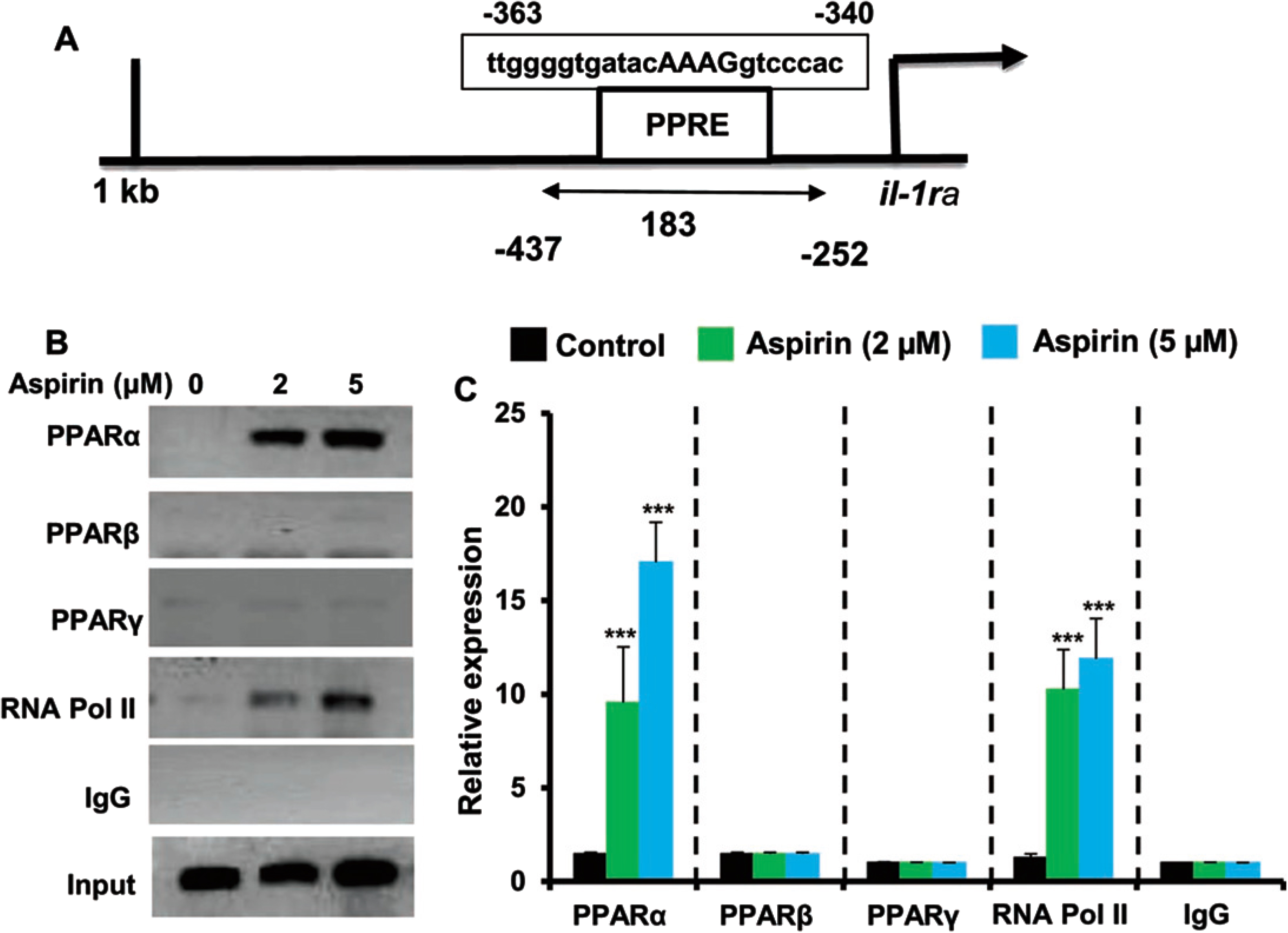
Since aspirin upregulated IL-1Ra in glial cells via PPARα, next to understand whether PPARα was directly involved in aspirin-mediated transcription of IL-1Ra gene, we examined the recruitment of PPARα to the IL-1Ra gene promoter by ChIP assay. Using Mat-Inspector V2.2 search machinery, we found one consensus PPRE in the IL-1Ra promoter (Fig. 5A). After immunoprecipitation of aspirin-treated astrocytic chromatin fragments using antibodies against PPARα, we were able to amplify 183 bp fragment from the IL-1Ra promoter (p < 0.001 versus control) corresponding to the PPRE (Fig. 5B, C). In contrast, we did not find any amplification for either PPARβ or PPARγ (Fig. 5B, C), indicating that aspirin induces the recruitment of PPARα, but neither PPARβ nor PPARγ, to the PPRE of the IL-1Ra gene promoter. Consistent to the recruitment of PPARα to the PPRE, aspirin was able to recruit RNA polymerase II (p < 0.001 versus control) to the PPRE (Fig. 5B, C). However, we did not find any amplification with control IgG, indicating the specificity.
Fig. 5
Aspirin induces the recruitment of PPARα to the IL-1Ra gene promotor. A) MAP of the PPRE in the IL-1Ra gene promoter and the region to be amplified for ChIP assay. Primary astrocytes isolated from WT mice were treated with different concentrations of aspirin for 30 min under serum-free condition followed by monitoring the recruitment of PPARα, PPARβ, PPARγ, and RNA Polymerase II to the PPRE by ChIP assay (B, PCR; C, real-time PCR). Results are mean±SD of three independent cell preparations. ***p < 0.001 versus control. Significance of mean between control and aspirin-treated cells was analyzed by a two-tailed paired t-test.
Oral administration of aspirin upregulates IL-1Ra in vivo in the cortex of WT and PPARβ–/–, but not PPARα –/–, mice
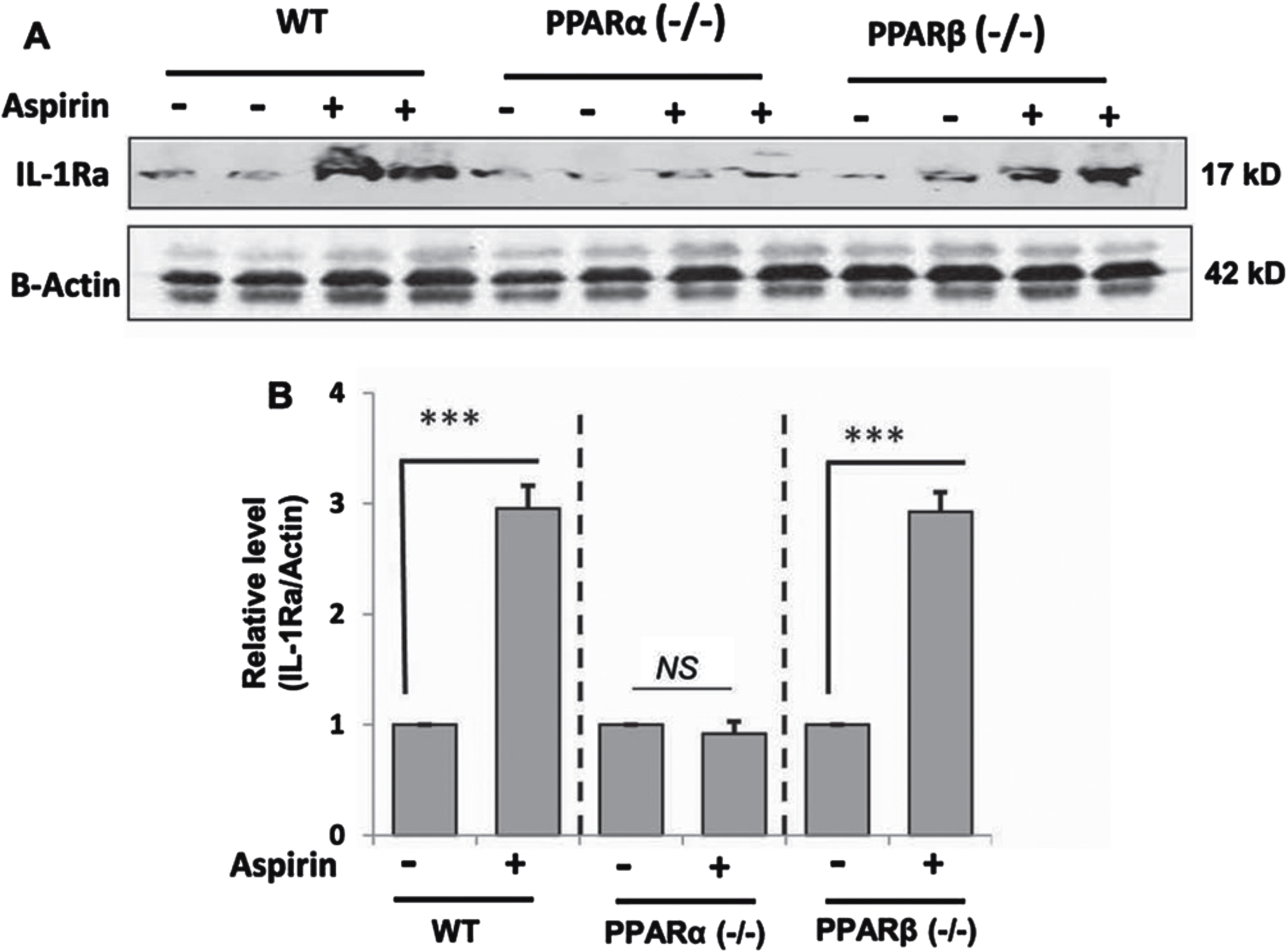
Next, we examined whether our cell culture finding was replicated in vivo in the brain. Therefore, WT, PPARα–/– and PPARβ–/– mice were treated with aspirin at a dose of 2 mg/kg/d for 7 days followed by monitoring the level of IL-1Ra in the cortex by western blot. While aspirin treatment led to about three-fold increase in IL-1Ra protein in the cortex of WT mice, aspirin remained unable to increase IL-1Ra in the cortex of PPARα–/– mice (Fig. 6A, B). On the other hand, the level of induction of IL-1Ra in the cortex of aspirin-treated PPARβ–/– mice was almost similar to that seen in WT mice (Fig. 6A, B). These results suggest that aspirin increases the level of IL-1Ra in vivo in the brain of mice via PPARα.
Fig. 6
Oral aspirin increases IL-1Ra protein in vivo in the cortex via PPARα. WT, PPARα–/–, and PPARα–/– mice (n = 6 in each group; 8–10 week old) were treated with aspirin (2 mg/kg/day) orally for 7 days via gavage followed by monitoring the protein level of IL-1Ra in the cortex by western blot (A). Actin was run as a loading control. Bands were scanned and values (IL-1Ra/Actin) presented as relative to untreated control in each group (B). Results are mean±SEM of six mice per group. ***p < 0.001; NS, not significant. Significance of mean between control and aspirin-treated groups was analyzed with a two-tailed paired t-test.
Oral administration of aspirin upregulates IL-1Ra in vivo in the cortex of 5xFAD mice via PPARα
Our next step was to determine if aspirin treatment was capable of increasing the level of IL-1Ra in vivo in the brain of 5xFAD mice, an animal model of AD. Since we found the involvement of PPARα in aspirin-mediated upregulation of IL-1Ra, we generated 5xFAD mice lacking PPARα. Therefore, 5xFAD and 5xFAD/PPARα–/– mice were subdivided into groups and some of them were fed aspirin. Aspirin-fed 5xFAD mice had increased levels of IL-1Ra immunoreactivity, indicating upregulation of IL-1Ra in the cortex of aspirin-fed 5xFAD mice (Fig. 7A). Double-labeling immunofluorescence showed that aspirin-induced IL-1Ra was present in both GFAP-positive astrocytes (Fig. 7) and Iba-1-positive microglia (Fig. 8). Mean fluorescence intensity (MFI) was performed to quantify the level of immunostained IL-1Ra (Fig. 7B). The MFI value difference between the 5xFAD and 5xFAD+ aspirin groups was significant at p < 0.001. Next, the number of IL-1Ra-positive cells per millimeter square was counted (Fig. 7C) and it was found that the number of IL-1Ra-positive cells was significantly (p < 0.001) higher in aspirin-treated 5xFAD mice than normal 5xFAD mice. In contrast, aspirin treatment remained unable to upregulate IL-1Ra in the cortex of 5xFAD/PPARα–/– mice, indicating that aspirin increases IL-1Ra in the brain of 5xFAD mice via PPARα.
Fig. 7
Aspirin treatment increases IL-1Ra in vivo in cortical astrocytes of 5XFAD mice via PPARα. Six-month old 5xFAD and 5xFAD/PPARα–/– mice (n = 5 or 6 per group) were treated with aspirin (2 mg/kg body weight/d) orally via gavage for 30 d followed by double-labeling of cortical sections for IL-1Ra and GFAP (A). Mean Fluorescence Intensity (MFI) of IL-1Ra was calculated in one section (two images per section) of each of five or six mice per group (B). IL-1Ra positive cells were counted in one section (two images per section) of each of five or six mice per group (C) and results were analyzed by one-way ANOVA. ***p < 0.001.
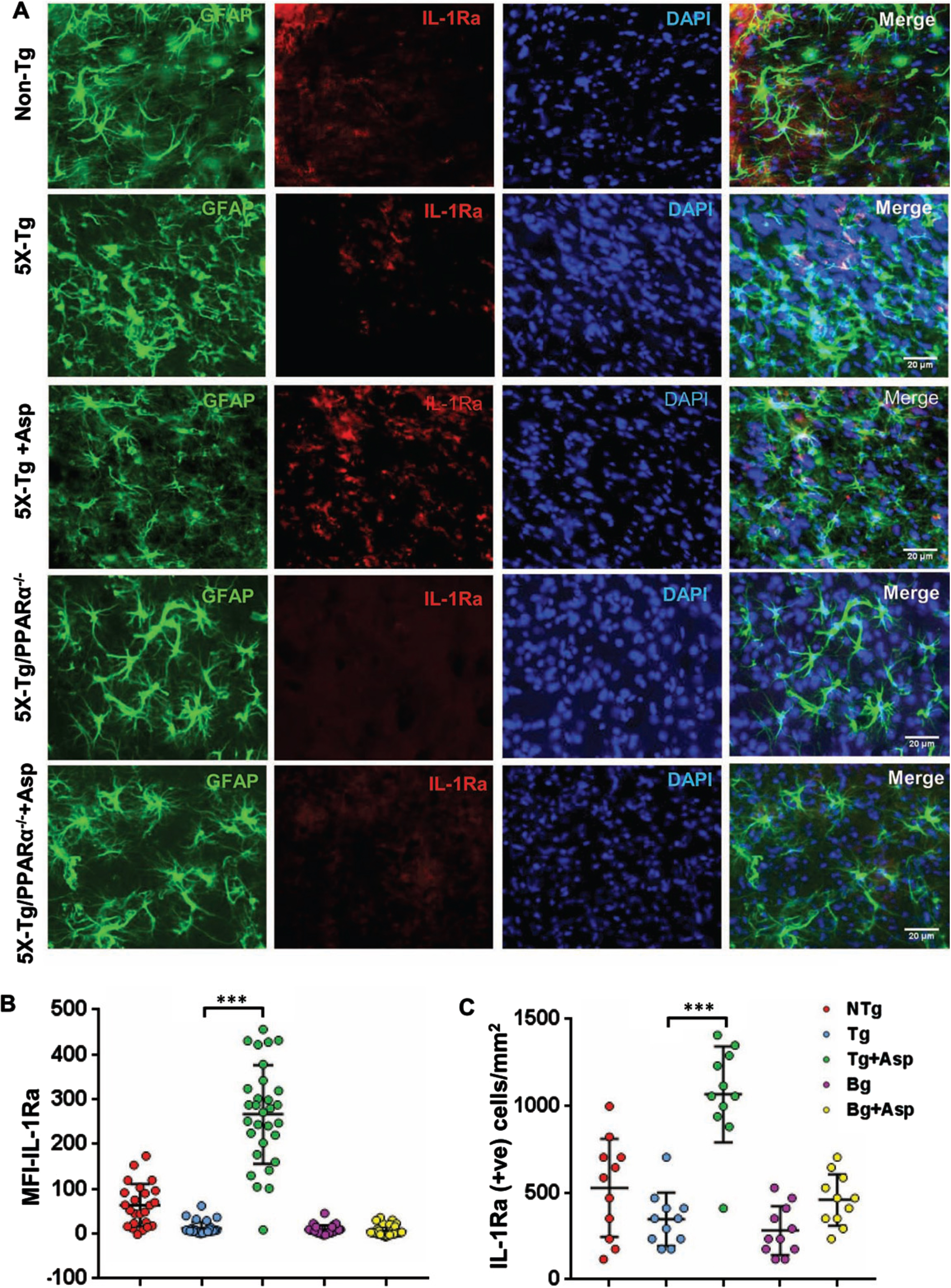
Fig. 8
Aspirin upregulates IL-1Ra in vivo in cortical microglia of 5XFAD mice via PPARα. Six-month old 5xFAD and 5xFAD/PPARα–/– mice (n = 5 or 6 per group) were treated with aspirin (2 mg/kg body weight/d) orally via gavage for 30 d followed by double-labeling of cortical sections for IL-1Ra and Iba1 (A). GFAP positive cells (B) and Iba1 positive cells (C) were counted in one section (two images per section) of each of five or six mice per group and results were analyzed by one-way ANOVA. ***p < 0.001.
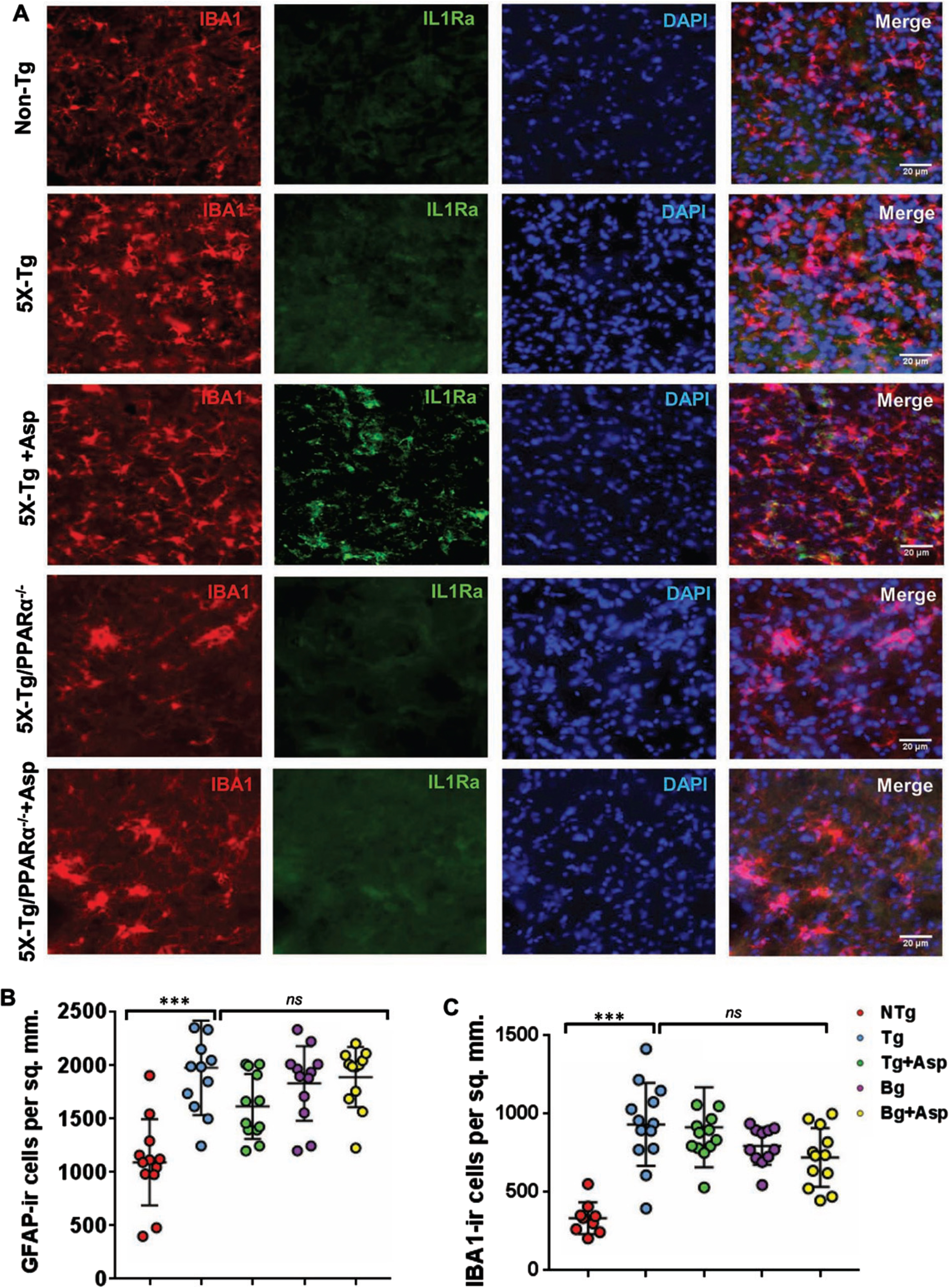
Consistent to that seen in different neurodegenerative disorders including AD and associated animal models, we found increased number of astrocytes (Fig. 7A and 8B) and microglia (Fig. 8A, C) in the cortex of 5xFAD mice as compared to age-matched non-transgenic mice. Knockdown of PPARα from 5xFAD mice did not modulate the number of either GFAP-positive astrocytes (Fig. 7A and 8B) or Iba-1-positive microglia (Fig. 8A, C) between 5xFAD and 5xFAD/PPARα–/– groups, suggesting that gliosis in 5xFAD mice may be independent on PPARα. However, despite upregulating IL-1Ra, oral aspirin remained unable to modulate either GFAP-positive astrocytes (Fig. 7A and 8B) or Iba-1-positive microglia (Fig. 8A, C) in the cortex of 5xFAD mice.
DISCUSSION
Interleukin 1 receptor antagonist (IL-1Ra) is a member of the interleukin 1 (IL-1) cytokine family that acts as a modulator of IL-1-related immune and inflammatory responses. IL-1Ra exerts anti-inflammatory activity by blocking IL-1 receptors and thereby preventing signal transduction from proinflammatory IL-1α and IL-1β [34]. IL-1Ra proteins exert their function by receptor blockers and other possible functions may exist [34]. Most intracellular components are participating in the cellular response to IL-1 as well as in the response to other cytokines like IL-18 and IL-33, which activate the heterodimeric IL-18α/β [35]. By inhibiting “danger” signals that activate the innate immune system, in particular, attenuate the inflammatory response preventing it from being sustained and damaging to nearby healthy tissue [36]. Therefore, it is important to study the pathway for the upregulation of IL-1Ra.
Salicylic acid has been used for millennia as a treatment for pain and its modern formulary of aspirin has widespread availability and utility for several ailments beyond pain management. A multitude of research has found that aspirin has the potential to combat neurodegenerative disorders including AD [37]. There are several possible therapeutic pathways that aspirin utilizes to protect against neurodegeneration [38]. Here we show that aspirin upregulates IL-1Ra in astrocytes and microglia. By increasing anti-inflammatory signaling it may reduce the inflammatory response that is found in different neurodegenerative disorders. In neurodegenerative conditions, the inflammatory response is sustained and prolonged leading to a multitude of secondary effects that are detrimental including accelerated cell death, aberrant cell signaling, and exaggerated oxidative stress [39]. When inflammation occurs, it is normally in response to an adverse event and then relaxes as the event is limited. In neurodegenerative disorders, the inflammatory response does not subside and continuously acts as if responding to a foreign pathogen. Microglia that are the resident macrophage-derived cells in the brain release cytokines and chemokines such as IL-1β and TNFα, among others [40]. Additionally, there is increased infiltration of peripheral monocytes and macrophages that exacerbate the inflammatory state [41]. Upregulation of IL-1Ra would act as a counter to the pro-inflammatory signaling released by activated microglia and astrocytes.
Signaling pathways required for the induction of IL-1Ra are poorly defined. The prototype function of peroxisome proliferator-activated receptor α (PPARα) is to regulate fatty acid metabolism. Most of the genes (e.g., acyl-CoA oxidase, thiolase, etc.) responsible for the β-oxidation of fatty acid harbor peroxisome proliferator response element (PPRE) for transcriptional regulation by PPARα [42, 43]. Here, we have demonstrated that aspirin increases IL-1Ra via PPARα. Our conclusion is based on the following: First, the IL-1Ra gene promoter harbors one consensus PPRE. Second, aspirin upregulates the expression of IL-1Ra in WT and PPARβ–/– astrocytes, but not PPARα–/– astrocytes. Third, oral administration of aspirin increases IL-1Ra in vivo in the cortex of WT and PPARβ–/–, but not PPARα–/–, mice. Fourth, aspirin treatment upregulated the expression of IL-1Ra in vivo in the cortex of 5xFAD, but not 5xFAD/PPARα–/–, mice. Fifth, aspirin treatment induced the recruitment of PPARα, but neither PPARβ nor PPARγ, to the PPRE of the IL-1Ra gene promoter in astrocytes. This is the first demonstration that the lipid-lowering transcription factor PPARα could be involved in the transcription of IL-1Ra gene.
Although aspirin is a century old medication, there was no known receptor for this widely-used drug. Recently, we have seen that aspirin is capable of activating PPARα and that aspirin binds to the ligand-binding domain of PPARα for its activation [14, 44]. Further analysis showed that binding to tyrosine 314 residue of PPARα plays the critical for aspirin-mediated activation of PPARα [14]. In this study, upregulation of IL-1Ra by aspirin in lenti-FL-PPARα-transduced, but not lenti-Y314D-PPARα-transduced, PPARα–/– astrocytes indicates the importance of Y314 residue of PPARα in aspirin-mediated upregulation of IL1-Ra. Since aspirin upregulates IL-1Ra via PPARα, it would be interesting to see if aspirin exhibits its prototype function of suppressing inflammation via PPARα.
Recent epidemiological investigations have shown a beneficial role for aspirin in AD [45, 46]. People who regularly take high doses of aspirin exhibit better cognition and lower incidence rates of AD [37]. A systematic investigation found that aspirin can lower the risk of getting AD later in life [47, 48]. Using aspirin as a treatment for AD also produced protective effects despite not reaching statistical significance [49, 50]. These findings are not without controversy. Other investigations have reported that aspirin has is no effect on cognition and does not lower the risk for AD [51]. While it remains an openly debated topic, there are limitations such as reporting inaccuracies and frequent comorbidities alongside AD that complicate and impair treatment.
Although mouse results are not always translated to humans, our studies indicate that low-dose aspirin can increase IL-1Ra in the brain via PPARα and that that low-dose aspirin may be considered to stimulate anti-inflammation in the CNS of patients with AD and other neurodegenerative disorders.
ACKNOWLEDGMENTS
This study was supported by a merit award from Veteran Affairs (I01BX002174) and grants (AG050431, AT010980 and NS108025) from NIH to KP. Moreover, KP is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the Department of Veterans Affairs.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Chen WW , Zhang X , Huang WJ ((2016) ) Role of neuroinflammation in neurodegenerative diseases (Review). Mol Med Rep 13: , 3391–3396. |
[2] | Jha MK , Jeon S , Suk K ((2012) ) Glia as a link between neuroinflammation and neuropathic pain. Immune Netw 12: , 41–47. |
[3] | Saha RN , Pahan K ((2006) ) Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal 8: , 929–947. |
[4] | Saha RN , Pahan K ((2006) ) Signals for the induction of nitric oxide synthase in astrocytes. Neurochem Int 49: , 154–163. |
[5] | Sofroniew MV , Vinters HV ((2010) ) Astrocytes: biology and pathology. Acta Neuropathol 119: , 7–35. |
[6] | Saha RN , Liu X , Pahan K ((2006) ) Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J Neuroimmune Pharmacol 1: , 212–222. |
[7] | Kettenmann H , Hanisch UK , Noda M , Verkhratsky A ((2011) ) Physiology of microglia. Physiol Rev 91: , 461–553. |
[8] | Jana M , Palencia CA , Pahan K ((2008) ) Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol 181: , 7254–7262. |
[9] | Steinkasserer A , Spurr NK , Cox S , Jeggo P , Sim RB ((1992) ) The human IL-1 receptor antagonist gene (IL1RN) maps to chromosome 2q14-q21, in the region of the IL-1 alpha and IL-1 beta loci. Genomics 13: , 654–657. |
[10] | Sims JE , March CJ , Cosman D , Widmer MB , MacDonald HR , McMahan CJ , Grubin CE , Wignall JM , Jackson JL , Call SM , et al. ((1988) ) cDNA expression cloning of the IL-1 receptor, a member of the immunoglobulin superfamily. Science 241: , 585–589. |
[11] | Reinhart WH ((1999) ) [Aspirin]. Ther Umsch 56: , 713–717. |
[12] | Yeomans ND ((2011) ) Aspirin: old drug, new uses and challenges. J Gastroenterol Hepatol 26: , 426–431. |
[13] | Choi HW , Tian M , Manohar M , Harraz MM , Park SW , Schroeder FC , Snyder SH , Klessig DF ((2015) ) Human GAPDH is a target of aspirin’s primary metabolite salicylic acid and its derivatives. PLoS One 10: , e0143447. |
[14] | Patel D , Roy A , Kundu M , Jana M , Luan CH , Gonzalez FJ , Pahan K ((2018) ) Aspirin binds to PPARalpha to stimulate hippocampal plasticity and protect memory. Proc Natl Acad Sci U S A 115: , E7408–E7417. |
[15] | Chandra S , Jana M , Pahan K ((2018) ) Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARalpha. J Neurosci 38: , 6682–6699. |
[16] | Mondal S , Jana M , Dasarathi S , Roy A , Pahan K ((2018) ) Aspirin ameliorates experimental autoimmune encephalomyelitis through interleukin-11-mediated protection of regulatory T cells. Sci Signal 11: , eaar8278. |
[17] | Pahan S , Pahan K ((2019) ) Mode of action of aspirin in experimental autoimmune encephalomyelitis. DNA Cell Biol 38: , 593–596. |
[18] | Rangasamy SB , Dasarathi S , Pahan P , Jana M , Pahan K ((2019) ) Low-dose aspirin upregulates tyrosine hydroxylase and increases dopamine production in dopaminergic neurons: implications for Parkinson’s disease. J Neuroimmune Pharmacol 14: , 173–187. |
[19] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26: , 10129–10140. |
[20] | Modi KK , Roy A , Brahmachari S , Rangasamy SB , Pahan K ((2015) ) Cinnamon and its metabolite sodium benzoate attenuate the activation of p21rac and protect memory and learning in an animal model of Alzheimer’s disease. PLoS One 10: , e0130398. |
[21] | Rangasamy SB , Corbett GT , Roy A , Modi KK , Bennett DA , Mufson EJ , Ghosh S , Pahan K ((2015) ) Intranasal delivery of NEMO-binding domain peptide prevents memory loss in a mouse model of Alzheimer’s disease. J Alzheimers Dis 47: , 385–402. |
[22] | Corbett GT , Gonzalez FJ , Pahan K ((2015) ) Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci U S A 112: , 8445–8450. |
[23] | Ghosh A , Pahan K ((2012) ) Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders. J Biol Chem 287: , 27189–27203. |
[24] | Khasnavis S , Jana A , Roy A , Mazumder M , Bhushan B , Wood T , Ghosh S , Watson R , Pahan K ((2012) ) Suppression of nuclear factor-kappaB activation and inflammation in microglia by physically modified saline. J Biol Chem 287: , 29529–29542. |
[25] | Jana M , Liu X , Koka S , Ghosh S , Petro TM , Pahan K ((2001) ) Ligation of CD40 stimulates the induction of nitric-oxide synthase in microglial cells. J Biol Chem 276: , 44527–44533. |
[26] | Pahan K , Sheikh FG , Liu X , Hilger S , McKinney M , Petro TM ((2001) ) Induction of nitric-oxide synthase and activation of NF-kappaB by interleukin-12 p40 in microglial cells. J Biol Chem 276: , 7899–7905. |
[27] | Khasnavis S , Pahan K ((2012) ) Sodium benzoate, a metabolite of cinnamon and a food additive, upregulates neuroprotective Parkinson disease protein DJ-1 in astrocytes and neurons. J Neuroimmune Pharmacol 7: , 424–435. |
[28] | Corbett GT , Roy A , Pahan K ((2012) ) Gemfibrozil, a lipid-lowering drug, upregulates IL-1 receptor antagonist in mouse cortical neurons: implications for neuronal self-defense. J Immunol 189: , 1002–1013. |
[29] | Jana A , Modi KK , Roy A , Anderson JA , van Breemen RB , Pahan K ((2013) ) Up-regulation of neurotrophic factors by cinnamon and its metabolite sodium benzoate: therapeutic implications for neurodegenerative disorders. J Neuroimmune Pharmacol 8: , 739–755. |
[30] | Khasnavis S , Pahan K ((2014) ) Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9: , 569–581. |
[31] | Brahmachari S , Jana A , Pahan K ((2009) ) Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. J Immunol 183: , 5917–5927. |
[32] | Dutta D , Majumder M , Paidi RK , Pahan K ((2021) ) Alleviation of Huntington pathology in mice by oral administration of food additive glyceryl tribenzoate. Neurobiol Dis 153: , 105318. |
[33] | Ghosh A , Corbett GT , Gonzalez FJ , Pahan K ((2012) ) Gemfibrozil and fenofibrate, Food and Drug Administration-approved lipid-lowering drugs, up-regulate tripeptidyl-peptidase 1 in brain cells via peroxisome proliferator-activated receptor alpha: implications for late infantile Batten disease therapy. J Biol Chem 287: , 38922–38935. |
[34] | Arend WP , Malyak M , Guthridge CJ , Gabay C ((1998) ) Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol 16: , 27–55. |
[35] | Dinarello CA ((2009) ) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27: , 519–550. |
[36] | Martinon F , Mayor A , Tschopp J ((2009) ) The inflammasomes: guardians of the body. Annu Rev Immunol 27: , 229–265. |
[37] | Nilsson SE , Johansson B , Takkinen S , Berg S , Zarit S , McClearn G , Melander A ((2003) ) Does aspirin protect against Alzheimer’s dementia? A study in a Swedish population-based sample aged>or=80 years. Eur J Clin Pharmacol 59: , 313–319. |
[38] | Moro MA , De Alba J , Cardenas A , De Cristobal J , Leza JC , Lizasoain I , Diaz-Guerra MJ , Bosca L , Lorenzo P ((2000) ) Mechanisms of the neuroprotective effect of aspirin after oxygen and glucose deprivation in rat forebrain slices. Neuropharmacology 39: , 1309–1318. |
[39] | Amor S , Peferoen LA , Vogel DY , Breur M , van der Valk P , Baker D , van Noort JM ((2014) ) Inflammation in neurodegenerative diseases–an update. Immunology 142: , 151–166. |
[40] | Smith JA , Das A , Ray SK , Banik NL ((2012) ) Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull 87: , 10–20. |
[41] | Varvel NH , Neher JJ , Bosch A , Wang W , Ransohoff RM , Miller RJ , Dingledine R ((2016) ) Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc Natl Acad Sci U S A 113: , E5665–5674. |
[42] | Reddy JK , Mannaerts GP ((1994) ) Peroxisomal lipid metabolism. Annu Rev Nutr 14: , 343–370. |
[43] | Roy A , Pahan K ((2015) ) PPARalpha signaling in the hippocampus: crosstalk between fat and memory. J Neuroimmune Pharmacol 10: , 30–34. |
[44] | Patel D , Roy A , Pahan K ((2020) ) PPARalpha serves as a new receptor of aspirin for neuroprotection. J Neurosci Res 98: , 626–631. |
[45] | in ‘t Veld BA , Launer LJ , Breteler MM , Hofman A , Stricker BH ((2002) ) Pharmacologic agents associated with a preventive effect on Alzheimer’s disease: a review of the epidemiologic evidence. Epidemiol Rev 24: , 248–268. |
[46] | Chandra S , Roy A , Patel DR , Pahan K ((2019) ) PPARalpha between aspirin and plaque clearance. J Alzheimers Dis 71: , 389–397. |
[47] | Zhang C , Wang Y , Wang D , Zhang J , Zhang F ((2018) ) NSAID exposure and risk of Alzheimer’s disease: an updated meta-analysis from cohort studies. Front Aging Neurosci 10: , 83. |
[48] | Chakrabarti S , Roy A , Prorok T , Patel D , Dasarathi S , Pahan K ((2019) ) Aspirin up-regulates suppressor of cytokine signaling 3 in glial cells via PPARalpha. J Neurochem 151: , 50–63. |
[49] | Etminan M , Gill S , Samii A ((2003) ) Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327: , 128. |
[50] | Benito-Leon J , Contador I , Vega S , Villarejo-Galende A , Bermejo-Pareja F ((2019) ) Non-steroidal anti-inflammatory drugs use in older adults decreases risk of Alzheimer’s disease mortality. PLoS One 14: , e0222505. |
[51] | Waldstein SR , Wendell CR , Seliger SL , Ferrucci L , Metter EJ , Zonderman AB ((2010) ) Nonsteroidal anti-inflammatory drugs, aspirin, and cognitive function in the Baltimore longitudinal study of aging. J Am Geriatr Soc 58: , 38–43. |


