
Stimulation of Dopamine Production by Sodium Benzoate, a Metabolite of Cinnamon and a Food Additive1
Abstract
Background:
Parkinson’s disease (PD) is one of the most important neurodegenerative disorders in human in which recovery of functions could be achieved by improving the survival and function of residual dopaminergic neurons in the substantia nigra pars compacta. Tyrosine hydroxylase (TH) is the rate-limiting enzyme in the dopamine (DA) biosynthesis pathway.
Objective:
Earlier our laboratory has shown that sodium benzoate (NaB), a metabolite of cinnamon and an FDA-approved drug against urea cycle disorders and glycine encephalopathy, increases neuroprotective molecules and protects dopaminergic neurons in a mouse model of PD. Here, we examined whether NaB could stimulate the production of DA in dopaminergic neurons.
Methods:
We employed PCR, real-time PCR, western blot, immunostaining, and HPLC to study the signature function of dopaminergic neurons. Locomotor functions were monitored in mice by open-field.
Results:
NaB increased the mRNA and protein expression of TH to produce DA in mouse MN9D dopaminergic neuronal cells. Accordingly, oral feeding of NaB increased the expression of TH in the nigra, upregulated striatal DA, and improved locomotor activities in striatum of normal C57/BL6 and aged A53T-α-syn transgenic mice. Rapid induction of cAMP response element binding (CREB) activation by NaB in dopaminergic neuronal cells and the abrogation of NaB-induced expression of TH by siRNA knockdown of CREB suggest that NaB stimulates the transcription of TH in dopaminergic neurons via CREB.
Conclusion:
These results indicate a new function of NaB in which it may be beneficial in PD via stimulation of DA production from residual dopaminergic neurons.
INTRODUCTION
Parkinson’s disease (PD) or paralysis agitans is a common age-associated neurodegenerative movement disorder characterized by progressive loss of dopaminergic neurons in the ventral midbrain [1, 2]. Clinically PD is characterized by resting tremor, bradykinesia, rigidity, and postural instability [1, 2]. Generally, the nigral neurons communicate with neurons in basal ganglia by releasing a specific neurotransmitter called dopamine (DA), and this biochemical interaction is essential for fine tuning of our movements. In PD, nigral neurons progressively degenerate, resulting in the loss of DA required for neurotransmission in the corpus striatum. Meanwhile, clinical studies evidenced that significant number of dopaminergic neurons still remains in the SN when motor signs first appear in PD patients. It has been reported that the remaining SN neurons could be around 50% in patients at the onset of their motor symptoms [3, 4]. On the other hand, few studies report the presence of 30–40% SN neurons in patients during their first visit to the clinic [5, 6]. Since it is not possible to bring back dead dopaminergic neurons, looking for pathways and drugs to protect these neurons and increase their function are of paramount importance.
Although catecholamine biosynthesis pathway begins with the hydroxylation of phenylalanine, tyrosine hydroxylase (TH) is the regulatory enzyme of this pathway. TH catalyzes the hydroxylation of tyrosine to L-DOPA, a precursor of DA, in dopaminergic neurons. Therefore, upregulation of TH is an important step for increasing the function of dopaminergic neurons. Sodium benzoate (NaB) is metabolite of commonly used spice cinnamon and a widely used food preservative. Being a component of Ucephan and Ammonul, NaB is an FDA-approved drug for the treatment of hepatic metabolic defects associated with hyperammonemia, such as urea cycle disorder in children [7, 8]. Until now, NaB is also the only FDA-approved drug for nonketotic hyperglycinemia or glycine encephalopathy. Earlier, our laboratory has delineated that NaB is anti-inflammatory [9], immunomodulatory [10], Treg booster [11], neurotrophic [12, 13], and anti-oxidant [14]. We have also demonstrated that NaB is capable of increasing the expression of DJ-1 [15] and protecting dopaminergic neurons and striatal neurotransmitters in an animal model of PD [16].
In this study, we examined the effect of NaB on the catecholamine biosynthetic pathway in dopaminergic neuronal cells. Here, we demonstrated that NaB stimulated the expression of TH and increased the production of DA in mouse MN9D dopaminergic neuronal cells via activation of cAMP response element binding (CREB). Oral administration of NaB also stimulated the level of TH in the nigra, increased the level of DA in the striatum and improved locomotor activities in normal as well as A53T α-synuclein (α-syn) transgenic mice, highlighting possible beneficial effects of NaB in PD.
MATERIALS AND METHODS
Reagents
Cell culture materials (DMEM/F-12, L-Glutamine, Hank’s balanced salt solution, 0.05% trypsin, and antibiotic-antimycotic) were purchased from Mediatech (Washington, DC). Fetal bovine serum (FBS) was obtained from Atlas Biologicals. We purchased anti-TH antibody (cat# P40101-150) from PelFreez Biologicals. Anti-actin antibody (cat# ab6276) was obtained from Abcam. Anti-CREB antibody (cat# CS203204) was obtained from Millipore-Sigma. Alexa-fluor antibodies used in immunostaining were obtained from Jackson ImmunoResearch and IR-dye-labeled reagents used for immunoblotting were from Li-Cor Biosciences.
Preparation of NaB
Powdered benzoic acid was dissolved in sterile water and the resulting solution was titrated in pH 7.0 using sodium hydroxide as described before [9, 10].
MN9D cell maintenance and NaB treatment
MN9D cells provided by Dr. Alfred Heller (University of Chicago) were cultured in high-glucose DMEM supplemented with 10% FBS and 50μg/ml each penicillin and streptomycin. These cells were maintained at 37°C in a humidified 5% CO2 incubator in their undifferentiated state and were used for experiments only below passage 20. During differentiation, cells were allowed to differentiate in neurobasal media containing 2% B27 and 1% antibiotic antimycotic mixture for 2 d. Subsequently, cells were treated with different concentrations of NaB in either neurobasal media without B27 or serum-free DMEM.
Animals and NaB treatment
A53T α-syn transgenic [B6;C3-Tg(Prnp-SNCA*A53T)83Vle/J] mice were purchased from the Jackson Laboratory. Animal maintenance and experiments were in accordance with National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use committee of the Rush University Medical Center, Chicago, IL. NaB was solubilized in water and 9-month-old male A53T transgenic mice were treated with NaB (50 mg/kg body wt/d) via gavage for 30 d. Similarly, in another set, 8–10-week-old C57/BL6 male mice (Harlan Sprague Dawley, Indianapolis, IN) were also treated with NaB. Earlier we have seen neuroprotection in this particular dose of NaB and duration of treatment [17].
Semi-quantitative RT-PCR analysis
Total RNA was isolated from cells by using the RNeasy mini kit (Qiagen, Valencia, CA) following manufacturer’s protocol. Contaminating genomic DNA was removed from total RNA by DNase digestion. Semi-quantitative RT-PCR was carried out as described earlier [10, 18, 19] using a RT-PCR kit from Clontech (Mountain View, CA). Briefly, 1μg of total RNA was reverse transcribed using oligo(dT)12–18 as primer and MMLV reverse transcriptase (Clontech) in a 20μL reaction mixture. The resulting cDNA was diluted as needed followed by amplification of diluted cDNA using Titanium Taq DNA polymerase. Amplified products were electrophoresed on a 1.8% agarose gels and visualized by ethidium bromide staining.
TH: Sense: 5’-CAGGACATTGGACTTGCATCTCTG-3’
Antisense: 5’-ATAGTTCCTGAGCTTGTCCTTGGC-3’
GAPDH: Sense: 5’-GGTGAAGGTCGGTGTGAACG3’
Antisense: 5’-TTGGCTCCACCCTTCAAGTG-3’
Real-time PCR analysis
The mRNA quantification was performed using the ABI-Prism7700 sequence detection system (Applied Biosystems, Foster City, CA) using SYBR Select master mix (Applied Biosystems) as described earlier [20–22]. The mRNA expression of the targeted genes was normalized to the level of Gapdh mRNA and data was processed by the ABI Sequence Detection System 1.6 software.
Immunoblotting
Western blotting was conducted as described earlier [20, 21]. Briefly, cells were scraped in lysis buffer, transferred to microfuge tubes and spun into pellet. The supernatant was collected and analyzed for protein concentration via the Bradford method (Bio-Rad). SDS sample buffer was added to protein samples and boiled for 5 min. Denatured samples were electrophoresed on NuPAGE® Novex® 4–12% Bis-Tris gels (Invitrogen) and proteins transferred onto a nitrocellulose membrane (Bio-Rad) using the Thermo-Pierce Fast Semi-Dry Blotter. The membrane was then washed for 15 min in TBS plus Tween 20 (TBST) and blocked for 1 h in TBST containing BSA. Next, membranes were incubated at 4°C under shaking conditions with primary antibodies (Table 1) followed by washing of membranes in TBST for 1 h. Membranes were then incubated in secondary antibodies for 1 h at room temperature, washed for one more hour, and visualized under the Odyssey® Infrared Imaging System (Li-COR, Lincoln, NE). Protein blots were analyzed using ImageJ (NIH, Bethesda, MD) and bands were normalized to their respective β-actin loading controls.
Table 1
Antibodies, sources, applications, and dilutions used in this paper
| Target | Antibody (Clone) | Epitope/immunogen | Application/dilution | Source; Catalog |
| Actin | Mouse monoclonal (AC-15) | a.a. 1-15 of Xenopus laevis β-actin | WB- 1 : 5000 | Abcam (Cambridge, MA, USA) |
| # ab6276 | ||||
| Tyrosine- | Rabbit polyclonal | SDS-denatured rat tyrosine | Immunoflourescense | PelFreez Biologicals (Rogers, AR, USA) |
| Hydroxylase | hydroxylase, purified from | 1 : 1500 | # P40101-150 | |
| Pheochromocytoma | WB- 1 : 2000 | |||
| Tyrosine | Mouse monoclonal | TH purified from ra t | Immunohistochemistry | Sigma |
| Hydroxylase | PC12 cells/LNC1clone | 1 : 10,000 | # 22941 |
Immunofluorescence analysis
It was performed as described earlier [9, 15]. Briefly, cover slips containing 100–200 cells/mm2 were fixed with 4% paraformaldehyde followed by treatment with cold ethanol and two rinses in phosphate-buffered saline (PBS). Samples were blocked with 3% bovine serum albumin (BSA) in PBS-Tween-20 (PBST) for 30 min and incubated in PBST containing 1% BSA and primary antibodies (Table 1). After three washes in PBST (15 min each), slides were further incubated with Cy2 (Jackson ImmunoResearch Laboratories, Inc.). For negative controls, a set of culture slides was incubated under similar conditions without the primary antibodies. The samples were mounted and observed under an Olympus IX81 fluorescence microscope.
Measurement of mean fluorescence intensity (MFI) of TH
MFI was measured using the “measurement module” of the microsuite V Olympus software. Briefly, images were opened in their green channel in order to monitor MFI of TH in the SNpc region. After that, measurement module was opened followed by the selection of two parameters including perimeter and MFI. Rectangular box tool was used to outline perimeter and then associated MFI in that given perimeter was automatically calculated.
Immunohistochemistry
Mice were anesthetized with ketamine-xylazine injectables and perfused with PBS followed by dissection of half brain from each mouse for biochemical assays and half for immunohistochemistry [16, 23]. Briefly, half brains were incubated in 4% paraformaldehyde (w/v) followed by 30% sucrose overnight at 4°C. Brains were then embedded, sectioned (30μm thick), and processed for tyrosine hydroxylase (TH) staining as described previously [23, 24].
HPLC analysis for measurement of DA and its metabolite levels
Levels of DA, DOPAC (3, 4-dihydroxyphenylacetic acid) and HVA (homovanillic acid) were quantified in striatum and supernatants of MN9D cells as described earlier [23–27]. Briefly, mice were sacrificed by cervical dislocation after treatment and their striata were collected and immediately frozen in dry ice and stored at –80°C until analysis. On the day of the analysis, tissues were sonicated in 0.2 M perchloric acid containing isoproterenol and resulting homogenates were centrifuged at 20,000 x g for 15 min at 4°C. After pH adjustment and filtration, 10μl of supernatant was injected onto a Eicompak SC-3ODS column (Complete Stand-Alone HPLC-ECD System EiCOMHTEC-500 from JM Science Inc., Grand Island, NY) and analyzed following manufacturer’s protocol. In case of supernatants of MN9D cells, 10μl supernatant was directly loaded onto a Eicompak SC-3ODS column.
Behavioral analyses
After treatment, animals were placed individually into an open field arena made of plexiglass cage [42 cm (L)×42 cm (W)×32 cm (H)] as described before [23, 28]. In this procedure, the open field environment is much larger than that of the home cage and is unfamiliar to the animal. Therefore, in the beginning of experiment, animals were allowed to adapt with the environment for 5 min. After that, animal’s open field behavior was recorded. Each animal was monitored for three times with 5 min acquisition and 5 min of interval time. Motor activity was monitored in the Ethovision XT 13.0 Open Field Activity System (Noldus). Here, the plexiglass cage was closely tracked by a computer-assisted camera continuously monitoring the animal’s movement. Test cage was cleaned with alcohol after replacing each animal. In the open-field experiment, various parameters, such as horizontal activity, total distance, number of movements, movement time, rest time, stereotypy counts, rearing, mean distance, meantime, center distance, and center time, were measured from collected videos accordingly.
For rotarod testing, animals were permitted to run on a motorized retrograde moving platform (Med associates Inc.) at a constant speed of 24-rpm with cut-off time 5 min. To eliminate stress and fatigues, mice were given a 5 min rest interval. Each animal was recorded three times.
Statistical analysis
Statistical analyses were performed with Student’s t test (for two-group comparisons) and one-way ANOVA, followed by Tukey’s multiple-comparison tests, as appropriate (for multiple groups comparison), using Prism 7 (GraphPad Software). Data represented as mean±SD or mean±SEM as stated in figure legends. A level of p < 0.05 was considered statistically significant.
RESULTS
NaB upregulates the expression of TH and stimulates DA synthesis in mouse MN9D dopaminergic neuronal cells
Since there are no effective drugs/molecules to upregulate TH and increase the production of DA in dopaminergic neurons, we examined whether NaB, an FDA-approved drug for urea cycle disorders and glycine encephalopathy, could stimulate the expression of TH in MN9D dopaminergic neuronal cells. It is evident from RT-PCR (Fig. 1A) and real-time PCR (Fig. 1B) that 2 h incubation with NaB at serum-free condition dose-dependently stimulated the mRNA expression of TH in MN9D cells with the highest increase in TH observed at 200μM. However, fold induction of TH mRNA decreased at 300μM NaB (Fig. 1A, B). Next, we examined if NaB could also upregulate the level of TH protein in MN9D neuronal cells. Similar to the increase in TH mRNA, NaB significantly increased the level of TH protein and again, the maximum increase was seen at 200μM NaB (Fig. 1C, D). To confirm these results further, we also performed double-label immunofluorescence for TH and actin (Fig. 1E) followed by analysis of mean fluorescence intensity (MFI) (Fig. 1F). As evident from Fig. 1E and F, NaB treatment increased the level of TH without modulating the status of actin. These results were specific as we did not find any such increase in TH by sodium formate, a negative control of NaB (Fig. 1E, F). These results suggest that NaB upregulates the expression of both TH mRNA and protein in MN9D neuronal cells.
Fig. 1
NaB increases tyrosine hydroxylase (TH) in mouse MN9D neuronal cells in dose- and time-dependent manner. A) Mouse MN9D neuronal cells were treated with different concentrations of NaB under serum-free conditions for 2 h followed by monitoring the mRNA expression of TH by semi-quantitative RT-PCR (A) and real-time PCR (B). The protein level of TH was monitored by western blot (C). Actin was run as a loading control. Bands were scanned and values (TH/actin) presented as relative to control (D). Results are mean±SD of different experiments. Cells were treated with 200μM NaB for 2 h followed by double-label immunofluorescence with antibodies against TH and actin (E). Sodium formate (NaFO) was used as a negative control for NaB. Mean fluorescence intensity (MFI) of TH and actin was calculated in 20 different cells (F) and presented as MFI-TH/MFI-actin. Results are mean±SEM of 20 different cells per group. G) Cells were treated with 200μM NaB for different time periods followed by measuring the level of DA, DOPAC and HVA in supernatants by HPLC. Results are mean±SD of at least three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 versus control. One-way ANOVA with Tukey’s multiple comparison test shows that time-dependent increase in DA by NaB is significant (p < 0.0001).
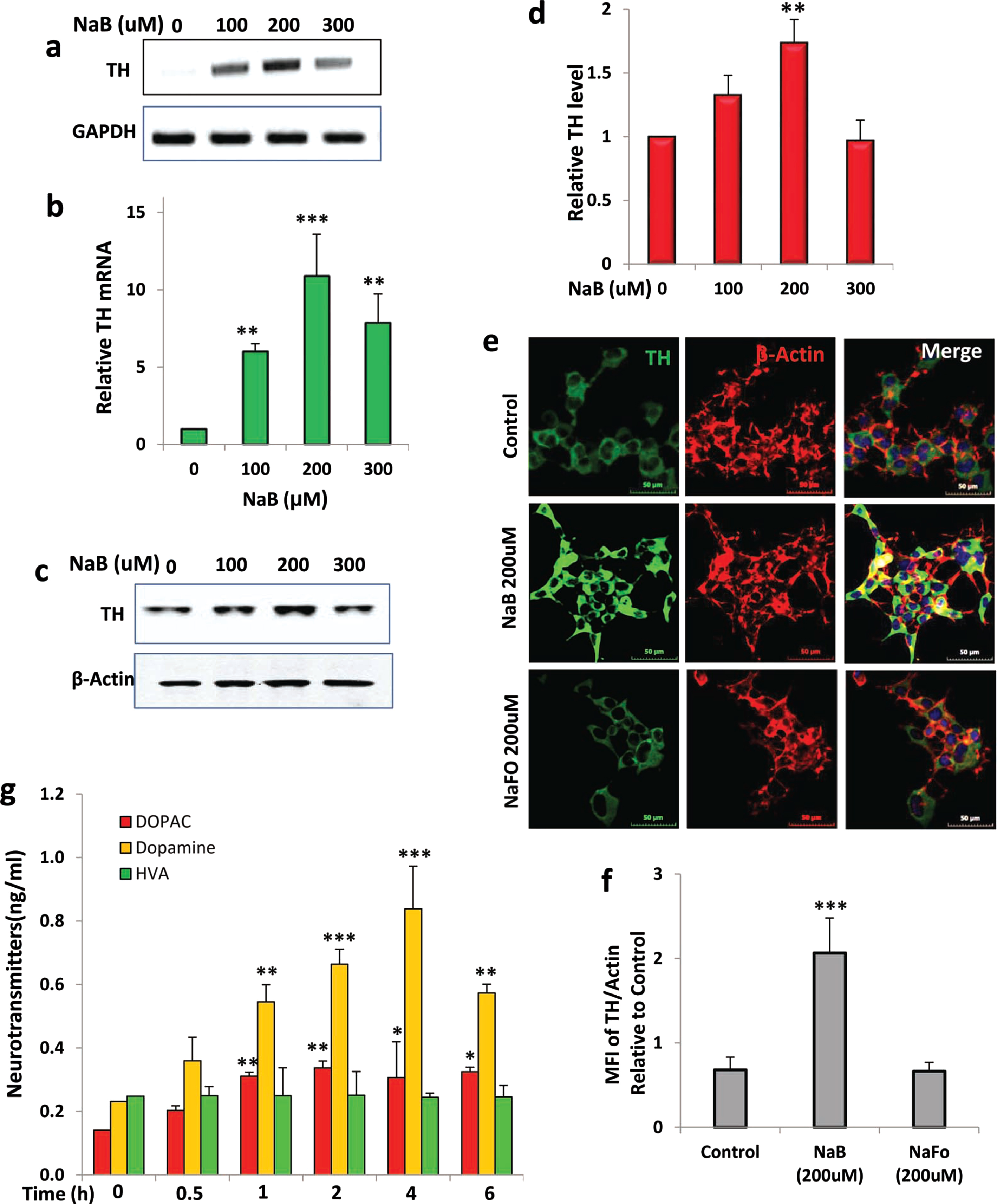
Since TH is known to catalyze the formation of DA, to understand the functional significance of NaB-mediated TH upregulation, we quantified the level of DA from the supernatant of NaB-treated neuronal cells by HPLC. NaB did not increase DA production significantly within 30 min of stimulation (Fig. 1G). However, marked increase in DA was seen at 1 h of treatment with the highest production of DA noticed at 4 h of treatment (Fig. 1G). Although we did not find any increase in HVA, the level of DOPAC was higher at different hours of NaB treatment (Fig. 1G), which could be due to the degradation of DA.
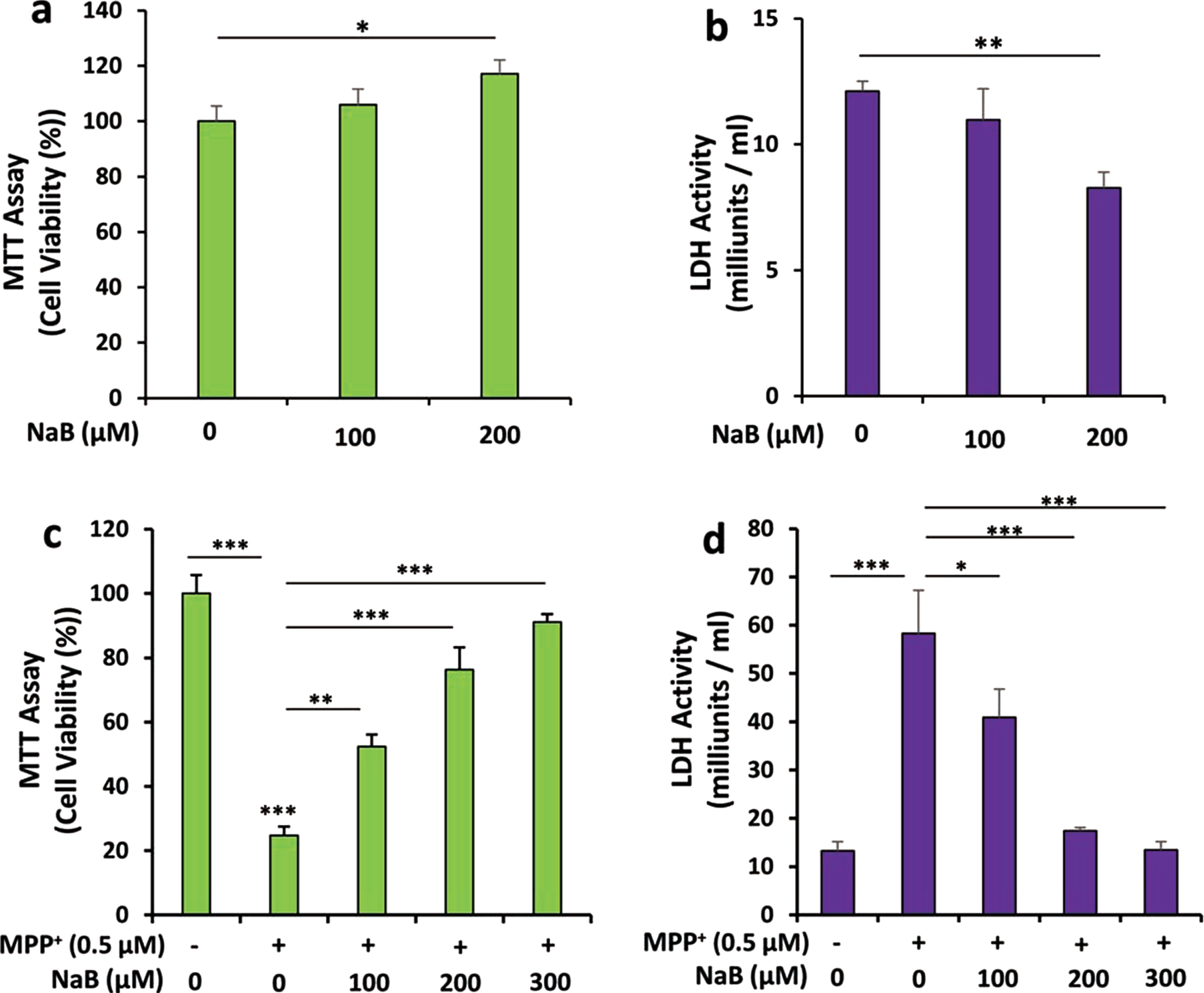
Next, we examined if NaB could increase the expression of TH and upregulate the production of DA in differentiated MN9D neuronal cells. We carried out a time-dependent study using 200μM NaB and found that similar to undifferentiated cells, NaB upregulated TH mRNA significantly starting from 1 h of incubation with maximum stimulation observed at 4 h followed by a decrease in subsequent hour of incubation (Fig. 2A, B). This was also supported by double-label immunofluorescence analysis of TH and β-actin (Fig. 2C, D). Again, we did not notice any increase in TH by NaFO (Fig. 2C, D). Similar to that observed in undifferentiated cells (Fig. 1G), NaB also increased the production of DA in differentiated MN9D neuronal cells and the highest increase was observed at 4 h of stimulation (Fig. 2E). NaB was not toxic to MN9D neuronal cells. In fact, at a concentration of 200μM, NaB increased cell viability as evident from increase in MTT metabolism (Fig. 3A) and decrease in LDH release (Fig. 3B). Moreover, 1-methyl-4-phenylpyridinium (MPP+) is a neurotoxicant and as expected, MPP+ insult markedly increased death of MN9D neuronal cells (Fig. 3C, D). However, NaB pretreatment dose-dependently increased MTT metabolism (Fig. 3C) and decreased LDH release (Fig. 3D) in MPP+-intoxicated cells, indicating that NaB protects MN9D neuronal cells from MPP+ toxicity.
Fig. 2
NaB increases TH and dopamine in differentiated mouse MN9D neuronal cells. A) Mouse MN9D neuronal cells were allowed to differentiate in neurobasal media containing B27 for 2 d following treatment with 200μM NaB for different time periods in neurobasal media without B27. The protein level of TH was monitored by western blot (A). Bands were scanned and values (TH/actin) presented as relative to control (B). Results are mean±SD of different experiments. *p < 0.05 and ***p < 0.001 versus control. Cells were treated with NaB and NaFO for 2 h followed by double-label immunofluorescence with antibodies against TH and actin (C). Mean fluorescence intensity (MFI) of TH and actin was calculated in 20 different cells (D) and presented as MFI-TH/MFI-actin. Results are mean±SEM of 20 different cells per group. ***p < 0.001 versus control; ns, not significant. E) Cells were treated with 200μM NaB for different time periods followed by measuring the level of DA, DOPAC, and HVA in supernatants by HPLC. Results are mean±SD of at least three independent experiments. ***p < 0.001 versus control. One-way ANOVA with Tukey’s multiple comparison test shows that time-dependent increase in DA by NaB is significant (p < 0.0001).
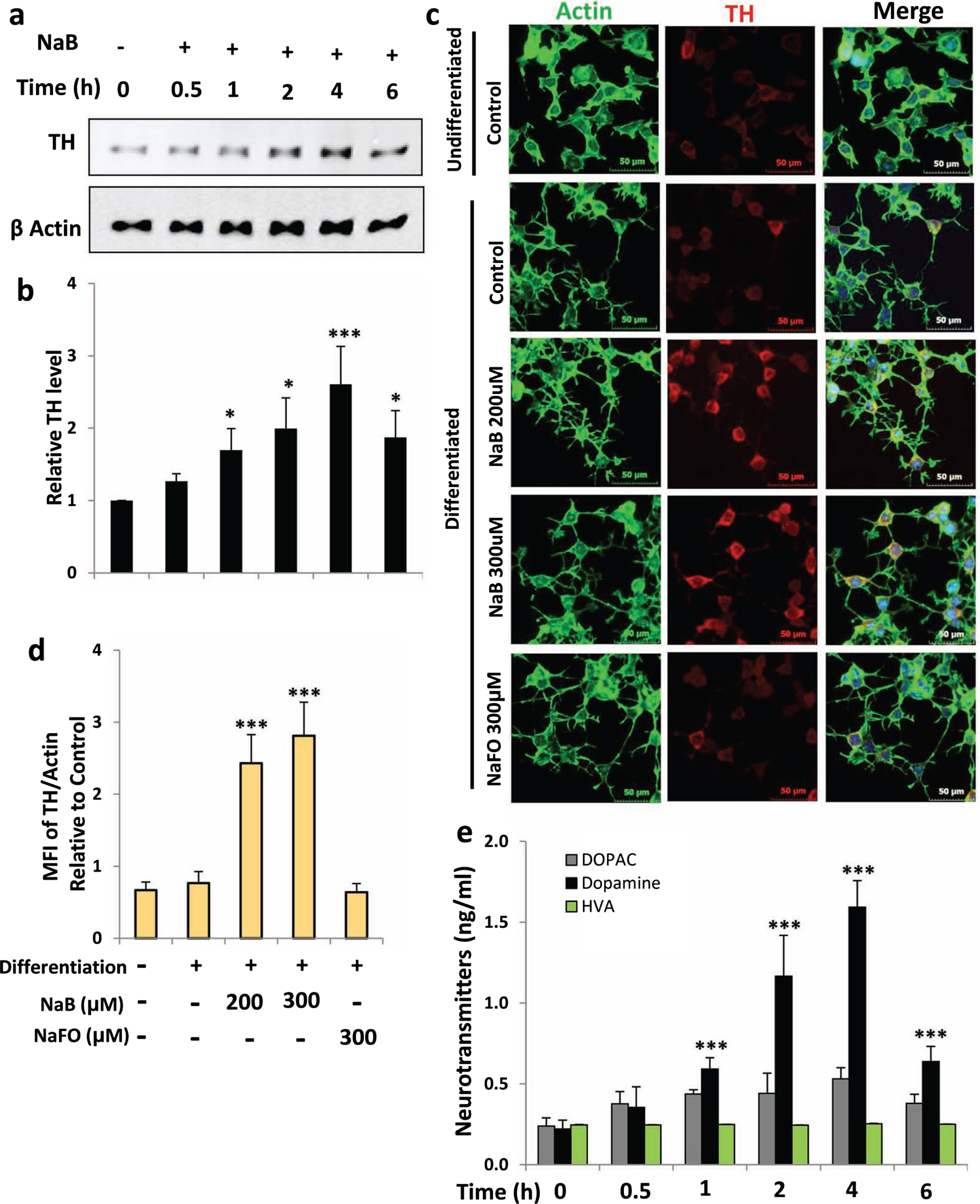
Fig. 3
NaB stimulates cell viability and protects MN9D neuronal cells from MPP+-induced insult. Mouse MN9D neuronal cells were treated with different concentrations of NaB for 18 h under serum-free condition followed by measuring cell viability by MTT (A) and LDH (B). Cells pretreated with different concentrations of NaB for 2 h were insulted with 0.5μM MPP+ under serum-free condition. After 18 h, cell viability was measured by MTT (C) and LDH (D). Results are mean±SD of three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001.
NaB upregulated TH in MN9D dopaminergic neuronal cells via activation of CREB
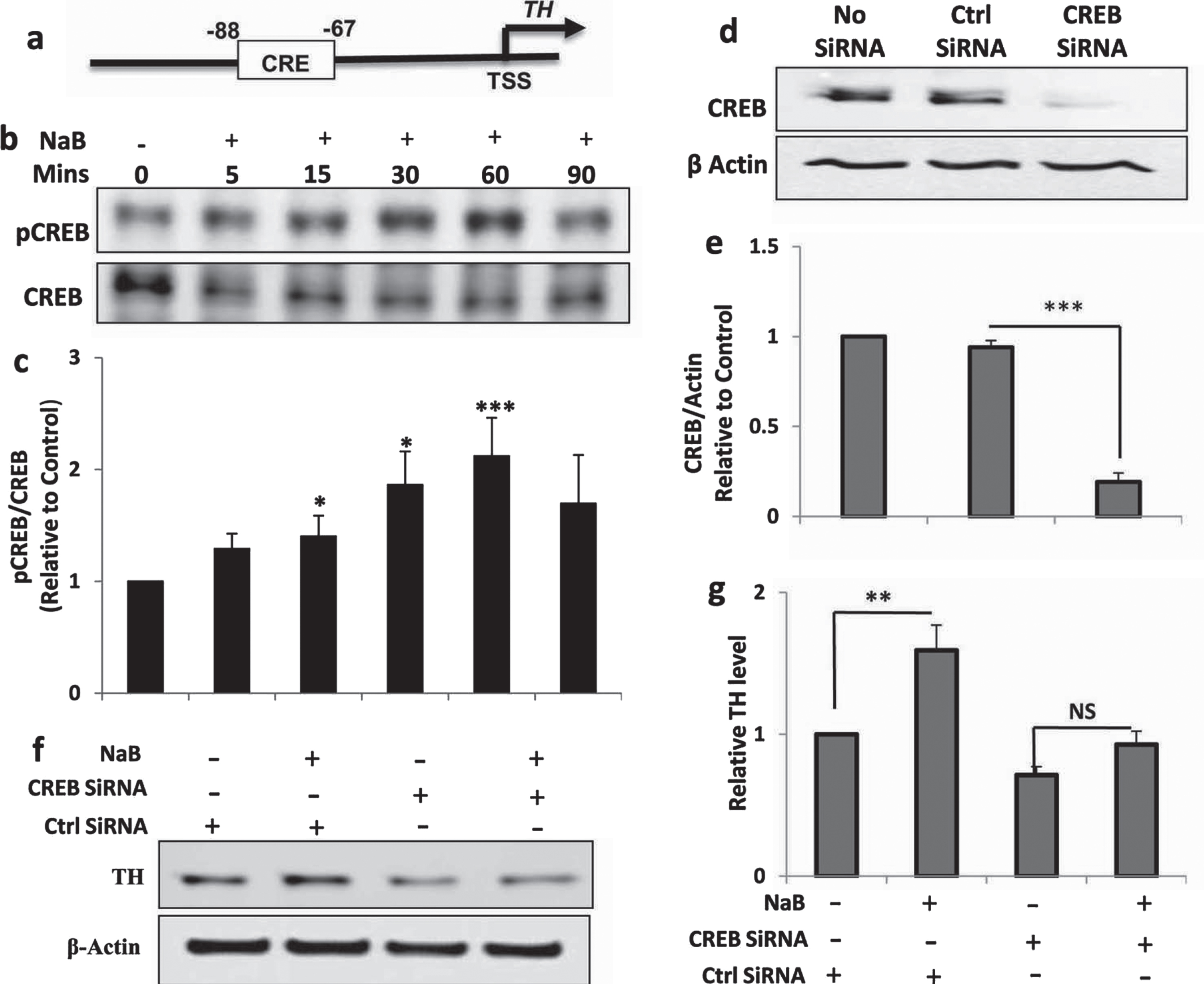
Next, we investigated mechanism by which NaB upregulated TH in neuronal cells. Earlier we have described that NaB is capable of inducing the activation of CREB in neurons [12]. Since the TH gene promoter harbors a consensus CRE (Fig. 4A), we examined if NaB required CREB for increasing the level of TH in MN9D neuronal cells. At first, we checked the activation of CREB by monitoring the levels of phospho-CREB and total CREB in NaB-stimulated neurons. Upregulation of phospho-CREB, but not total CREB, by NaB (Fig. 4B, C) suggests the activation of CREB in MN9D neuronal cells. Next, to study the involvement of CREB, we employed CREB siRNA. Downregulation of CREB protein by CREB siRNA, but not control siRNA, in neuronal cells indicates the efficacy of CREB siRNA (Fig. 4D, E). Next, we examined the effect of CREB siRNA on NaB-mediated upregulation of TH. Upregulation of TH by NaB in control siRNA-transfected, but not CREB siRNA-transfected, neuronal cells (Fig. 4F, G) suggests that NaB requires CREB for increasing TH in neuronal cells.
Fig. 4
NaB increases the expression of TH in MN9D neuronal cells via CREB. A) TH gene promoter harbors a consensus CRE. MN9D neuronal cells were treated with 200μM NaB for different min under serum-free conditions followed by monitoring the level of phospho-CREB (pCREB) by western blot (B). Graph represents densitometric analysis of pCREB protein levels normalized to total CREB (loading control) (C). Results are mean±SD of three independent experiments. ***p < 0.001 versus control; *p < 0.05 versus control. D) Cells were transfected with either control or CREB siRNA. Forty-eight h after transfection, level of CREB was monitored by western blot. E) Graph represents densitometric analysis of CREB protein levels normalized to β-Actin (loading control). Results are mean±SD of three independent experiments. ***p < 0.001 versus control (no siRNA). F) Cells were transfected with either control siRNA or CREB siRNA. Forty-eight h after transfection, cells were treated with 200μM NaB for 2 h under serum-free condition followed by monitoring the level of TH by western blot. G) Graph represents densitometric analysis of TH protein levels normalized to β-Actin (loading control). Results are mean±SD of three independent experiments. **p < 0.01 versus control. NS, not significant.
Oral NaB increases the level of TH in the nigra of C57/BL6 and A53T α-syn transgenic mice
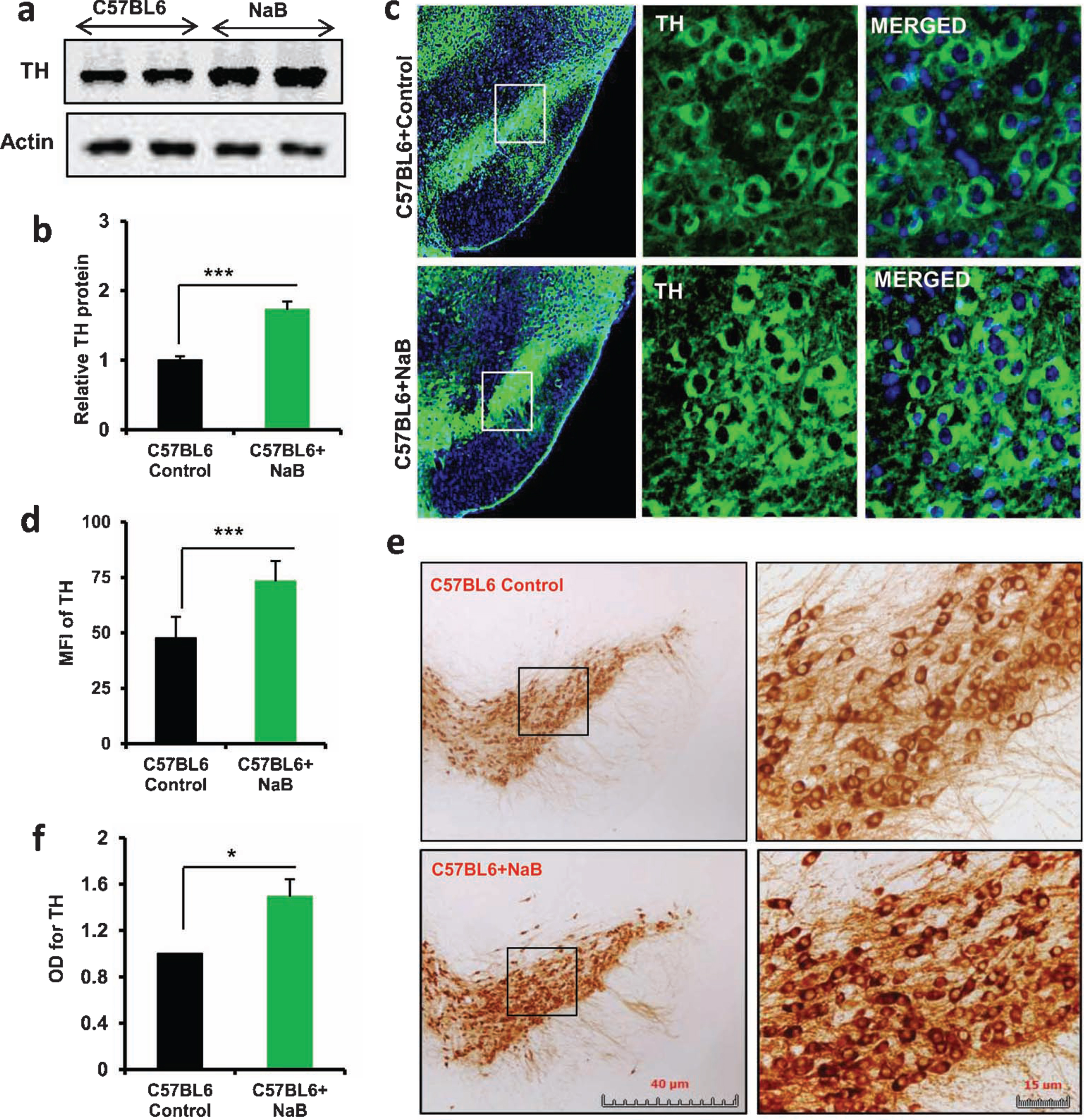
As NaB upregulated the expression of TH in MN9D neuronal cells, we next tested whether oral treatment with NaB could increase the level of TH in vivo in the nigra of normal C57/BL6 mice. As evident from western blot of nigral extracts, NaB was capable of increasing the expression of TH protein (Fig. 5A, B). Increase in TH in the nigra of NaB-treated C57/BL6 mice was also confirmed by immunofluorescence analysis (Fig. 5C) followed by MFI quantification (Fig. 5D). Furthermore, TH DAB staining followed by monitoring optical density of TH also indicated nigral upregulation of TH by oral NaB (Fig. 5E, F).
Fig. 5
Oral treatment with NaB increases the level of TH in vivo in the nigra of normal C57/BL6 mice. Male C57/BL6 mice (n = 5 per group) were treated with NaB (50 mg/kg body wt/d) mixed in 0.5% methylcellulose orally via gavage. Control mice received 0.5% methylcellulose as vehicle. After 30 d of treatment, the level of TH was monitored in the SNpc by western blot (A). Actin was run as loading control. Bands were scanned and values (TH/actin) presented as relative to control (B). Results are mean ± SEM of five mice per group. ***p < 0.001 versus control by two-tailed paired t-tests. The level of TH was monitored in ventral midbrain sections by immunofluorescence (C). MFI of TH (D) was calculated in two nigral sections of each of five mice per group. Results are mean±SEM of five mice per group. ***p < 0.001 versus control by two-tailed paired t-tests. The level of TH was monitored in ventral midbrain sections by DAB immunostaining (E). Optical density of TH (F) was calculated in two nigral sections of each of five mice per group. Results are mean±SEM of five mice per group. *p < 0.05 versus control by two-tailed paired t-tests.
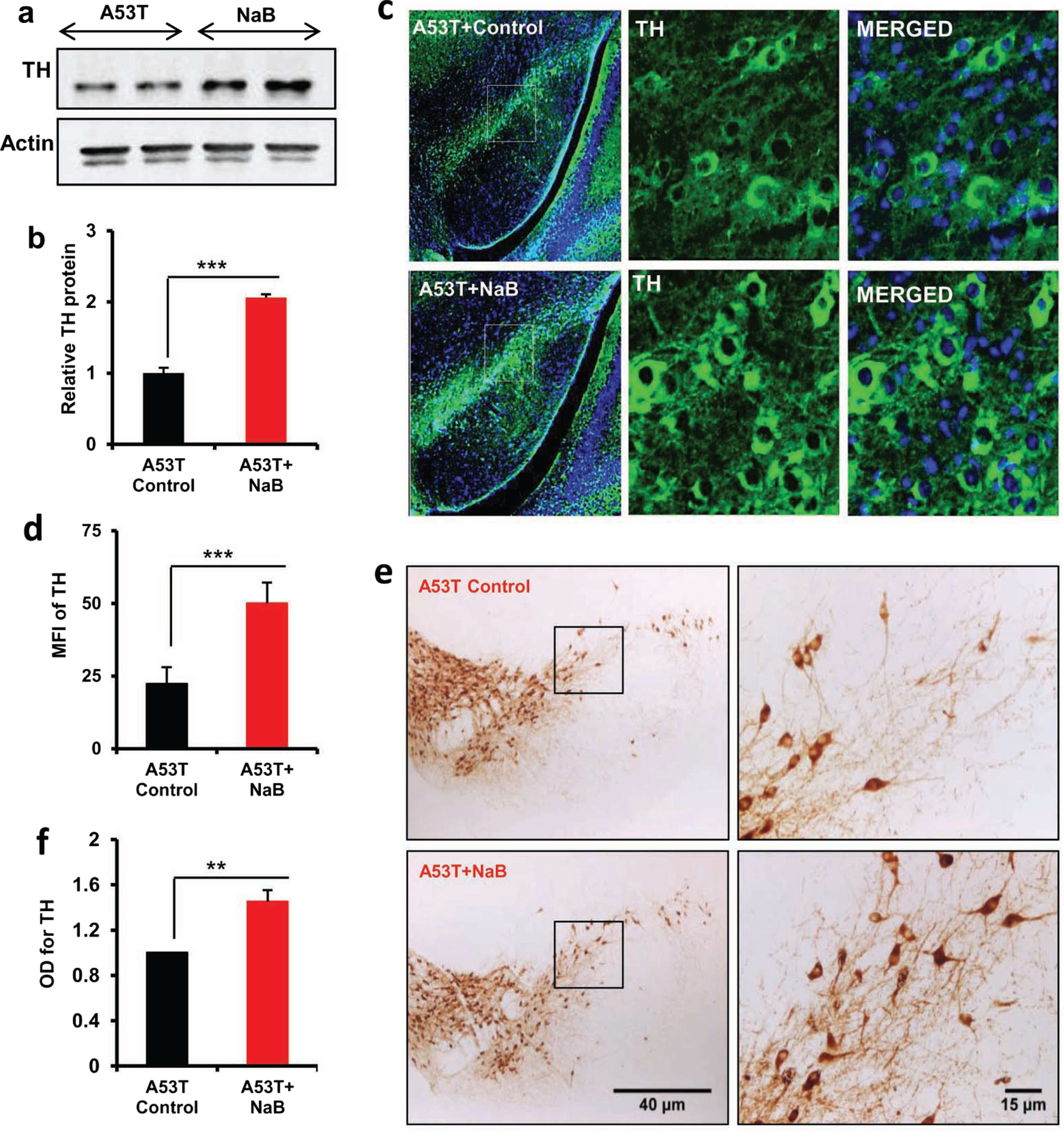
The presence of intracytoplasmic inclusions containing α-syn and the demise of dopaminergic neurons in the nigra are pathological hallmarks of PD [29]. Therefore, we also examined if NaB treatment could also increase the expression of TH in the nigra of A53T α-syn transgenic (A53T-Tg) mice. As observed in C57/BL6 mice (Fig. 5), western blot (Fig. 6A, B), immunofluorescence (Fig. 6C, D), and DAB immunostaining (Fig. 6E, F) results showed that oral NaB also upregulated the level of TH in the nigra of A53T-Tg mice.
Fig. 6
Oral NaB increases the level of TH in vivo in the nigra of A53T-Tg mice. Nine-month-old male A53T-Tg mice (n = 5 per group) were treated with NaB (50 mg/kg body wt/d) mixed in 0.5% methylcellulose orally via gavage. Control mice received 0.5% methylcellulose as vehicle. After 30 d of treatment, the level of TH was monitored in the SNpc by western blot (A). Actin was run as loading control. Bands were scanned and values (TH/actin) presented as relative to control (B). Results are mean ± SEM of five mice per group. ***p < 0.001 versus control by two-tailed paired t-tests. The level of TH was monitored in ventral midbrain sections by immunofluorescence (C). MFI of TH (D) was calculated in two nigral sections of each of five mice per group. ***p < 0.001 versus control by two-tailed paired t-tests. The level of TH was monitored in ventral midbrain sections by DAB immunostaining (E). Optical density of TH (F) was calculated in two nigral sections of each of five A53T-Tg mice per group. Results are mean±SEM of five mice per group. **p < 0.01 versus control by two-tailed paired t-tests.
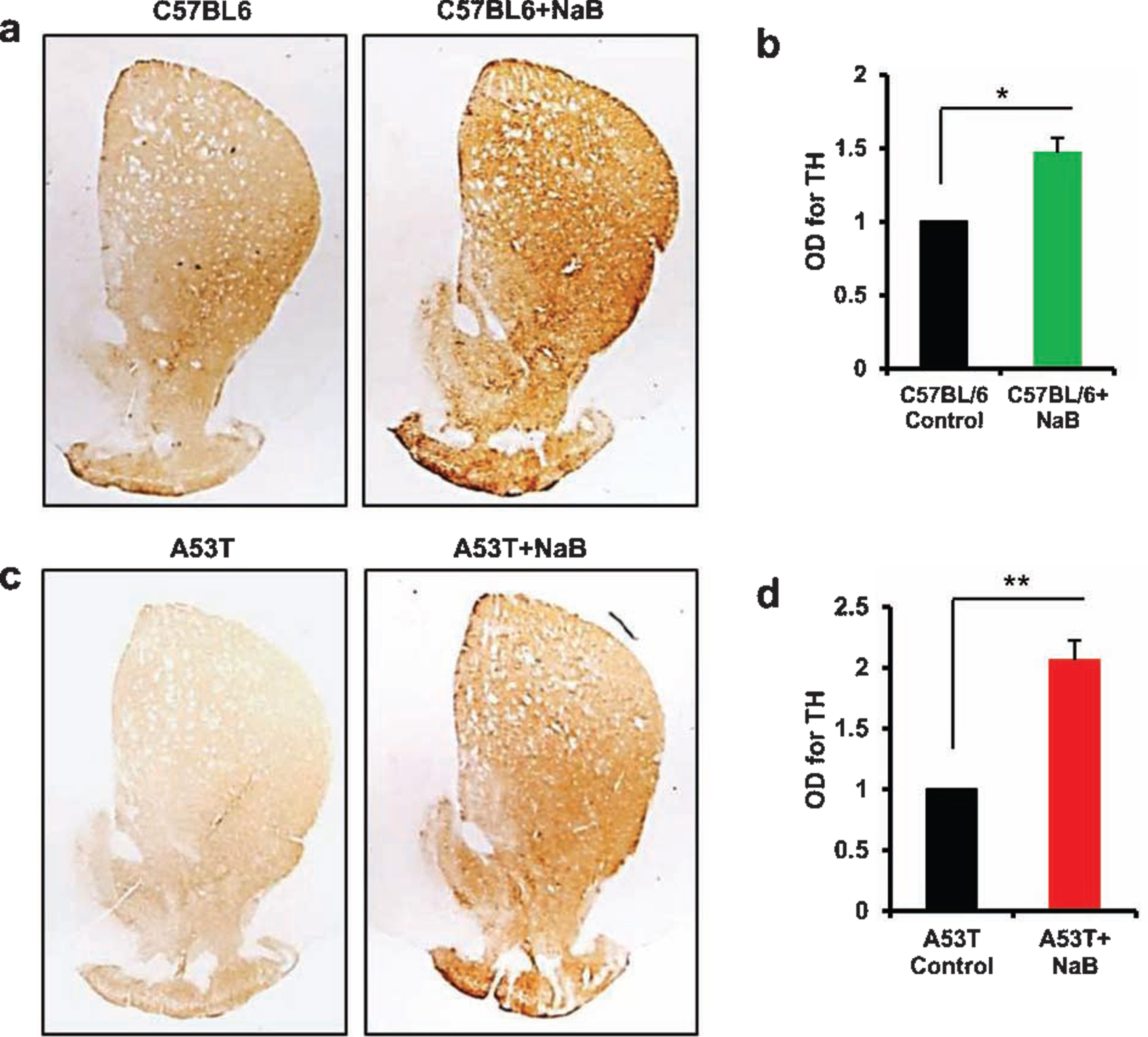
Oral NaB increases the density of striatal TH fibers in C57/BL6 and A53T α-syn transgenic mice
Since striatum is the main recipient of nigral fibers and loss of TH neurons in the SNpc leads to denervation of dopaminergic terminals in the striatum, we examined if oral NaB was capable of upregulating striatal TH fibers. Consistent to NaB-mediated increase in TH in cell bodies in the nigra (Figs. 5 and 6), we observed higher level of TH fibers in the striatum of NaB-treated C57/BL6 mice as compared to untreated mice (Fig. 7A, B). Similarly, oral NaB treatment also increased the density of TH fibers in the striatum of A53T-Tg mice (Fig. 7C, D).
Fig. 7
Oral NaB upregulates the density of TH fibers in vivo in the striatum of C57/BL6 and A53T-Tg mice. Male C57/BL6 (A, B) and A53T-Tg (C, D) mice (n = 5 per group) were treated with NaB (50 mg/kg body wt/d) mixed in 0.5% methylcellulose orally via gavage. Control mice received 0.5% methylcellulose as vehicle. After 30 d of treatment, striatal sections were immunostained for TH (A, C57/BL6; C, A53T-Tg). Optical density of TH fibers was quantified in two striatal sections of each of five mice per group (B, C57/BL6; D, A53T-Tg). Results are mean±SEM of five mice per group. *p < 0.05 versus control (B); **p < 0.01 versus control (D) by two-tailed paired t-tests.
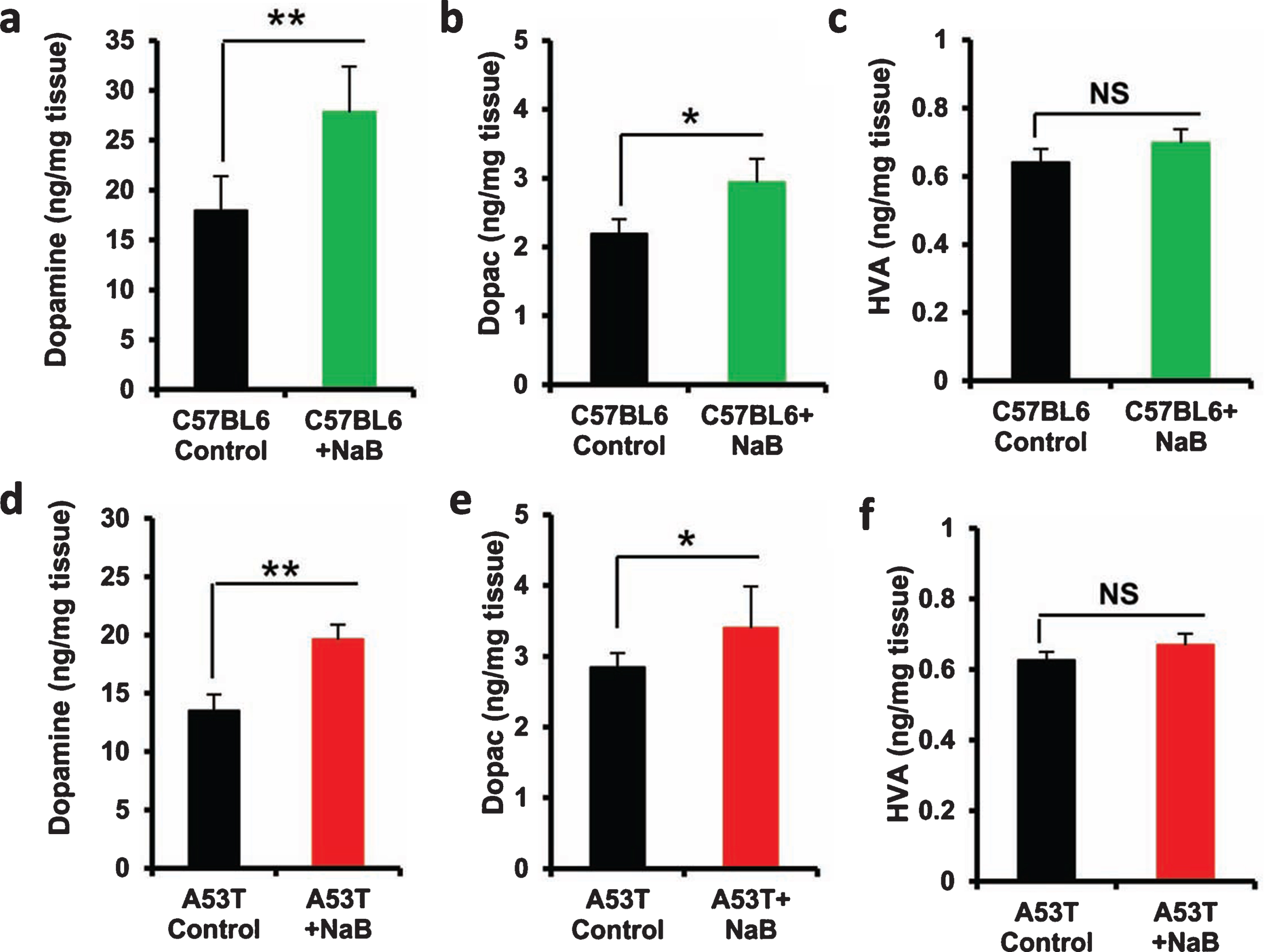
Oral treatment of NaB upregulates the level of DA in the striatum of C57/BL6 and A53T α-syn transgenic mice
Next, to examine whether oral NaB is also capable of increasing neurotransmitters in the striatum, we quantified the levels of DA, DOPAC, and HVA in striata after 30 d of NaB treatment. Oral NaB significantly increased the level of DA (Fig. 8A) and DOPAC (Fig. 8B), but not HVA (Fig. 8C), in striatum of C57/BL6 mice as compared to vehicle treatment. Similarly NaB treatment also augmented the level of DA (Fig. 8D) and DOPAC (Fig. 8E), but not and HVA (Fig. 8F), in the striatum of A53T-Tg mice.
Fig. 8
Oral NaB increases the level of DA in vivo in the striatum of C57/BL6 and A53T-Tg mice. Male C57/BL6 (A-C) and A53T-Tg (D-F) mice (n = 5 per group) were treated with NaB (50 mg/kg body wt/d) mixed in 0.5% methylcellulose orally via gavage. Control mice received 0.5% methylcellulose as vehicle. After 30 d of treatment, levels of DA (A, D), DOPAC (B, E), and HVA (C, F) were measured in striatum by HPLC. Results are mean±SEM of five mice per group. **p < 0.01 and *p < 0.05 versus control by two-tailed paired t-tests. NS, not significant.
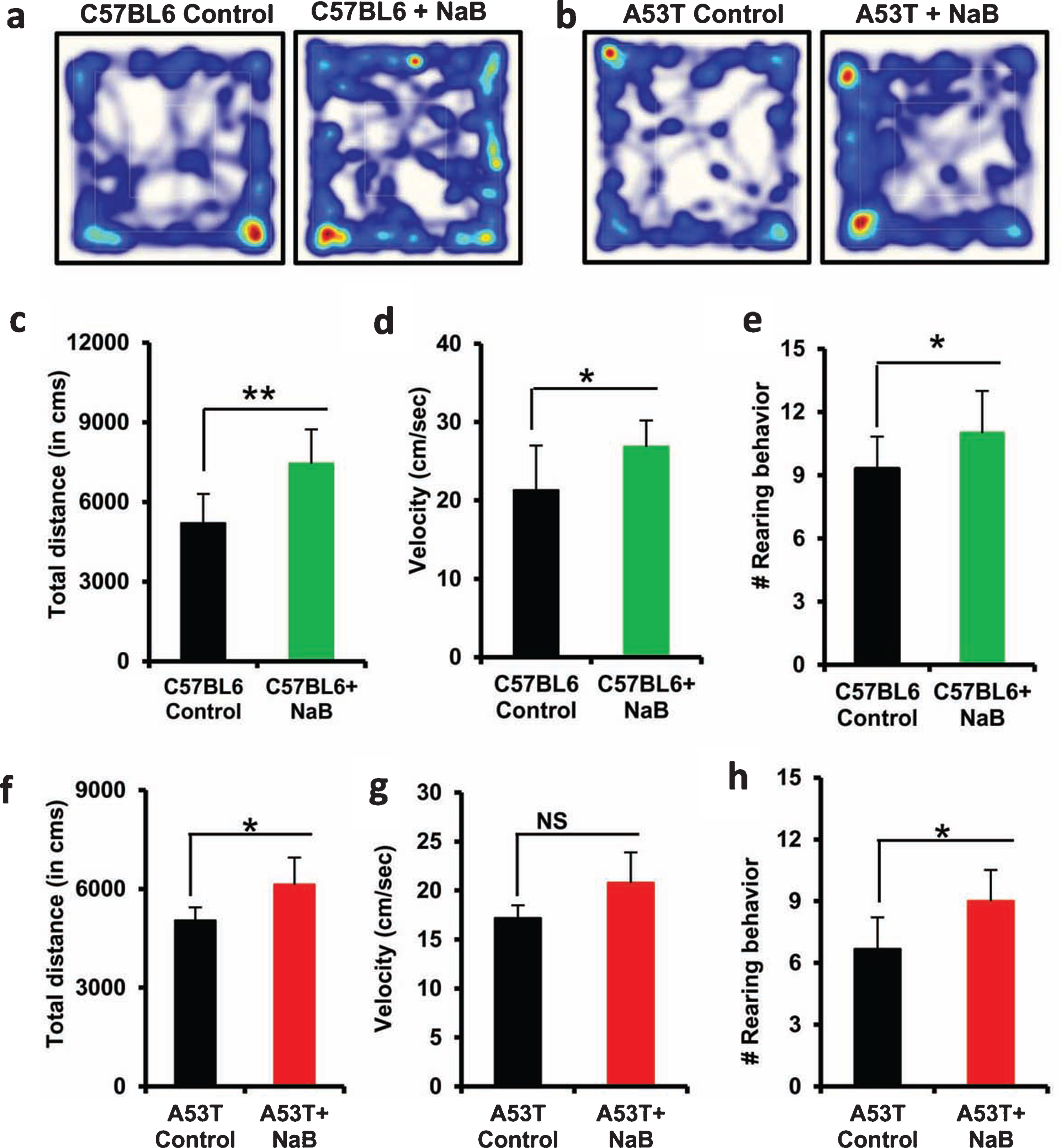
Oral NaB improves locomotor activities of C57/BL6 and A53T α-syn transgenic mice
Locomotor dysfunction is an early sign of PD and therefore, to examine whether oral NaB leads to only structural and neurotransmitter enhancement but also functional improvements, we monitored locomotor activities. Consistent to the upregulation of nigral neurons, striatal fibers, and striatal neurotransmitters, an increase in overall movement (Fig. 9A), distance traveled (Fig. 9C), velocity (Fig. 9D), and rearing (Fig. 9E) was seen in C57/BL6 mice after NaB treatment. Similarly NaB treatment also improved locomotor activities in A53T-Tg mice as evidenced by track plot (Fig. 9B), distance traveled (Fig. 9F), velocity (Fig. 9G), and rearing (Fig. 9H).
Fig. 9
Improvement of locomotor activities in C57/BL6 and A53T-Tg mice by oral NaB. Male C57/BL6 (A, C-E) and A53T-Tg (B, F-H) mice (n = 5 per group) were treated with NaB (50 mg/kg body wt/d) mixed in 0.5% methylcellulose orally via gavage. Control mice received 0.5% methylcellulose as vehicle. After 30 d of treatment, locomotor activities were monitored (A and B, track plot; C and F, distance traveled; D and G, velocity; E and H, rearing). Results are mean±SEM of five mice per group. **p < 0.01; *p < 0.05 versus control by two-tailed paired t-tests. NS, not significant.
DISCUSSION
Upregulation of TH in the nigra to increase the level of DA in the striatum is beneficial for PD. However, until now, there is no FDA-approved drug to increase the level of TH in residual dopaminergic neurons for upregulating striatal DA in PD patients. Therefore, describing new drugs or molecules to stimulate DA production in the brain of PD patients is an important area of research. Cinnamon, a natural product, is a commonly used spice and flavoring material for deserts, candies, chocolate, etc. After intake, cinnamaldehyde, the major component of cinnamon, is converted into cinnamic acid by oxidation. Then cinnamic acid undergoes β-oxidation in the liver to be converted into benzoate that exists as sodium salt (NaB) or benzoyl-CoA. Interestingly, NaB is a widely used food preservative due to its anti-microbial properties. Our laboratory has demonstrated that NaB inhibits the expression of proinflammatory molecules in activated astrocytes and microglia [9], upregulates regulatory T cells via increasing TGFβ [11], and protects mice from EAE via modification of T cells at various stages [10]. In addition, oral NaB is capable of improving memory and learning in an animal model of Alzheimer’s disease [14] and protecting dopaminergic neurons in MPTP mouse model of PD [16]. Several lines of evidence presented in this study clearly support the conclusion that NaB is capable of upregulating TH in mouse MN9D dopaminergic neuronal cells. Furthermore, oral treatment of NaB augmented the expression of TH in the nigra and upregulated the level of DA in striatum of normal mice as well as A53T α-syn transgenic mice. Accordingly, NaB treatment also improved locomotor activities in normal mice as well as in A53T α-syn transgenic mice. Here, we used 9-month-old A53T Tg mice because although these mice exhibit microglial activation and α-syn deposition at earlier age, no DA loss is seen even at the age of 6 months [30]. However, we have seen some loss of DA in A53T mice at the age of 9 months, which could be upregulated by oral NaB treatment. Therefore, NaB may have therapeutic importance for early and moderate stages PD when substantial number of dopaminergic neurons still remain in the nigra. Moreover, these results also suggest that NaB may find prophylactic use for delaying and/or preventing nigrostriatal degeneration.
The signaling mechanisms by which the TH gene is upregulated are poorly understood. Upon analysis of TH gene promotor using Genomatix Software Suite, we have found one cAMP response element (CRE) between 67 and 88 base pairs upstream of the TH open reading frame, indicating the possible involvement of CREB in the upregulation of TH gene. Accordingly, suppression of NaB-mediated expression of TH by siRNA knockdown of CREB shows that NaB upregulates TH in neurons via CREB. Earlier investigations from our laboratory have delineated that NaB upregulates the expression of CNTF via PKA-mediated activation of CREB in astrocytes and oligodendrocytes [13]. The cAMP-protein kinase A (PKA), one of the most important protein kinases in eukaryotic cells, is involved in the regulation of multiple signal transduction pathways including cell growth, differentiation, survival, and synaptic plasticity [31, 32]. Earlier studies from our laboratory demonstrated that NaB alone was capable of inducing the activation of CREB via PKA. While both protein kinase C (PKC) and protein kinase A (PKA) are known to phosphorylate and activate CREB, NaB specifically induces the activation of PKA [12]. Suppression of PKA by H-89 inhibited NaB-induced activation of CREB and expression of neurotrophic factors [12]. Therefore, it is likely that NaB upregulates TH in neuronal cells via PKA-CREB pathway.
Despite intense investigation, until now, no effective interdictive therapy is available for stopping the progression of PD. Although Sinemet, a combination of levodopa and carbidopa, is widely used throughout the world for treating PD, it has long term side effects as the patients develop dyskinesia following a few years of treatment. DA receptor agonists are also used as either monotherapy or together with Sinemet to treat PD patients. However, these medications show limited symptomatic relief and a number of side effects including hallucinations, nausea and postural hypotension. Deep brain stimulation (DBS) is definitely becoming popular for PD; however, a majority of PD patients do not meet the strict DBS criteria. Moreover, many PD patients suffer from seizure, confusion, headache, and stroke following DBS surgery. On the other hand, there are several advantages of NaB over available PD therapies. First, NaB is fairly nontoxic. It is water soluble and if consumed in excess, it is released through the urine. Cinnamon, which has been widely used as flavoring material and spice throughout the world for centuries, is metabolized to NaB. NaB is an FDA-approved drug against urea cycle disorders and glycine encephalopathy in children. Second, the least painful route of drug treatment is oral administration. It is nice to mention that NaB can be taken as an oral solution. We have demonstrated that NaB treatment of mice with relapsing-remitting experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis, via drinking water suppresses the disease process of EAE [10]. Further, we have found that oral administration of cinnamon powder exhibits neuroprotective effect in a mouse model of Alzheimer’s disease [14]. Third, NaB is very economical compared to the other existing anti-PD therapies. Fourth, after oral treatment, NaB rapidly diffuses through the blood-brain barrier. It has been found that upon treatment of patients with urea cycle disorders, NaB is known to combine with glycine to produce hippurate, a compound that is readily excreted in the urine. Simultaneous investigations of serum and cerebrospinal fluid samples of these patients exhibited comparable levels of hippurate and NaB in cerebrospinal fluid [33, 34], suggesting that NaB is capable of crossing the blood-brain barrier.
In summary, we have demonstrated that NaB, a metabolite of cinnamon, a commonly used food additive and an FDA-approved drug for urea cycle disorders and glycine encephalopathy, upregulates the expression of TH to increase the production of DA in dopaminergic neurons. Since NaB enters into the central nervous system, these results suggest that oral NaB may have therapeutic importance in early stage to moderate stage PD patients in which NaB may stimulate the production of DA from existing neurons.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This study was supported by merit awards (BX003033 and BX005002) from US Department of Veterans Affairs and a grant (NS108025) from NIH to KP. Moreover, KP is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the Department of Veterans Affairs.
REFERENCES
[1] | Vila M , Przedborski S ((2004) ) Genetic clues to the pathogenesis of Parkinson’s disease. Nat Med 10: (Suppl), S58–62. |
[2] | Olanow CW , Tatton WG ((1999) ) Etiology and pathogenesis of Parkinson’s disease. Annu Rev Neurosci 22: , 123–144. |
[3] | Marsden CD ((1990) ) Parkinson’s disease. Lancet 335: , 948–952. |
[4] | Ross GW , Petrovitch H , Abbott RD , Nelson J , Markesbery W , Davis D , Hardman J , Launer L , Masaki K , Tanner CM , White LR ((2004) ) Parkinsonian signs and substantia nigra neuron density in decendents elders without PD. Ann Neurol 56: , 532–539. |
[5] | Dauer W , Przedborski S ((2003) ) Parkinson’s disease: Mechanisms and models. Neuron 39: , 889–909. |
[6] | Lang AE , Lozano AM ((1998) ) Parkinson’s disease. First of two parts. N Engl J Med 339: , 1044–1053. |
[7] | Gropman AL , Summar M , Leonard JV ((2007) ) Neurological implications of urea cycle disorders. J Inherit Metab Dis 30: , 865–879. |
[8] | Misel ML , Gish RG , Patton H , Mendler M ((2013) ) Sodium benzoate for treatment of hepatic encephalopathy. Gastroenterol Hepatol (N Y) 9: , 219–227. |
[9] | Brahmachari S , Jana A , Pahan K ((2009) ) Sodium benzoate, a metabolite of cinnamon and a food additive, reduces microglial and astroglial inflammatory responses. J Immunol 183: , 5917–5927. |
[10] | Brahmachari S , Pahan K ((2007) ) Sodium benzoate, a food additive and a metabolite of cinnamon, modifies T cells at multiple steps and inhibits adoptive transfer of experimental allergic encephalomyelitis. J Immunol 179: , 275–283. |
[11] | Kundu M , Mondal S , Roy A , Martinson JL , Pahan K ((2016) ) Sodium benzoate, a food additive and a metabolite of cinnamon, enriches regulatory T cells via STAT6-mediated upregulation of TGF-beta. J Immunol 197: , 3099–3110. |
[12] | Jana A , Modi KK , Roy A , Anderson JA , van Breemen RB , Pahan K ((2013) ) Up-regulation of neurotrophic factors by cinnamon and its metabolite sodium benzoate: Therapeutic implications for neurodegenerative disorders. J Neuroimmune Pharmacol 8: , 739–755. |
[13] | Modi KK , Jana M , Mondal S , Pahan K ((2015) ) Sodium benzoate, a metabolite of cinnamon and a food additive, upregulates ciliary neurotrophic factor in astrocytes and oligodendrocytes. Neurochem Res 40: , 2333–2347. |
[14] | Modi KK , Roy A , Brahmachari S , Rangasamy SB , Pahan K ((2015) ) Cinnamon and its metabolite sodium benzoate attenuate the activation of p21rac and protect memory and learning in an animal model of Alzheimer’s disease. PLoS One 10: , e0130398. |
[15] | Khasnavis S , Pahan K ((2012) ) Sodium benzoate, a metabolite of cinnamon and a food additive, upregulates neuroprotective Parkinson disease protein DJ-1 in astrocytes and neurons. J Neuroimmune Pharmacol 7: , 424–435. |
[16] | Khasnavis S , Pahan K ((2014) ) Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol 9: , 569–581. |
[17] | Modi KK , Rangasamy SB , Dasarathi S , Roy A , Pahan K ((2016) ) Cinnamon converts poor learning mice to good learners: Implications for memory improvement. J Neuroimmune Pharmacol 11: , 693–707. |
[18] | Roy A , Pahan K ((2013) ) Myelin basic protein-primed T helper 2 cells suppress microglial activation via AlphaVBeta3 integrin: Implications for multiple sclerosis, J Clin Cell Immunol 7: , 158. |
[19] | Mondal S , Roy A , Pahan K ((2009) ) Functional blocking monoclonal antibodies against IL-12p40 homodimer inhibit adoptive transfer of experimental allergic encephalomyelitis. J Immunol 182: , 5013–5023. |
[20] | Corbett GT , Roy A , Pahan K ((2012) ) Gemfibrozil, a lipid-lowering drug, upregulates IL-1 receptor antagonist in mouse cortical neurons: Implications for neuronal self-defense. J Immunol 189: , 1002–1013. |
[21] | Ghosh A , Pahan K ((2012) ) Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: Implications for neurodegenerative disorders. J Biol Chem 287: , 27189–27203. |
[22] | Khasnavis S , Jana A , Roy A , Mazumder M , Bhushan B , Wood T , Ghosh S , Watson R , Pahan K ((2012) ) Suppression of nuclear factor-kappaB activation and inflammation in microglia by physically modified saline. J Biol Chem 287: , 29529–29542. |
[23] | Ghosh A , Roy A , Liu X , Kordower JH , Mufson EJ , Hartley DM , Ghosh S , Mosley RL , Gendelman HE , Pahan K ((2007) ) Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 104: , 18754–18759. |
[24] | Benner EJ , Mosley RL , Destache CJ , Lewis TB , Jackson-Lewis V , Gorantla S , Nemachek C , Green SR , Przedborski S , Gendelman HE ((2004) ) Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 101: , 9435–9440. |
[25] | Dasgupta S , Jana M , Liu X , Pahan K ((2003) ) Role of very-late antigen-4 (VLA-4) in myelin basic protein-primed T cell contact-induced expression of proinflammatory cytokines in microglial cells. J Biol Chem 278: , 22424–22431. |
[26] | Khasnavis S , Roy A , Ghosh S , Watson R , Pahan K ((2014) ) Protection of dopaminergic neurons in a mouse model of Parkinson’s disease by a physically-modified saline containing charge-stabilized nanobubbles. J Neuroimmune Pharmacol 9: , 218–232. |
[27] | Roy A , Fung YK , Liu X , Pahan K ((2006) ) Up-regulation of microglial CD11b expression by nitric oxide. J Biol Chem 281: , 14971–14980. |
[28] | Patel D , Roy A , Raha S , Kundu M , Gonzalez FJ , Pahan K ((2020) ) Upregulation of BDNF and hippocampal functions by a hippocampal ligand of PPARalpha. JCI Insight 5: , e136654. |
[29] | Surmeier DJ , Obeso JA , Halliday GM ((2017) ) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18: , 101–113. |
[30] | Lee MK , Stirling W , Xu Y , Xu X , Qui D , Mandir AS , Dawson TM , Copeland NG , Jenkins NA , Price DL ((2002) ) Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 –>Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A 99: , 8968–8973. |
[31] | Kandel ER ((2012) ) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5: , 14. |
[32] | Taylor SS , Kim C , Cheng CY , Brown SH , Wu J , Kannan N ((2008) ) Signaling through cAMP and cAMP-dependent protein kinase: Diverse strategies for drug design. Biochim Biophys Acta 1784: , 16–26. |
[33] | Leonard JV , Morris AA ((2002) ) Urea cycle disorders. Semin Neonatol 7: , 27–35. |
[34] | Scaglia F , Carter S , O’Brien WE , Lee B ((2004) ) Effect of alternative pathway therapy on branched chain amino acid metabolism in urea cycle disorder patients. Mol Genet Metab 81: (Suppl 1), S79–85. |


