
Sorting Out the Role of the Sortilin-Related Receptor 1 in Alzheimer’s Disease
Abstract
Sortilin-related receptor 1 (SORL1) encodes a large, multi-domain containing, membrane-bound receptor involved in endosomal sorting of proteins between the trans-Golgi network, endosomes and the plasma membrane. It is genetically associated with Alzheimer’s disease (AD), the most common form of dementia. SORL1 is a unique gene in AD, as it appears to show strong associations with the common, late-onset, sporadic form of AD and the rare, early-onset familial form of AD. Here, we review the genetics of SORL1 in AD and discuss potential roles it could play in AD pathogenesis.
INTRODUCTION
Sortilin-related receptor1 (SORL1, also known as LR11 or SORLA) encodes a hybrid receptor with multiple domains involved in intracellular sorting and trafficking of proteins into their respective subcellular compartments. The sorting of proteins is essential for normal cell function and defects in these pathways are thought to be important factors in the pathogenesis of Alzheimer’s disease (AD).
AD, a progressive neurodegenerative disorder, is the most common form of dementia. It is defined by progressive cognitive decline, widespread neurodegeneration and two proteinopathies: senile neuritic plaques [consisting of the product of the proteolytic processing of the amyloid-β protein precursor (AβPP), amyloid-β (Aβ)], and neurofibrillary tangles (consisting of hyperphosphorylated tau protein) (reviewed in [1]). AD can be classified into two broad subtypes defined by the age of disease onset: early-onset AD (<65 years of age, EOAD) and late onset AD (>65 years of age, LOAD). There can be both sporadic and familial varieties of both EOAD and LOAD. The majority of AD cases (~95%) belong to the LOAD subgroup, and these mainly arise sporadically. Conversely, ~5% of cases belong to the EOAD subgroup [2]. A portion of EOAD cases are caused by autosomal dominant variants in AβPP [3–7], presenilin 1 (PSEN1) [8–13], and presenilin 2 (PSEN2) [14–17] (EOfAD). Recently, variants in SORL1 have been recognized as likely causing EOfAD [18, 19]. Intriguingly, SORL1 is also recognized as a genetic risk factor for development of late-onset, sporadic AD (sAD) [20–22]. The exact role of SORL1 in the pathogenesis of AD is unclear since there are multiple pathways which link it to AD pathological mechanisms.
This review will discuss the normal expression of SORL1 and the genetic evidence which supports a role in LOAD and EOAD. We then discuss the possible roles which SORL1 may play in different pathologies observed in AD. SORL1 appears to be strongly associated with both EOAD and LOAD and provides a unique opportunity to understand the similarities and differences between these two subtypes of AD.
SORL1 HAS MULTIPLE TRANSCRIPT SPLICING PRODUCTS
SORL1 is located on chromosome 11q23.2-24.2 in humans and is widely expressed in the brain. It is particularly highly expressed in neurons of the hippocampus and some nuclei of the brainstem and Purkinje cells, and has slightly weaker expression in neurons of the thalamus and the hypothalamus [23]. It is also expressed in other tissue types such as the testes, ovaries, thyroid, and lymph nodes [24].
Alternative splicing occurs for SORL1, with at least 5 protein-coding transcripts arising from the gene in humans according to the ENSEMBL database [25] (Fig. 1). The shortest alternative splice product, SORL1-206, has a transcription initiation site in intron 30 and is expressed highly in the hippocampus and temporal lobe, moderately in the entorhinal cortex and frontal cortex and is undetectable in the testes or kidney [26]. Expression of this particular splice product is unchanged in the cerebellum of AD patients. However, total SORL1 mRNA levels are relatively constant in the cerebellum of AD brains [27], so this is not surprising. Grear et al. [28] reported two SORL1 mRNA isoforms expressed in the temporal lobe in both AD and non-AD samples: SORL1-Δ2 and SORL1-Δ9, which lack exon 2 and exon 19 coding sequences respectively. SORL1-Δ2 comprises of up to 5% of total SORL1 expression and SORL1-Δ19 comprises of less than 1% (and was not investigated further). The SORL1-Δ2 isoform lacks sequences coding for amino acid residues V96 to D134, the N-terminal proximal region of the VPS10 domain of SORL1 protein. It has increased abundance in white matter compared to grey matter, which suggests varying expression across cell types (as white and grey matter contain different ratios of cell types). However, expression levels of the SORL1-Δ2 isoform did not correlate with Braak staging of AD progression (while the expression of the longest SORL1 transcript splice form did show correlation). Finally, a non-coding RNA (ncRNA) termed 51A maps in anti-sense configuration to intron 1 of the SORL1 gene and drives the alternative splicing of SORL1 towards two splice products: 1) splice product B (SORL1-B), with a coding sequence beginning in exon 23 and ending with the stop codon used for translation of the full length protein and 2) splice product F (SORL1-F), with a coding sequence beginning with the start codon used in translation of the full length protein within exon 1 and ending in exon 14. Overexpression of the 51A ncRNA in a neuroblastoma cell line resulted in increased levels of Aβ peptides. Interestingly, 51A is upregulated in AD brains (although the authors observed substantial variation among individuals) [29].
Fig.1
SORL1 encodes a multi-domain containing protein and its transcripts are subject to alternative splicing. (A) depicts gene models for alternative SORL1 splice products from the ENSEMBL database (ENSG00000137642) and published literature. Exons are numbered and color-coded to indicate which protein domains they encode. Protein-coding domains are as given for the human SORL1 protein (Uniprot ID: Q92673) with SMART database annotations. VPS10, vacuolar protein sorting 10; LDLR, low density lipoprotein receptor; EGF, epidermal growth factor. (B) depicts a schematic of the full length SORL1 protein consisting of a pro-peptide sequence, a VPS10 domain, five LDLR class B repeats, an EGF-like domain, eleven LDLR class A repeats, six fibronectin-type (FN) repeats, a transmembrane domain (TMD) and a cytosolic intracellular domain (ICD) containing recognition motifs for cytosolic adaptors. Binding sites of amyloid-β protein precursor (AβPP), amyloid-β (Aβ), sorting nexin 27 (SNX27), Golgi-localizing, γ-adaptin ear homology domain ARF-interaction (GGA), clathrin adaptor protein 1/2 (AP1/2), and phosphofurin acidic cluster sorting protein 1 (PACS1) and the nuclear localization signal (NLS) are indicated.
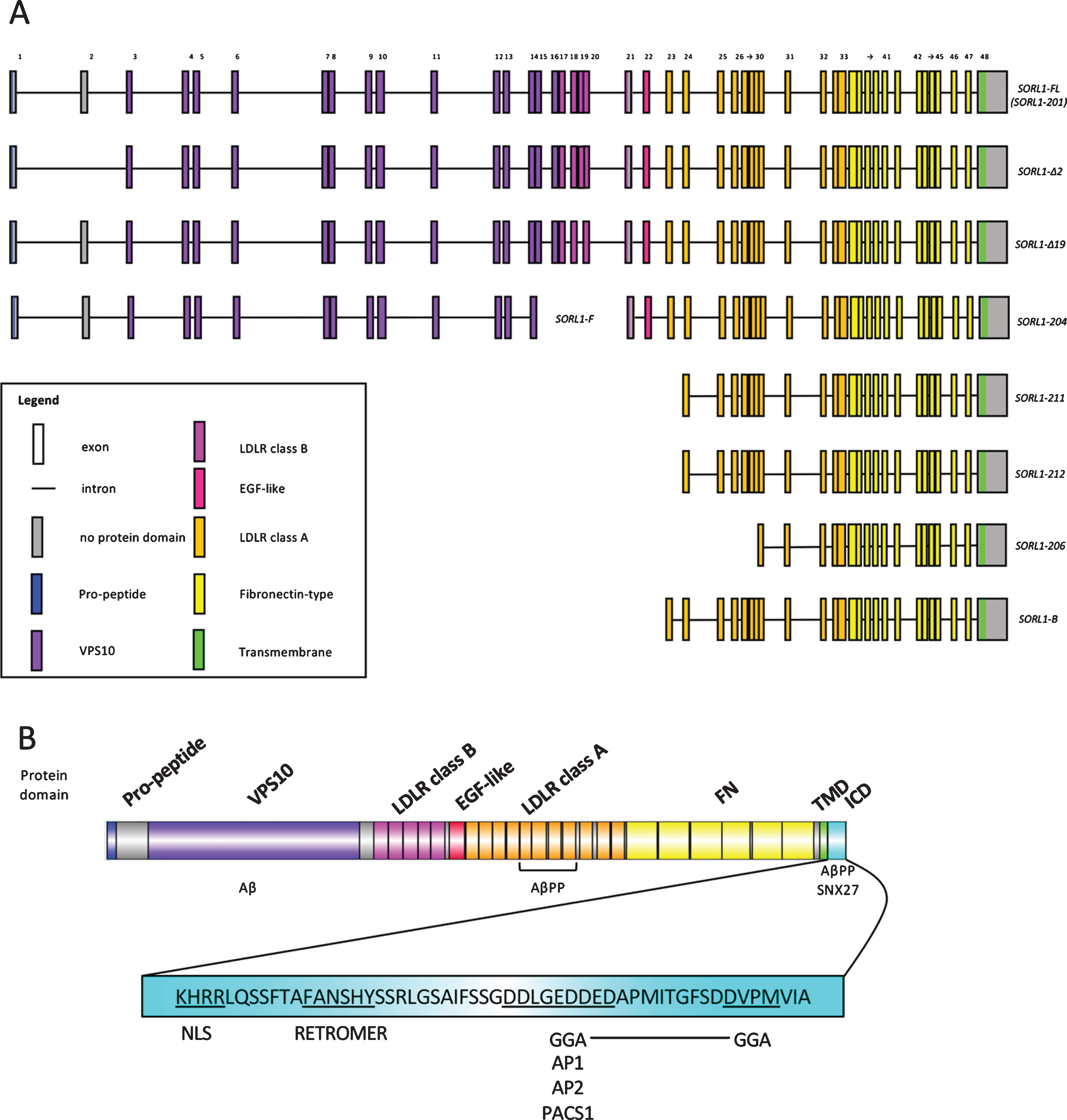
Together, these studies show the complexity of alternative splicing of SORL1 transcripts. However, the scientific literature lacks functional studies investigating the roles of these alternative splice products.
SORL1 ENCODES A MEMBRANE-BOUND RECEPTOR WITH MULTIPLE FUNCTIONAL DOMAINS
The full-length transcript of SORL1 encodes a 250 kDa, membrane-bound protein which primarily localizes in the endosomal and Golgi compartments [30]. The protein is comprised of multiple functional domains. These include a vacuolar protein sorting 10 (VPS10) domain, five low-density lipoprotein receptor (LDLR) class B repeats, an epidermal growth factor-like (EGF-like) domain, eleven LDLR class A repeats, six fibronectin-type (FN-type) repeats, a transmembrane domain and a cytosolic domain containing recognition motifs for cytosolic adaptors [31–34] (Fig. 1B).
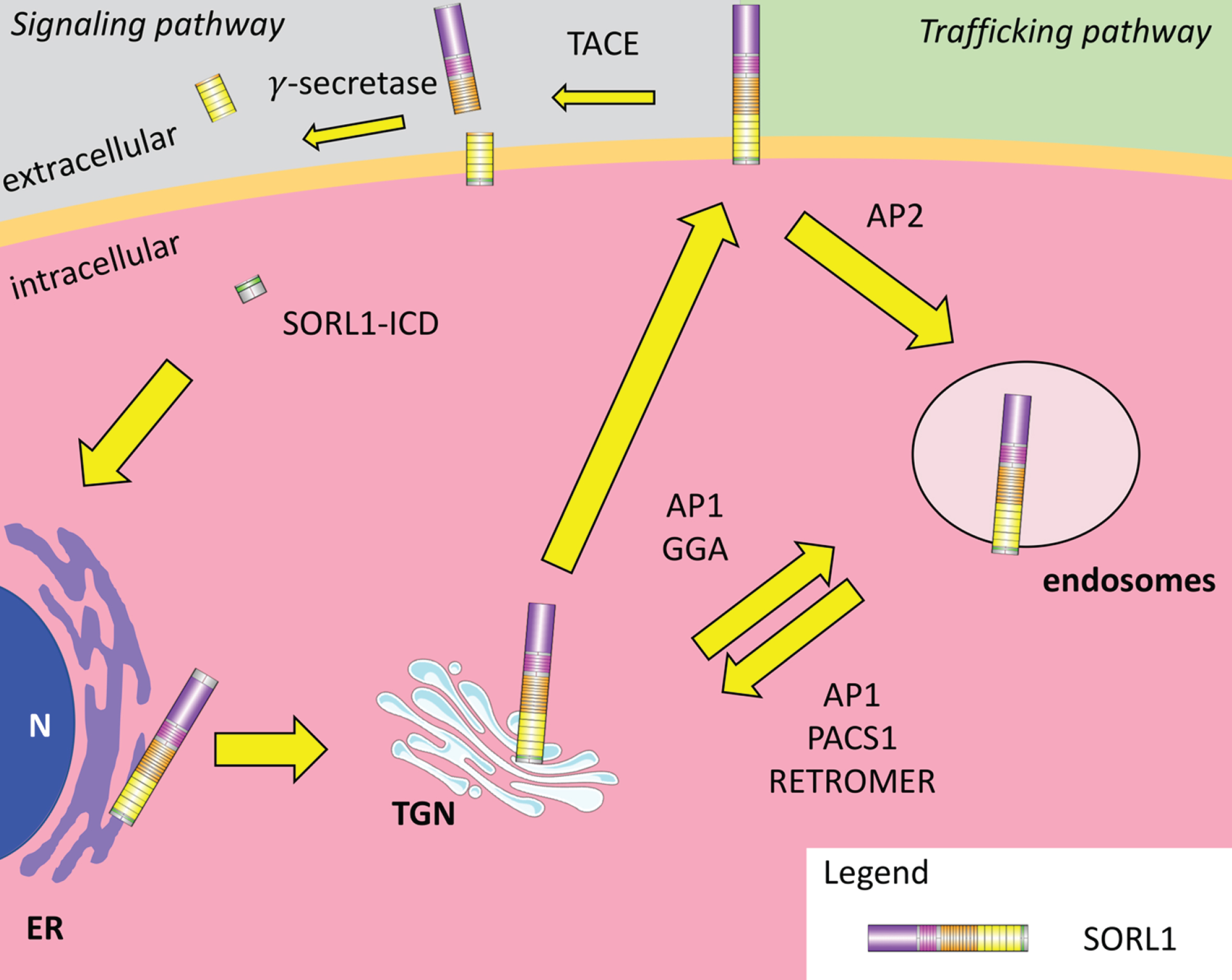
Nascent SORL1 proteins are generated in endoplasmic reticulum (ER) and are then transported through the trans-Golgi network (TGN) and eventually to the cell surface. They are initially inactive due to the pro-peptide at the NH2-terminal which is removed by furin-mediated cleavage in the TGN due to the RRKR furin recognition sequence [35]. This allows the receptor to be directed to the cell surface. Once at the cell surface, it appears that SORL1 can be utilized either in a signaling pathway or a trafficking pathway. In the signaling pathway, SORL1 is subject to ectodomain shedding by the α-secretase tumor necrosis factor-A containing enzyme (TACE/ADAM-17) [36–38]. The exact site at which TACE cleaves SORL1 is unclear. SORL1 is then processed further by γ-secretase at the plasma membrane, releasing a fragment of SORL1 into the extracellular space and a SORL1 intracellular domain (SORL1-ICD) into the cytosol [39, 40]. The SORL1-ICD contains a nuclear localization motif KHRR. It was demonstrated in a reporter assay that the SORL1-ICD can enter the nucleus to regulate transcription [39]. Whether the ICD has this function in reality and what genes it might regulate are yet to be determined.
As an alternative to processing at the cell surface for signaling, SORL1 can also enter a trafficking pathway if it is internalized by clathrin-mediated endocytosis employing the chaperone clathrin adaptor protein 2 (AP2) [34]. Subsequently, internalized SORL1 receptors generally shuttle between the TGN and endosomes, guided by cytosolic adaptor proteins which will be discussed later in this review in the context of AβPP trafficking (Fig. 2) (reviewed in [41, 42]).
Fig.2
SORL1 trafficking pathways. Nascent SORL1 peptides are generated in the endoplasmic reticulum (ER) and follow the constituent secretory pathway to the trans-Golgi network (TGN) where the pro-peptide is removed by furin-mediated cleavage. This allows the receptor to move to the plasma membrane where it can follow a signaling pathway (left) or a trafficking pathway (right). In the signaling pathway, SORL1 is cleaved by tumor necrosis factor-A converting enzyme (TACE) and then by γ-secretase, releasing luminal fragments of SORL1 and a cytosolic SORL1 intracellular domain (SORL1-ICD). SORL1-ICD can move to the nucleus (N) and regulate transcription of as yet unknown genes. In the trafficking pathway, SORL1 can be internalized via clathrin-mediated endocytosis utilizing the chaperone clathrin adaptor protein 2 (AP2). Internalized SORL1 receptors then shuttle between the TGN and the endosomes, guided by cytosolic adaptor proteins such as adaptor protein 1 (AP1), phosphofurin acidic cluster sorting protein (PACS1), Golgi-localizing, γ-adaptin ear homology domain ARF-interaction (GGA) and the retromer complex.
GENETIC EVIDENCE FOR A ROLE OF SORL1 IN AD
The first characterization of genetic variation of SORL1 in AD came from a candidate gene approach in 2007. Rogaeva et al. [21] investigated 29 single nucleotide polymorphisms (SNPs) associated with AD throughout the SORL1 locus in 6 cohorts of familial and sporadic forms of LOAD from different ethnicities. Associations of these individual SNPs with AD were modest, with odds ratios (ORs) ranging from 1.4–2.6 (compared to 14.9 for homozygosity for the ɛ4 allele of APOE [43]). Haplotype analysis using a sliding window covering 3 SNPs confirmed these associations by demonstrating that two clusters of SNPs in SORL1 were independently associated with AD: the 5’ cluster and the 3’ cluster. The 5’ cluster consists of SNPs 8, 9 and 10 which are located within intron 6 of SORL1. Possession of the T – A – T haplotype was associated with decreased risk of AD in Caribbean-Hispanic families (p = 0.0086) and in Israeli-Arab (p = 0.0037) and North European (p = 0.068) case-control cohorts. Conversely, the C – G – C haplotype for these SNPs was associated with increased risk for developing AD. The 3’ cluster consists of SNPs 22 – 25 and two overlapping haplotypes were associated with increased risk of AD: the C – T – T alleles at SNPs 22–24 and the T – T – C alleles at SNPs 23–25 in North European families and North European case-control cohorts. This region was also associated with AD in an African-American cohort. However, it was the A – C – T alleles for SNPs 23 – 24 (p = 0.0025) which showed the latter association.
Further evidence for the association of the SORL1 locus with LOAD comes from a large-scale meta-analysis of four published genome-wide association studies (GWAS) with a combined sample size of 74,046 individuals (25,580 LOAD cases and 48,466 controls). The SORL1 locus was one of the 14 genomic regions to be associated with LOAD (p < 5×10–8). The strongest signal in the SORL1 region was rs11218343, an intronic variant which was observed to be protective with an odds ratio of 0.77 (95% CI = 0.72 – 0.82) [44]. This study was extended to include 35,274 cases and 59,163 controls and SORL1 remained as one of the 25 genes reaching the threshold for genome-wide significance (p < 5×10–8) [45].
Due to the nature of GWAS, only common variants can be identified. With the advancement of next generation sequencing, whole exome sequencing (WES) experiments have identified rare variants in SORL1 associated with AD. Intriguingly, an early WES study performed on families showing autosomal dominant inheritance of EOAD but without pathological variants in AβPP, PSEN1, or PSEN2 found that 7/29 families carried putatively pathological variants in SORL1. This supported the idea that SORL1 could be a fourth autosomal dominant EOfAD gene alongside the presenilins and AβPP. These variants were found throughout the coding sequence of the gene and were either missense (Gly511Arg, Asn924Ser, Asn1358Ser, Gly1681Asp, and Tyr141Cys) or nonsense (Cys1478X and Trp1821X) [18] (Fig. 3). However, Campion et al. [46] noted that some of the affected individuals in these families possessed variants in SORL1 and other AD risk loci (e.g., APOE ɛ4, TREM2, and ABCA7). It is also worthy of note that the ages of disease onset of some of these reported patients were close to the conventional threshold of 65 years of age. Therefore, it is still somewhat uncertain whether SORL1 variants can cause EOfAD.
Fig.3
EOfAD variants in SORL1. Figure 3 depicts a schematic of the SORL1 full length protein, indicating the sites for early-onset, familial Alzheimer’s disease (EOfAD) variants for which pedigrees have been published to date. Red arrows indicate variants published in [18] and blue arrows indicate variants published from [19]. VPS10, vacuolar protein sorting 10; LDLR, low density lipoprotein receptor; EGF, epidermal growth factor; FN, fibronectin-type; TMD, transmembrane domain; ICD, intracellular domain.
![EOfAD variants in SORL1. Figure 3 depicts a schematic of the SORL1 full length protein, indicating the sites for early-onset, familial Alzheimer’s disease (EOfAD) variants for which pedigrees have been published to date. Red arrows indicate variants published in [18] and blue arrows indicate variants published from [19]. VPS10, vacuolar protein sorting 10; LDLR, low density lipoprotein receptor; EGF, epidermal growth factor; FN, fibronectin-type; TMD, transmembrane domain; ICD, intracellular domain.](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200177/adr-4-adr200177-g003.jpg)
Despite the observations of Campion et al. [46], numerous other studies have found that variants in SORL1 are associated with EOAD. An independent study found 3 additional variants in SORL1 segregating with EOAD in families (Arg416X, Gly1017-Glu1074del, and Arg1303Cys; Fig. 3), although some of the affected individuals had APOE genotype ɛ3/ɛ4 and had a later age of disease onset [19]. Nicolas et al. [47] found an enrichment of rare variants of SORL1 in AD cases compared to controls in a WES study in a French cohort (OR = 5.03, 95% CI = 2.02–14.99, p = 7.49 x 10–5). When the authors restricted the analysis to cases showing a family history of AD, the OR increased to 8.86 (95% CI = 3.35–27.31, p = 3.82 x 10–7). Other studies have shown variants in SORL1 to be associated with EOAD in different cohorts [48, 49]. However, these studies do not describe family histories of the EOAD variants, so no conclusions can be made about Mendelian inheritance.
SORL1 expression levels are also reduced in the sporadic form of AD. Gene expression profiling of lymphoblasts from LOAD patients and age-matched controls found that SORL1 was downregulated approximately 2-fold at the mRNA level, and 2.5-fold at the protein level [50]. Sager et al. [51] quantified SORL1 protein levels in frontal cortex of AD patients and found that approximately 30% of AD cases had reduced SORL1 protein levels. However, they did not find any significant differences between AD and control brains overall. Another independent study found that SORL1 protein levels were decreased in post mortem, sporadic AD brains but not in EOfAD brains [52]. These results suggest that SORL1 plays different roles in EOfAD and the sporadic forms of AD. Thus, the relationship between SORL1 and the different subtypes of AD is still unclear.
Taken together, the above genetic studies support that SORL1 is a unique gene in AD as, currently, no other genes are strongly implicated in both the early and late onset forms of the disease. Understanding the molecular and cellular changes occurring due to variation in SORL1 could help us illuminate the differences between EOfAD and LOAD.
FUNCTIONAL STUDIES OF SORL1 VARIANTS
Functional studies characterizing the effects of genetic variation in SORL1 have largely been based on variants’ effects on AβPP processing (AβPP processing and trafficking are discussed in detail later in this review). Cuccaro et al. [53] showed that the EOfAD variants T588I and T2134M of SORL1 reduce the binding affinity of SORL1 protein for AβPP, resulting in altered AβPP trafficking in HEK293 cell lines. Similarly, Vardarajan et al. [54] showed that the E270K, A528K, and T947M variants found in LOAD patients also have reduced binding affinities for AβPP and alter AβPP trafficking in HEK293 cell lines.
Young et al. [55] used human induced pluripotent stem cell (hIPSC)-derived neurons from LOAD patients to investigate the cellular effects of the risk (R) or protective (P) haplotype at the 5’ SNP cluster in SORL1. They found expression of SORL1 was variable among their hIPSC-derived neurons, and there were no significant differences between levels of SORL1 mRNAs due to SORL1 haplotype or disease state. This supported that the effect of this haplotype was likely not due to altered basal SORL1 expression levels. However, they did find that patients carrying the R/R haplotype generally do not increase SORL1 expression levels in response to brain-derived neurotrophic factor (BDNF, a known inducer of SORL1 expression [56, 57] that is also implicated in AD [58, 59]). This led to increased Aβ levels in R/R haplotype-containing hIPSC-derived neurons relative to hIPSC-derived neurons carrying the R/P or P/P haplotypes and, therefore, can partially explain the increased risk associated with the R/R haplotype. Having one copy of the protective haplotype appears to be sufficient to increase SORL1 expression in response to BDNF. Therefore, increasing SORL1 expression in response to BDNF is likely a protective mechanism.
SORL1 AND AβPP
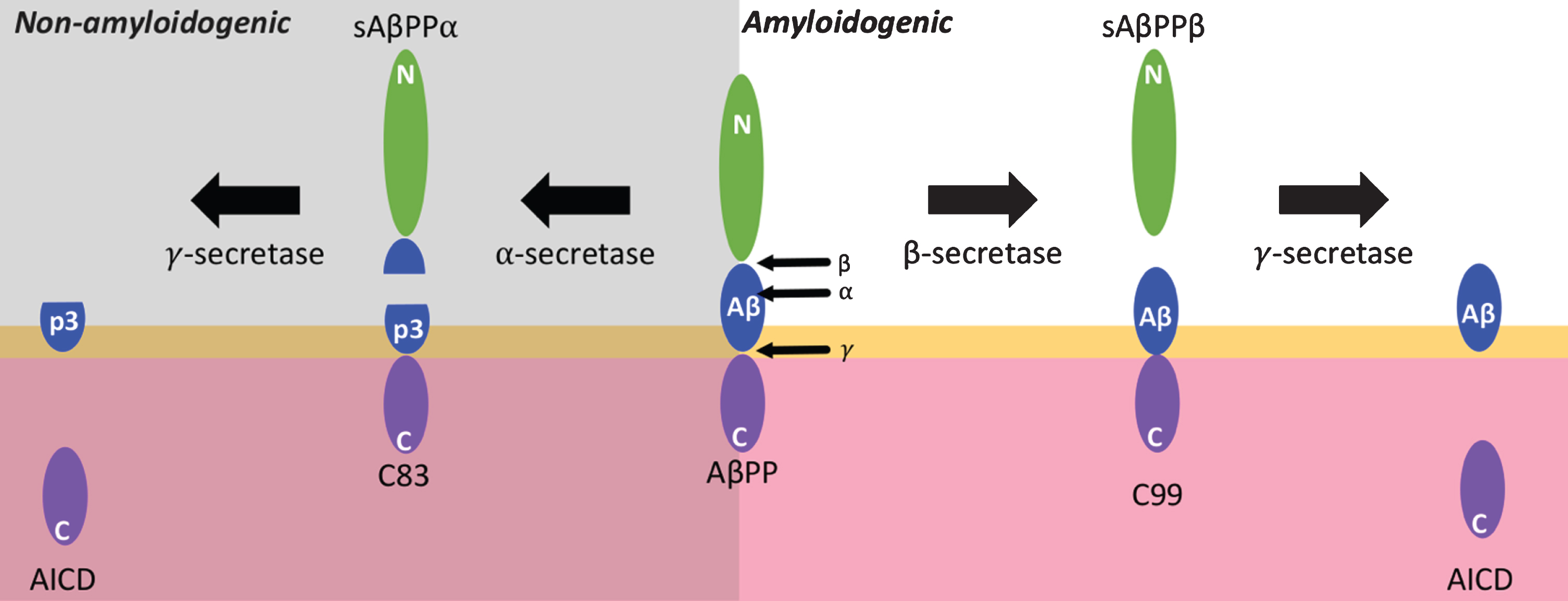
The most studied role of SORL1 in AD is its role in AβPP trafficking. AβPP is a single transmembrane-spanning protein which can be subjected to an amyloidogenic, or non-amyloidogenic proteolytic processing pathway. In the non-amyloidogenic pathway, AβPP is cleaved by a-secretase, giving two fragments: a secreted form of AβPP, sAβPPα, and C-terminal fragment C83. The ADAM (A disintegrin and metalloprotease domain) family of proteolytic enzymes are responsible for α-secretase cleavage of AβPP. ADAM-9 [60], ADAM-10 [60, 61], ADAM-17 [60, 62], and ADAM-19 [63] have been shown to contain α-secretase activity. However, ADAM-10 appears to be the main α-secretase [64]. Further processing of sAβPPα by γ-secretase (of which the PSENs are catalytic subunits (reviewed in [65])) gives a p3 fragment and an AβPP intracellular domain (AICD). This pathway is known as non-amyloidogenic as α-secretase cleaves within, and prevents the formation of, the Aβ peptide. In the amyloidogenic pathway, AβPP is cleaved by β-secretase (β-site AβPP cleaving enzyme 1, BACE1), giving sAβPPβ and C99 fragments. Then C99 is further processed by γ-secretase to give Aβ peptides of mainly 40-42 amino acids in length (Fig. 4) (reviewed in [66]).
Fig.4
Proteolytic processing of AβPP. Membrane bound amyloid-β protein precursor (AβPP) can be subjected to non-amyloidogenic or amyloidogenic processing. In the non-amyloidogenic pathway, AβPP is first cleaved by α-secretase within the amyloid-β (Aβ) sequence, producing a soluble sAβPPα fragment and a membrane-bound C83 fragment. C83 can be processed further by the γ-secretase complex to give a p3 fragment and an AβPP intracellular domain (AICD). In the amyloidogenic pathway, AβPP can be cleaved by β-secretase, giving the C99 and sAβPPβ fragments. Then C99 is cleaved by γ-secretase to give Aβ peptides and an AICD. β-secretase and γ-secretase may function together as a supramolecular complex. Yellow depicts a lipid bi-layer and pink depicts a cytosolic region.
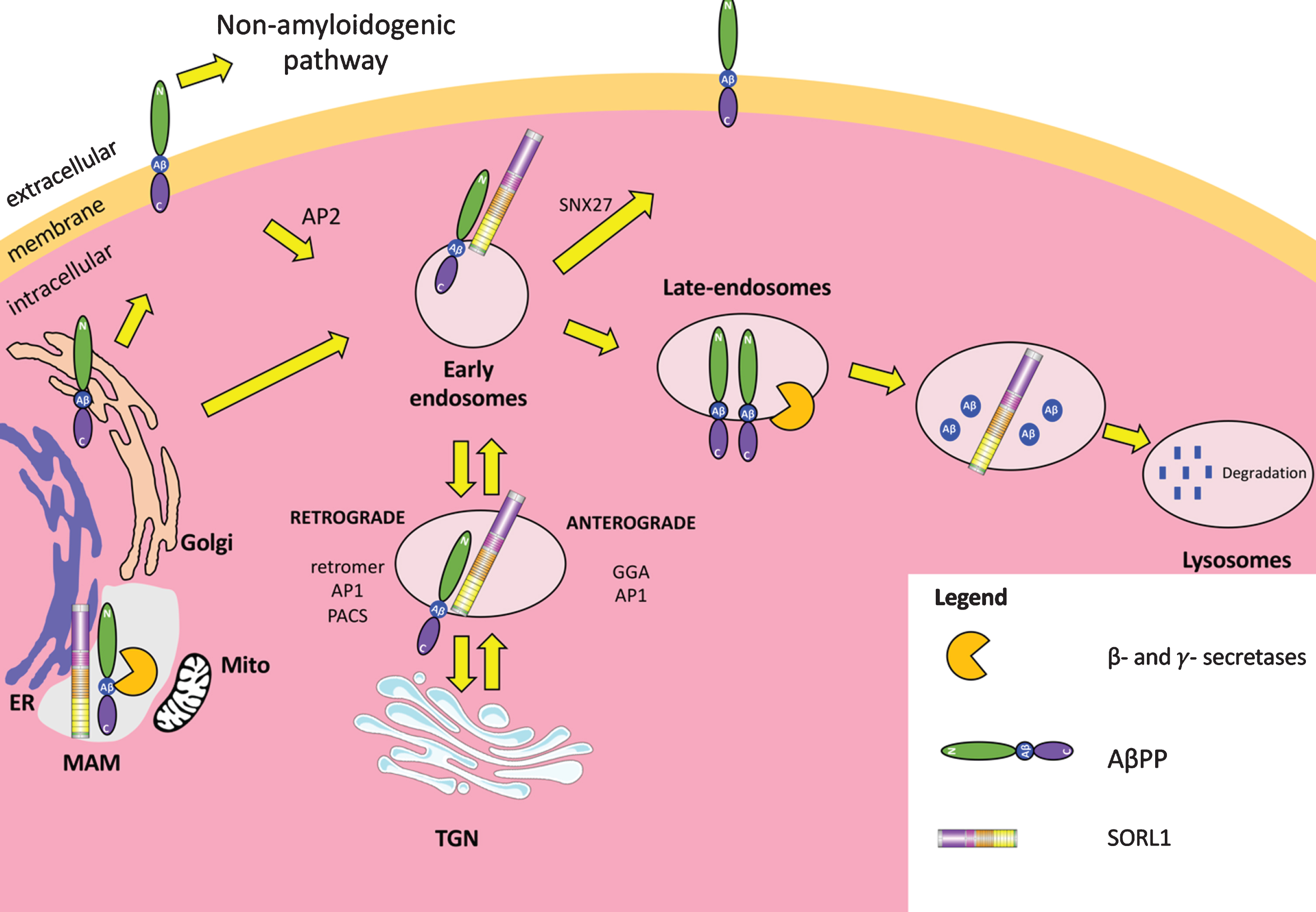
AβPP undergoes a series of trafficking steps within cells (Fig. 5). Nascent AβPP holoprotein is generated in the ER, before further processing in the Golgi. Some AβPP molecules can be transported through secretory vesicles to the plasma membrane, where the majority are cleaved by α-secretase and so are non-amyloidogenically processed [67]. Some surface AβPP may be internalized within endosomes by clathrin-mediated endocytosis. Internalization occurs due to interaction of the NPxY motif in the cytoplasmic tail of AβPP and the clathrin adaptor AP2 [68, 69]. Amyloidogenic processing of AβPP has been shown to proceed in many membranous organelles in the cell including the early and late-endosomal compartments [70, 71], the plasma membrane [71, 72], the TGN [73] (non-amyloidogenic processing is also thought to occur in the TGN [74]), the lysosomes [75, 76], and the ER [77, 78]. In regards to amyloidogenic processing at the ER, Area-Gomez et al. [79] showed that PSENs (i.e., γ-secretase) are particularly enriched in the mitochondrial associated membranes (MAMs) of the ER, which are lipid raft-like regions of the ER that form close associations with mitochondria and regulate their activity (reviewed in [80, 81]). BACE1 also is enriched in lipid rafts, and has been specifically detected in MAMs [82]. Therefore, the proteolytic cleavage of AβPP to form Aβ, which appears to occur in the ER, likely occurs in MAMs. Intriguingly, MAMs have been implicated in AD (see [83, 84] for excellent reviews on this topic).
Fig.5
SORL1 dependent trafficking of AβPP. AβPP is translated at the endoplasmic reticulum (ER) and is processed in the Golgi for direction to the cell surface. Some AβPP is cleaved in the non-amyloidogenic pathway by α-secretase and some AβPP is internalized by clathrin-mediated endocytosis via the chaperone clathrin adaptor protein 2 (AP2). SORL1 is present in early endosomes and can guide AβPP throughout different pathways in the cell by interacting with different adaptor proteins. SORL1 and AβPP can move directly back to the plasma membrane (orange) mediated by sorting nexin 27 (SNX27). They can also move retrogradely to the trans-Golgi network (TGN) mediated by the retromer complex, clathrin adaptor protein 1 (AP1), and/or phosphofurin acidic cluster sorting protein (PACS1). They can also move anterogradely from the TGN to the early endosomes mediated by Golgi-localizing, γ-adaptin ear homology domain ARF-interaction (GGA) proteins. Without SORL1, AβPP can move to late endosomal compartments where some β- and γ-secretase activities are thought to be located and can be proteolytically cleaved to form Aβ. SORL1 can also bind newly-formed Aβ and direct it to the lysosome for degradation. SORL1, AβPP, β- and γ-secretases are also present in the mitochondrial associated membranes (MAMs) of the ER.
An interesting observation is that SORL1 forms a complex with BACE1 in perinuclear regions in neurons [85]. BACE1 and γ-secretase have also been shown to form a complex, which sequentially cleaves AβPP holoprotein to form Aβ and this also occurs in perinuclear regions of the cell [86]. MAMs also have a perinuclear subcellular distribution [79, 87] and, as mentioned above, are enriched in PSENs and contain the other components of γ-secretase [79]. We have also detected SORL1 protein in the MAM [88]. Taken together, these results support the idea that SORL1, BACE1, and γ-secretase may form a complex in MAMs which proteolytically cleaves AβPP holoprotein to form Aβ. However, direct evidence for this SORL1/AβPP/BACE1/γ-secretase complex has not been observed. As mentioned previously, SORL1 is also cleaved by γ-secretase [39, 40] and this could also occur in this complex. This idea is intriguing, as it links, functionally, all four EOfAD loci (if SORL1 indeed is an EOfAD locus). Further characterization of the interactions between these AD-related proteins is required.
SORL1 can act to modulate the distribution of AβPP within cells. It has been shown that overexpression of SORL1 results in an accumulation of AβPP in the Golgi and prevents it from being sorted to the late endosomal membranes where some Aβ is produced [27, 30, 89]. Conversely, Sorl1 deficiency results in increased Aβ levels compared to controls [30, 90]. SORL1 interacts with the carbohydrate-linked domain of AβPP through its LDLR class A domain (repeats 5-8 appear to be crucial region for binding) [30, 91, 92], as well as with the cytoplasmic domain of AβPP through its intracellular domain [93] (Fig. 1B). There is evidence supporting that this interaction between SORL1 and AβPP prevents oligomerization of AβPP, which is required for the γ-secretase-cleavage of AβPP [94]. It has also been shown that SORL1 acts as an adaptor between AβPP and other intracellular sorting proteins (including the retromer complex, AP1, GGA, and PACS1) which guide AβPP through the endosomal/secretory pathways, keeping it away from subcellular compartments where Aβ is thought to form (such as in endosomes). These adaptors are discussed below and are summarized in Fig. 5. Binding sites of these adaptors in the SORL1 cytoplasmic tail are shown in Fig. 1B.
The retromer complex is part of the endocytic machinery in cells and is responsible for retrograde transport of cargo from endosomes to the TGN as well as recycling of cargo from endosomes to the plasma membrane. It is composed of two subcomplexes of proteins encoded by genes from the vacuolar protein sorting (VPS) family. The subcomplex responsible for cargo selection consists of the proteins VPS35, VPS29 and VPS26 while the subcomplex responsible for vesicle formation consists of VPS5 and VPS17 (reviewed in [95, 96]). SORL1 interacts with the VPS26 subunit of the retromer complex through a FANSHY motif (Fig. 1B) in SORL1’s cytoplasmic domain and loss of this binding motif in SORL1 resulted in aberrant subcellular localization of SORL1 and an accumulation of AβPP in late endosomes [31]. Loss of retrograde transport of SORL1 back to the TGN would result in retention of SORL1 in endosomes. This would, in turn, increase retention of AβPP in endosomes where some Aβ is formed. This is supported by results from a cell culture study, where siRNA knockdown of the retromer complex caused impaired AβPP trafficking to the TGN [97]. Interestingly, gene expression profiling of the entorhinal cortex and the dentate gyrus of AD brains found that expression levels of genes encoding retromer components VPS35 and VPS26 were decreased in AD patients (a mix of EOAD and LOAD) compared to age-matched controls [98]. Also a GWAS showed that SNPs in retromer-associated genes were associated with sporadic AD in a Caucasian cohort [99]. In animal models, retromer activity knockdown in the mouse brain resulted in memory defects, synaptic dysfunction and increased Aβ levels, which is reminiscent of AD pathology [100, 101]. Taken together, these results indicate the possibility that loss of retromer function could also play a role in sporadic forms of AD.
Golgi-localizing, γ-adaptin ear homology domain ARF-interaction (GGA) proteins are responsible for anterograde transport from the TGN to endosomes. GGA binds to SORL1 via recognition of a DVPM motif (Fig. 1B) and an acidic cluster of amino acid residues in the cytoplasmic domain of SORL1 [32, 33]. Loss of the recognition motif for GGA proteins results in an inability of SORL1 to return to the TGN so that it accumulates in early endosomes and at the plasma membrane. This also results in increased sAβPPα products as AβPP also accumulates in the organelles in which α-secretase resides [33, 102, 103].
Phosphofurin acidic cluster sorting protein 1 (PACS1) is another sorting protein which mediates both retrograde and anterograde transport between the Golgi and endosomes [104]. The furin binding region of PACS1 interacts with SORL1 by recognizing the acid cluster motif DDLGEDDED in the SORL1 cytoplasmic domain [33, 34] (Fig. 1B). Loss of PACS1 activity has a similar outcome to loss of GGA proteins, where AβPP accumulates in early endosomes, likely due to aberrant subcellular localization of SORL1 [105].
Clathrin adaptor protein 1 (AP1) binds to an overlapping region in the cytoplasmic domain of SORL1 (DDLGEDDE) (Fig. 1B) and knockdown of AP1 also results in aberrant subcellular localization of SORL1 [34].
Sorting nexin 27 (SNX27) is a sorting protein involved in retrograde transport from the endosomes to the plasma membrane. SNX27 mediates the transport of AβPP via binding to the membrane-proximal region of the SORL1 cytoplasmic tail. Knockdown of SNX27 resulted in a reduced amount of cell surface SORL1 and AβPP protein levels. Conversely, overexpression of SNX27 enhanced distribution of AβPP and SORL1 to the cell surface. An increase in AβPP protein levels at the cell surface also resulted in increased α-secretase cleavage of AβPP to give sAβPPα [106]. However, SNX27 has been shown to interact with PSEN1 protein and this appears to reduce γ-secretase activity (i.e., reduced cleavage of AβPP and Notch) [107] so this could also explain the increased levels of sAβPPα.
SORL1 also plays a role in trafficking of Aβ. It interacts with Aβ through its VPS10 domain as shown by fluorescence polarization assays. Overexpression of SORL1 in a neuronal cell line also expressing a human AβPP isoform resulted in faster turnover of intracellular Aβ and accumulation of Aβ in lysosomes. Furthermore, an EOfAD variant in the VPS10 domain of SORL1 disrupts the ability for SORL1 to bind Aβ [108, 109]. This process also appears to be mediated by GGA proteins [103]. These results indicate that SORL1 directs Aβ to lysosomes for degradation.
In summary, SORL1 plays an important and central role in the complex trafficking pathways of AβPP within cells. Therefore, it is no surprise that the loss of SORL1 activity in sporadic and some familial forms of AD leads to AβPP-related pathologies.
OTHER POTENTIAL ROLES OF SORL1 IN AD
SORL1 and lipoprotein metabolism
When AD was first described by Alois Alzheimer over a century ago, he observed a third pathological hallmark of ‘adipose inclusions’ and ‘lipid granules’ in the glia of his deceased patient Auguste Deter [110]. Subsequently, these observations were largely overlooked, but it is now well established that aberrant lipid metabolism occurs in AD brains. Lipids in the brain include glycerophospholipids, sphingolipids and cholesterol. These lipids play important roles such as in the structure of the myelin sheath which insulates axons and in formation of the plasma membrane. Cholesterol is one of the most well studied lipids and its homeostasis is implicated in AD. Indeed, there is an enrichment of cholesterol in cell membranes in AD patients compared to controls and there appears to be a positive correlation between cholesterol level and disease severity [111]. It is thought that cholesterol in the CNS is synthesized by glial cells and then imported into neurons as APOE-containing lipoproteins. However, neurons have also been shown to express genes involved in cholesterol synthesis [112] so the reality of brain cholesterol metabolism may be more complex. SORL1 mediates cholesterol intake into neurons by acting as a receptor for APOE. SORL1 has preferential binding affinity for each of the isoforms of APOE which corresponds with the relative risk that each APOE allele presents for development of AD. SORL1’s highest affinity is with the ɛ4 isoform, followed by ɛ3 then ɛ2 [113]. Each isoform of APOE differs by cysteine (Cys) and arginine (Arg) content at positions 112 and 158: ɛ4 (Arg, Arg), ɛ3 (Cys, Arg), and ɛ2 (Cys, Cys). These amino acid residue changes result in conformational changes to the protein structure of APOE (reviewed in [114]) and could explain the differential affinities between each of the APOE isoforms and SORL1.
Overexpression of SORL1 increases cellular uptake of APOE ɛ4 and ɛ3 (but not ɛ2, which is protective for AD (reviewed in [115])) as shown by a cell culture study [113]. Additionally, neural stem cells isolated from an AD patient with genotype ɛ4/ɛ4 had lower SORL1 expression after 5 weeks in culture than neural stem cells from AD and control subjects with the other possible APOE genotypes [116]. However, this study only analyzed one homozygous ɛ4 patient. Collectively, these results indicate that SORL1 and APOE are functionally related.
SORL1 and insulin signaling
It has been proposed that AD may represent a ‘type 3 diabetes’ (reviewed in [117]) and that insulin resistance/deficiency underlies AD pathology. The insulin signaling pathway has been shown to be perturbed in AD brains [118, 119]. Since insulin signaling stimulates glucose metabolism, this could partly explain the aberrant glucose metabolism observed in AD brains in FDG-PET studies (reviewed in [120]).
SORL1 has been shown to act as a sorting receptor for the insulin receptor in adipocytes. It directs internalized insulin receptor back to the cell surface, preventing lysosomal catabolism and, thereby amplifying insulin signals [121]. Loss of SORL1 activity could decrease the amount of insulin receptor at the cell surface and perturb the insulin signaling pathway. It is unknown whether this also occurs in the CNS and further investigation is required.
Is SORL1 involved in iron homeostasis?
There is strong evidence that perturbation of iron homeostasis may play a role in AD (reviewed in [122]). To our knowledge, SORL1 has not been linked directly to iron homeostasis. However, AβPP is thought to play a role in maintaining iron homeostasis and so SORL1 may have indirect effects.
Iron content is increased in both living [123] and post-mortem [124, 125] AD brains relative to controls. Iron dyshomeostasis may also be linked to many of the pathologies observed in AD such as mitochondrial dysfunction, vascular pathologies, changes in energy metabolism, and inflammation (reviewed in [122]).
A recent, elegant study by Yambire et al. [126] showed that acidification of the endo-lysosomal system was critical for ferric iron (Fe3+) to be reduced to the more reactive ferrous (Fe2+) form and released into the cytosol. Inhibition of endo-lysosomal acidification was shown to lead to a cellular ferrous iron deficiency and a pseudo-hypoxic response (as HIF-1α, a master regulator of cellular responses to hypoxia, is normally inhibited from acting under normoxia by a degradative mechanism requiring Fe2+). Mitochondrial biogenesis and function were also disturbed and inflammation markers were increased. It has been shown that the C99 fragment of AβPP (and interestingly, also the PSEN1 holoprotein [127]) is required for acidification of the endo-lysosomal system [128]. Also, AβPP-/- mice are observed to have accumulation of intraneuronal iron [129, 130]. This demonstrates that interference with AβPP activity affects cellular iron homeostasis. If SORL1 is required for the proper subcellular localization of AβPP, then the loss of SORL1 activity observed in sporadic AD and some EOfAD cases may also indirectly affect iron homeostasis via AβPP. Further investigation of the role of SORL1 in iron homeostasis is required.
SORL1 and estrogens
One hypothesis to explain the female bias observed in AD incidence [141, 142], is the reduced estrogen levels in females due to menopause. This may increase the susceptibility of post-menopausal females to AD. Indeed, estrogen levels have been shown to be reduced in female AD brains relative to healthy, age-matched, female controls [143].
Estrogens have been shown to have neuroprotective effects in the brain. Estrogen administration reduces the severity of lesions in the brain after either permanent or transient ischemia [144]. This is relevant to AD as hypoxia/reduced blood flow is evident in AD brains [145, 146]. It has also been shown that estrogens are protective against Aβ toxicity in vitro [147, 148].
SORL1 expression appears to be stimulated by estrogens. Ratnakumar et al. [149] found that SORL1 was one of the 504 genes significantly differentially expressed in laser-dissected, serotonergic neurons from female, adult, ovariectomized Rhesus macaques treated with estrogen. Interestingly, AβPP and APOE were also among these genes significantly differentially expressed (p < 0.05, log fold change > 2). SORL1 has also been shown to be differentially expressed in various cancer cell lines in response to estrogen treatments [150, 151]. Intriguingly, some studies have found an association between SORL1 variants and AD in a sex-specific manner in various cohorts [152, 153]. However, a detailed investigation of the relationship between estrogens and SORL1 in the context of AD is yet to be performed.
CONCLUSION
It is no surprise that variation at the SORL1 locus is associated with AD, as SORL1 plays roles in many of the cellular processes that have been linked to AD pathologies. SORL1 shows potential to illuminate mechanistic similarities and differences between rare EOfAD and the much more common sporadic forms of LOAD. However, more family-based studies analyzing the segregation of variants in SORL1 with EOAD are required to confirm whether SORL1 can be regarded as a causative EOfAD locus alongside the PSENs and AβPP.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
ACKNOWLEDGMENTS
The authors declare no conflicts of interest, financial or otherwise, in publication of this paper. KB is supported by an Australian Government Research Training Program Scholarship. MN is supported by National Health and Medical Research Council (NHMRC) Project Grant APP1061006. ML is an academic employee of the University of Adelaide. ML is also supported by the NHMRC (APP1045507 and APP1061006).
REFERENCES
[1] | Blennow K , de Leon MJ , Zetterberg H ((2006) ) Alzheimer’s disease. Lancet 368: , 387–403. |
[2] | Masters CL , Bateman R , Blennow K , Rowe CC , Sperling RA , Cummings JL ((2015) ) Alzheimer’s disease. Nat Rev Dis Primers 1: , 15056. |
[3] | Mullan M , Crawford F , Axelman K , Houlden H , Lilius L , Winblad B , Lannfelt L ((1992) ) A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N–terminus of β–amyloid. Nat Genet 1: , 345–347. |
[4] | Kamino K , Orr HT , Payami H , Wijsman EM , Alonso ME , Pulst SM , Anderson L , O’Dahl S , Nemens E , White JA , et al. ((1992) ) Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am J Hum Genet 51: , 998–1014. |
[5] | Kumar-Singh S , De Jonghe C , Cruts M , Kleinert R , Wang R , Mercken M , De Strooper B , Vanderstichele H , Lofgren A , Vanderhoeven I , Backhovens H , Vanmechelen E , Kroisel PM , Van Broeckhoven C ((2000) ) Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum Mol Genet 9: , 2589–2598. |
[6] | Murrell J , Farlow M , Ghetti B , Benson MD ((1991) ) A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 254: , 97–99. |
[7] | Goate A , Chartier-Harlin MC , Mullan M , Brown J , Crawford F , Fidani L , Giuffra L , Haynes A , Irving N , James L , et al. ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: , 704–706. |
[8] | Campion D , Flaman JM , Brice A , Hannequin D , Dubois B , Martin C , Moreau V , Charbonnier F , Didierjean O , Tardieu S , et al. ((1995) ) Mutations of the presenilin I gene in families with early-onset Alzheimer’s disease. Hum Mol Genet 4: , 2373–2377. |
[9] | Hutton M , Busfield F , Wragg M , Crook R , Perez-Tur J , Clark RF , Prihar G , Talbot C , Phillips H , Wright K , Baker M , Lendon C , Duff K , Martinez A , Houlden H , Nichols A , Karran E , Roberts G , Roques P , Rossor M , Venter JC , Adams MD , Cline RT , Phillips CA , Goate A , et al. ((1996) ) Complete analysis of the presenilin 1 gene in early onset Alzheimer’s disease. Neuroreport 7: , 801–805. |
[10] | Crook R , Ellis R , Shanks M , Thal LJ , Perez-Tur J , Baker M , Hutton M , Haltia T , Hardy J , Galasko D ((1997) ) Early-onset Alzheimer’s disease with a presenilin-1 mutation at the site corresponding to the Volga German presenilin-2 mutation. Ann Neurol 42: , 124–128. |
[11] | Lanoiselee HM , Nicolas G , Wallon D , Rovelet-Lecrux A , Lacour M , Rousseau S , Richard AC , Pasquier F , Rollin-Sillaire A , Martinaud O , Quillard-Muraine M , de la Sayette V , Boutoleau-Bretonniere C , Etcharry-Bouyx F , Chauvire V , Sarazin M , le Ber I , Epelbaum S , Jonveaux T , Rouaud O , Ceccaldi M , Felician O , Godefroy O , Formaglio M , Croisile B , Auriacombe S , Chamard L , Vincent JL , Sauvee M , Marelli-Tosi C , Gabelle A , Ozsancak C , Pariente J , Paquet C , Hannequin D , Campion D , collaborators of the CNR-MAJ project ((2017) ) APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med 14: , e1002270. |
[12] | Lemere CA , Lopera F , Kosik KS , Lendon CL , Ossa J , Saido TC , Yamaguchi H , Ruiz A , Martinez A , Madrigal L , Hincapie L , Arango JC , Anthony DC , Koo EH , Goate AM , Selkoe DJ , Arango JC ((1996) ) The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med 2: , 1146–1150. |
[13] | Ishikawa A , Piao Y-S , Miyashita A , Kuwano R , Onodera O , Ohtake H , Suzuki M , Nishizawa M , Takahashi H ((2005) ) A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer’s disease. Ann Neurol 57: , 429–434. |
[14] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K , et al. ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[15] | Finckh U , Alberici A , Antoniazzi M , Benussi L , Fedi V , Giannini C , Gal A , Nitsch RM , Binetti G ((2000) ) Variable expression of familial Alzheimer disease associated with presenilin 2 mutation M239I. Neurology 54: , 2006–2008. |
[16] | Jayadev S , Leverenz JB , Steinbart E , Stahl J , Klunk W , Yu CE , Bird TD ((2010) ) Alzheimer’s disease phenotypes and genotypes associated with mutations in presenilin 2. Brain 133: , 1143–1154. |
[17] | Rogaev EI , Sherrington R , Rogaeva EA , Levesque G , Ikeda M , Liang Y , Chi H , Lin C , Holman K , Tsuda T , et al. ((1995) ) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376: , 775–778. |
[18] | Pottier C , Hannequin D , Coutant S , Rovelet-Lecrux A , Wallon D , Rousseau S , Legallic S , Paquet C , Bombois S , Pariente J , Thomas-Anterion C , Michon A , Croisile B , Etcharry-Bouyx F , Berr C , Dartigues JF , Amouyel P , Dauchel H , Boutoleau-Bretonniere C , Thauvin C , Frebourg T , Lambert JC , Campion D , PHRC GMAJ Collaborators ((2012) ) High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry 17: , 875–879. |
[19] | Thonberg H , Chiang H-H , Lilius L , Forsell C , Lindström A-K , Johansson C , Björkström J , Thordardottir S , Sleegers K , Van Broeckhoven C , Rönnbäck A , Graff C ((2017) ) Identification and description of three families with familial Alzheimer disease that segregate variants in the SORL1 gene. Acta Neuropathol Commun 5: , 43. |
[20] | Wen Y , Miyashita A , Kitamura N , Tsukie T , Saito Y , Hatsuta H , Murayama S , Kakita A , Takahashi H , Akatsu H , Yamamoto T , Kosaka K , Yamaguchi H , Akazawa K , Ihara Y , Kuwano R ((2013) ) SORL1 is genetically associated with neuropathologically characterized late-onset Alzheimer’s disease. J Alzheimers Dis 35: , 387–394. |
[21] | Rogaeva E , Meng Y , Lee JH , Gu Y , Kawarai T , Zou F , Katayama T , Baldwin CT , Cheng R , Hasegawa H , Chen F , Shibata N , Lunetta KL , Pardossi-Piquard R , Bohm C , Wakutani Y , Cupples LA , Cuenco KT , Green RC , Pinessi L , Rainero I , Sorbi S , Bruni A , Duara R , Friedland RP , Inzelberg R , Hampe W , Bujo H , Song Y-Q , Andersen OM , Willnow TE , Graff-Radford N , Petersen RC , Dickson D , Der SD , Fraser PE , Schmitt-Ulms G , Younkin S , Mayeux R , Farrer LA , St George-Hyslop P ((2007) ) The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet 39: , 168–177. |
[22] | Liu G , Sun JY , Xu M , Yang XY , Sun BL ((2017) ) SORL1 variants show different association with early-onset and late-onset Alzheimer’s disease risk. J Alzheimers Dis 58: , 1121–1128. |
[23] | Motoi Y , Aizawa T , Haga S , Nakamura S , Namba Y , Ikeda K ((1999) ) Neuronal localization of a novel mosaic apolipoprotein E receptor, LR11, in rat and human brain. Brain Res 833: , 209–215. |
[24] | Jacobsen L , Madsen P , Moestrup SK , Lund AH , Tommerup N , Nykjaer A , Sottrup-Jensen L , Gliemann J , Petersen CM ((1996) ) Molecular characterization of a novel human hybrid-type receptor that binds the alpha2-macroglobulin receptor-associated protein. J Biol Chem 271: , 31379–31383. |
[25] | Frankish A , Vullo A , Zadissa A , Yates A , Thormann A , Parker A , Gall A , Moore B , Walts B , Aken BL , Cummins C , Girón CG , Ong CK , Sheppard D , Staines DM , Murphy DN , Zerbino DR , Ogeh D , Perry E , Haskell E , Martin FJ , Cunningham F , Riat HS , Schuilenburg H , Sparrow H , Lavidas I , Loveland JE , To JK , Mudge J , Bhai J , Taylor K , Billis K , Gil L , Haggerty L , Gordon L , Amode M R , Ruffier M , Patricio M , Laird MR , Muffato M , Nuhn M , Kostadima M , Langridge N , Izuogu OG , Achuthan P , Hunt SE , Janacek SH , Trevanion SJ , Hourlier T , Juettemann T , Maurel T , Newman V , Akanni W , McLaren W , Liu Z , Barrell D , Flicek P ((2017) ) Ensembl 2018. Nucleic Acids Res 46: , D754–D761. |
[26] | Blechingberg J , Poulsen ASA , Kjølby M , Monti G , Allen M , Ivarsen AK , Lincoln SJ , Thotakura G , Vægter CB , Ertekin-Taner N , Nykjær A , Andersen OM ((2018) ) An alternative transcript of the Alzheimer’s disease risk gene SORL1 encodes a truncated receptor. Neurobiol Aging 71: , 266.e211–266.e224. |
[27] | Offe K , Dodson SE , Shoemaker JT , Fritz JJ , Gearing M , Levey AI , Lah JJ ((2006) ) The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci 26: , 1596–1603. |
[28] | Grear KE , Ling IF , Simpson JF , Furman JL , Simmons CR , Peterson SL , Schmitt FA , Markesbery WR , Liu Q , Crook JE , Younkin SG , Bu G , Estus S ((2009) ) Expression of SORL1 and a novel SORL1 splice variant in normal and Alzheimer’s disease brain. Mol Neurodegener 4: , 46. |
[29] | Ciarlo E , Massone S , Penna I , Nizzari M , Gigoni A , Dieci G , Russo C , Florio T , Cancedda R , Pagano A ((2013) ) An intronic ncRNA-dependent regulation of SORL1 expression affecting Aβ formation is upregulated in post-mortem Alzheimer’s disease brain samples. Dis Model Mech 6: , 424. |
[30] | Andersen OM , Reiche J , Schmidt V , Gotthardt M , Spoelgen R , Behlke J , von Arnim CA , Breiderhoff T , Jansen P , Wu X , Bales KR , Cappai R , Masters CL , Gliemann J , Mufson EJ , Hyman BT , Paul SM , Nykjaer A , Willnow TE ((2005) ) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 102: , 13461–13466. |
[31] | Fjorback AW , Seaman M , Gustafsen C , Mehmedbasic A , Gokool S , Wu C , Militz D , Schmidt V , Madsen P , Nyengaard JR , Willnow TE , Christensen EI , Mobley WB , Nykjaer A , Andersen OM ((2012) ) Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. J Neurosci 32: , 1467–1480. |
[32] | Jacobsen L , Madsen P , Nielsen MS , Geraerts WPM , Gliemann J , Smit AB , Petersen CM ((2002) ) The sorLA cytoplasmic domain interacts with GGA1 and -2 and defines minimum requirements for GGA binding. FEBS Lett 511: , 155–158. |
[33] | Schmidt V , Sporbert A , Rohe M , Reimer T , Rehm A , Andersen OM , Willnow TE ((2007) ) SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem 282: , 32956–32964. |
[34] | Nielsen MS , Gustafsen C , Madsen P , Nyengaard JR , Hermey G , Bakke O , Mari M , Schu P , Pohlmann R , Dennes A , Petersen CM ((2007) ) Sorting by the cytoplasmic domain of the amyloid precursor protein binding receptor SorLA. Mol Cell Biol 27: , 6842–6851. |
[35] | Jacobsen L , Madsen P , Jacobsen C , Nielsen MS , Gliemann J , Petersen CM ((2001) ) Activation and functional characterization of the mosaic receptor SorLA/LR11. J Biol Chem 276: , 22788–22796. |
[36] | Guo L ((2002) ) A proteomic approach for the identification of cell-surface proteins shed by metalloproteases. Mol Cell Proteomics 1: , 30–36. |
[37] | Hampe W , Riedel IB , Lintzel J , Bader CO , Franke I , Schaller HC ((2000) ) Ectodomain shedding, translocation and synthesis of SorLA are stimulated by its ligand head activator. J Cell Sci 113 Pt 24: , 4475–4485. |
[38] | Hermey G , Sjøgaard SS , Petersen CM , Nykjaer A , Gliemann J ((2006) ) Tumour necrosis factor alpha-converting enzyme mediates ectodomain shedding of Vps10p-domain receptor family members. Biochem J 395: , 285–293. |
[39] | Böhm C , Seibel NM , Henkel B , Steiner H , Haass C , Hampe W ((2006) ) SorLA signaling by regulated intramembrane proteolysis. J Biol Chem 281: , 14547–14553. |
[40] | Nyborg AC , Ladd TB , Zwizinski CW , Lah JJ , Golde TE ((2006) ) Sortilin, SorCS1b, and SorLA Vps10p sorting receptors, are novel γ-secretase substrates. Mol Neurodegener 1: , 3. |
[41] | Willnow TE , Andersen OM ((2013) ) Sorting receptor SORLA–a trafficking path to avoid Alzheimer disease. J Cell Sci 126: , 2751–2760. |
[42] | Schmidt V , Subkhangulova A , Willnow TE ((2017) ) Sorting receptor SORLA: cellular mechanisms and implications for disease. Cell Mol Life Sci 74: , 1475–1483. |
[43] | Farrer LA , Cupples LA , Haines JL , Hyman B , Kukull WA , Mayeux R , Myers RH , Pericak-Vance MA , Risch N , van Duijn CM ((1997) ) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278: , 1349–1356. |
[44] | Lambert J-C , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , Jun G , DeStefano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thornton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin C-F , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau M-T , Choi S-H , Reitz C , Pasquier F , Hollingworth P , Ramirez A , Hanon O , Fitzpatrick AL , Buxbaum JD , Campion D , Crane PK , Baldwin C , Becker T , Gudnason V , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Morón FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fiévet N , Huentelman MJ , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuinness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossù P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Naranjo MCD , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F , European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology, Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannfelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O’Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley TH Jr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RFAG , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JSK , Boerwinkle E , Riemenschneider M , Boada M , Hiltunen M , Martin ER , Schmidt R , Rujescu D , Wang L-S , Dartigues J-F , Mayeux R , Tzourio C , Hofman A , Nöthen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452. |
[45] | Kunkle BW , Grenier-Boley B , Sims R , Bis JC , Damotte V , Naj AC , Boland A , Vronskaya M , Van Der Lee SJ , Amlie-Wolf A , Bellenguez C , Frizatti A , Chouraki V , Martin ER , Sleegers K , Badarinarayan N , Jakobsdottir J , Hamilton-Nelson KL , Moreno-Grau S , Olaso R , Raybould R , Chen Y , Kuzma AB , Hiltunen M , Morgan T , Ahmad S , Vardarajan BN , Epelbaum J , Hoffmann P , Boada M , Beecham GW , Garnier J-G , Harold D , Fitzpatrick AL , Valladares O , Moutet M-L , Gerrish A , Smith AV , Qu L , Bacq D , Denning N , Jian X , Zhao Y , Del Zompo M , Fox NC , Choi S-H , Mateo I , Hughes JT , Adams HH , Malamon J , Sanchez-Garcia F , Patel Y , Brody JA , Dombroski BA , Naranjo MCD , Daniilidou M , Eiriksdottir G , Mukherjee S , Wallon D , Uphill J , Aspelund T , Cantwell LB , Garzia F , Galimberti D , Hofer E , Butkiewicz M , Fin B , Scarpini E , Sarnowski C , Bush WS , Meslage S , Kornhuber J , White CC , Song Y , Barber RC , Engelborghs S , Sordon S , Voijnovic D , Adams PM , Vandenberghe R , Mayhaus M , Cupples LA , Albert MS , De Deyn PP , Gu W , Himali JJ , Beekly D , Squassina A , Hartmann AM , Orellana A , Blacker D , Rodriguez-Rodriguez E , Lovestone S , Garcia ME , Doody RS , Munoz-Fernadez C , Sussams R , Lin H , Fairchild TJ , Benito YA , Holmes C , Karamujić-Čomić H , Frosch MP , Thonberg H , Maier W , Roshchupkin G , Ghetti B , Giedraitis V , Kawalia A , Li S , Huebinger RM , Kilander L , Moebus S , Hernández I , Kamboh MI , Brundin R , Turton J , Yang Q , Katz MJ , Concari L , Lord J , Beiser AS , Keene CD , Helisalmi S , Kloszewska I , Kukull WA , Koivisto AM , Lynch A , Tarraga L , Larson EB , Haapasalo A , Lawlor B , Mosley TH , Lipton RB , Solfrizzi V , Gill M , Longstreth WT , Montine TJ , Frisardi V , Diez-Fairen M , Rivadeneira F , Petersen RC , Deramecourt V , Alvarez I , Salani F , Ciaramella A , Boerwinkle E , Reiman EM , Fievet N , Rotter JI , Reisch JS , Hanon O , Cupidi C , Andre Uitterlinden AG , Royall DR , Dufouil C , Maletta RG , De Rojas I , Sano M , Brice A , Cecchetti R , George-Hyslop PS , Ritchie K , Tsolaki M , Tsuang DW , Dubois B , Craig D , Wu C-K , Soininen H , Avramidou D , Albin RL , Fratiglioni L , Germanou A , Apostolova LG , Keller L , Koutroumani M , Arnold SE , Panza F , Gkatzima O , Asthana S , Hannequin D , Whitehead P , Atwood CS , Caffarra P , Hampel H , Quintela I , Carracedo Á , Lannfelt L , Rubinsztein DC , Barnes LL , Pasquier F , Frölich L , Barral S , McGuinness B , Beach TG , Johnston JA , Becker JT , Passmore P , Bigio EH , Schott JM , Bird TD , Warren JD , Boeve BF , Lupton MK , Bowen JD , Proitsi P , Boxer A , Powell JF , Burke JR , Kauwe JSK , Burns JM , Mancuso M , Buxbaum JD , Bonuccelli U , Cairns NJ , McQuillin A , Cao C , Livingston G , Carlson CS , Bass NJ , Carlsson CM , Hardy J , Carney RM , Bras J , Carrasquillo MM , Guerreiro R , Allen M , Chui HC , Fisher E , Masullo C , Crocco EA , Decarli C , Bisceglio G , Dick M , Ma L , Duara R , Graff-Radford NR , Evans DA , Hodges A , Faber KM , Scherer M , Fallon KB , Riemenschneider M , Fardo DW , Heun R , Farlow MR , Kölsch H , Ferris S , Leber M , Foroud TM , Heuser I , Galasko DR , Giegling I , Gearing M , Hüll M , Geschwind DH , Gilbert JR , Morris J , Green RC , Mayo K , Growdon JH , Feulner T , Hamilton RL , Harrell LE , Drichel D , Honig LS , Cushion TD , Huentelman MJ , Hollingworth P , Hulette CM , Hyman BT , Marshall R , Jarvik GP , Meggy A , Abner E , Menzies GE , Jin L-W , Leonenko G , Real LM , Jun GR , Baldwin CT , Grozeva D , Karydas A , Russo G , Kaye JA , Kim R , Jessen F , Kowall NW , Vellas B , Kramer JH , Vardy E , Laferla FM , Jöckel K-H , Lah JJ , Dichgans M , Leverenz JB , Mann D , Levey AI , Pickering-Brown S , Lieberman AP , Klopp N , Lunetta KL , Wichmann HE , Lyketsos CG , Morgan K , Marson DC , Brown K , Martiniuk F , Medway C , Mash DC , Nöthen MM , Masliah E , Hooper NM , McCormick WC , Daniele A , McCurry SM , Bayer A , McDavid AN , Gallacher J , McKee AC , Van Den Bussche H , Mesulam M , Brayne C , Miller BL , Riedel-Heller S , Miller CA , Miller JW , Al-Chalabi A , Morris JC , Shaw CE , Myers AJ , Wiltfang J , O’Bryant S , Olichney JM , Alvarez V , Parisi JE , Singleton AB , Paulson HL , Collinge J , Perry WR , Mead S , Peskind E , Cribbs DH , Rossor M , Pierce A , Ryan NS , Poon WW , Nacmias B , Potter H , Sorbi S , Quinn JF , Sacchinelli E , Raj A , Spalletta G , Raskind M , Caltagirone C , Bossù P , Orfei MD , Reisberg B , Clarke R , Reitz C , Smith AD , Ringman JM , Warden D , Roberson ED , Wilcock G , Rogaeva E , Bruni AC , Rosen HJ , Gallo M , Rosenberg RN , Ben-Shlomo Y , Sager MA , Mecocci P , Saykin AJ , Pastor P , Cuccaro ML , Vance JM , Schneider JA , Schneider LS , Slifer S , Seeley WW , Smith AG , Sonnen JA , Spina S , Stern RA , Swerdlow RH , Tang M , Tanzi RE , Trojanowski JQ , Troncoso JC , Van Deerlin VM , Van Eldik LJ , Vinters HV , Vonsattel JP , Weintraub S , Welsh-Bohmer KA , Wilhelmsen KC , Williamson J , Wingo TS , Woltjer RL , Wright CB , Yu C-E , Yu L , Saba Y , Pilotto A , Bullido MJ , Peters O , Crane PK , Bennett D , Bosco P , Coto E , Boccardi V , De Jager PL , Lleo A , Warner N , Lopez OL , Ingelsson M , Deloukas P , Cruchaga C , Graff C , Gwilliam R , Fornage M , Goate AM , Sanchez-Juan P , Kehoe PG , Amin N , Ertekin-Taner N , Berr C , Debette S , Love S , Launer LJ , Younkin SG , Dartigues J-F , Corcoran C , Ikram MA , Dickson DW , Nicolas G , Campion D , Tschanz J , Schmidt H , Hakonarson H , Clarimon J , Munger R , Schmidt R , Farrer LA , Van Broeckhoven C , C. O’Donovan M , Destefano AL , Jones L , Haines JL , Deleuze J-F , Owen MJ , Gudnason V , Mayeux R , Escott-Price V , Psaty BM , Ramirez A , Wang L-S , Ruiz A , Van Duijn CM , Holmans PA , Seshadri S , Williams J , Amouyel P , Schellenberg GD , Lambert J-C , Pericak-Vance MA ((2019) ) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat Genet 51: , 414–430. |
[46] | Campion D , Charbonnier C , Nicolas G ((2019) ) SORL1 genetic variants and Alzheimer disease risk: a literature review and meta-analysis of sequencing data. Acta Neuropathol 138: , 173–186. |
[47] | Nicolas G , Charbonnier C , Wallon D , Quenez O , Bellenguez C , Grenier-Boley B , Rousseau S , Richard AC , Rovelet-Lecrux A , Le Guennec K , Bacq D , Garnier JG , Olaso R , Boland A , Meyer V , Deleuze JF , Amouyel P , Munter HM , Bourque G , Lathrop M , Frebourg T , Redon R , Letenneur L , Dartigues JF , Génin E , Lambert JC , Hannequin D , Campion D ((2016) ) SORL1 rare variants: a major risk factor for familial early-onset Alzheimer’s disease. Mol Psychiatry 21: , 831–836. |
[48] | Verheijen J , Van den Bossche T , van der Zee J , Engelborghs S , Sanchez-Valle R , Lladó A , Graff C , Thonberg H , Pastor P , Ortega-Cubero S , Pastor MA , Benussi L , Ghidoni R , Binetti G , Clarimon J , Lleó A , Fortea J , de Mendonça A , Martins M , Grau-Rivera O , Gelpi E , Bettens K , Mateiu L , Dillen L , Cras P , De Deyn PP , Van Broeckhoven C , Sleegers K ((2016) ) A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol 132: , 213–224. |
[49] | Bellenguez C , Charbonnier C , Grenier-Boley B , Quenez O , Le Guennec K , Nicolas G , Chauhan G , Wallon D , Rousseau S , Richard AC , Boland A , Bourque G , Munter HM , Olaso R , Meyer V , Rollin-Sillaire A , Pasquier F , Letenneur L , Redon R , Dartigues JF , Tzourio C , Frebourg T , Lathrop M , Deleuze JF , Hannequin D , Genin E , Amouyel P , Debette S , Lambert JC , Campion D ; CNR MAJ collaborators ((2017) ) Contribution to Alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol Aging 59: , 220.e1–220.e9. |
[50] | Scherzer CR , Offe K , Gearing M , Rees HD , Fang G , Heilman CJ , Schaller C , Bujo H , Levey AI , Lah JJ ((2004) ) Loss of apolipoprotein e receptor lr11 in Alzheimer disease. Arch Neurol 61: , 1200–1205. |
[51] | Sager KL , Wuu J , Herskowitz JH , Mufson EJ , Levey AI , Lah JJ ((2012) ) Neuronal LR11 expression does not differentiate between clinically-defined Alzheimer’s disease and control brains. PLoS One 7: , e40527. |
[52] | Dodson SE , Gearing M , Lippa CF , Montine TJ , Levey AI , Lah JJ ((2006) ) LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J Neuropathol Exp Neurol 65: , 866–872. |
[53] | Cuccaro ML , Carney RM , Zhang Y , Bohm C , Kunkle BW , Vardarajan BN , Whitehead PL , Cukier HN , Mayeux R , St George-Hyslop P , Pericak-Vance MA ((2016) ) SORL1 mutations in early- and late-onset Alzheimer disease. Neurol Genet 2: , e116. |
[54] | Vardarajan BN , Zhang Y , Lee JH , Cheng R , Bohm C , Ghani M , Reitz C , Reyes-Dumeyer D , Shen Y , Rogaeva E , St George-Hyslop P , Mayeux R ((2015) ) Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 77: , 215–227. |
[55] | Young JE , Boulanger-Weill J , Williams DA , Woodruff G , Buen F , Revilla AC , Herrera C , Israel MA , Yuan SH , Edland SD , Goldstein LS ((2015) ) Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16: , 373–385. |
[56] | Rohe M , Carlo A-S , Breyhan H , Sporbert A , Militz D , Schmidt V , Wozny C , Harmeier A , Erdmann B , Bales KR , Wolf S , Kempermann G , Paul SM , Schmitz D , Bayer TA , Willnow TE , Andersen OM ((2008) ) Sortilin-related Receptor with A-type Repeats (SORLA) affects the amyloid precursor protein-dependent stimulation of ERK signaling and adult neurogenesis. J Biol Chem 283: , 14826–14834. |
[57] | Rohe M , Synowitz M , Glass R , Paul SM , Nykjaer A , Willnow TE ((2009) ) Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J Neurosci 29: , 15472–15478. |
[58] | Ng TKS , Ho CSH , Tam WWS , Kua EH , Ho RC-M ((2019) ) Decreased serum brain-derived neurotrophic factor (BDNF) levels in patients with Alzheimer’s disease (AD): a systematic review and meta-analysis. Int J Mol Sci 20: , E257. |
[59] | Weinstein G , Beiser AS , Choi SH , Preis SR , Chen TC , Vorgas D , Au R , Pikula A , Wolf PA , DeStefano AL , Vasan RS , Seshadri S ((2014) ) Serum brain-derived neurotrophic factor and the risk for dementia: the Framingham Heart Study. JAMA Neurol 71: , 55–61. |
[60] | Asai M , Hattori C , Szabó B , Sasagawa N , Maruyama K , Tanuma S-i , Ishiura S ((2003) ) Putative function of ADAM9, ADAM10, and ADAM17 as APP α-secretase. Biochem Biophys Res Commun 301: , 231–235. |
[61] | Lammich S , Kojro E , Postina R , Gilbert S , Pfeiffer R , Jasionowski M , Haass C , Fahrenholz F ((1999) ) Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A 96: , 3922–3927. |
[62] | Buxbaum JD , Liu KN , Luo Y , Slack JL , Stocking KL , Peschon JJ , Johnson RS , Castner BJ , Cerretti DP , Black RA ((1998) ) Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem 273: , 27765–27767. |
[63] | Tanabe C , Hotoda N , Sasagawa N , Sehara-Fujisawa A , Maruyama K , Ishiura S ((2007) ) ADAM19 is tightly associated with constitutive Alzheimer’s disease APP α-secretase in A172 cells. Biochem Biophys Res Commun 352: , 111–117. |
[64] | Kuhn P-H , Wang H , Dislich B , Colombo A , Zeitschel U , Ellwart JW , Kremmer E , Roßner S , Lichtenthaler SF ((2010) ) ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J 29: , 3020–3032. |
[65] | Tolia A , De Strooper B ((2009) ) Structure and function of γ-secretase. Semin Cell Dev Biol 20: , 211–218. |
[66] | Haass C , Kaether C , Thinakaran G , Sisodia S ((2012) ) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2: , a006270. |
[67] | Sisodia SS ((1992) ) Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci U S A 89: , 6075–6079. |
[68] | Lai A , Sisodia SS , Trowbridge IS ((1995) ) Characterization of sorting signals in the β-amyloid precursor protein cytoplasmic domain. J Biol Chem 270: , 3565–3573. |
[69] | Boll W , Rapoport I , Brunner C , Modis Y , Prehn S , Kirchhausen T ((2002) ) The mu 2 subunit of the clathrin adaptor AP-2 binds to FDNPVY and YppO sorting signals at distinct sites. Traffic 3: , 590–600. |
[70] | Vassar R , Bennett BD , Babu-Khan S , Kahn S , Mendiaz EA , Denis P , Teplow DB , Ross S , Amarante P , Loeloff R , Luo Y , Fisher S , Fuller J , Edenson S , Lile J , Jarosinski MA , Biere AL , Curran E , Burgess T , Louis JC , Collins F , Treanor J , Rogers G , Citron M ((1999) ) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: , 735–741. |
[71] | Frykman S , Hur J-Y , Frånberg J , Aoki M , Winblad B , Nahalkova J , Behbahani H , Tjernberg LO ((2010) ) Synaptic and endosomal localization of active γ-secretase in rat brain. PLoS One 5: , e8948. |
[72] | Chyung JH , Raper DM , Selkoe DJ ((2005) ) Gamma-secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J Biol Chem 280: , 4383–4892. |
[73] | Choy RW-Y , Cheng Z , Schekman R ((2012) ) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in thetrans-Golgi network. Proc Natl Acad Sci U S A 109: , E2077–E2082. |
[74] | Tan JZA , Gleeson PA ((2019) ) The trans-Golgi network is a major site for α-secretase processing of amyloid precursor protein in primary neurons. J Biol Chem 294: , 1618–1631. |
[75] | Pasternak SH , Bagshaw RD , Guiral M , Zhang S , Ackerley CA , Pak BJ , Callahan JW , Mahuran DJ ((2003) ) Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem 278: , 26687–26694. |
[76] | Sannerud R , Esselens C , Ejsmont P , Mattera R , Rochin L , Tharkeshwar Arun K , De Baets G , De Wever V , Habets R , Baert V , Vermeire W , Michiels C , Groot Arjan J , Wouters R , Dillen K , Vints K , Baatsen P , Munck S , Derua R , Waelkens E , Basi Guriqbal S , Mercken M , Vooijs M , Bollen M , Schymkowitz J , Rousseau F , Bonifacino Juan S , Van Niel G , De Strooper B , Annaert W ((2016) ) Restricted location of PSEN2/γ-secretase determines substrate specificity and generates an intracellular Aβ pool. Cell 166: , 193–208. |
[77] | Cook DG , Forman MS , Sung JC , Leight S , Kolson DL , Iwatsubo T , Lee VMY , Doms RW ((1997) ) Alzheimer’s Aβ(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med 3: , 1021–1023. |
[78] | Hartmann T , Bieger SC , Bruhl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease A[beta]40/42 amyloid peptides. Nat Med 3: , 1016–1020. |
[79] | Area-Gomez E , de Groof AJ , Boldogh I , Bird TD , Gibson GE , Koehler CM , Yu WH , Duff KE , Yaffe MP , Pon LA , Schon EA ((2009) ) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175: , 1810–1816. |
[80] | Vance JE ((2014) ) MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim Biophys Acta 1841: , 595–609. |
[81] | Raturi A , Simmen T ((2013) ) Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim Biophys Acta 1833: , 213–224. |
[82] | Del Prete D , Suski JM , Oulès B , Debayle D , Gay AS , Lacas-Gervais S , Bussiere R , Bauer C , Pinton P , Paterlini-Bréchot P , Wieckowski MR , Checler F , Chami M ((2016) ) Localization and processing of the amyloid-β protein precursor in mitochondria-associated membranes. J Alzheimers Dis 55: , 1549–1570. |
[83] | Paillusson S , Stoica R , Gomez-Suaga P , Lau DHW , Mueller S , Miller T , Miller CCJ ((2016) ) There’s something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci 39: , 146–157. |
[84] | Schon EA , Area-Gomez E ((2010) ) Is Alzheimer’s disease a disorder of mitochondria-associated membranes? J Alzheimers Dis 20 Suppl 2: , S281–292. |
[85] | Spoelgen R , von Arnim CA , Thomas AV , Peltan ID , Koker M , Deng A , Irizarry MC , Andersen OM , Willnow TE , Hyman BT ((2006) ) Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and beta-secretase beta-site APP-cleaving enzyme. J Neurosci 26: , 418–428. |
[86] | Liu L , Ding L , Rovere M , Wolfe MS , Selkoe DJ ((2019) ) A cellular complex of BACE1 and γ-secretase sequentially generates Aβ from its full-length precursor. J Cell Biol 218: , 644–663. |
[87] | Newman M , Wilson L , Verdile G , Lim A , Khan I , Moussavi Nik SH , Pursglove S , Chapman G , Martins RN , Lardelli M ((2014) ) Differential, dominant activation and inhibition of Notch signalling and APP cleavage by truncations of PSEN1 in human disease. Hum Mol Genet 23: , 602–617. |
[88] | Lim AHL ((2015) ) Analysis of the subcellular localization of proteins implicated in Alzheimer’s disease (thesis). Department of Genetics and Evolution, University of Adelaide, p. 235. |
[89] | Herskowitz JH , Offe K , Deshpande A , Kahn RA , Levey AI , Lah JJ ((2012) ) GGA1-mediated endocytic traffic of LR11/SorLA alters APP intracellular distribution and amyloid-β production. Mol Biol Cell 23: , 2645–2657. |
[90] | Dodson SE , Andersen OM , Karmali V , Fritz JJ , Cheng D , Peng J , Levey AI , Willnow TE , Lah JJ ((2008) ) Loss of LR11/SORLA enhances early pathology in a mouse model of amyloidosis: evidence for a proximal role in Alzheimer’s disease. J Neurosci 28: , 12877–12886. |
[91] | Mehmedbasic A , Christensen SK , Nilsson J , Rüetschi U , Gustafsen C , Poulsen ASA , Rasmussen RW , Fjorback AN , Larson G , Andersen OM ((2015) ) SorLA complement-type repeat domains protect the amyloid precursor protein against processing. J Biol Chem 290: , 3359–3376. |
[92] | Andersen OM , Schmidt V , Spoelgen R , Gliemann J , Behlke J , Galatis D , McKinstry WJ , Parker MW , Masters CL , Hyman BT , Cappai R , Willnow TE ((2006) ) Molecular dissection of the interaction between amyloid precursor protein and its neuronal trafficking receptor SorLA/LR11. Biochemistry 45: , 2618–2628. |
[93] | Spoelgen R , von Arnim CAF , Thomas AV , Peltan ID , Koker M , Deng A , Irizarry MC , Andersen OM , Willnow TE , Hyman BT ((2006) ) Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and β-secretase β-site APP-cleaving enzyme. J Neurosci 26: , 418. |
[94] | Schmidt V , Baum K , Lao A , Rateitschak K , Schmitz Y , Teichmann A , Wiesner B , Petersen CM , Nykjaer A , Wolf J , Wolkenhauer O , Willnow TE ((2012) ) Quantitative modelling of amyloidogenic processing and its influence by SORLA in Alzheimer’s disease. EMBO J 31: , 187–200. |
[95] | Seaman MN ((2012) ) The retromer complex - endosomal protein recycling and beyond. J Cell Sci 125: , 4693–4702. |
[96] | Zhang H , Huang T , Hong Y , Yang W , Zhang X , Luo H , Xu H , Wang X ((2018) ) The retromer complex and sorting nexins in neurodegenerative diseases. Front Aging Neurosci 10: , 79. |
[97] | Vieira SI , Rebelo S , Esselmann H , Wiltfang J , Lah J , Lane R , Small SA , Gandy S , Da Cruz E Silva EF , Da Cruz E Silva OA ((2010) ) Retrieval of the Alzheimer’s amyloid precursor protein from the endosome to the TGN is S655 phosphorylation state-dependent and retromer-mediated. Mol Neurodegener 5: , 40. |
[98] | Small SA , Kent K , Pierce A , Leung C , Kang MS , Okada H , Honig L , Vonsattel J-P , Kim T-W ((2005) ) Model-guided microarray implicates the retromer complex in Alzheimer’s disease. Ann Neurol 58: , 909–919. |
[99] | Vardarajan BN , Bruesegem SY , Harbour ME , George-Hyslop PS , Seaman MNJ , Farrer LA ((2012) ) Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol Aging 33: , 2231.e2215–2231.e2233. |
[100] | Muhammad A , Flores I , Zhang H , Yu R , Staniszewski A , Planel E , Herman M , Ho L , Kreber R , Honig LS , Ganetzky B , Duff K , Arancio O , Small SA ((2008) ) Retromer deficiency observed in Alzheimer’s disease causes hippocampal dysfunction, neurodegeneration, and Abeta accumulation. Proc Natl Acad Sci U S A 105: , 7327–7332. |
[101] | Wen L , Tang F-L , Hong Y , Luo S-W , Wang C-L , He W , Shen C , Jung J-U , Xiong F , Lee D-H , Zhang Q-G , Brann D , Kim T-W , Yan R , Mei L , Xiong W-C ((2011) ) VPS35 haploinsufficiency increases Alzheimer’s disease neuropathology. J Cell Biol 195: , 765–779. |
[102] | Von Arnim CAF , Spoelgen R , Peltan ID , Deng M , Courchesne S , Koker M , Matsui T , Kowa H , Lichtenthaler SF , Irizarry MC , Hyman BT ((2006) ) GGA1 acts as a spatial switch altering amyloid precursor protein trafficking and processing. J Neurosci 26: , 9913–9922. |
[103] | Dumanis SB , Burgert T , Caglayan S , Fuchtbauer A , Fuchtbauer EM , Schmidt V , Willnow TE ((2015) ) Distinct functions for anterograde and retrograde sorting of SORLA in amyloidogenic processes in the brain. J Neurosci 35: , 12703–12713. |
[104] | Wan L , Molloy SS , Thomas L , Liu G , Xiang Y , Rybak SL , Thomas G ((1998) ) PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94: , 205–216. |
[105] | Burgert T , Schmidt V , Caglayan S , Lin F , Fuchtbauer A , Fuchtbauer EM , Nykjaer A , Carlo AS , Willnow TE ((2013) ) SORLA-dependent and -independent functions for PACS1 in control of amyloidogenic processes. Mol Cell Biol 33: , 4308–4320. |
[106] | Huang TY , Zhao Y , Li X , Wang X , Tseng IC , Thompson R , Tu S , Willnow TE , Zhang YW , Xu H ((2016) ) SNX27 and SORLA interact to reduce amyloidogenic subcellular distribution and processing of amyloid precursor protein. J Neurosci 36: , 7996–8011. |
[107] | Wang X , Huang T , Zhao Y , Zheng Q , Robert , Bu G , Zhang Y-W , Hong W , Xu H ((2014) ) Sorting nexin 27 regulates Aβ production through modulating γ-secretase activity. Cell Rep 9: , 1023–1033. |
[108] | Caglayan S , Takagi-Niidome S , Liao F , Carlo A-S , Schmidt V , Burgert T , Kitago Y , Füchtbauer E-M , Füchtbauer A , Holtzman DM , Takagi J , Willnow TE ((2014) ) Lysosomal sorting of amyloid-β by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci Transl Med 6: , 223ra220. |
[109] | Kitago Y , Nagae M , Nakata Z , Yagi-Utsumi M , Takagi-Niidome S , Mihara E , Nogi T , Kato K , Takagi J ((2015) ) Structural basis for amyloidogenic peptide recognition by sorLA. Nat Struct Mol Biol 22: , 199–206. |
[110] | Foley P ((2010) ) Lipids in Alzheimer’s disease: A century-old story. Biochim Biophys Acta 1801: , 750–753. |
[111] | Cutler RG , Kelly J , Storie K , Pedersen WA , Tammara A , Hatanpaa K , Troncoso JC , Mattson MP ((2004) ) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101: , 2070–2075. |
[112] | Valdez CM , Smith MA , Perry G , Phelix CF , Santamaria F ((2010) ) Cholesterol homeostasis markers are localized to mouse hippocampal pyramidal and granule layers. Hippocampus 20: , 902–905. |
[113] | Yajima R , Tokutake T , Koyama A , Kasuga K , Tezuka T , Nishizawa M , Ikeuchi T ((2015) ) ApoE-isoform-dependent cellular uptake of amyloid-beta is mediated by lipoprotein receptor LR11/SorLA. Biochem Biophys Res Commun 456: , 482–488. |
[114] | Frieden C , Garai K ((2012) ) Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci U S A 109: , 8913–8918. |
[115] | Suri S , Heise V , Trachtenberg AJ , Mackay CE ((2013) ) The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE varepsilon2. Neurosci Biobehav Rev 37: , 2878–2886. |
[116] | Zollo A , Allen Z , Rasmussen HF , Iannuzzi F , Shi Y , Larsen A , Maier TJ , Matrone C ((2017) ) Sortilin-related receptor expression in human neural stem cells derived from Alzheimer’s disease patients carrying the APOE epsilon 4 allele. Neural Plast 2017: , 1892612. |
[117] | de la Monte SM ((2014) ) Type 3 diabetes is sporadic Alzheimer’s disease: mini-review. Eur Neuropsychopharmacol 24: , 1954–1960. |
[118] | Lee S , Tong M , Hang S , Deochand C , de la Monte S ((2013) ) CSF and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late Alzheimer’s disease. J Alzheimers Dis Parkinsonism 3: , 128. |
[119] | Rivera EJ , Goldin A , Fulmer N , Tavares R , Wands JR , de la Monte SM ((2005) ) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis 8: , 247–268. |
[120] | Mosconi L ((2005) ) Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging 32: , 486–510. |
[121] | Schmidt V , Schulz N , Yan X , Schurmann A , Kempa S , Kern M , Bluher M , Poy MN , Olivecrona G , Willnow TE ((2016) ) SORLA facilitates insulin receptor signaling in adipocytes and exacerbates obesity. J Clin Invest 126: , 2706–2720. |
[122] | Lumsden AL , Rogers JT , Majd S , Newman M , Sutherland GT , Verdile G , Lardelli M ((2018) ) Dysregulation of neuronal iron homeostasis as an alternative unifying effect of mutations causing familial Alzheimer’s disease. Front Neurosci 12: , 533–533. |
[123] | Du L , Zhao Z , Cui A , Zhu Y , Zhang L , Liu J , Shi S , Fu C , Han X , Gao W , Song T , Xie L , Wang L , Sun S , Guo R , Ma G ((2018) ) Increased iron deposition on brain quantitative susceptibility mapping correlates with decreased cognitive function in Alzheimer’s disease. ACS Chem Neurosci 9: , 1849–1857. |
[124] | Lovell MA , Robertson JD , Teesdale WJ , Campbell JL , Markesbery WR ((1998) ) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158: , 47–52. |
[125] | Samudralwar DL , Diprete CC , Ni BF , Ehmann WD , Markesbery WR ((1995) ) Elemental imbalances in the olfactory pathway in Alzheimer’s disease. J Neurol Sci 130: , 139–145. |
[126] | Yambire KF , Rostosky C , Watanabe T , Pacheu-Grau D , Torres-Odio S , Sanchez-Guerrero A , Senderovich O , Meyron-Holtz EG , Milosevic I , Frahm J , West AP , Raimundo N ((2019) ) Impaired lysosomal acidification triggers iron deficiency and inflammation in vivo. Elife 8: , e51031. |
[127] | Lee JH , McBrayer MK , Wolfe DM , Haslett LJ , Kumar A , Sato Y , Lie PP , Mohan P , Coffey EE , Kompella U , Mitchell CH , Lloyd-Evans E , Nixon RA ((2015) ) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12: , 1430–1444. |
[128] | Jiang Y , Sato Y , Im E , Berg M , Bordi M , Darji S , Kumar A , Mohan PS , Bandyopadhyay U , Diaz A , Cuervo AM , Nixon RA ((2019) ) Lysosomal dysfunction in Down syndrome is APP-dependent and mediated by APP-βCTF (C99). J Neurosci 39: , 5255–5268. |
[129] | Duce JA , Tsatsanis A , Cater MA , James SA , Robb E , Wikhe K , Leong SL , Perez K , Johanssen T , Greenough MA , Cho H-H , Galatis D , Moir RD , Masters CL , McLean C , Tanzi RE , Cappai R , Barnham KJ , Ciccotosto GD , Rogers JT , Bush AI ((2010) ) Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142: , 857–867. |
[130] | Needham BE , Ciccotosto GD , Cappai R ((2014) ) Combined deletions of amyloid precursor protein and amyloid precursor-like protein 2 reveal different effects on mouse brain metal homeostasis. Metallomics 6: , 598–603. |
[131] | Waldvogel-Abramowski S , Waeber G , Gassner C , Buser A , Frey BM , Favrat B , Tissot J-D ((2014) ) Physiology of iron metabolism. Transfus Med Hemother 41: , 213–221. |
[132] | Gunshin H , Mackenzie B , Berger UV , Gunshin Y , Romero MF , Boron WF , Nussberger S , Gollan JL , Hediger MA ((1997) ) Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 388: , 482–488. |
[133] | Jenkitkasemwong S , Wang C-Y , Mackenzie B , Knutson MD ((2012) ) Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals 25: , 643–655. |
[134] | Richardson DR , Ponka P ((1997) ) The molecular mechanisms of the metabolism and transport of iron in normal and neoplastic cells. Biochim Biophys Acta 1331: , 1–40. |
[135] | Sargent PJ , Farnaud S , Evans RW ((2005) ) Structure/function overview of proteins involved in iron storage and transport. Curr Med Chem 12: , 2683–2693. |
[136] | Arosio P , Levi S ((2010) ) Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim Biophys Acta 1800: , 783–792. |
[137] | Wong BX , Tsatsanis A , Lim LQ , Adlard PA , Bush AI , Duce JA ((2014) ) β-amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 9: , e114174. |
[138] | Dlouhy AC , Bailey DK , Steimle BL , Parker HV , Kosman DJ ((2019) ) Fluorescence resonance energy transfer links membrane ferroportin, hephaestin but not ferroportin, amyloid precursor protein complex with iron efflux. J Biol Chem 294: , 4202–4214. |
[139] | Ji C , Steimle BL , Bailey DK , Kosman DJ ((2018) ) The ferroxidase hephaestin but not amyloid precursor protein is required for ferroportin-supported iron efflux in primary hippocampal neurons. Cell Mol Neurobiol 38: , 941–954. |
[140] | Lane DJR , Merlot AM , Huang MLH , Bae DH , Jansson PJ , Sahni S , Kalinowski DS , Richardson DR ((2015) ) Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim Biophys Acta 1853: , 1130–1144. |
[141] | Gao S , Hendrie HC , Hall KS , Hui S ((1998) ) The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Arch Gen Psychiatry 55: , 809–815. |
[142] | Andersen K , Launer LJ , Dewey ME , Letenneur L , Ott A , Copeland JR , Dartigues JF , Kragh-Sorensen P , Baldereschi M , Brayne C , Lobo A , Martinez-Lage JM , Stijnen T , Hofman A ((1999) ) Gender differences in the incidence of AD and vascular dementia: The EURODEM Studies. EURODEM Incidence Research Group. Neurology 53: , 1992–1997. |
[143] | Yue X , Lu M , Lancaster T , Cao P , Honda SI , Staufenbiel M , Harada N , Zhong Z , Shen Y , Li R ((2005) ) Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci U S A 102: , 19198–19203. |
[144] | Gibson CL , Gray LJ , Murphy SP , Bath PM ((2006) ) Estrogens and experimental ischemic stroke: a systematic review. J Cereb Blood Flow Metab 26: , 1103–1113. |
[145] | Iturria-Medina Y , Sotero R , Toussaint P , Mateos-Pérez J , Evans A , Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7: , 11934. |
[146] | Rivera-Rivera LA , Schubert T , Turski P , Johnson KM , Berman SE , Rowley HA , Carlsson CM , Johnson SC , Wieben O ((2017) ) Changes in intracranial venous blood flow and pulsatility in Alzheimer’s disease: A 4D flow MRI study. J Cereb Blood Flow Metab 37: , 2149–2158. |
[147] | Marin R , Guerra B , Hernández-Jiménez JG , Kang XL , Fraser JD , López FJ , Alonso R ((2003) ) Estradiol prevents amyloid-β peptide-induced cell death in a cholinergic cell line via modulation of a classical estrogen receptor. Neuroscience 121: , 917–926. |
[148] | Nilsen J , Chen S , Irwin RW , Iwamoto S , Brinton RD ((2006) ) Estrogen protects neuronal cells from amyloid beta-induced apoptosis via regulation of mitochondrial proteins and function. BMC Neurosci 7: , 74. |
[149] | Ratnakumar A , Zimmerman SE , Jordan BA , Mar JC ((2019) ) Estrogen activates Alzheimer’s disease genes. Alzheimers Dement (N Y) 5: , 906–917. |
[150] | Zhang S-t , Zuo C , Li W-n , Fu X-q , Xing S , Zhang X-p ((2016) ) Identification of key genes associated with the effect of estrogen on ovarian cancer using microarray analysis. Arch Gynecol Obstet 293: , 421–427. |
[151] | Shen M , Cao J , Shi H ((2018) ) Effects of estrogen and estrogen receptors on transcriptomes of HepG2 cells: a preliminary study using RNA sequencing. Int J Endocrinol 2018: , 5789127. |
[152] | Cellini E , Tedde A , Bagnoli S , Pradella S , Piacentini S , Sorbi S , Nacmias B ((2009) ) Implication of sex and SORL1 variants in italian patients with Alzheimer disease. Arch Neurol 66: , 1260–1266. |
[153] | Casingal CR , Daroy MLG , Mapua CA , Florendo DJA , Natividad FF , Dominguez JC ((2014) ) Polymorphisms in the Sortilin-Related Receptor 1 gene are associated with cognitive impairment in Filipinos. Asian J Neurosci 2014: , 1–7. |


