
Apolipoprotein A1 in Cerebrospinal Fluid Is Insufficient to Distinguish Alzheimer’s Disease from Other Dementias in a Naturalistic, Clinical Setting
Abstract
Apolipoprotein A1 (ApoA1) is the major protein component of the high-density lipoprotein and involved in cholesterol transport. Disruption of cholesterol homeostasis has been identified as a contributing factor for Alzheimer’s disease (AD). Moreover, polymorphisms of ApoA1 have been associated with higher risk of disease onset and cognitive decline. Therefore, ApoA1 has been suggested as a biomarker in AD. Here, we tested a small cohort of AD and non-AD dementia patients and measured levels of ApoA1 in cerebrospinal fluid. Our results indicate that ApoA1 might not be applicable to distinguish AD from other forms of dementia.
INTRODUCTION
ApoA1 is an important component of high-density lipoprotein (HDL) and is responsible for reverse transport of cholesterol from tissues to the liver as well as for excretion of cholesterol by promoting efflux from other tissues. This protein has been associated with premature coronary artery disease, hepatosplenomegaly, or recurrent peripheral neuropathy within HDL deficiency (e.g., reviewed in [1]). Amyloidosis 8, a hereditary generalized amyloidosis, is caused by mutations in the human ApoA1 gene [2]. Therefore, it seems plausible to assume also contribution of ApoA1 to other amyloidoses such as Aβ aggregation in Alzheimer’s disease. This is further strengthened by the observation that regulation of amyloid-β protein precursor (AβPP) processing is based on cholesterol content of neuronal cells (for example [3]). Together with its neuroprotective properties, such as anti-aggregational function towards amyloid-β (Aβ), it is tempting to speculate that ApoA1 could serve as an AD biomarker.
Antibodies raised against ApoA1 occasionally stained senile plaques and amyloid angiopathies in the cortex of AD patients [4]. However, by comparing 11–13 samples of AD cases and controls within the same investigation, no difference in the amount was observed in brain tissue by immunoblot. Upon examination of pooled cerebrospinal fluid (CSF) samples obtained from AD patients and non-demented control subjects (both n = 43) using 2-dimensional gel electrophoresis, ApoA1 levels together with cathepsin D and transthyretin were identified as being significantly reduced in CSF from AD cases [5]. Nonetheless, an alternative study using the ELISA technique, found no difference in ApoA1 CSF levels between early and late onset AD or healthy controls [6]. More recently, a study comparing healthy subjects with subjective cognitive decline and mild cognitively impaired patients (MCI) revealed higher CSF and lower plasma ApoA1 levels to be associated with an increased risk of clinical progression in APOE4 carriers [7]. This might indicate an important role for ApoA1 at an early stage of disease development. In manifested AD (median Mini-Mental State Examination (MMSE) score of 23, [8]), a decrease in CSF ApoA1 has been found and ApoA1 was described to be associated with disease status. By comparing AD cases with other types of dementia [vascular dementia, dementia with Lewy bodies, or frontotemporal dementia (FTD)], no conclusive difference was obtained and it is worthy to note that ApoA1 levels correlated with MMSE throughout the study population, including healthy controls. Despite all these clinical studies, it has not been demonstrated whether ApoA1 levels can discriminate AD from other forms of dementia nor if it is a generalized indicator for cognitive function in CSF. In order to investigate this, we analyzed the amount of ApoA1 in gender- and age-matched groups of AD patients and patients with other types of dementia syndromes.
METHODS
Participants
The participants of the study were recruited from daily clinical work after having given written consent (§14 AVB, University Medical Center Johannes Gutenberg University Mainz) that their surplus material (e.g., CSF) taken for clinical routine or diagnosis can be used for research in an anonymous way. This allowed investigating samples from a naturalistic clinical setting.
Collection of CSF samples
Lumbar puncture at the L3/L4 or L4/L5 interspace was performed before noon, with the patient in a sitting position. CSF samples were immediately frozen at –80°C in polypropylene tubes until examination. Biomarkers were assessed after anonymization of samples in the clinical central laboratory of the University Medical Center by using dementia marker assays (Innotest, Fujirebio Gent, Belgium). All values were measured in technical replicates despite p-Tau. From 14 samples of both, AD and non-AD dementia group, one sample had to be excluded due to non-plausible values (for demographic data see Table 1).
Table 1
Demographic data of patients. PD, Parkinson’s disease; FTD, frontotemporal dementia; nph, normal pressure hydrocephalus; nd, no final diagnosis but AD excluded
| AD | Non-AD dementia | ||||
| female | male | female | male | ||
| n | 7 | 6 | 7 | 6 | |
| Age | 72.1±6.3 | 72±6.7 | 72.9±3.8 | 74.2±10.9 | |
| Diagnosis | AD | AD | 2 PD | 1 PD | |
| 1 FTD | 2 FTD | ||||
| 1 nph | 1 vascular | ||||
| 1 vascular | 2 nd | ||||
| 2 nd | |||||
| Biomarker (mean±SEM) | p | ||||
| Aβ42 [pg/ml] | 646±72 | 777±72 | 0.209 | ||
| Aβ40 [pg/ml] | 9498±1228 | 8682±932 | 0.601 | ||
| p-Tau [pg/ml] | 77±14 | 57±6 | 0.264 | ||
| Tau [pg/ml] | 412±78 | 268±48 | 0.081 | ||
| Aβ42*10/ Aβ40 | 0.72±0.06 | 0.98±0.11 | 0.044 | ||
| Aβ42/p-Tau | 10.6±1.8 | 15.6±2.0 | 0.077 | ||
Western blotting
Protein content of samples was assessed using the Nanoquant reagent (Roth, Karlsruhe, Germany). 7 μg of protein were separated on a 10% SDS acrylamide/bisacrylamide gel and transferred to a nitrocellulose membrane. Antibodies used were: anti-ApoA1 (Thermo Fisher Scientific, Karlsruhe, Germany), anti-human serum albumin (Abcam, Cambridge, UK), and appropriate secondary HRP-labelled antibodies (Thermo Fisher, Karlsruhe, Germany). Recombinant ApoA1 (Chemicon International, Temecula, CA, USA) was used for specificity control of the respective antibody (not shown). Development of the signals was conducted using the SuperSignal West Femto chemiluminescent substrate (Thermo Scientific, Karlsruhe, Germany) and a CCD-camera imaging system (Raytest, Straubenhardt, Germany). Quantitative analysis was carried out using the AIDA image analyzer 4.26 software (Raytest, Straubenhardt, Germany).
Statistical analyses
Statistical analyses were performed using the statistical software GraphPad Prism 6.07 (GraphPad Software, La Jolla, USA). Differences between group means were analyzed via the Student’s t-test or Mann-Whitney test if non-Gaussian distribution was observed. A Welsh correction was applied if significantly different variances occurred. A p-value < 0.05 was considered statistically significant.
RESULTS
We included 13 CSF samples from routine clinical sampling for AD and for non-AD dementia patients (see Table 1). This generated a naturalistic cohort of patients without considering any further inclusion or exclusion criteria. Gender composition was 54% females and age comparable between the two groups (One Way ANOVA with Tukey’s multiple comparison test, minimal p of group-wise comparison: p = 0.94). Non-AD-dementia patients included those suffering from Parkinson’s disease (PD), FTD, vascular dementia, or hydrocephalus. Four patients were not ultimately diagnosed at the time-point of CSF collection but AD had been excluded. Combination of Aβ1–42, T-tau, and P-tau181 has the highest diagnostic power to discriminate between AD and cognitively healthy controls [9]; however, discrimination of non-AD dementia and AD is difficult: for example Aβ42 and T-tau display intermediate levels between AD and healthy controls in FTD or vascular dementia. Other biomarkers such as Aβ42/40 ratio or Aβ42/p-Tau seem more promising to distinguish between both conditions [10, 11]. In our small cohort, Aβ42/40 reached statistical significance when comparing both groups and p-value was close to significance level for Aβ42/p-Tau (p = 0.077; Table 1).
ApoA1 levels in patient CSF were assessed by immunoblot and normalized to albumin detection signal (Fig. 1A). In samples from AD patients, ApoA1 increased to 142 % of the mean measurement from non-AD-dementia patients. However, this did not reach statistical significance (Fig. 1B; p = 0.39). Re-analyzing subsets of males and females in separate groups also did not lead to statistically significant differences (p = 0.47 for males, p = 0.32 for females).
Fig.1
ApoA1 expression in non-AD- and AD dementia patients. A) Detection of ApoA1 in CSF by western blotting. ApoA1 from 7 μg total protein of CSF was quantified by western blotting. Representative western blots are shown for both, male and female patient samples. B) Quantitative analysis of ApoA1. Values for ApoA1 were normalized to albumin levels. The mean of non-AD samples was set to 100% (separately for male and female patients) and all other values normalized to this mean. Data are presented including the median; open circles and squares show data for male patients, filled ones show data for female patients. Statistical analysis was performed by using the Mann-Whitney test (p = 0.39). C) Correlation of biomarker measures with relative ApoA1 levels. Linear regressions from all patients (male and female) are presented. None of the correlations was statistically significant (e.g., Aβ42*10/Aβ40 and ApoA1: p = 0.065; Aβ42/p-Tau and ApoA1: p = 0.096).
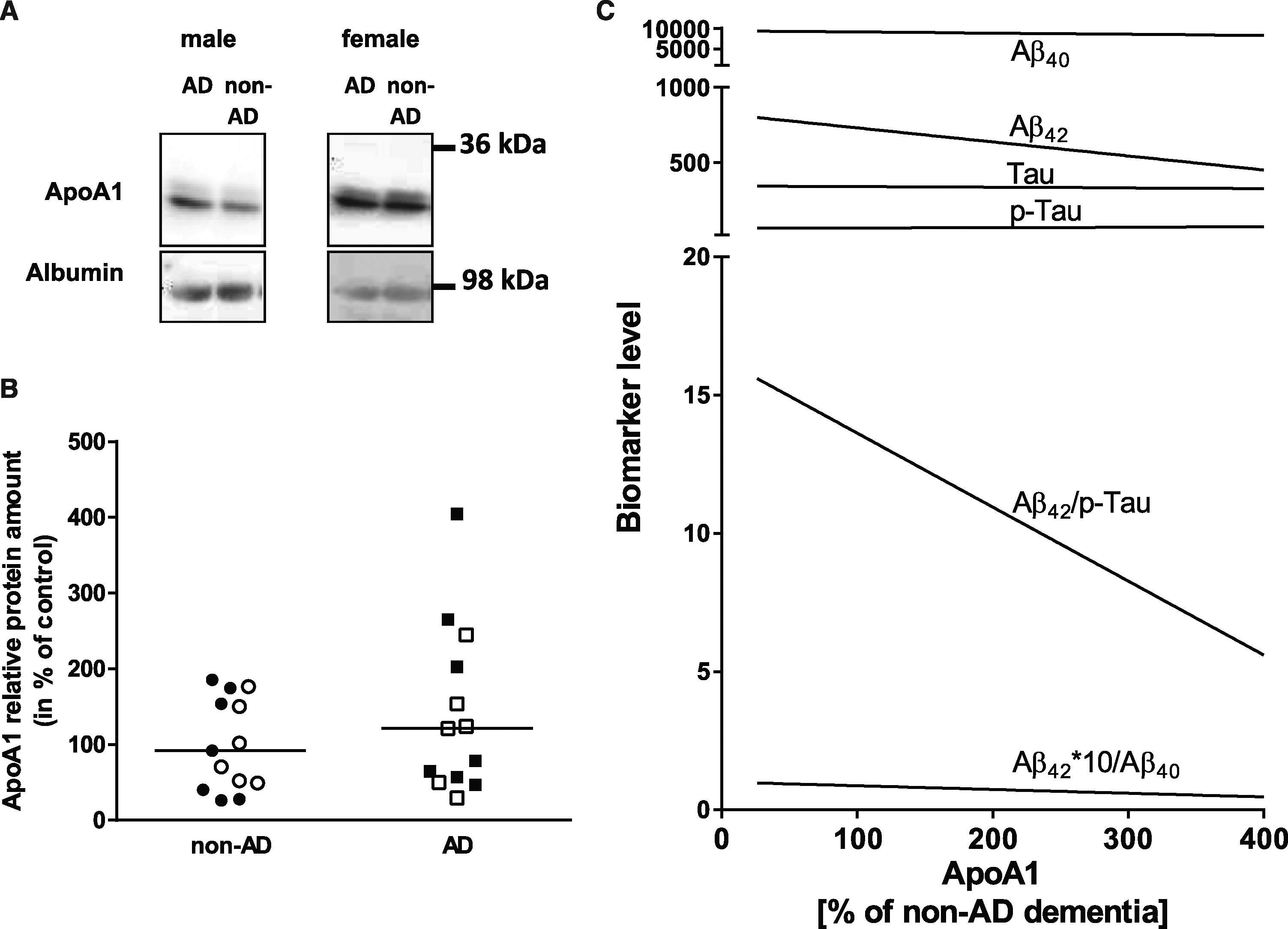
As Tau or p-Tau levels are used for neurodegeneration markers, we also investigated whether ApoA1 levels correlate with those. None of the correlations reached statistical significance; only Aβ42/Aβ40 and Aβ42/p-Tau correlation with ApoA1 showed a trend of dependency (Fig. 1C).
DISCUSSION
In this short report, we show that it is not possible to discriminate between AD-affected patients and other demented individuals by ApoA1 quantitation from CSF derived by clinical routine sampling even if groups are well balanced for sex and age.
ApoA1 expression and circulation is affected by nutritional components such as Vitamin C [12] or food products containing added sugar such as chocolate (negatively correlated with ApoA1 blood levels [13]). This connotes that ApoA1 can be massively affected by lifestyle, veiling disease-related changes. In a clinical setting, a biomarker should be as independent from such factors as possible. For AD, changes in food preference have been observed including cravings for sweet food [14]. The latter has also been reported for FTD (e.g., [15]) which already might corroborate their diagnostic separation via ApoA1 if one of the diseases leads to direct changes in ApoA1 levels.
Furthermore, discriminating against healthy controls is less of an interest when the biomarker is required for identification of AD patients in a cohort of demented individuals as would be the case for example in a memory clinic. For other diseases such as PD or schizophrenia altered ApoA1 expression also has been reported: elevated ApoA1 levels, for example, have been found in CSF of a small cohort of PD patients while the AD comparison group was indifferent from healthy controls [16]. By using different techniques, ApoA1 could be demonstrated to be reduced in CSF from schizophrenia patients but also in liver and red blood cells [17]. Another publication reports an increase in ApoA1 levels during the active state of meningitis while finding no changes in PD or AD [18]. HDLs are known to have antimicrobial properties against parasites and bacteria, therefore making them part of the immune response (reviewed in [19]). Thus, diseases such as AD that are accompanied by different inflammatory events should have an impact on the proteinaceous component of HDL. In the future, a more detailed analysis of ApoA1 and its proteoforms would be required: Fania and colleagues for example reported that only the proteoform precipitating at pH 5.2 was changed in AD versus control and idiopathic normal pressure hydrocephalus [20]. Potentially, other biological sources such as hepatic or intestinal tissue could deliver ApoA1 amounts with better discrimination potential as they present the production site of ApoA1/HDL particles. Our own study is clearly limited by lack of information on APOE genotype, MMSE of patients, and nutritional status as well as intake of lipid-lowering drugs. Moreover, numbers were small and not derived from longitudinal sampling. However, this would be the scenario at the beginning of a diagnostic procedure. Interestingly, the only correlations that came close to statistical significance level in our study were Aβ42/Aβ40 and Aβ42/p-Tau ratios with ApoA1. By combining these new biomarkers with ApoA1, a better discrimination against other types of dementia may be achieved, yet this has to be proven in independent and larger cohorts of patients.
Despite all these difficulties and controversies in establishing ApoA1 as a biomarker, it nevertheless might have value in regard to therapeutic approaches. An acute treatment with reconstituted HDL significantly reduced soluble brain Aβ40 and Aβ42 levels in APP/PS1 model mice while prolonged treatment had no effect [21]. Interestingly, no effect on total Aβ or deposited Aβ was observed in ApoA1 overexpressing APP/PS1 mice; nevertheless, preservation of cognitive function, attenuated neuroinflammation and cerebral amyloid angiopathy was found [22].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was funded by the Stiftung Innovation Rhineland-Palatinate. We thank all patients and clinical staff involved in lumbar puncture for their generosity and help. We are grateful to H. May-Simera (Johannes Gutenberg-University Mainz) for help with the manuscript.
REFERENCES
[1] | von Eckardstein A ((2006) ) Differential diagnosis of familial high density lipoprotein deficiency syndromes. Atherosclerosis 186: , 231–239. |
[2] | Joy T , Wang J , Hahn A , Hegele RA ((2003) ) APOA1 related amyloidosis: A case report and literature review. Clin Biochem 36: , 641–645. |
[3] | Kojro E , Gimpl G , Lammich S , Marz W , Fahrenholz F ((2001) ) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci U S A 98: , 5815–5820. |
[4] | Harr SD , Uint L , Hollister R , Hyman BT , Mendez AJ ((1996) ) Brain expression of apolipoproteins E, J, and A-I in Alzheimer’s disease. J Neurochem 66: , 2429–2435. |
[5] | Castano EM , Roher AE , Esh CL , Kokjohn TA , Beach T ((2006) ) Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol Res 28: , 155–163. |
[6] | Saito K , Seishima M , Heyes MP , Song H , Fujigaki S , Maeda S , Vickers JH , Noma A ((1997) ) Marked increases in concentrations of apolipoprotein in the cerebrospinal fluid of poliovirus-infected macaques: Relations between apolipoprotein concentrations and severity of brain injury. Biochem J 321 (Pt 1): , 145–149. |
[7] | Slot RE , Van Harten AC , Kester MI , Jongbloed W , Bouwman FH , Teunissen CE , Scheltens P , Veerhuis R , van der Flier WM ((2017) ) Apolipoprotein A1 in cerebrospinal fluid and plasma and progression to Alzheimer’s disease in non-demented elderly. J Alzheimers Dis 56: , 687–697. |
[8] | Johansson P , Almqvist EG , Bjerke M , Wallin A , Johansson JO , Andreasson U , Blennow K , Zetterberg H , Svensson J ((2017) ) Reduced cerebrospinal fluid concentration of apolipoprotein A-I in patients with Alzheimer’s disease. J Alzheimers Dis 59: , 1017–1026. |
[9] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S , Crutch S , Engelborghs S , Frisoni GB , Fox NC , Galasko D , Habert MO , Jicha GA , Nordberg A , Pasquier F , Rabinovici G , Robert P , Rowe C , Salloway S , Sarazin M , Epelbaum S , de Souza LC , Vellas B , Visser PJ , Schneider L , Stern Y , Scheltens P , Cummings JL ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[10] | Maddalena A , Papassotiropoulos A , Muller-Tillmanns B , Jung HH , Hegi T , Nitsch RM , Hock C ((2003) ) Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to beta-amyloid peptide42. Arch Neurol 60: , 1202–1206. |
[11] | Struyfs H , Van Broeck B , Timmers M , Fransen E , Sleegers K , Van Broeckhoven C , De Deyn PP , Streffer JR , Mercken M , Engelborghs S ((2015) ) Diagnostic accuracy of cerebrospinal fluid amyloid-beta isoforms for early and differential dementia diagnosis. J Alzheimers Dis 45: , 813–822. |
[12] | Kim DS , Burt AA , Rosenthal EA , Ranchalis JE , Eintracht JF , Hatsukami TS , Furlong CE , Marcovina S , Albers JJ , Jarvik GP ((2014) ) HDL-3 is a superior predictor of carotid artery disease in a case-control cohort of 1725 participants. J Am Heart Assoc 3: , e000902. |
[13] | Frondelius K , Borg M , Ericson U , Borne Y , Melander O , Sonestedt E ((2017) ) Lifestyle and dietary determinants of serum apolipoprotein A1 and apolipoprotein B concentrations: Cross-sectional analyses within a Swedish cohort of 24,984 individuals. Nutrients 9: , E211. |
[14] | Mungas D , Cooper JK , Weiler PG , Gietzen D , Franzi C , Bernick C ((1990) ) Dietary preference for sweet foods in patients with dementia. J Am Geriatr Soc 38: , 999–1007. |
[15] | Bathgate D , Snowden JS , Varma A , Blackshaw A , Neary D ((2001) ) Behaviour in frontotemporal dementia, Alzheimer’s disease and vascular dementia. Acta Neurol Scand 103: , 367–378. |
[16] | Wang ES , Sun Y , Guo JG , Gao X , Hu JW , Zhou L , Hu J , Jiang CC ((2010) ) Tetranectin and apolipoprotein A-I in cerebrospinal fluid as potential biomarkers for Parkinson’s disease. Acta Neurol Scand 122: , 350–359. |
[17] | Huang JT , Wang L , Prabakaran S , Wengenroth M , Lockstone HE , Koethe D , Gerth CW , Gross S , Schreiber D , Lilley K , Wayland M , Oxley D , Leweke FM , Bahn S ((2008) ) Independent protein-profiling studies show a decrease in apolipoprotein A1 levels in schizophrenia CSF, brain and peripheral tissues. Mol Psychiatry 13: , 1118–1128. |
[18] | Song H , Seishima M , Saito K , Maeda S , Takemura M , Noma A , Kondo A , Manabe M , Urakami K , Nakashima K ((1998) ) Apo A-I and apo E concentrations in cerebrospinal fluids of patients with acute meningitis. Ann Clin Biochem 35 (Pt 3): , 408–414. |
[19] | Montecucco F , Favari E , Norata GD , Ronda N , Nofer JR , Vuilleumier N ((2015) ) Impact of systemic inflammation and autoimmune diseases on apoA-I and HDL plasma levels and functions. Handb Exp Pharmacol 224: , 455–482. |
[20] | Fania C , Arosio B , Capitanio D , Torretta E , Gussago C , Ferri E , Mari D , Gelfi C ((2017) ) Protein signature in cerebrospinal fluid and serum of Alzheimer’s disease patients: The case of apolipoprotein A-1 proteoforms. PLoS One 12: , e0179280. |
[21] | Robert J , Stukas S , Button E , Cheng WH , Lee M , Fan J , Wilkinson A , Kulic I , Wright SD , Wellington CL ((2016) ) Reconstituted high-density lipoproteins acutely reduce soluble brain Abeta levels in symptomatic APP/PS1 mice. Biochim Biophys Acta 1862: , 1027–1036. |
[22] | Lewis TL , Cao D , Lu H , Mans RA , Su YR , Jungbauer L , Linton MF , Fazio S , LaDu MJ , Li L ((2010) ) Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem 285: , 36958–36968. |


