
BACE1 Translation: At the Crossroads Between Alzheimer’s Disease Neurodegeneration and Memory Consolidation
Abstract
Human life unfolds not only in time and space, but also in the recollection and interweaving of memories. Therefore, individual human identity depends fully on a proper access to the autobiographical memory. Such access is hindered under pathological conditions such as Alzheimer’s disease, which affects millions of people worldwide. Unfortunately, no effective cure exists to prevent this disorder, the impact of which will rise alarmingly within the next decades. While Alzheimer’s disease is largely considered to be the outcome of amyloid-β (Aβ) peptide accumulation in the brain, conceiving this complex disorder strictly as the result of Aβ-neurotoxicity is perhaps a too straight-line simplification. Instead, complementary to this view, the tableau of molecular disarrangements in the Alzheimer’s disease brain may be reflecting, at least in part, a loss of function phenotype in memory processing. Here we take BACE1 translation and degradation as a gateway to study molecular mechanisms putatively involved in the transition between memory and neurodegeneration. BACE1 participates in the excision of Aβ-peptide from its precursor holoprotein, but plays a role in synaptic plasticity too. Its translation is governed by eIF2α phosphorylation: a hub integrating cellular responses to stress, but also a critical switch in memory consolidation. Paralleling these dualities, the eIF2α-kinase HRI has been shown to be a nitric oxide-dependent physiological activator of hippocampal BACE1 translation. Finally, beholding BACE1 as a representative protease active in the CNS, we venture a new perspective on the cellular basis of memory, which may incorporate neurodegeneration in itself as a drift in memory consolidating systems.
INTRODUCTION
The threshold between normality and disease is not—and has not—always been clear; senile dementia, for instance, was still conceived as a normal process of the aging mind when the seminal paper by Alois Alzheimer appeared in 1907. In his “Über eine eigenartige Erkrankung der Hirnrinde” (About a peculiar disease of the cerebral cortex) [1], the German psychiatrist achieved to correlate the cognitive deterioration of one of his patients, Auguste D., with distinctive histopathological features present in her brain after postmortem examination. Alzheimer described an atrophic brain with unusual deposits in the cortex and prominent neurofibrillary pathology, which in our days are referred to as senile plaques and neurofibrillary tangles, respectively, and still constitute the two histopathological hallmarks of choice in the postmortem diagnosis of Alzheimer’s disease (AD). Though, more importantly, such findings marked a starting point: they proved the existence of an actual organic disease that could potentially be treated.
In the late 1980s, studies of the molecular pathology underlying AD led to important initial discoveries. Amyloid-β (Aβ) peptide was found to be the main component of senile plaques [2, 3] and the Aβ coding-gene, later on named Amyloid Precursor Protein (AβPP), was cloned and localized in chromosome 21 [4–7]. The observation that trisomy 21 (Down’s syndrome, DS) invariably caused AD dementia [8] culminated in the formulation of the Amyloid Cascade Hypothesis [9–12], according to which the initial deposition of Aβ peptide in the brain parenchyma was the central event driving AD pathogenesis. One decade later, in the late 1990s, the “Beta-site AβPP cleaving enzyme-1” (BACE1) was identified as the protease catalyzing the rate-limiting step in the excision of Aβ peptide from its precursor holoprotein [13–17]. Thus, the aspartyl protease BACE1 entered the scene at the epicenter of AD pathophysiology, making the thorough comprehension of its biological regulation a scientific endeavor of prime importance in order to solve the riddle of AD neurodegeneration.
More than a century has elapsed since Alzheimer’s pioneering discovery at the turn of the twentieth century and the disease that bears his name continues to be a major burden for the health of the elderly, affecting ∼35 million of people worldwide—an estimative which is predicted to increase to over 100 million people by 2050 [18]. The Amyloid Cascade Hypothesis, which emerged as a synthesis of genetic and histopathological observations, has dominated research over the last two decades. However, all the Aβ-centric approaches that have reached phase III clinical trials have failed [19–21], and the quest for new therapies to prevent the disease, delay its onset, slow its progression or ameliorate its symptoms remains largely unsuccessful. Therefore, it is timely to put the focus on the shadows of the amyloid cascade hypothesis so that the light of new ideas can contribute to bring new impulses in the field of AD research. Indeed, pursuing non-amyloid research approaches in AD is gradually being felt as a deep and urgent need for the field [22].
In this regard, it is generally acknowledged that Aβ plaque burden does not correlate with cognitive impairment [23–25] and that the deposition of Aβ peptide neither correlates with the presence of neurofibrillary tangles (made of hyperphosphorylated microtubule-associated protein tau), cell loss nor dementia [26]. Mutations in genes related to Aβ peptide generation do cause AD in a dominant autosomal inheritable fashion, but the total number of early onset familiar AD cases is very low (∼5%) compared with sporadic AD (SAD), which is the major form in which the disease manifests and is triggered by other mechanisms than mutations in AβPP or its processing enzymes. In addition, the Amyloid Cascade Hypothesis cannot provide a satisfactory link between the number of amyloid plaques and tau lesions, failing to incorporate within its conceptual framework one of the most crucial issues in AD pathophysiology, i.e., the precise mechanistic and temporal relationship connecting tau pathology with Aβ peptide deposition. Bringing these facts into full consciousness, some authors have raised the criticism that a role for Aβ peptide in all AD cases might have been unduly extrapolated from genetic evidences suggesting a role for an altered AβPP processing in early onset familiar AD (FAD) [27].
In parallel, research in the biology of BACE1 has unveiled other substrates for this protease beyond AβPP. For instance, BACE1 modulates peripheral nerve myelinization through the shedding of neuregulin-1 (NGL-1), a surface receptor involved in neuron-glia signaling [28, 29]. BACE1 also cleaves the voltage gated sodium channels β2 [30] and β4 [31] subunits, regulating membrane excitability [30] and neurite length [31]. Recently, more systematic approaches for the identification of novel BACE1 substrates have been carried out [32–36], uncovering 34 neuronal membrane proteins requiring BACE1 for their shedding [33], which corresponds to 19% of the total 183 shed membrane proteins in neurons. These novel results have definitely changed the previous view that BACE1 was a sheddase for only a few specific proteins. On the contrary, according to the new compelling evidences, BACE1 emerges as a major contributor to the shedding process in neurons, side by side with other proteases considered to be major sheddases in the CNS, such as the metalloproteases ADAM10 or ADAM17. Furthermore, the newly identified BACE1 substrates point to a central function of this protease in neurite outgrowth and synapse formation, a finding in frame with previous neurophysiological observations linking BACE1 activity with long-term potentiation and memory acquisition [37, 38].
As AD is a pathology which primarily compromises memory, and taking into account the stagnation of the purely amyloid paradigm, it seems not reckless to seek for common nexus connecting the molecular mechanisms of AD and memory formation; treating them not as two unrelated phenomena taking place in parallel, but as two sides of the same coin. Why not bridging, whenever possible, neurodegeneration with memory acquisition will help us gain new insights into fundamental memory processes putatively disrupted in AD, which can eventually inspire new therapeutic approaches?
From this perspective, translational control of gene expression can contribute a significant share in the unification of these two fields, as the control of protein translation is a process chiefly involved in both sides of the spectrum. On the one hand, local control of protein synthesis at synapses is crucial for synaptic plasticity [39–43] and memory processing [44–55] (reviewed in [56–68]) and, on the other hand, disrupted protein translation—and more specifically, undesirable BACE1 translational activation—is a mechanism suggested to be involved in the triggering of SAD [69–71].
Our first aim in this review has been to update the current knowledge about the molecular mechanisms governing BACE1 translation and its implications for cellular physiology. A quick overview to BACE1 transcript immediately reveals the presence of prominent untranslated regions (UTRs) flanking the open reading frame, which are indicative per se of a tightly regulated translation (Fig. 1). BACE1 3’UTR is very long (∼3,8 Kb) and contains sequences that facilitate mRNA localization in dendrites by the CPE-binding protein (CPEB). But here we draw our attention preferentially to the 5’UTR, a ∼500 bp-long sequence that gates BACE1 translation initiation intertwining two levels of complexity: 1) structured secondary conformation arising from a high GC content, and 2) the presence of upstream open reading frames (uORFs) interspersed in the sequence.
Fig.1
BACE1 open reading frame flanked by its 5’ and 3’ untranslated regions (UTRs). Schematic representation depicting BACE1 transcript structure. Note how BACE1 3’UTR is almost 2.5 times larger than the protein coding region (ORF). In addition, BACE1 3’UTR contains two cytoplasmic polyadenylation (CPE) motifs (UUUUAU) and accessory sequences (AAUAA) that facilitate dendritic mRNA sorting by the CPE-binding protein (CPEB) [307]. BACE1 5’UTR, in turn, gates translation initiation combining the inhibitory effect of 1) upstream initiation codons (uAUGs) and 2) stable secondary structure deriving from a high GC-content (∼75 %).
![BACE1 open reading frame flanked by its 5’ and 3’ untranslated regions (UTRs). Schematic representation depicting BACE1 transcript structure. Note how BACE1 3’UTR is almost 2.5 times larger than the protein coding region (ORF). In addition, BACE1 3’UTR contains two cytoplasmic polyadenylation (CPE) motifs (UUUUAU) and accessory sequences (AAUAA) that facilitate dendritic mRNA sorting by the CPE-binding protein (CPEB) [307]. BACE1 5’UTR, in turn, gates translation initiation combining the inhibitory effect of 1) upstream initiation codons (uAUGs) and 2) stable secondary structure deriving from a high GC-content (∼75 %).](https://content.iospress.com:443/media/adr/2019/3-1/adr-3-1-adr180089/adr-3-adr180089-g001.jpg)
In particular, as we will later discuss, the regulation of BACE1 translation initiation by uORFs provides a molecular foundation upon which: 1) Stress-driven BACE1 expression in pathological conditions; 2) BACE1 physiological expression in the context of nitric oxide signaling; 3) The balance in the phosphorylation state of translation initiation factors required for memory consolidation; 4) A putative mechanistic link between insulin resistance, BACE1 translation, and tau phosphorylation – all this can be integrated resting on a single molecular mechanism.
Nonetheless, complex biological phenomena such as AD or memory cannot be traced to single isolated molecular mechanisms, as a general rule; thus, to avoid falling prey of such a reductionist temptation, we include a chapter in which we discuss other interesting aspects of BACE1 biology that have recently come to light. In this chapter, we review the recent advances made in the elucidation of intracellular degradative pathways that, altered as they are in SAD, disrupt the normal balance of BACE1 activity in neurons.
As mentioned before, synaptic plasticity and memory acquisition have been proposed over the last years as biological activities in which BACE1 might be involved physiologically. However, as BACE1 activity was originally identified when reconstructing the “criminal scenario” of Aβ deposition in the AD brain, we wanted to open this review with a chapter dedicated to describe the details of BACE1 discovery and the relevance of β-secretase activity in AD pathogenesis. This allows us to describe how the amyloid cascade hypothesis was originated, and the good reasons that led this scientific paradigm to be the dominant hypothesis driving research in the AD field in the last decades.
Finally, we close the review with a chapter devoted to do some speculation on the issues covered throughout the review. Speculation in science can be dangerous or misleading, but it is nevertheless the fuel needed to spark the fire of further developments. As the English poet William Blake (1757–1827) once put it, “all that has been proved, was first imagined”. Hence, we make use of our imagination to pull all the threads at our hands originating from the molecular mechanisms minutely discussed throughout the text. In this regard, deep reflection on the role played by proteases such as BACE1(both in AD and in synaptic plasticity) led us to venture a new perspective on the neurobiological basis of learning and memory, according to which not unilateral growth and synaptic strengthening, but also dismantling and destabilizing processes, might be required as biological substrates supporting cognition.
Such an assertion might be shocking for the un-expecting reader. But, if visual perception arises from a continuous self-renewing destruction of the visual pigments in the retina, why thinking (and the ensuing cognitive capacities) could not be related to its neuronal substrate in a similar manner?
THE DISCOVERY OF BACE1 IN THE CONTEXT OF THE AMYLOID CASCADE HYPOTHESIS
Notwithstanding the weaknesses mentioned before, the amyloid cascade hypothesis continues so far to be the best encompassing conceptual framework we have at our disposal to explain the etiology of AD. Hence, newer perspectives on the etiology of this devastating disorder will necessarily have to incorporate the solid genetic basis in which the amyloid hypothesis is built, as well as the large body of experimental evidences it has brought to the surface in the last twenty-five years of research.
Indeed, the discovery that AD could be inherited in an autosomal dominant fashion was a turning point in AD research [72]. The mutation was found in the gene coding for AβPP, the integral transmembrane precursor holoprotein from which Aβ peptide is generated via sequential cleavages at two specific sites, which were then named as γ- and β-secretase sites respectively. The term “secretase” was the operative nomination used to designate those enzymatic activities responsible for AβPP cleavage, while the ultimate identities of the actual proteases active upon AβPP awaited to be discovered. In the case of the protease behind β-secretase activity, the quest for its identification crystallized at the turn of the millennium, in the biennium 1999-2000, when the work of five independent groups converged in the identification of BACE1 as the β-secretase active in the CNS [13–17]. These works combined three different approaches, including conventional protein purification [14], expression cloning [13], and bioinformatics [15–17].
Meanwhile, the Amyloid Cascade Hypothesis (originally inspired by the phenotypical semblance between AD and DS premature dementia), had been articulated on the basis of the newly localized AD-causative genetic mutations in AβPP gene [9–12, 73] (Fig. 2A). Indeed, this initiative was to receive a further enthusiastic impulse with the discovery that the catalytic subunit of the multimeric protein complex responsible for γ-secretase activity was no other than presenilin-1 [74]: a gene in which, together with its homologue presenilin-2, mutations causative of inheritable FAD had been previously identified [75–77].
Fig.2
Point mutations in AβPP and its processing by BACE1. A)“Framing β -amyloid”. Snapshot from an article by Prof. John Hardy [Framing β -amyloid, Nature Genetics 1, 233–234 (1992)] showing the initial AD-causative mutations (labelled as “AD” in the figure) that were discovered in AβPP. Today more than 15 disease-causing mutations are known to impact AβPP. For a recent personal account by Prof. Hardy on the story of how AD-causative mutations in AβPP were discovered, and how this led to the formulation of the amyloid hypothesis, you can see [84]. Image reproduced with permission. B) BACE1 participation in A β PP processing. Amyloid-β (Aβ) peptide is excised out of the amyloid precursor protein (AβPP) via sequential cleavages catalyzed by BACE1 and the γ-secretase complex, respectively. Inhibition (of both proteases) is an actively prosecuted goal as a pharmacological strategy in AD therapy. Nevertheless, all of the synthesized inhibitors to date have repeatedly failed in clinical trials. C) Scheme showing AD pathogenic mutations on AβPP affecting the β-secretase cleavage (in red), the β’-secretase cleavage (in blue), the α-secretase cleavage (in purple), and the γ-secretase cleavage (in orange) of AβPP. Those mutations affecting the aggregation of the peptide are highlighted with a “*” symbol, while those mutations affecting the degradation of the peptide are indicated with a “**” symbol. The symbol # is used to indicate the mutations that are pathogenic for cerebral amyloid angiopathy, while the symbol “?” indicates that the mechanisms of pathogenesis remains unknown for that particular mutation.
![Point mutations in AβPP and its processing by BACE1. A)“Framing β
-amyloid”. Snapshot from an article by Prof. John Hardy [Framing β
-amyloid, Nature Genetics 1, 233–234 (1992)] showing the initial AD-causative mutations (labelled as “AD” in the figure) that were discovered in AβPP. Today more than 15 disease-causing mutations are known to impact AβPP. For a recent personal account by Prof. Hardy on the story of how AD-causative mutations in AβPP were discovered, and how this led to the formulation of the amyloid hypothesis, you can see [84]. Image reproduced with permission. B) BACE1 participation in A
β
PP processing. Amyloid-β (Aβ) peptide is excised out of the amyloid precursor protein (AβPP) via sequential cleavages catalyzed by BACE1 and the γ-secretase complex, respectively. Inhibition (of both proteases) is an actively prosecuted goal as a pharmacological strategy in AD therapy. Nevertheless, all of the synthesized inhibitors to date have repeatedly failed in clinical trials. C) Scheme showing AD pathogenic mutations on AβPP affecting the β-secretase cleavage (in red), the β’-secretase cleavage (in blue), the α-secretase cleavage (in purple), and the γ-secretase cleavage (in orange) of AβPP. Those mutations affecting the aggregation of the peptide are highlighted with a “*” symbol, while those mutations affecting the degradation of the peptide are indicated with a “**” symbol. The symbol # is used to indicate the mutations that are pathogenic for cerebral amyloid angiopathy, while the symbol “?” indicates that the mechanisms of pathogenesis remains unknown for that particular mutation.](https://content.iospress.com:443/media/adr/2019/3-1/adr-3-1-adr180089/adr-3-adr180089-g002.jpg)
During the following years, the observation that AβPP locus duplication on chromosome 21 was linked to autosomal dominant early-onset FAD and cerebral amyloid angiopathy contributed to further establish the importance of AβPP gene dosage in AD [78]. In parallel, newer mutations causative of early-onset FAD were being discovered in a yearly basis, to the point that more than 200 distinct disease-causing mutations are known today [19]. The totality of these mutations impact either AβPP or one of the two presenilin genes, influencing Aβ metabolism in one way or another. Paradigmatically, mutations in AβPP are located within the region codifying for Aβ-peptide or in the vicinity of γ- and β-secretase cleaving sites. For instance, some mutations located in the middle domain of the Aβ region have been shown to increase the degree of Aβ-peptide aggregation (Dutch and Arctic mutations) [79–81]. Other mutations make AβPP a more efficient substrate for BACE1 cleavage, thus increasing the levels of Aβ production (Swedish mutation) [82]. However, the vast majority of AβPP mutations and all of the mutations found in presenilins drift Aβ metabolism toward the production of the more aggregant 42-aa isoform of the peptide (Aβ1–42) [83]. In summary, this recollection of genetic evidences seemed to point clearly toward Aβ production and aggregation as crucial events driving the disease, thereby placing BACE1 activity as a main proximal event in AD triggering (Fig. 2B).
Further research revealed the nature of the neurotoxic effects derived from A βpeptide production (Fig. 3). Soluble Aβ turns neurotoxic when misfolded in β-sheet fibrils, which were found to be a source of oxidative stress [85]. The lumen of Aβ protofibrils forms a suitable microenvironment for the production of hydrogen peroxide [86–88], an upstream generator of oxidative stress in the intracellular milieu through a series of redox chained reactions. Moreover, the highly diffusible nature of hydrogen peroxide, which penetrates freely through biological membranes, made it likely that Aβ toxicity could be in this way spread away from the original focus of Aβ-fibril deposition [89]. These findings were aligned with the previous notion that oxidative stress was intimately linked to AD pathology [90–92]. Interestingly enough, oxidative stress induces BACE1 expression both at the transcriptional [93] and translational levels [70], thus promoting a positive feed-back loop between amyloidogenesis and oxidative stress.
Fig.3
Cascade of neurotoxic effects deriving from Aβ peptide aggregation. Soluble monomers of Aβ peptide aggregate into different species (oligomers, protofibrils, larger fibrils). Neurotoxic effects attributed to each aggregative state are indicated in capital letters in the right column of the figure. Up to the present day, the relative contribution of each Aβ-species to the overall neurotoxicity-effect in AD remains a subject of intense debate. Little arrows indicate the presence of protofibrils in the middle transmission electron microscopy (TEM) panel. Image magnification: × 40,000, for oligomers and larger fibrils; for protofibrils, lower right scale bar equals 200 nM.
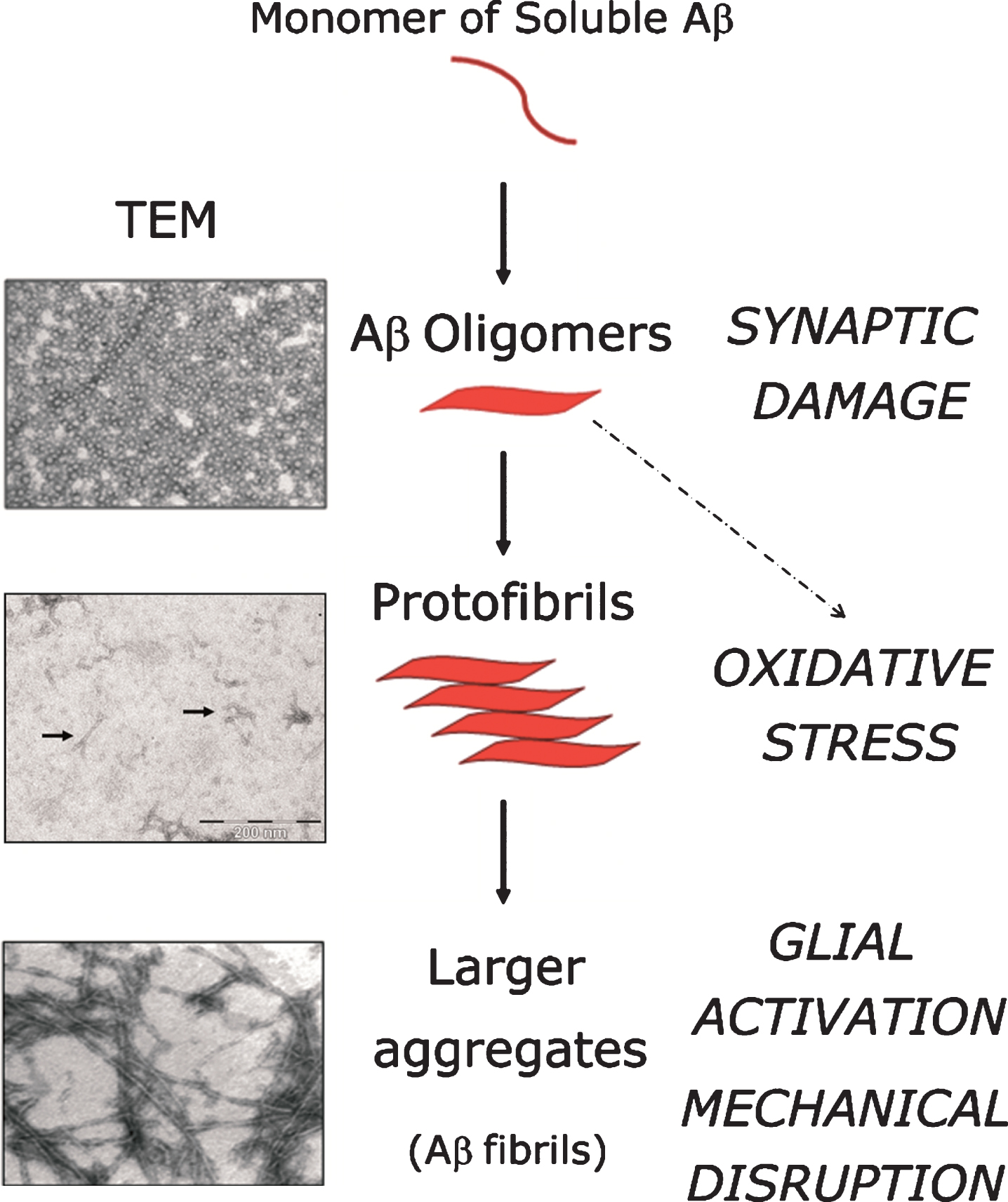
However, not fibrillar Aβ — but its oligomeric forms — are thought to be more decisive determinants of Aβ-mediated neurotoxicity. Aβ-oligomers cause synaptic dysfunction (Fig. 3), a phenomenon believed to be at the base of the neurological symptoms affecting AD sufferers [94]. Interestingly, Aβ-oligomers trigger a burst in oxidative stress through the activation of N-methyl-D-aspartate (NMDA) receptors, causing synaptic damage [95]. In this regard, the fact that synaptic loss strongly correlates with the cognitive impairment occurring in AD [96] (reviewed in [97]), whereas fibrilar Aβ deposition correlates poorly with cognitive phenotypes, further contributes to support the notion of the preeminence of Aβ-oligomers to neuronal toxicity in comparison to its larger aggregates. Nonetheless, this issue is not empty of controversy, as the exact molecular composition of oligomers and their precise mechanism of action remains elusive. Some authors claim that, despite the fact that soluble oligomers induce several features of AD pathology, this does not necessarily imply that Aβ-aggregates and plaques are devoid of neurotoxic effects significant to the overall neurodegenerative process. From a purely mechanical perspective Aβ-aggregates are able to disrupt axonal and dendritic trafficking, therefore contributing to neuronal toxicity [98]. Large Aβ-aggregates can also activate the microglia and astrocytes surrounding the plaques, leading to pro-inflammatory responses comprising the release of excitatory aminoacids, contributing thus to induce exitotoxicity in neighboring cells, as well as the release of oxidative species which, as mentioned above, stimulate BACE1 transcription [93] and translation [70]. Hence, while the relative contribution of oligomers, protofibrils and larger Aβ-aggregates remains a subject of intense debate, the relevance of BACE1 is not affected by this controversy, being the activity of this protease, together with that of the γ-secretase complex, the proximal cause of Aβ peptide production and all its ensuing aggregative forms.
Therefore, BACE1 occupies a central place in AD molecular pathology, constituting, from a strictly logical standpoint, a suitable target for Aβ-lowering therapies designed to treat AD [99]. At the same time, however, careful simultaneous attention should be drawn to the myriad of physiological substrates cleaved by BACE1 in order to prevent unwanted side-effects resulting from BACE1 inhibition [100–103]. Furthermore, an important fact must be borne in mind, which is of special interest for this review: in the brain of SAD sufferers there is an increase in BACE1 protein, which is not paralleled by a rise in BACE1 transcript levels [104–107]. In other words, when coming into terms with BACE1 in SAD we enter the realm of either abnormally increased protein translation or abnormally decreased protein degradation.
The details of the molecular and cellular mechanisms controlling these two aspects of BACE1 biology will be covered throughout the following chapters.
MOLECULAR MECHANISMS GOVERNING BACE1 TRANSLATION
The observation that BACE1 protein expression was upregulated in brains of SAD patients [104, 105] independently of the levels of its preexisting mRNA clearly suggested the existence of translational mechanisms at work with a potential influence in the pathogenesis of AD, stimulating therefore an active research in the field directed to uncover the molecular determinants governing BACE1 translation from its transcript.
In particular, BACE1 5’untranslated region (5’UTR) presented homologous features when compared with the 5’UTRs of genes (such as proto-oncogenes or cytokines) whose translation was known to be strictly regulated in accordance with an optimal physiological performance of these gene-products in the context of delicate cellular equilibriums. In this regard, whereas most vertebrate transcripts bear short (less than 200 nt) and unstructured 5’UTRs, BACE1 5’UTR on the contrary was long (∼500 nucleotides), had a rich GC content (∼75%), contained upstream initiation codons (uAUGs) and was highly conserved among species. All these features were indicative of an important regulatory role of BACE1 5’UTR over the ensuing translation of that protease so intimately linked to Aβ-peptide production. Therefore, the mysteries of BACE1 translational activation caught the attention of molecular biologists active in the AD field, in a quest that was foreseen to be of pivotal importance for attaining a better comprehension of AD pathogenesis.
In this chapter, we first describe the molecular mechanisms intervening in translation initiation, with a special emphasis on those molecular players that are relevant to a specific model proposed for BACE1 translation initiation. We continue describing the original works that revealed how the molecular determinants present in BACE1 5’UTR governed BACE1 translation, an exciting scientific race that culminated in the quatriennium 2004-2007, when the results of this enterprise (widely based on extensive molecular biology approaches) came gradually to the public domain. Noteworthy, as in any active research field with different contributors spread around the world, the results of these works were not unanimously aligned: controversies arouse specially in the attribution of the relative importance of uAUGs as mediators of BACE1 translational control.
Hence, after describing the current state of the art regarding the knowledge about BACE1 5’UTR role in regulating translation, we pay special attention to the “uAUGs controversy” and its possible interpretations. Then, we move forward following the thread leading from the mechanistic implicit in uAUGs as molecular determinants influencing BACE1 translation to its wider connection with cellular physiopathological scenarios that converge on the modulation of ternary complex availability, which are tripartite molecular assemblies crucially involved in translation initiation.
Translation initiation at a glance
This chapter recapitulates some fundamental aspects of translation initiation, which will enable the non-translation expert to follow easily the implications beyond the leaky scanning & reinitiation model proposed for BACE1 translation initiation (see next section).
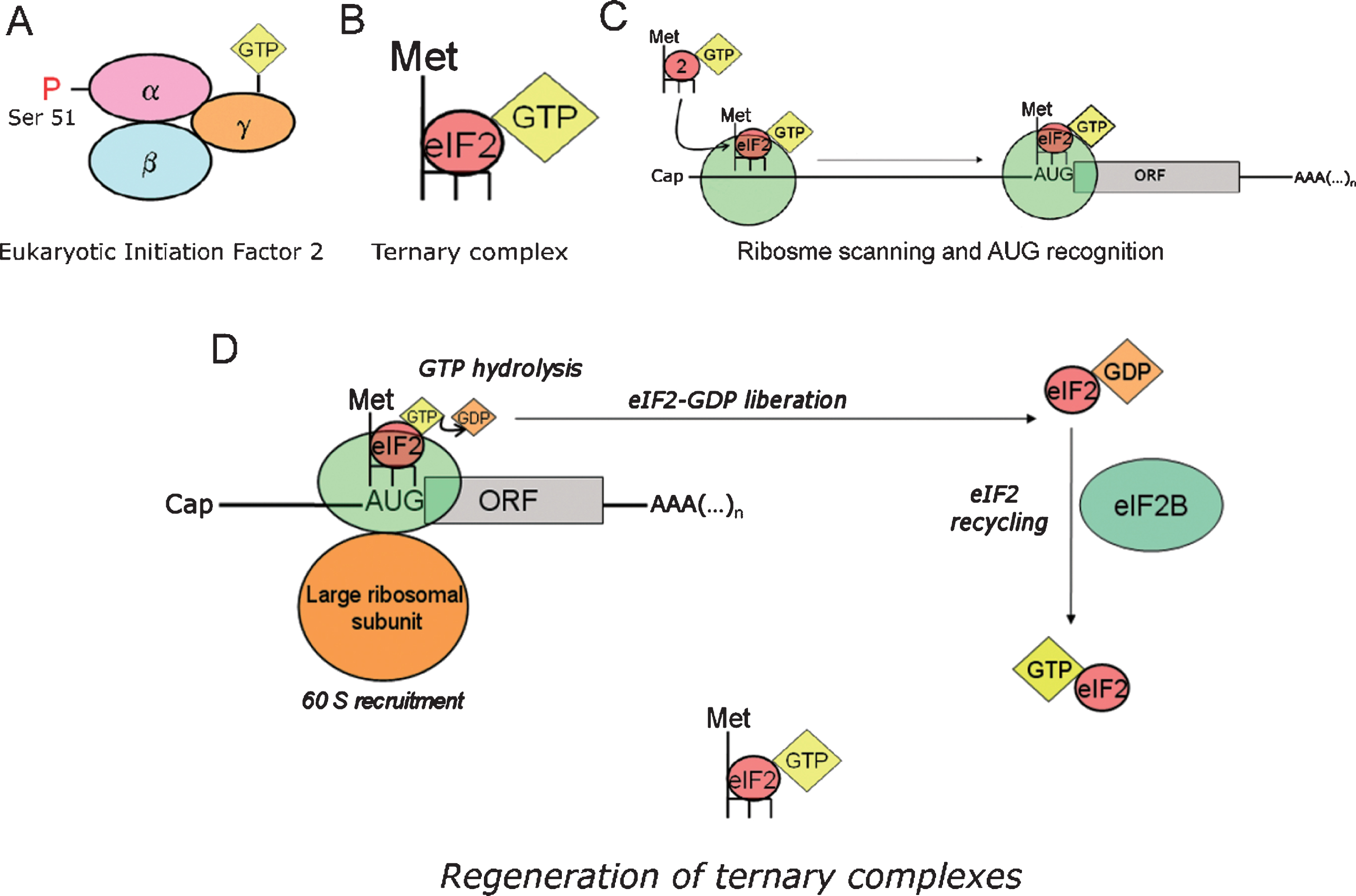
To initiate protein translation, the first Methionine of the nascent polypeptide chain (initiator Methionine, Meti) is loaded into an initiator transfer RNA (tRNAi). The latter provides the molecular basis (anticodon) for recognizing the initiation codon (AUG) in the mRNA through base-pairing. In initiation codon recognition, a tripartite interaction takes place between the small ribosomal subunit (40S), the RNA sequence (belonging to the transcript to be translated) and a ternary complex containing Met-tRNAi,eukaryotic initiation factor-2 (eIF2) and GTP (Fig. 4A–C). The formation of the ternary complex is thermodynamically favored by eIF2-GTP, whereas other initiation factors intervene favoring the loading of eIF2-GTP onto the 40S ribosomal subunit. For AUG recognition and commitment of the ribosome to complete the initiation pathway, the consumption of the energy contained in the third phosphate bond of the GTP molecule is necessary, a process rendering a low energy eIF2-GDP complex as an end-product. Thus, to maintain protein synthesis as an active metabolic process, a third player enters the scene: the guanine-exchange factor eIF2B, which restores the lost GTP molecule to eIF2 so that fresh active ternary complexes can be formed anew to foster translation initiation (Fig. 4D).
Fig.4
Role of ternary complexes in translation initiation. eIF2 recycling & ternary complexes regeneration. A) Eukaryotic initiation factor 2 (eIF2) is a heterotrimeric protein consisting in three subunits (α, β, γ). eIF2 binds a GTP molecule in its γ-subunit, while phosphorylation at serine 51 of the α-subunit modulates its interaction with eIF2B. B) eIF2 forms a ternary complex with the initiator transfer RNA coupled to the corresponding Methionine (Met-tRNAi), and a GTP molecule docked at the γ-subunit of eIF2. Note that tRNAi appears depicted in the diagram as a dark backbone finishing with three tips symbolizing the anticodon. C) Ternary complexes (the formation of which is thermodynamically favored by eIF2-GTP) are loaded in to the 40S ribosomal thanks to the intervention of other translation factors. D) Translation initiation is a GTP-dependent process. The energy resulting from the hydrolysis of the eIF2-bound GTP is required for initiation codon recognition and commitment of the ribosome to complete the initiation pathway. As a result, a low energy eIF2-GDP complex is released after each round of translation initiation. eIF2B is the guanine exchange factor (GEF) refueling eIF2 with new energy-rich GTP molecules. This process yields new active ternary complexes ready to engage in new cycles of translation initiation.
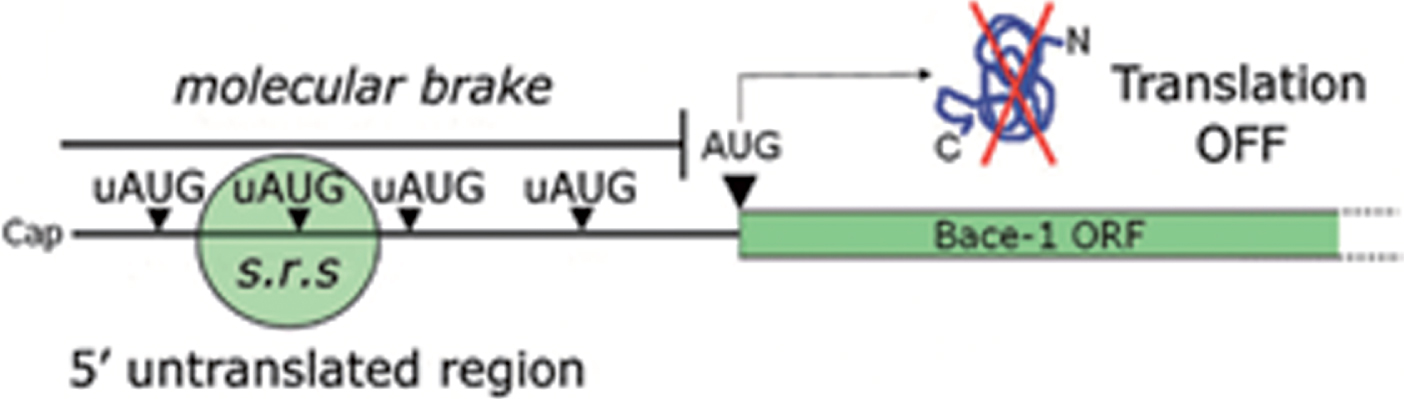
Anticipating what will be thoroughly discussed in the next chapter, efficient recognition of upstream AUGs (uAUGs, i.e., initiation codons located upstream of the main AUG) in the BACE1 5’UTR prevents the translational machinery from reaching the main BACE1 open reading frame (ORF), thereby shutting down protein translation (Fig. 5). The question, however, is how this repression may be eventually bypassed: which is the mechanistic basis accounting for BACE1 translation de-repression?
Fig.5
uAUGs contribution in gating BACE1 translation. BACE1 transcript leader, alternatively named 5’untranslated region (5’UTR), hinders BACE1 translation initiation under basal conditions. Upstream initiation codons (uAUGs) present in BACE1 5’UTR “seduce” the small ribosomal subunit and prevent it from reaching the main open reading frame (ORF). As a consequence, BACE1 translation will be kept at a low, insignificant rate. This situation will be reversed only following a drop in ternary complex availability.
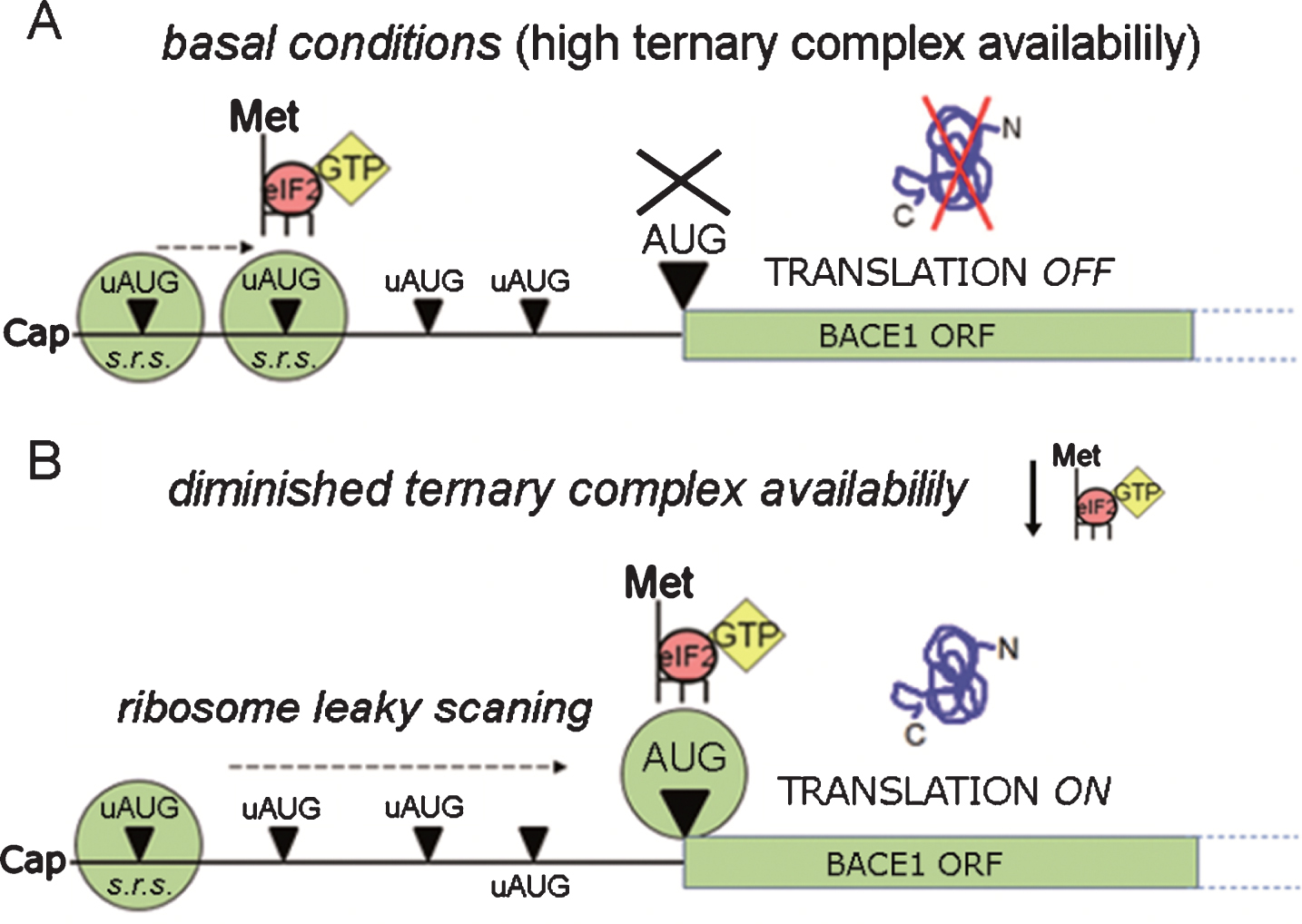
Summarizing in just a few words, such translational repression as that of BACE1 can be overcome when there is a drop in ternary complex availability. The former assumption implies, nonetheless, the preeminence of a leaky scanning & reinitiation model applied to BACE1 translation (see next section). In this particular scenario, the ribosome has a lower probability of encountering an active ternary complex when scanning through the uAUGs, and therefore has an enhanced probability to skip them, reaching thereby the main AUG to start protein synthesis: this is how the leaky scanning & reinitiation model accounts for the activation of the otherwise repressed BACE1 protein synthesis (Fig. 6).
Fig.6
BACE1 translation facilitation. BACE1 translational repression can be by-passed by ribosomal leaky scanning & reinitiation, a condition requiring a lowering in ternary complex availability. As depicted in the diagram, when ternary complex availability is high (A, basal conditions) translation initiation can occur at high frequency at the uAUGs, preventing translation of BACE1 from its main ORF. Conversely, when the availability of ternary complexes diminishes (B) there is a drop in the formation of active ribosomal complexes, leading to a decreased recognition of upstream AUGs (ribosome leaky scanning) and to a and more frequent recognition of the main ORF allowing BACE1 protein synthesis to start. s.r.s., small ribosomal subunit.
Hence, BACE1 protein expression is facilitated in conditions that lower the availability of ternary complexes. Such thing happens when the regenerating activity of the guanine exchange factor eIF2B is blocked, which can occur through two alternative paths (Fig. 7): directly, by glycogen synthase kinase 3 beta (GSK3β)-mediated inhibitory phosphorylation [108], or indirectly, through competitive blockade by phosphorylated eIF2α [109]. Furthermore, as eIF2α is expressed at much higher levels than its counterpart eIF2B, modest shifts in eIF2α phosphorylation suffice to block eIF2B activity considerably.
Fig.7
Intracellular signaling converging in eIF2B inhibition. The guanine-exchange activity of eIF2B can be modulated in response to stimuli conveyed by two alterative pathways. On the one hand, phosphorylation of the eukariotic initiation 2-alpha (eIF2α) is mediated by four different stress-activated eIF2α kinases (PERK, PKR, HRI, and GCN2), resulting in a competitive blockade of eIF2B. On the other hand, the otherwise constitutively inhibited Glycogen synthase kinase 3β (GSK3β), when released from its inhibition, catalyzes a direct inhibitory phosphorylation upon eIF2B. s.r.s., small ribosomal subunit.
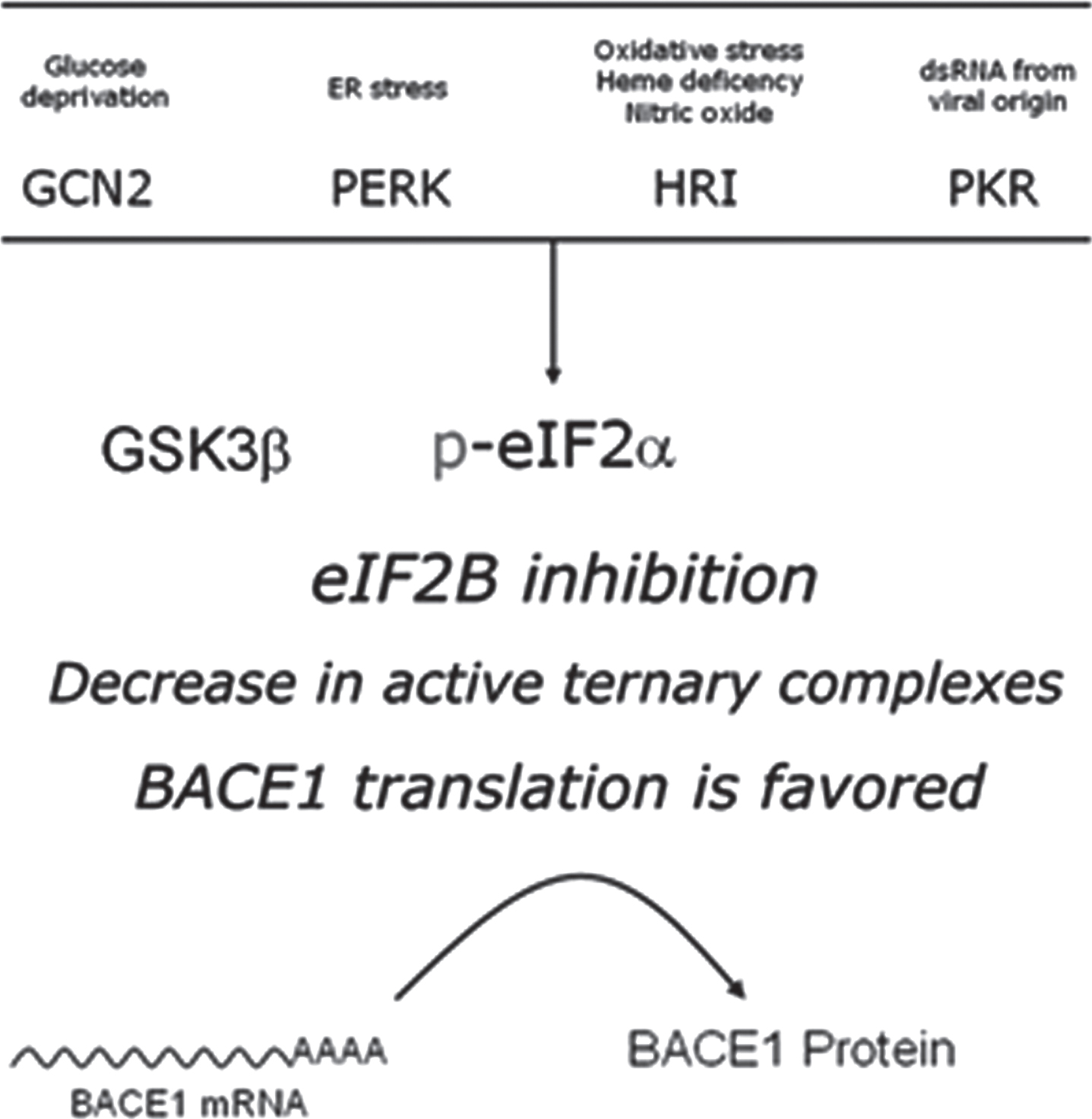
Therefore, intracellular signaling cascades culminating in either GSK3β activation or eIF2α phosphorylation have the potential of facilitating BACE1 protein synthesis from its translationally dormant transcript (Fig. 7). Remarkably enough, GSK3β is one of the main kinases responsible for protein tau phosphorylation[110] (reviewed in [111]); therefore, it is worth noticing how GSK3β activity may constitute a node mechanistically connecting amyloidogenesis (through BACE1 translational activation) and neurofibrillary tangles (through tau phosphorylation).
The discovery of translation initiation determinants present in BACE1 5’UTR
Two pioneering works devoted to the study of BACE1 transcript leader run their course with a striking chronological coincidence in the northern and southern sides of the Alps mountain range, a casual geographical symmetry that was added to the synchrony in time and scope of these two seminal papers on BACE1 5’UTR, which were carried out in laboratories located at the cities of Milano and Munich respectively [112, 113].
The first of these two reports by De Pietri Tonelli and colleagues [112] appeared published on-line at the beginning of March 2004. The author’s initial observation was that one of the uAUGs in BACE1 5’UTR (the second one) was in a good context for translation initiation, which meant that this uAUG could potentially be recognized by the ribosome with at least the same efficiency as the main AUG, leading thereby to translational inhibition of the main BACE1 ORF (Fig. 5). By working with reporter genes, the authors demonstrated that—as predicted—the second uAUG was efficiently recognized by the translational machinery and exerted a prominent repression on translation, while mutations of the three other uAUGs had an additive effect in releasing the translational block imposed by the BACE1 5’UTR. In accordance with these findings, the Milano group hypothesized that ribosome skipping through the uAUGs (leaky scanning) was the putative mechanism by which the main BACE1 ORF was eventually reached, being the second uAUG, which as a matter of fact was positioned in a better Kozak context for translational initiation, the main responsible for the overall inhibitory effect on BACE1 translation. Importantly, De Pietri Tonelli et al. were the first to propose, in line with the core of their original discovery, that eIF2α phosphorylation could be a mechanism underlying BACE1 translational de-repression through the avoidance of ribosomal recognition of the second uAUG.
A second report by a group in Munich came to light soon after, appearing in its on-line version at the end of May 2004. In this work, Lammich et al. [113] also investigated the putative inhibitory effect of BACE1 5’UTR over translation initiation. In agreement with their colleagues in Milano, they too brought forth compelling evidences that BACE1 long transcript leader caused translational repression, showing in addition that the 3’UTR did not have any effect on translation. However, Lammich et al. approached the study of BACE1 5’UTR directing their attention primarily to the fact that, besides the presence of uORFs in good context for translation initiation and its potential inhibitory effect on translation, the high GC content found in the long BACE1 5’UTR could induce its folding into stable secondary RNA structures that would compromise translation initiation. By mutagenesis approaches targeting the uAUGs followed by transfection into a HEK293-AβPP cell line, the authors showed that the main cause of BACE1 protein expression repression in this model was the presence of the integral 5’UTR sequence, whereas mutation of the single uAUGs (either individually, or combined) contributed only modestly to rescue BACE1 protein expression, which was never found to reach the high levels of expression detected by immunoblot when the empty plasmid lacking the 5’UTR was transfected. This result supported the hypothesis that the GC-rich sequence in BACE1 5’UTR formed a tightly folded secondary structure which was the major element determining translational repression. At this point, to further investigate this phenomenon, Lammich and colleagues proceeded to inflict ablations upon larger regions comprised within the BACE1 5’UTR, determining thereby that wider regions of the sequence—and not single upstream initiation codons—caused the largest part of the translational block exerted over BACE1 expression when exogenously transfected. Thus, they concluded that the main responsible of suppressing BACE1 protein expression were not the uAUGs, but the GC-rich region interspersed throughout the 5’UTR, which formed a constitutive translation barrier able by itself to prevent the ribosome from effectively translating BACE1 mRNA, while the uAUGs accounted only to a minor extent in repressing BACE1 translation.
To add more complexity to this intricate issue, a third report had appeared (also at the beginning of March 2004) by Rogers et al. [114] pointing toward yet another different mechanism, in addition to eIF2α phosphorylation, by which the translational repression imposed by BACE1 5’UTR could be overcome: ribosomal shunting, a mechanism originally described in viral RNAs by which the ribosome skips large regions of mRNA containing stable secondary RNA structures and/or uORFs to land upstream of the main initiator AUG to start translation. Two years later however, Zhou and Song published a new report about BACE1 translation [115] in which they discarded ribosomal shunting considering it to be a rare mechanism in mammals. Bringing back the argument to the point originally raised by De Pietri Tonelli and colleagues [112], Zhou et al. reported that the fourth uAUG of the human BACE1 5’UTR could function efficiently as a translation initiation codon and had an inhibitory effect on translation, while the rest of the uAUGs were skipped over as translation initiation codons by leaky ribosomal scanning. The fourth uAUG in Zhou et al. work corresponded to the second uAUG pointed out to be the main responsible for BACE1 translational inhibition in De Pietro Tonelli et al. work two years before [112]. The reason for this unmatched numbering of uAUGs was to be found in the fact that the recent work published by Zhou et al. had been carried out using a larger BACE1 5’UTR (transcribed from a ∼200 nucleotides upstream tsp) which contained two extra uAUGs. Thus, based on their new results, Zhou et al. concluded that leaky scanning in the 5’UTR and reinitiation at the physiological BACE1 AUG site were responsible for the low levels of expression of BACE1 under basal conditions, and therefore the original hypothesis by De Pietri Tonelli et al. on the preponderant effect of the second/fourthu AUG on translational repression regained momentum.
One year later, in 2007, the intellectual challenge brought about by Lammich and colleagues, who had stressed the preeminence of the GC-rich translational barrier over the inhibitory role of uAUGs [113], was responded by the Milano group with a work in which an extensive mutational analysis of uAUGs had been carried out. In this report, Mihailovich and colleagues [116] convincingly showed, mainly through the use of reporter constructs, that all four uAUGs affected translation, but to a different extent, being the second uAUG efficiently recognized whereas the others were probably skipped by leaky scanning. This was then the third report highlighting the second uAUG as the main inhibitory element in BACE1 5’UTR, while, according to the author’s report, mutation of the other uAUGs contributed to fully de-repress translation.
Although the results obtained by this approach were compelling, an ulterior concern about its interpretation arouse. The use of reporter constructs (in which the native sequence of BACE1 ORF was replaced by that of firefly or renilla luciferases) was regarded as potentially misleading because, it was thought, an exogenous sequence cloned immediately downstream BACE1 5’UTR might abrogate, or at least alter, the formation of stable secondary structures in the vicinity of the 5’UTR with the ORF, underrating therefore its barrier effect on translation.
Nonetheless, a rather similar criticism could be directed to the transfection experiments by Lammich et al. [113] seemingly showing a predominant inhibitory effect of the GC-rich translational barrier. In the presentation of these experiments, the reference value for BACE1 protein expression was set to be the empty plasmid lacking the 5’UTR, which indeed reached very high levels of expression indicative of the inhibitory role exerted by the full-length 5’UTR. From this primal observation, the authors proceeded to evaluate the possibility to rescue BACE1 expression by mutating the uAUGs, showing that the very high levels yielded by the empty plasmid were never reached when the uAUGs had been mutated. However, it should be noted that this setting does not correspond with the real physiological scenario, in which the transcript encoding for BACE1 always bears the full length 5’UTR [116]. Therefore, to assess the relative importance of the inhibitory effect of AUGs on translation, one must definitely take into account the presence of the full-length 5’UTR itself, a condition that is irremediably lost by the ablation approaches carried out by Lammich and colleagues [113].
Indeed, defining the specific relevance of the uAUGs is not a simple issue. The use of analytical molecular biology techniques, reporter constructs, mutagenesis, deletions, necessarily fails to grasp the wholeness of the phenomenon, i.e., the actual occurring interplay among all molecular determinants present in BACE1 5’UTR, and its biological significance in repressing the expression of this protease. Probably, the central question is not whether the folding into stable secondary structures predominates over the inhibitory role exerted by the uAUGs or vice versa, but to which extent these two determinants complement themselves to govern the rate of BACE1 translation initiation. Most likely, translation factors with helicase activity may be differentially modulated in different cell types and/or physiological states, thereby unwinding secondary structures along BACE1 5’UTR to different extents, which in turn would let BACE1 translation activation rest upon mechanisms related to uAUGs leaky scanning (such as eIF2α phosphorylation). This would give BACE1 translational activation a certain plasticity of output according to subtle changes in different pathophysiological cellular states.
In addition, however, a strong hint about the importance of the uAUGs emerges from their evolutionary conservation among orthologues, which is suggestive per se of a relevant biological function associated to these upstream initiation codons. Furthermore, the mutational analysis carried out by Lammich and colleagues, which was exempt of the putative confounding effect on folding produced by exogenous ORFs in reporter-based studies, in fact showed that uAUGs did repress translation when compared with the levels of BACE1 protein expression yielded not by the empty plasmid, but by the plasmid containing the full length 5’UTR [113], which in fact was the plasmid encoding for the naturally occurring version of BACE1 transcript.
In any case though, admitting that the uAUGs contribution to the overall inhibitory effect of BACE1 5’UTR might be modest in quantitative terms does not mean it is irrelevant (Fig. 5). Indeed, a modest facilitation on BACE1 translation might be crucial, either in the physiological scenarios in which this protease is involved, or in the critical catalysis of AβPP cleavage to produce the neurotoxic Aβ-peptide. In particular, the importance of uAUGs gains importance before our eyes when we direct our gaze to the intracellular signaling cascades diminishing ternary complex availability, enabling ribosomal leaky scanning and there by facilitating BACE1 translation initiation; in other words, when we draw the picture of the stimuli able to induce eukaryotic initiation factor 2 B (eIF2B) inhibition, either directly, or indirectly, through competitive blockade by phosphorylated eIF2α (Fig. 7).
Indeed, contemplating this diagram we are faced with a panorama in which a myriad of cellular stresses can be transmuted into a cellular/molecular state in which BACE1 translation is favored. But ... are really all of these potential BACE1 activators just cellular stressors?
No!
Nitric oxide, a neurotransmitter and neuromodulator [117–123] involved in memory processing [124–127] (reviewed in [125, 128]), activates the Heme-regulated eIF2α-kinase (HRI) in an analogue manner in which it activates the guanyate cyclase: by binding to the heme motifs present as prosthetic groups in this fascinating eIF2α-kinase [129–133].
Nitric oxide signaling as an activator of BACE1 translation: Role in synaptic plasticity and memory consolidation
This story began in a laboratory by the sea, in the ebullient Mediterranean city of Barcelona. An observation was made at the turn of the years 2005-2006: stimulating neuroblastoma cells with physiological concentrations of nitric oxide, BACE1 protein expression was upregulated in the order of minutes. Such rapid response suggested a translational activation from a pre-existing mRNA, rather than de novo gene-transcription. With this premise in mind, inquiries were made to check out whether cyclic GMP, the canonical downstream mediator of nitric oxide signaling, could activate protein translation in any possible manner. Unfortunately, though, nothing convincing was found in the literature. This initial frustration, however, was soon to be compensated, and it was with an enthusiastic joy that the works on the molecular mechanisms controlling BACE1 translation, which had been published little before [112–114], were received. These brand-new scientific reports wholly devoted to reveal the mechanistic underlying BACE1 translational activation were indeed the right food needed at that very moment, when the attempt was being made to connect nitric oxide signaling with mechanisms that could potentially account for BACE1 translational activation.
In this regard, the contribution by De Pietri Tonelliand colleagues [112] was particularly inspiring, for the authors suggested eIF2α phosphorylation as a putative mechanism to trigger BACE1 translation, an assertion that converged with the simultaneous confirmation that one of the eIF2α-kinases, the HRI kinase, could be directly activated by nitric oxide binding on its heme-groups [131]. Therefore, a three-step pathway (NO/HRI/p-eIF2α) was envisaged as a putative signaling cascade driving BACE1 translation (Fig. 8).
Fig.8
Three-step pathway model for NO-induced BACE1 translation activation. The heme-regulated eIF2α kinase (HRI), a nitric oxide sensor, appeared to be a plausible node connecting nitric oxide (NO) stimulation with BACE1 expression, through phospho-eIF2α-mediated BACE1 translational de-repression. Other uAUG-bearing transcripts such as GluN2B [149] respond in a similar way to NO/HRI induced eIF2-phosphorylation, opening the possibility that NO behaves as a general translational facilitator for uAUG-bearing transcripts.
![Three-step pathway model for NO-induced BACE1 translation activation. The heme-regulated eIF2α kinase (HRI), a nitric oxide sensor, appeared to be a plausible node connecting nitric oxide (NO) stimulation with BACE1 expression, through phospho-eIF2α-mediated BACE1 translational de-repression. Other uAUG-bearing transcripts such as GluN2B [149] respond in a similar way to NO/HRI induced eIF2-phosphorylation, opening the possibility that NO behaves as a general translational facilitator for uAUG-bearing transcripts.](https://content.iospress.com:443/media/adr/2019/3-1/adr-3-1-adr180089/adr-3-adr180089-g008.jpg)
The fact that the initial experiments carried out on neuroblastoma cells were designed trying to emulate neuronal nitric oxide synthase (nNOS) physiological production of nitric oxide [134, 135] (by adapting the concentration of nitric oxide to the nanomolar range, and the exposure time to the order of minutes), led to the hypothesis that nitric oxide-mediated eIF2α phosphorylation could actually be driving BACE1 translation physiologically.
At this point, however, an intriguing question arouse. Intracellular cascades converging in eIF2α phosphorylation were in fact a cellular adaptive response to environmental stress, leading either to alleviate cellular injury managing stress conditions (securing metabolic resources by shutting down protein synthesis), or directly promoting apoptosis when the crisis caused by stress could not be halted [136]. Was it then possible that such a stress–triggered signaling cascade could have evolved to accomplish a differentiated physiological role in neurons? Taking into account the fact that the phosphorylation of this translation factor was known to be strictly related to stress adaptation and cellular death... could any genuine CNS function such as synaptic plasticity or memory consolidation could rest upon eIF2α phosphorylation?
This indeed stressing question had just started to be brilliantly answered from the other side of the Atlantic, in a laboratory located in Canada, at the French-speaking city of Montr
However, while the involvement of eIF2α phosphorylation in synaptic plasticity and memory consolidation seemed quite clear from the experimental evidences brought about by the just mentioned works, the ultimate role played by this factor in the cellular/molecular events related to the so-called cellular phase of memory consolidation remained less clear. According to their results, Costa-Mattioli and colleagues working idea was the notion that memory consolidation required eIF2α dephosphorylation to allow local protein synthesis at synapses [a process known to be required for memory consolidation [63]]. However, to be dephosphorylated a protein needs to be phosphorylated first. Therefore, such view supporting a “decreased eIF2α phosphorylation” as a critical process underlying memory formation necessarily implied a previous phosphorylation event targeting eIF2α, an issue that was not tackled in these two initial reports [137, 138]. (See [67, 139–141] for additional reviews discussing eIF2-alpha dephosphorylation in LTM).
Following the same line of reasoning about the relevance of eIF2α dephosphorylation in promoting LTM, two years later, in 2009, it was proposed that activity–dependent changes in synaptic strength would increase amino acid import into neurons inactivating thereby GCN2 [67] (an eIF2α-kinase which is conventionally activated by unloaded tRNAs lacking its amino acidic part). This hypothesis synthesized the original findings on the GCN2–/–mice (published in 2005[137]) with the pharmacogenetic work published two years before, in 2007, which had so strongly contributed to postulate eIF2α dephosphorylation as a pivotal mechanism in LTM formation [138].
However, while perfectly possible in speculative terms, this hypothesishad a major drawback: it presupposed a resting state in synapses in which eIF2α phosphorylation would be constitutively high, while synaptic activity would “release the brake” dephosphorylating eIF2α through the inactivation of GCN2, allowing thereby protein synthesis to take place. Accordingly, as a direct consequence of this hypothesis, the basal state at synapses would be that of total protein synthesis inhibition, something indeed possible (for in biology, impossible is nothing) but rather unlikely as it would permanently prevent protein turnover of locally translated proteins at resting synapses all over the brain.
Analternative view to these matters, not denying any of the previous findings, but incorporating them under another interpretative framework, was the possibility that following glutamatergic stimulation, synaptically derived nitric oxide would activate the HRI kinase causing thereby a dynamic phosphorylation of eIF2α which would in turn promote the timely synthesis of uORF-containing 5’UTR-bearing transcripts (such as BACE1). Then, only afterwards, such a fast response would rapidly return to the basal resting state through eIF2α dephosphorylation. Furthermore, due to the highly diffusible nature of nitric oxide, in this model specific local protein synthesis would be activated homogeneously both in the post- and pre-synaptic compartments (Fig. 9).
Fig.9
Proposed model for eIF2α-mediated translational control at the synapse. In a resting state exempt of cellular stress, nitric oxide (NO)-HRI signaling induces a peak in eIF2α phosphorylation levels, which are rapidly turned back to their basal state through the action of eIF2α-phosphatases. Such transient rise in eIF2α levels triggers the timely translation of synaptically polarized uAUG-bearing mRNAs, while temporarily arresting general protein translation. Importantly, such signaling model can only be effective when cellular stress does not interfere with eIF2α phosphorylation through the action of other eIF2-kinases (PERK, PKR, GCN2) or even an eventually stress-activated HRI.
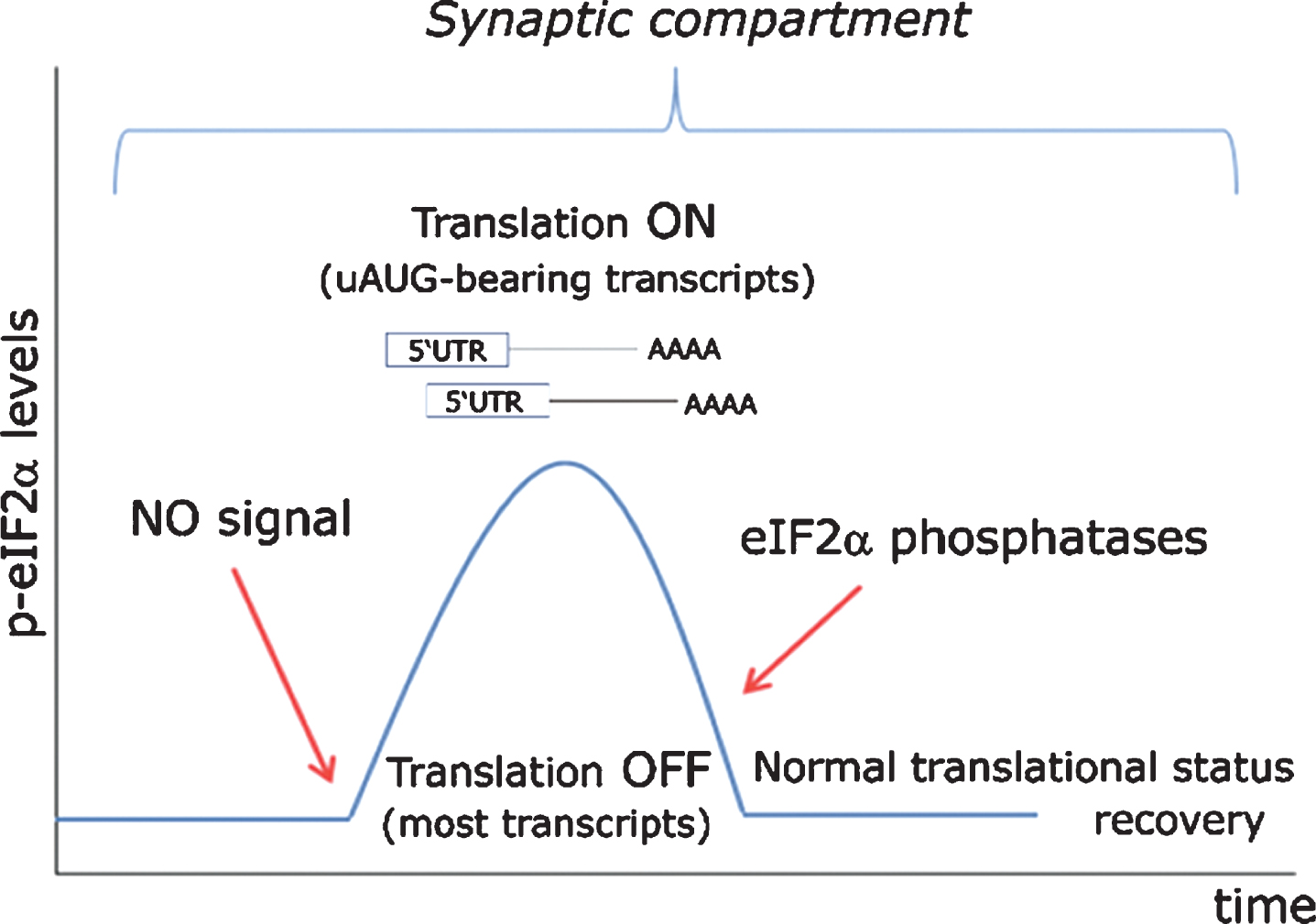
Such alternative view received its first experimental support from a group based in Porto Alegre, the energetic southernmost capital of Brazil in which all virtually existing human types are harmoniously blended. Issued from this austral city, a short communication appeared in the spring of 2013 demonstrating the requirement of HRI kinase activity in the hippocampus for the consolidation of object recognition memory [142]. By a classical neuropharmacological approach, the authors showed that eIF2α was actively phosphorylated in the CA1 region of the dorsal hippocampus after behavioral training, an effect seemingly subordinated to HRI activity. As expected, this phenomenon was accompanied by an increased expression of BACE1 and ATF4, two proteins synthesized from uAUGs-containing mRNAs. Stereotaxical microinjection of AMI, a specific HRI inhibitor, hindered such molecular response and abrogated memory retention.
However, a certain fringe still remained to be deepened, as HRI—a kinase originally identified in rabbit reticulocytes [143] and mainly studied in the context of hemoglobin synthesis regulation [144]—had never been detected in the CNS before. Such experimental evidences were only published two years later, in 2015, when the original findings from the Barcelona group (pursued since almost one decade before) came finally to the public domain [145]. In this report, the authors presented compelling evidences that HRI kinase was localized at hippocampal synapses, where it regulated BACE1 translation through the activation of eIF2α phosphorylation upon nitric oxide signaling. Working with synaptosomes, which are nuclei-free preparations suitable for studying transcription-independent events, they showed that BACE1 expression becomes upregulated in response to glutamate stimulation, in an effect depending simultaneously on 1) nNOS, 2) HRI, and 3) eIF2α phosphorylation. Furthermore, the previous finding that HRI was required for memory consolidation in rats [142] was extended to a mice model with the same positive conclusion [145].
In fact, not only HRI, but in particular the other three eIF2α-kinases PERK, PKR and GCN2, have been a subject of intense research as mediators of both general and gene-specific translational control in the context of synaptic plasticity and memory formation. This effort has brought forth a wealth of experimental data resulting from genetic, molecular, electrophysiological and pharmacological studies (see [146–148] for reviews on this topic). Nonetheless, there is still very little information concerning whether these kinases are activated under normal physiological conditions in neurons. Indeed, a major unresolved goal in the field is the investigation of the biochemical signaling cascades that couple neurotransmitter and neurotrophin receptors to the activation/inactivation of eIF2α kinases, a slippery fish-like issue that has remained a gap in knowledge since at least the last decade [61].
In this regard, nitric oxide activation of HRI could be one possible biochemical pathway by which NMDA receptor activation is coupled to eIF2α phosphorylation to promote specific translation of uAUGs-bearing mRNAs (see [149] for a report on GluN2B translation by the NO/HRIpathway in cortical neurons). At the same time, however, little doubt can be harbored that additional complex regulatory events (probably involving the unwinding of secondary mRNA structures and a wider network interplaying with other translation initiation factors) sum up to play a decisive role in determining the final translational output overeach synaptically localized transcript, being perhaps the NO/HRI pathway a mean by which ribosomal skipping in uAUGs-bearing mRNAs is facilitated.
Concerning the interpretation of BACE1 translation being driven by synaptic activity in memory consolidation processes, one must readily admit that this finding stays counter intuitively with the widely held notion that newly synthesized proteins contribute to the building-up of new structures (for example, growth or formation of new synapses) that are ultimately responsible for the storage of the memory trace [150, 151]. BACE1 is a protease; and proteases catalyze the breaking-down of proteins, promoting thereby dismantling rather than building-up processes.
Notwithstanding this apparent contradiction, it should be noted that proteases play a significant role in cell-cell communication, tissue homeostasis and development through the shedding of membrane-bound proteins [reviewed in [152]] and, in the particular case of brain development, a well-orchestrated proteolytic activity is required for proper neurogenesis [153, 154] and axon guidance [155, 156] (reviewed in [157]). In addition, it is becoming increasingly clear that protases intervene in learning and synaptic plasticity [158–160]. ADAM10, for instance, is required for synaptogenesis [161], whereas Matrix Metalloproteinase-9 (MMP9) has a pivotal role in regulating synaptic plasticity, learning and memory when locally translated upon synaptic activation [162]; these findings are aligned with a role for BACE1 local translation in promoting synaptogenesis [145], and share the same underlying interpretation, namely: regulated cleavage of cell adhesion proteins at synapses would enable the structural remodeling necessary for establishing those persistent modifications of synapse structure and function required for learning and memory processes [145, 161, 162].
However, another interpretation is still possible according to which not unilateral synaptic growth, but also breaking-down processes, might be required for thinking, learning and memory processing. Such “physiological death”, when excessive or not duly repaired afterwards, would result in the triggering of neurodegeneration. This would be, perhaps, an interesting bridge to explore: a sort of communicating vessel connecting cognition (thinking, learning and memory processes) with neurodegeneration, and its entailing potential therapeutic implications.
In the particular case of BACE1, such intimate connection among destruction and cognitive performance becomes evident when one realizes that the wiring of stimuli converging in the translational activation of this protease (through eIF2α phosphorylation) defines a tragic setting in which physiologically-controlled proteolysis can only occur in the total absence of cellular stress. Hence, such an intrinsic vulnerability of the system marks a fatal transition between physiological and stress-driven BACE1 expression (Fig. 10).
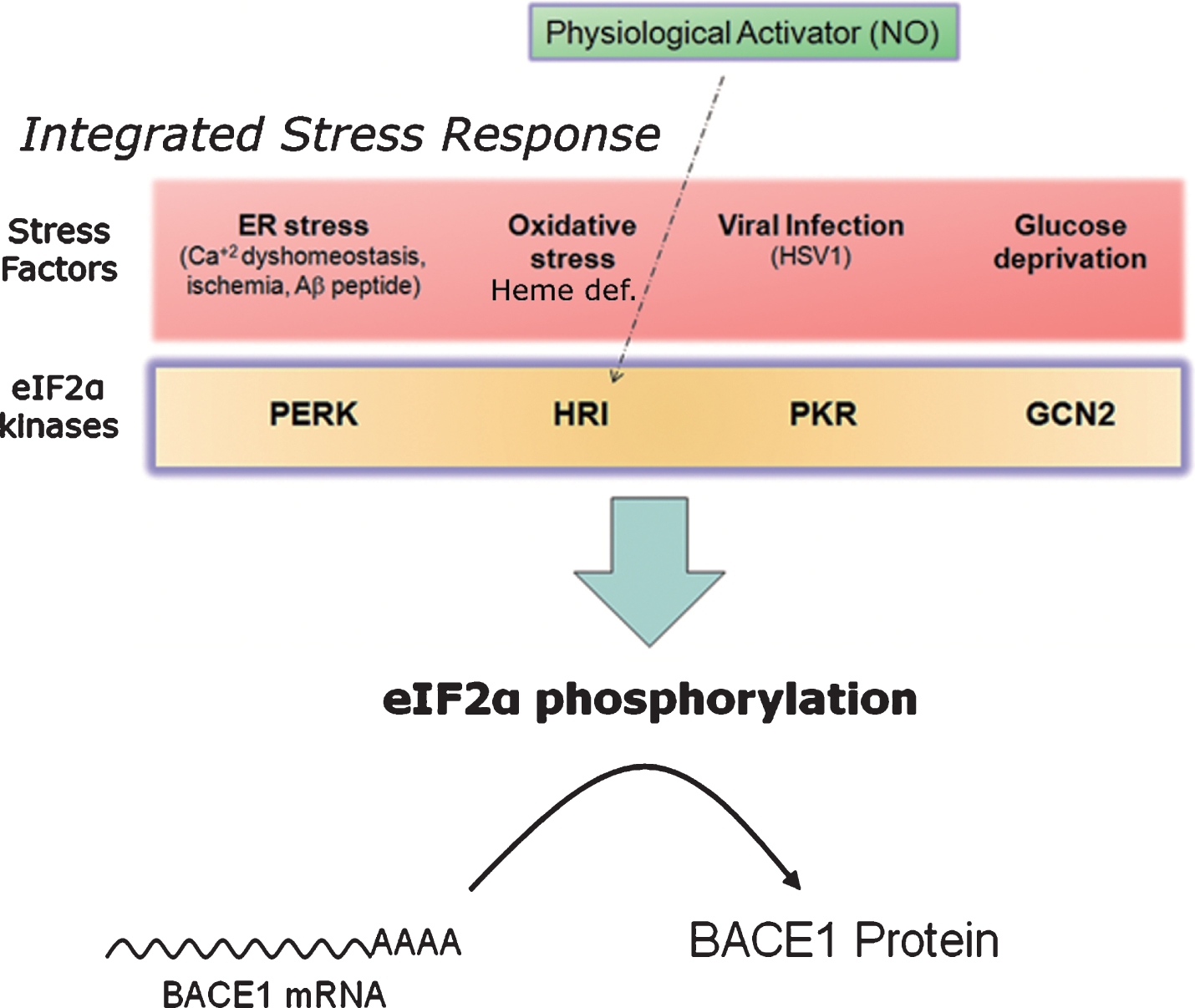
Fig.10
Nitric oxide: a master physiological mediator hovering above the integrated stress response? The four existing eIF2α-kinases (PERK, PKR, HRI, GCN2) share a common catalytic-domain and harbor different activator-domains endowing each kinase with a differential sensitivity to stress. Following stress sensing, eIF2α-kinases phosphorylate eIF2α and shut down protein translation in a process known as integrated stress response. This process aims, for example, at securing metabolic resources under energy deprivation conditions, alleviating protein load in the endoplasmic reticulum under missfolding protein conditions, or at avoiding the translation of exogenous proteins from viral origin. In the absence of stress, nitric oxide (NO) signals through the Heme-regulated eIF2α-kinase (HRI), activating thereby BACE1 translation from its mRNA in a physiological setting.
Stress-driven BACE1 translation: eIF2α phosphorylation
As previously mentioned, eIF2α-kinases are, above all, stress sensors [163]. Deriving from this fact, a kind of road map can be traced leading from different cellular stressors, sensed by the four existing eIF2α-kinases PERK, PKR, HRI and GCN2, to amyloidogenesis, in a straight-line sequential pathway involving: 1) stress sensing, 2) kinase activation, 3) eIF2α phosphorylation, and 4) BACE1 translational de-repression (Figs. 7 and 10).
All these stresses are related in one way or another to brain aging processes. Therefore, after the discovery of the molecular determinants governing BACE1 translation [112–116], an appealing connection emerged between age-related stress and BACE1 translational activation in relation with SAD triggering (see [164] for a review on the role of eIf2α kinases in AD pathogenesis).
This research line was inaugurated in 2008, when O’Connor and colleagues published a report showing that energy deprivation induced eIF2α phosphorylation, BACE1 expression and Aβ peptide production [69]. Surprisingly, they did not find GCN2 to be the mediator-kinase of this effect (something that could have been expected as this kinase is activated by aminoacid deprivation). Instead, they found PERK to be the nexus between energy deprivation and eIF2α phosphorylation, suggesting that ATP depletion probably impairedprotein folding in the ER, leading thereby to PERK activation [69].
Two years later, Devi and Ohno showed a correlation between increased BACE1 expression and p-eIF2α levels in a mouse model of AD [165]. By crossing BACE1 heterozygousmice (BACE1(+/–)) with an aggressive amyloid model (Tg6799 strain, which co-overexpressesAβPP/PS1 and harborsfive FAD mutations), and analyzing BACE1 content at different age stages, they showed that a reduction in BACE1 gene dosage was not enough to diminish BACE1 upregulation in advance age, a phenomenon seemingly related to increased eIF2α-phosphorylation. However, the ultimate nature of the stressing stimuli and the kinases involved in eIF2α phosphorylation were not identified in this work.
Another stress factor correlated to AD is Herpes Simplex Virus Type-1 (HSV1) (reviewed in [166–168]). HSV1 is present in the brains of elderly humans [169], confers risk of AD in APOE ɛ4 carriers [170], causes Aβ accumulation [171] and abnormal tau phosphorylation [172], and colocalizes with amyloid plaques in AD brains [172]. Interestingly, the chief stimulus activating PKR is double-strand RNA (dsRNA) from viral origin [173], suggesting a connection between viral infection and BACE1 expression through the activation of this kinase. In this regard, a report appeared in 2011 showing a relation between PKR activation and increased BACE1 protein translation in the context of HSV1 infection [71]. In this work, the authors found striking PKR activation upon HSV1 infection in mouse dorsal ganglion root neurons, together with neuronal positive PKR staining in brain sections from AD individuals. In addition, by making use of a synthetic dsRNA analog, poly (I:C), they provided evidences that PKR activation induced eIF2α phosphorylation, BACE1 translational de-repression and Aβ production in cultured cells.
As commented before (see above), a stressing factor of major relevance in AD etiology is oxidative stress [90–92], which has been known for some years to induce BACE1 mRNA transcription [93]. Connecting for the first time oxidative stress with BACE1 translational activation through eIF2α phosphorylation, a pioneering report appeared in 2012 by Mouton-Liger and colleagues, which placed PKR as the upstream activated kinase in response to this particular kind of cellular stress of such broad implications in AD etiology [70]. By using AD brain sections, a mice model of AD and hydrogen peroxide as a source to induce oxidative stress in human neuroblastoma cells, the authors showed a correlation between p-eIF2α levels, activated PKR and increased BACE1 levels. In their study, hydrogen peroxide-mediated BACE1 increase in neuroblastoma cells was attenuated by inhibiting the PKR/p-eIF2α pathway.
An evident source of cellular stress in AD is Aβ accumulation itself, which induces a plethora of neurotoxic effects including oxidative stress [95], endoplasmic reticulum (ER) stress [174] and calcium dyshomehostasis [175, 176]. The observation that Aβ accumulation induces BACE1 elevation in dystrophic neurites surrounding plaques [177–179], which in turn accelerates Aβ generation in 5XFAD mouse and human AD brains, led to the hypothesis that eIF2α kinases (mostly PERK and PKR) might actually behave as mediators of amyloid plaque growth in a self-amplifying feedback mechanism. Nevertheless, a report published by Sadleir and colleagues in 2014 [180] showed that genetic inhibition of eIF2α phosphorylation in a mouse model of AD did not prevent Aβ-dependent elevation of BACE1 nor reduced amyloid pathology. In this study, the authors used three different genetic strategies designed to circumvent the effect of p-eIF2α phosphorylation over BACE1 translational activation, dissecting thereby the hierarchical dependence of BACE1 expression upon eIF2α phosphorylation. Using this approach, they showed that Aβ oligomers are able to upregulate BACE1 expression independently of eIF2α phosphorylation [180].
Revisiting the original notion by O’Connor and colleagues that energy-deprivation induced BACE1 expression through eIF2α phosphorylation [69], Mouton-Liger et al. showed that disruption of brain thiamine homeostasis, which has a prominent role in energy metabolism and has a reduced activity in AD, is related to an altered BACE1 expression through the eIF2α pathway [181]. In a mouse thiamine deficiency-model, they demonstrated increased BACE1 expression, p-eIF2α an Aβ peptide production, something that led to motor deficits and neurodegeneration in a PKR-dependent manner [181]. In addition, the protagonist role of PERK in mediating BACE1 upregulation through eIF2α phosphorylation (an issue also initially raised by O’Connor et al. [69]) received additional support in a report by Devi and Ohno [182], in which genetic reduction of PERK was found to mitigate AD-neuropathology (including elevated BACE1 expression) and cognitive impairments in a mouse model of AD [182]. At the same time, however, genetic downregulation of PKR has been shown to protect lypopolysaccharide (LPS)-treated mice from neuroinflammation [183], in an effect likely to be mediated through the suppression of BACE1 expression in PKR–/– mice.
Evidences from plant compounds capable of modulating PERK and PKR pathways in AD pathogenesis have recently come to light [184–186]. In a report by Yan et al. [184], the broad-spectrum anti-cancer compound Icaraside II (extracted from the Chinese medicinal plant Herba Epimedium [187]) was administered to a double transgenic mice model of AD (AβPPsw/PS1 dbKO). As a result, learning increased and plaque burden diminished in an effect likely to be mediated through the inhibition of PERK-mediated eIF2α phosphorylation and its subsequent downregulatory effect on BACE1 expression. Even though, other targets such as ADAM10, peroxisome proliferator-activated receptor-γ (PPARγ) or phosphodiesterase-5A (PDE5A) could also stay behind the beneficial behavioral effects [184]. Another report by Li et al. showed how an ethanol extract of Thamnolia Vermicularis (a plant widely used in Chinese medicine for its antioxidant and antioncogenic properties [188]) ameliorated learning in AβPPsw/PS1 transgenic mice and induced a drop in Aβ production and protein tau phosphorylation in neuroblastoma cells [185]. The signaling cascades involved in the latter effect included: 1) stimulation of PI3K and AMPK pathways, 2) suppression of PERK/eIF2α-mediated BACE1 expression, and 3) inhibition of GSK3β [185]. While the authors proposed the extract of T. Vermicularis to be a multi target drug hitting Aβ and tau simultaneously, we note here that BACE1 expression and tau phosphorylation could also be mechanistically connected through GSK3β activity (see Fig. 7). Beyond the PERK pathway, however, the PKR/eIF2α pathway has also received attention as a putative target of plant compounds as stated above. In a work by Zhang et al. [186], the phenolic glucoside gastrodin (extracted from the Chinese herbal medicine Gastrodia elata Blume) suppressed BACE1 expression by inhibiting the PKR/eIF2α pathway both in a transgenic mice model (Tg2576, carrying the AβPPsw mutation) and in human neuroblastoma cells.
Conversely to the potentially beneficial effects of plant compounds as therapeutic modulators of the PERK-PKR/eIF2α pathways, unwanted side effects of antiretroviral drugs have been recently linked to BACE1 translational upregulation through PERK activation [189]. Antiretroviral drugs (ARV) are being pointed as the potential culprits of human immunodeficiency virus (HIV)-associated neurocognitive disorders [190]. Given the ability of ARV to induce ER stress, Gannon et al. [189] hypothesized that ARV-mediated ER stress in the CNS would result in an activation of the unfolded protein response (UPR), which involves PERK activation as one of its main effector branches [191–193] (Figs. 7 and 10). Using in vitro and in vivo models the authors showed how a specific ARV class known as “HIV protease inhibitors” induced neuronal damage, ER stress and subsequent PERK-mediated BACE1 translational activation through eIF2α phosphorylation. As expected, this effect was coupled to an increase in Aβ production measured in vitro as well as to neuronal damage triggering in invivo models (for a recent review of UPR and PERK as a new therapeutic target in AD, see [194]).
In summary, a total of twelve research papers have appeared in the last decade (2008-2018) investigating the relationship between neuronal stress, eIF2α phosphorylation and BACE1 expression. Nonetheless, despite the intense activity dedicated to investigate these pathways hitherto, there might still be unexplored paths to be walked in this “roadmap”, which may eventually shed more light about possible mechanisms driving BACE1 expression through eIF2α phosphorylation.
For instance, disrupted calcium homeostasis is thought to play a central role in AD pathogenesis, as increased intracellular calcium triggers Aβ accumulation, tau hyperphosphorylation, and neuronal death. In addition, the most significant signal-transduction cascades associated with AD are mediated by disruption of calcium regulation in the ER, while FAD-causing mutations have been linked to intracellular calcium signaling pathways [195] (reviewed in [196, 197]). Interestingly, the ER-localized eIF2α-kinase PERK is activated by ER stress and, more specifically, by Ca+2 dyshomeostasis [198]. Hence, a suggestive connection exists between destabilization of calcium signaling in AD and PERK activation. In this regard, in the early 1990s of last century the observation was made that calcium release from the ER enhanced Aβ production [199, 200]. Thus, in an historical context where the role of Aβ peptide as the upstream event triggering AD pathogenesis had not yet become the leading trend in AD research, such evidences linking calcium and Aβ led to the hypothesis that the β- and γ-secretase activities could be modified by calcium-binding proteins, or that AβPP processing could be directly carried out by calcium-dependent proteases [201]. The ulterior identification of the proteases responsible for β- and γ-secretase activities ruled out the possibility that they were calcium-dependent proteases in a direct manner, for neither BACE1 nor proteins of the γ-secretase complex harbored any calmodulin-binding domain. However, recuperating this notion twenty years laterin the light of the achievements made in the field of BACE1 translational control, the possibility remains open that β-secretase is, to some extent, a calcium-dependent protease but in an indirect way, through Ca+2/PERK/eIF2α-mediated translational activation. In addition, γ-secretase activity could also be modulated by this pathway. Nicastrin, a scaffolding protein required for the stability of the γ-secretase complex, is translated from an mRNA containing five uAUGs, which could potentially be responsive to a drop in ternary complex availability in a similar way than BACE1 uAUGs are (for a recent update on the “Calcium hypothesis” of AD, see [202]).
Another main research avenue to be pursued in the “p-eIF2α-mediated Stress-Driven-BACE1-translation pathway” is the one stemming from HRI activation, a kinase that has been largely ignored in the field because it was considered to be only expressed in little amounts in the CNS [139, 148]. It might be true that, in quantitative terms, HRI is expressed at lower levels in neurons when compared to other eIF2α-kinases, but the fact that HRI has been recently localized at synaptic sites [145] makes no longer possible to continue ignoring the pathophysiological consequences of HRI-mediated eIF2α phosphorylation in neurons. The physiological implications of HRI activation by nitric oxide werealready described in a previous chapter. Nonetheless, it should be noted here that HRI can also be activated by stress factors intimately related to AD pathogenesis. To begin with, HRI is activated by oxidative stress [203], whose pivotal importance in AD pathology has also been discussed above. But the chief stress factor activating HRI is heme deficiency: this is the mechanism by which this kinase prevents the synthesis of defective hemoglobin in the absence of heme in reticulocytes [144]. In fact, the discovery of HRI activity originated from the primal observation that reticulocyte lysates, which were the eukaryotic nonnucleated cell model of choice for the study of protein translation during the 1980s, were required to be exogenously supplemented with heme (or hemin) for protein synthesis. Such reticulocytes, lacking mitochondria, which are the natural sources of heme in cells, started protein synthesis at a high rate but decreased sharply within a few minutes (through fast eIF2α-phosphorylation) unless hemin was added to the system, a phenomenon which led to hypothesize the existence of a “heme-controlled inhibitor” (HCI) of protein synthesis (reviewed in [204]). HRI kinase (standing for heme-regulated inhibitor) was later identified as the kinase responsible for HCI activity [143]. Intriguingly, evidence of heme deficiency has been reported in AD brains [205], which express significantly reduced levels of rate-limiting enzymes in the heme biosynthetic pathway [206]. Indeed, heme is a common factor linking several metabolic perturbations in AD [207], including iron metabolism, mitochondrial complex IV, heme oxygenase, and bilirubin. Furthermore, oxidative stress induces the expression of heme oxygenase-1 (HO-1) [208–210], an enzyme that degrades heme to generate bilirubin, a potent antioxidant. Remarkably, pronounced HO-1 immunoreactivity is detected in neurofibrillary tangles and in neurites surrounding senile plaques [211], and the level of bilirubin is increased in AD patients [212]. All these evidences seem compelling enough to consider HRI kinase integrity at synapses, putatively compromised in AD due to chronical heme deficiency, also as an appealing pathway for future research in the interesting unexplored wild territories at the border between memory consolidation and AD neurodegeneration.
INTRACELLULAR DEGRADATIVE PATHWAYS IN SAD: PRESENILIN AND AβPP MUTATIONS DISRUPT BACE1 PROTEIN HOMEOSTASIS
One of the early pathological manifestations in AD patients (before the appearance of amyloid plaques) is the enlargement of endosomal compartments and the accumulation of autophagic vacuoles [213]. Interestingly, FAD-linked AβPP and presenilin mutations not only alter the species of Aβ generated (Aβ42/Aβ40 ratio), but affect biological pathways that may be responsible, at least in part, for the onset of AD.
Presenilin-1, apart from its role as the catalytic subunit of the γ-secretase complex, is required for the proper translocation of the Vacuolar-type H+-ATPase (V-ATPase) to lysosomes and consequently, the correct acidification of these organelles. Lysosomes are terminal compartments with a low pH in the lumen for the proper activity of the hydrolases responsible for the degradation of many proteins coming from multivesicular bodies (MVBs) or autophagosomes. MVBs (late endosomal compartments) are formed during the maturation of early endosomes (EE) by invagination of the endosomal membrane towards the lumen and the consecutive scission, giving rise to intraluminal vesicles (ILVs) (Fig. 11).
Fig.11
Alteration of intracellular compartments in AD. Left panel: Intracellular compartments in a healthy neuron. AβPP is endocyted from the plasma membrane and cleaved to AβPP-βCTF by BACE in the membrane of early endosomes (EEs). EEs mature to multivesicular bodies (MVBs) when the rim of the endosome invaginates toward the lumen forming the intraluminal vesicles (ILVs). AβPP-βCTF can be cleaved to Aβ by γ-secretase on the membrane of the endosome or be incorporated to the ILVs where can also be processed by γ-secretase to Aβ. After ILVs are degraded after fusion of MVBs to lysosomes or secreted in form of exosomes. A utophagosomes are also degraded after fusion to lysosomes. Right panel: In AD, the loss of function of lysosomes decreases the degradation of autophagosomes and MVBs, which accumulated and become enlarged. In addition, accumulation of AβPP-βCTF in endosomes enhance the enlargement by recruiting the APPL1 effector. To compensate the lack of degradation of ILVs and their content (including Aβ), there is an increased secretion of ILVs in form of exosomes upon fusion of MVBs to the plasma membrane. Aβ is secreted from the lumen of MVBs or bound to exosomes. The secreted Aβ may work as a glue for proteins, lipids and nucleic acids destined to be degraded by the activated microglia. Exosomes can also be removed from the brain through the blood-brain barrier.
ILVs and their content are degraded upon fusion of MVBs to lysosomes, or alternatively, they can be released to the extracellular space after fusion of the MVBs to the plasma membrane in form of exosomes (extracellular lipid bilayer enclosed membrane vesicles with a size ranging from 50 to 100 nm [214] (Fig. 11). On the other hand, authophagy is a supportive pathway for the degradation of macromolecules and organelles activated upon cellular stress and the accumulation of misfolded proteins or when conditions requiring recycling of cellular components are met (e.g., under starvation). The autophagosome is formed de novo by the elongation of a spherical double membrane bilayer. Proteins to be degraded are recruited to the nascent autophagosome and processed after fusion to lysosomes. Interestingly, neuronal deficiency of presenilin-1 triggers the accumulation of autophagosomes [215, 216]. Indeed, it was shown that mutations linked to FAD on presenilin-1 affect the proteolysis by lysosomes and the authophagic pathway [217, 218]. Interestingly, carriers of the p.Leu (381) Phe mutation on presenilin-1, which causes early-onset AD, show lysosomal inclusions in peripheral fibroblasts, resembling those found in Kufs disease (KD), the major adult form of neural ceroid lipofucinosis [219]. KD patients suffer from dementia and voluntary movement symptoms. The disease is caused by mutations in genes related to lipid degradation/excretion that lead to the accumulation of lipopigments in lysosomal compartments, linking lysosome function to dementia. Thus, the fact that the FAD presenilin mutant p.Leu (381) Phe induces a KD-like phenotype in peripheral fibroblasts suggests that lysosomal dysfunction may be also an early trigger of dementia in AD.
On the other hand, the AβPP-βCTF (β-secretase C-terminal fragment) released upon cleavage of AβPP by BACE1 can also alter the function of endolysosomal compartments. It was reported that the AβPP Y682G mutation affects the interaction with sortilin-related receptor (SorLA), a protein that controls the trafficking of AβPP between the trans-Golgi network (TGN) and EEs [220] and whose low mRNA levels have been associated to SAD [221, 222]. Disruption of the interaction between SorLA and AβPP by the Y682G mutation leads to lysosomal dysfunction in the hippocampus of mice [223]. Altered levels of the AβPP holoprotein also produce abnormalities in endosomal compartments (Fig. 11). Both full-length wild-type AβPP and AβPP-βCTF induce endosomal enlargement and disrupt NGF signaling and axonal trafficking in cultured rat basal forebrain cholinergic neurons [224]. Interestingly, induced pluripotent stem cells-derived neurons from two patients with FAD caused by duplication of the AβPP gene show rab5-enlarged endosomes compared to controls (rab5 is a protein enriched in EE) [225]. In addition, studies of individuals with DS link AβPP levels to the presence of abnormal endosomal compartments. People with DS are a paradigmatic case of increased AβPP gene dosage due to the trisomy of chromosome 21 where the AβPP gene is located. Moreover, DS individuals are known to develop AD in the adulthood. A segmental trisomy 16 mouse model of DS present endosomal abnormalities and impaired NGF axonal transport, similar to the findings in AβPP-over expressing basal forebrain cholinergic neurons [224, 226, 227]. These endosomal abnormalities in DS are reverted with a BACE1 inhibitor, what points to the involvement of AβPP-βCTF as the main mediator of these alterations, thereby placing BACE1 activity (together with AβPP and PS1/2) at the stage of degradative pathways putatively disrupted in AD [228, 229].
BACE1 interplaying with AβPP and presenilins in endosomal degradative pathways
AβPP is internalized from the plasma membrane and processed in EE to AβPP-βCTF by BACE1 [230, 231]. ABPP-βCTF can be cleaved by γ-secretase in endosomes to produce Aβ or be sorted to ILVs during the maturation of EE to MVBs and degraded by the lysosome after fusion of MVBs to this organelle. Conditions impairing the lysosomal/autophagosomal degradative pathways or the fusion of endosomes to lysosomes lead to an increase in the levels of AβPP-βCTF and BACE1 in endosomal compartments (BACE1 is also degraded by lysosomes) (Fig. 11). This results in higher production of Aβ after cleavage by γ-secretase [232, 233] and the enlargement of endosomes due to the recruitment by AβPP-βCTF of the cytosolic rab5-effector APPL1 (Adaptor protein phosphotyrosine interacting with PH domain and leucine zipper 1), which stabilizes the GTP-rab5 in the active state and leads to endosome swelling [234].
Thus, an impairment of the degradative pathways caused by FAD mutations on AβPP and presenilins (or aging-related molecular changes in the case of SAD [235]) may lead to elevated AβPP-βCTF levels and BACE1 in endosomal compartments. This in turn leads to an enlargement of these organelles after recruitment of APPL1 and provides more substrate for Aβ generation, which can be produced in the ILVs. ILVs containing Aβ (most probably bound to the outer membrane) are then secreted to the extracellular space in form of exosomes. Thus, if one hypothesizes that lysosomal/autophagosomal dysfunction is at the base of AD and causes AβPP-CTFs accumulation and Aβ generation, it is logical that abnormalities in the endocytic pathways are observed earlier than amyloid plaques [235]. Loss of intracellular lysosomal degradation may be compensated by increased secretion of misfolded or toxic proteins via exosomes after fusion of MVBs to the plasma membrane and release of the ILVs that cannot be degraded intracellulary [236–240]. AβPP-CTFs and Aβ have been detected in exosomes [233, 241, 242] and interestingly neural-derived exosomes isolated from the blood of AD patients contain increased levels of Aβ42 and the lysosomal proteins cathepsin D and LAMP1 (Lysosomal-associated membrane protein-1), up to 10 years before the clinical manifestation of the disease [243, 244]. It has been shown that treating cells with bafilomycine A1, an inhibitor of the V-ATPase required for the acidification and the function of lysosomes, increases the secretion of exosomes and Aβ42 [245, 246]. Interestingly the Aβ42/Aβ40 ratio is elevated in exosomes and it is plausible that the consequent enhanced secretion of Aβ-bearing exosomes caused by impairment of the lysosome contribute to the formation of amyloid plaques [246]. This agrees with the observation that inhibition of the neutral sphingomyelinase 2 (nSMase 2), an enzyme required for exosomes formation, decrease the pathological phenotype of an AD mouse model (5xFAD) [247].
Extracellular Aβ-aggregates can trap other toxic or misfolded proteins secreted by exosomes, activating microglia for their removal. Indeed, aggregation of Aβ42 stimulates its uptake [248] and could work as a stimulus to remove misfolded proteins extracellularly. Finally, some genes found to increase the risk of developing AD may do so by increasing the convergence of AβPP and BACE1 in EE (Bin1, CD2AP) [249], regulating the degradation of AβPP-CTFs via autophagy (PICALM) [250, 251], or affecting lysosomal/autophagosomal systems (PLD3) [252].
The above studies demonstrate the key role of presenilins and AβPP in the regulation of lysosomal and autophagosomal compartments and the control of protein degradation and homeostasis, a critical process for long-lived neurons in which BACE1 seems to play also a decisive role. The impairment of these homeostatic mechanisms by FAD mutations on presenilins and AβPP (FAD) or by age-related molecular changes (SAD), is an early pathological event in AD and may be at the base of this disorder. These changes may lead to a later accumulation of Aβ and axonal transport deficits in AD patients as a consequence of AβPP-βCTF and BACE1 accumulation in endosomes.
Summarizing, FAD-associated mutations, beyond its effects in Aβ-peptide generation, are related to lysosomal/autophagosomal dysfunction; this dysfunction presents as an early phenotype prior to the onset of the disease, and is connected to BACE1 protein homeostasis which, in turn, is connected to memory processing.
CONCLUSIONS AND PERSPECTIVES
The frame of mind instituted by the amyloid cascade hypothesis nearly 25 years ago seems to have clashed with the gloomy reality of repeated clinical failures of anti-amyloid therapies [19, 20, 253–255]. Recently, the failures in phase II/III trials of two compounds into which many hopes were laid (“solanezumab Expedition 3”, a monoclonal antibody against soluble Aβ-peptide, and “verubecestat”, a BACE1 inhibitor) [21], seem to corroborate the notion that lowering Aβ-peptide load in the brain (either blocking its production or promoting its clearance) might not be the answer leading to an effective therapy for AD.
Aβ peptide, however, is not only sticky in its very physical aggregative properties, but it also sticks persistently into our minds as a central phenomenon in AD pathogenesis; and rightly so, because, no matter how modest is the incidence of FAD in quantitative terms, mutations in AβPP are sufficient to cause AD. This genetic evidence cannot be overlooked.
It is the gap between genetics and physiology what needs to be fulfilled. Establishing causal relationships in physiopathological scenarios is not easy. When seeing a man falling into a lake and drowning, for instance, the most immediate reaction is to think that he died of suffocation. But how can we be sure that he did not suffer a heart attack first, died of it, and then, only afterwards, fell inside the lake? For if this would be the case, suffocation signs would still appear in the corpse of the dead man, but they would be the consequence, rather than the cause of this figurative accidental death. Hence, what about Aβ? Is it the cause, or the consequence of AD? Or is it just an epiphenomenon?
Again, genetic evidences make us think of Aβ as bearing a strong causal effect over the development of the disease. But still, an alternative way of interpreting the facts is possible, which can bring conciliation among genetic evidences (FAD genetic evidences) and a secondary, perhaps anecdotic, role for Aβ in AD etiology. Such an alternative view consists in conceiving the proteolytic cleavage upon AβPP as an integral part of a wider proteolytic system necessary for memory formation. Under this alternative framework, FAD mutations in AβPP and PS appear before us under a different light, not just as Aβ-generating molecules, but as memory-processing enzymes whose physiological performance has been impacted by the mutations. Conversely, cellular stress, aging, and other similar insults can be looked at as capable of inflicting external aggressions to those memory-consolidating systems, leading to sporadic memory-loss phenotypes.
Not few evidences have accumulated over the years about the participation of AD molecular players (AβPP, secretases) in synaptic plasticity, learning and memory formation. For instance, BACE1 activity is necessary for synaptic and cognitive functions [37] and, more specifically, BACE1-mediated cleavage of AβPP is required for memory formation, learning and synaptic plasticity [38]. Synaptic activity induces Aβ secretion [256] (induces therefore AβPP proteolysis, we could equally say). Even endogenous Aβ has been found to mediate learning and memory [257]. More recently, BACE1 inhibition was found to impair synaptic plasticity and cognitive functions [258]. Similarly, in the disappointing context of failed γ-secretase clinical trials (avagacestat, tarenfurbil) [255], we have learned how γ-secretase inhibition has actually led to cognitive deterioration.
Hence, the relationship between what we could call “molecular AD players” (i.e., those intervening in the amyloid cascade) and synaptic plasticity, learning and memory has been firmly established since at least the last decade; and from a number of different angles. What remains to be determined, however, is whether these memory-associated phenotypes can stay in the same relation to AD as the heart failure was in the imaginary example mentioned above, about a drowning man. Is it a primal loss in memory-processing phenotypes, among which AβPP proteolysis would play an essential part, what is making sufferers fall in the fatal slope of AD neurodegeneration?
In this review, we have focused on molecular mechanisms pivoting around BACE1 protein homeostasis. We have tried to highlight, in the first place, how signaling cascades modulating BACE1 translation are connected to synaptic plasticity and memory formation. We have also described how, when cellular conditions are not the optimal and stress make its appearance (virtually all kinds of stresses, from oxidative stress to ER stress or metabolic deficiency) these very same signaling pathways are exogenously impacted. Finally, we have described how FAD-associated mutations disrupt lysosomal/autophagosomal function prior to the onset of the disease, and how this phenomenon alters synaptic BACE1 availability.
But many issues remained that claimed for a deeper discussion; issues ranging from specific molecular mechanisms, to speculation on the role proteolysis might be playing in cognition.
Can AβPP cleavage be understood as branch of a wider system modulating cellular adhesion?
The function of AβPP in the CNS is not fully elucidated. Yet, it is known that AβPP is a potent cell adhesion protein [259, 260]. A somewhat amusing observation gives a hint on how AβPP cleavage might take part in the modulation of cellular adhesion. Mouse embryonic fibroblasts that are double knockout for presenilins (PS–/– dbKO MEFs) are so strongly adhered to the culture vessels, that one can hear scientists literally smashing the flasks against the bench with all their forces when doing passages of this cell line in the tissue culture room. Otherwise, the cells won’t detach! Conversely, HEK cells overexpressing AβPP harboring the Swedish mutation (AβPPsw, which facilitates AβPP cleavage by BACE1) are so loosely attached to the flask surface that experimentalists, when manipulating such cultures, are forced to move as felines do when approaching the prey, with very gentle, extremely calculated moves, because they know well that the least tremor will suffice to detach large patches of the culture layer doing away with their experiment. One even fears that laying too strong a look on the AβPPsw cells would be enough to detach them!
Of course, beyond AβPP, presenilins cleave other substrates that could account for the modified adhesion phenotype in PS–/– dbKO MEFs. But in HEK-AβPPsw cells we have a clear indication on how specific BACE1-mediated AβPP cleavage influences adhesion. Besides, it would be unlikely that AβPP-cleavage was left alone in modulating cell adhesion. On the contrary, if we admit that macroscopic biological responses rarely rely just in one gene, we should rather expect AβPP-cleavage to be a particular example belonging to a more general, wider system of cell adhesion proteins with its corresponding cleaving proteolytic partners.
In this regard, the literature offers an ever-increasing number of works describing how adhesion proteins (other than AβPP) are cleaved by secretases; and also how proteases (other than secretases) cleave adhesion proteins (or extracellular matrix proteins), including AβPP, in the context of synaptic plasticity, learning, and memory. For example,on the basis of previous screening identification [33], Pigoni and colleagues have recently demonstrated how BACE1 cleaves two transmembrane proteins, Seizure Protein 6 (Sez6) and Sez6-like(Sez6L) [261], which seem to have cell adhesion functions [262]. Interestingly, Sez6 proteins are also processed by γ-secretase [261]. BACE1 also cleaves the postsynaptic cell adhesion protein Neuregulin-1 (NGL-1) in the context of myelination [28, 29]. In turn, NGL-1 is also cleaved by ADAM10, MMP9, and neuropsin. ADAM10 is the α-secretase, the protease responsible of AβPP-cleavage in the non-amyloidogenic pathway; Suzuky et al. [263] showed how, after NMDA receptor activation, ADAM10 cleaves NGL-1 and, as it happens withAβPP-cleavage, a further step of intra-membrane proteolytical processing by γ-secretase follows [263]. MMP9, on the other hand, cleaves cell adhesion proteins such as Intracellular Adhesion Molecule 5 (ICAM5) and integrins (specially β1-integrins), besides NGL-1. Both ADAM10 and MMP9 participate in synaptic plasticity: ADAM10 is required for synaptogenesis [161] and MMP9, when locally released, sculpts dendritic spine morphology [264–266]. Another protease cleaving NGL-1 is Neuropsin, which in turn cleaves the cell adhesion protein NCAML1 [267] in the context of neural plasticity. Finally, the presynaptic adhesion molecule Neurexin 3β is processed either by α- and γ-secretases [268]. Neurexins are the presynaptic partners of Neuroligins, and their bipartite cell-adhesion-to-cell-adhesion protein interaction supports synaptic structure: Neurexins and Neuregulins “shake hands” to build the synapse. This means, inevitably, that cleavage of such adhesion proteins by proteases like BACE1, MMP9, Neuropsin, ADAM10, or γ-secretase must dismantle the synapse, or at least destabilize it.
Thus, from this perspective it seems clear that AβPP-cleavage is integrated into a wider network of cell adhesion proteins which, together with its associated processing proteases, is involved in the modulation of cell adhesion to the extracellular matrix (ECM); and that this activity-induced modulation is something crucial in synaptic plasticity.
Parsimoniously with the general belief that cellular consolidation of long-term memory implies the growth of new connections and the rearrangement of the existing ones [151], it is possible to conceive the active modulation in adherence-strength of spines and synapses to the ECM as a pre-requisite for subsequent structural rearrangements. Indeed, the vast majority of proteins found in the post-synaptic density are in fact adhesion molecules [269]; and a multiple-pathway analysis from two genome wide association studies points to cellular adhesion molecules pathways as major contributors to AD [270]. This seems to support an initial memory-loss phenotype for AD triggering, connected with the adhesion properties of AβPP and the activity of its associated proteases (secretases).
However, if AβPP is just a component of a larger web modulating cell adhesion, why is it so that AβPP is the only protein (together with PSs, which are AβPP-cleaving enzymes) targeted by pathogenic FAD mutations? And if Aβ-accumulation in the brain of non-demented individuals is accepted as a criticism to the amyloid cascade hypothesis, why such a “miss processing” of AβPP would not similarly have to result, necessarily, in a memory deficit phenotype?
Once again, thinking in AβPP-cleavage as a member of a larger network supporting memory-related plasticity processes may help to imagine how such a system (with all its components) is able to resist a certain number of hits, having nodes which are more vulnerable than others, etc. Without underestimating Aβ-derived neurotoxicity, which, as a result of miss regulated AβPP cleavage, might constitute the second hit (after the loss of activity-modulated cell adhesion) knocking down the system.
HRI-kinase: A new effector arm in NO-signaling?
Nitric oxide (NO) is a highly diffusible, gaseous free radical involved in a plethora of physiological responses, including macrophage activation, vascular tone regulation or memory processing. Two main canonical pathways have been related to NO-physiological signaling hitherto: 1) cyclic GMP (cGMP) production, through the intermediary activation of guanylate cyclase, and 2) protein nitrosylation, consisting in the direct covalent binding of NO into amino acidic residues of proteins. Recently, two reports have brought forth compelling evidences supporting a role for the HRI kinase as a downstream mediator of NO-signaling; by exploring the same underlying molecular mechanism, these works showed how neuronal NO activates the translation of BACE1 [145] and GluN2B [149], respectively, via HRI kinase-mediated eIF2α phosphorylation [145, 149].
Thus, the existence of a novel third branch in NO-signaling might be suggested. In this alternative pathway, NO would behave as a translation facilitator for uAUG-bearing transcripts (Figs. 7 and 8).
Nevertheless, very little is known about the function of HRI in the nervous system yet. In 2013, Viader and colleagues found HRI to be the effector kinase in a model of peripheral neuropathy secondary to mitochondrial dysfunction [271]. Concerning, however, what might be a physiological role for HRI kinase in neurons, and to the best of our knowledge, just three works have been published to date [142, 145, 149], which will need further confirmation by other groups. Intriguingly enough, although the vast majority of known NO-dependent responses in neurons rely on cGMP production, other transduction pathways that signal through redox events are thought to be possible. NO participates in the wiring of the CNS by regulating growth cone motility, filopodial dynamics, and neurite outgrowth-retraction [272]. Cyclic GMP is involved in all these processes. But it is nevertheless interesting to note how this dynamics growth versus retraction is also coherent with a putative translational activation of either BACE1 [145] (a dismantling protease) and GluN2B [149] (a NMDA-receptor subunit linked to neurite outgrowth and synaptic strengthening [273]). Furthermore, studies using the cGMP inhibitor [1H-1, 2, 4] oxadiazolo-[4, 3-a]quinoxalin-1-one] (ODQ) suggest that “the oxidation of one or more redox-sensitive proteins, such as hemo-proteins, is likely involved in mediating the effect of ODQ on neurite outgrowth and retraction” [274]. ODQ is commonly used as a specific cGMP inhibitor, although ODQ is, in fact, a non-selective oxidizer of heme groups thus able to target other heme-proteins besides sGC [275]. Therefore, the heme-protein HRI kinase [129–133] could be, in principle, a good candidate for mediating such orphan NO-depending effects in neurons.
However, the signaling properties of NO were not appreciated in neurons in the first place, but in the vascular wall. Furchgott and Zawadzki postulated, in their pioneering works during the 1980s, the existence of an endothelial-derived relaxing factor (EDRF) that would depart from the endothelial cells towards the underlying vascular smooth muscle cells, inducing miorrelaxation [276]. Later on EDRF was shown to be NO [277, 278]; and a substance with “EDRF activity” was described in the cerebellum as being the node linking NMDA receptor activation with cGMP production in the postsynaptic compartment [117].
Here we propose to take a small detour the other way around. Because we found HRI transcript to be expressed as well in smooth vascular cells (Fig. 11B), and given the tremendous importance of vascular tone regulation for human health (including brain performance), we decided to briefly discuss the potential role of HRI kinase as a NO sensor in the vascular wall.
Vascular implications
If HRI is to behave, through its heme-prosthetic groups, as a NO-sensor involved in translational facilitation in neurons, it seems reasonable it could play a similar role in the vasculature as well, where NO signaling plays such an important part. Specially, if we take into account that, beyond cGMP production, from which the majority of NO-derived physiological functions in the vasculature derive [279–286], there exist two cationic channels (known to play a major role in the control of vascular tone and reactivity) which are translated from mRNAs harboring uAUGs-bearing 5’UTRs. Thus, hypothetically, these two channels could respond positively to the NO/HRI/p-eIF2α pathway to promote smooth muscle cell (SMC) relaxation (Fig. 12). These two genes are KCNMB1 (Potassium Calcium-Activated Channel Subfamily M Regulatory Beta Subunit 1, also known as “BK channel subunit- β1”) and KCNN3 (Potassium Calcium-Activated Channel Subfamily N Member 3, also known as SK3). Although the mere presence of uAUGs in the 5’UTRs, in silico detected, does not necessarily imply a translational facilitation of these mRNAs in response to eIF2α-phosphorylation, it is nevertheless a necessary prerequisite: the sequence-foundation needed for mRNA translation activation in response to a potential NO/HRI induced ribosomal skipping mediated via eIF2α phosphorylation (Fig. 12C).
Fig.12
Heme-regulated eIF2α-kinase (HRI) activation by nitric oxide (NO) (A), HRI mRNA expression in different cellular types (B), and hypothetical model for NO/HRI-induced translational activation of cationic channels at the vascular wall (C). A) Diagram depicting NO-mediated signaling, including HRI kinase activation. HRI kinase activation by nitric oxide may constitute a third branch of NO-mediated intracellular signaling, by which translation initiation in uAUG-bearing transcripts becomes facilitated. B) Profile of HRI mRNA expression in different cellular types. HRI mRNA expression was detected in all the cellular types assayed, including human aortic vascular smooth cells (HA-VSMCs) and human cerebral vascular smooth cells (HC-VSMCs). (–), negative control without input RNA. C) Hypothetical model for NO/HRI-induced translational activation of cationic channels at the vascular wall. After NO is synthesized in the endothelial cells, it diffuses to the adjacent VSMC where, theoretically, it could activate the VSMC-resident HRI kinase; this would in turn facilitate the translation of uAUG-bearing transcripts such as those coding for the β1 subunit of the BK channel (KCNMB1), and the SK3 channel (KCNN3), respectively. Both cationic channels depicted in the figure (KCNMB1 and KCNN3) are calcium activated potassium channels (K Ca). eNOS, endothelial nitric oxide synthase; nNOS, neuronal nitric oxide synthase; mtNOS, mitochondrial nitric oxide synthase; iNOS, inducible nitric oxide synthase; Cell lines: Raw, macrophages; BV2, microglia; SH-SY5Y: human neuroblastoma; LCFSN: traqueal epithelial. Primers used for semi-quantitative RT-PCR amplification: AGGAACAAGCGGAGCCG(mHRI_F); CCGACCAGTCCTTACGCC (mHRI_R). [446 bp amplicon].
![Heme-regulated eIF2α-kinase (HRI) activation by nitric oxide (NO) (A), HRI mRNA expression in different cellular types (B), and hypothetical model for NO/HRI-induced translational activation of cationic channels at the vascular wall (C). A) Diagram depicting NO-mediated signaling, including HRI kinase activation. HRI kinase activation by nitric oxide may constitute a third branch of NO-mediated intracellular signaling, by which translation initiation in uAUG-bearing transcripts becomes facilitated. B) Profile of HRI mRNA expression in different cellular types. HRI mRNA expression was detected in all the cellular types assayed, including human aortic vascular smooth cells (HA-VSMCs) and human cerebral vascular smooth cells (HC-VSMCs). (–), negative control without input RNA. C) Hypothetical model for NO/HRI-induced translational activation of cationic channels at the vascular wall. After NO is synthesized in the endothelial cells, it diffuses to the adjacent VSMC where, theoretically, it could activate the VSMC-resident HRI kinase; this would in turn facilitate the translation of uAUG-bearing transcripts such as those coding for the β1 subunit of the BK channel (KCNMB1), and the SK3 channel (KCNN3), respectively. Both cationic channels depicted in the figure (KCNMB1 and KCNN3) are calcium activated potassium channels (K
Ca). eNOS, endothelial nitric oxide synthase; nNOS, neuronal nitric oxide synthase; mtNOS, mitochondrial nitric oxide synthase; iNOS, inducible nitric oxide synthase; Cell lines: Raw, macrophages; BV2, microglia; SH-SY5Y: human neuroblastoma; LCFSN: traqueal epithelial. Primers used for semi-quantitative RT-PCR amplification: AGGAACAAGCGGAGCCG(mHRI_F); CCGACCAGTCCTTACGCC (mHRI_R). [446 bp amplicon].](https://content.iospress.com:443/media/adr/2019/3-1/adr-3-1-adr180089/adr-3-adr180089-g012.jpg)
The channels codified by KCNMB1 and KCNN3 are both members calcium-activated potassium channels (KCa), which comprises also intermediate conductance channel (IK) [287]. The opening of KCa hyperpolarizes the membrane of vascular SMC, inducing a closure of voltage-dependent calcium channels, and opposing vasoconstriction [288]. Both cited channels are found in the vascular wall, showing a very specific subcellular localization.
BK channels are virtually present in all vascular SMC [287], while they are scarce in endothelial cells [289–291]. The BK channel is formed by α subunits, forming the pore, and β, regulatory, each one with different roles in modulating the pharmacology and activation of this channel. However, the β1 is the predominant subunit in vascular smooth muscle cells (VSMCs) and seems to be responsible for the regulation of physiological calcium regulation [292, 293]. The deletion of this subunit leads to an important decrease in calcium sensitivity in BK channels, inducing a more depolarized membrane potential and finally to increase in arterial blood pressure [294–296]. Similarly, the blockade of BK channels by Iberotoxin induces membrane depolarization, higher intracellular calcium and vasoconstriction [294, 297, 298].
KCNN3 in turn, encodes the small-conductance calcium-activated potassium channel SK3. This channel is mainly expressed in the vascular endothelium, participating in hyperpolarization and contributing to vasodilatation [287]. There are few evidences supporting a functional role for these channels in the VSMC. However, this is a controversial subject as it was demonstrated the presence of apamine-sensitive channels (apamine is a SK3 blocker) in VSMC from several vascular beds as aorta, renal and coronary arteries [299–301]. Moreover, in transgenic mice, the suppression of SK3 depolarizes endothelial cells and VSMC, reducing the diameter of resistance vessels and elevating arterial pressure [302].
The first indication relating NO signaling and potassium channels comes from the 1990’s, when Archer and colleagues (1994) [303] observed that patch-clamp of rat VSMC showed an enhancement in potassium current, induced by NO and cGMP, after using a phosphatase inhibitor (okadaic acid). This inhibitor delays dephosphorylation and was able to increase the potassium current in cells sensitive to NO, indicating that the stabilization of protein phosphorylation has an important role in the activity of these channels. These facts demonstrated an important role of phosphorylation in calcium-activated potassium channels responsive to NO. In the same year, Bolotina et al. (1994) demonstrated that NO, coming from endothelium or exogenously added, could directly activate KCa in VSMC, not requiring cGMP [283]. However, the nature of the mechanisms by which the maintenance of phosphorylation could be leading to potassium current enhancement, or how was KCa being directly activated by NO, was not elucidated. Interestingly, more recent reports using a polyclonal antibody against BK channels have shown BK-channel expression to be induced by NO in the supra-optic nucleic and neural lobe of rats [304, 305], opening the door for speculation about a possible contribution of the HRI kinase in this effect.
Hence, given the properties of HRI as a NO sensor, its expression in VSMCs (Fig. 11B), and taking into consideration its potential target mRNAs, the possibility cannot be ruled out that HRI inhibitors and/or activators could eventually be exploitable as therapeutic tools for hypertension treatment.
Involution and retraction: biological substrates of cognition?
Ever since the Spanish neuroscientist Ramon y Cajal introduced the concept at the close of XIXth century [306], it has become an undebated matter-of-course assumption that the neuroanatomical substrate of cognition rests in growth, strengthening & sprouting phenomena in neuron terminals. Nonetheless, present day evidences on a prominent synaptically-summoned proteolytic apparatus [145, 263–267] invites us to reconsider whether, beyond being a mere “tinkering-agent”in spine remodeling, proteolytical processing (involving dismantling, destabilization, and even circumscribed physiological death processes) can constitute, by itself, the physical substrate out of which cognition arises.
This would not be a novelty in physiology: examples exist in which biological function arises out of decomposition processes. For example, in the retina (Cajal’s favorite organ) light-sensitive cells (cons and rods) undergo a permanent self-renewal decomposition of photopigments, out of which visual perception arises.
Could a similar process occur in the brain? If this would be the case, neurodegeneration could be understood as a drift in cognitive-memory processes: The incapacity to rebuild the system once physiological-decomposition has taken place.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
FXG and GIR wish to express their gratitude to Dr. Francisco José Muñoz López, from the Universitat Pompeu Fabra, Barcelona, Catalonia; for his teachings on Alzheimer’s disease and oxidative stress. GIR would like to express his gratefulness also to Dr. Martin Cammarota, from the Brain Institute, Federal University of Rio Grande do Norte, Brazil; for his teachings on memory. FXG would like to thank Prof. Carlos G. Dotti for his support. This work was supported by the European Union (grants No. 267264 of the Seventh Programme for Research-FP7, and H2020-MSCA-IF-2015-708152) and from the Coordination for the Improvement of Higher Education Personnel (CAPES) agency of Brazil.
REFERENCES
[1] | Alzheimer A ((1907) ) über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Z Psychiatrie Psychisch-Gerichtliche Med 64: , 146–148. |
[2] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: , 885–890. |
[3] | Masters CL , Simms G , Weinman NA , Multhaup G , McDonald BL , Beyreuther K ((1985) ) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82: , 4245–4249. |
[4] | Kang J , Lemaire HG , Unterbeck A , Salbaum JM , Masters CL , Grzeschik KH , Multhaup G , Beyreuther K , Muller-Hill B ((1987) ) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325: , 733–736. |
[5] | Goldgaber D , Lerman MI , McBride OW , Saffiotti U , Gajdusek DC ((1987) ) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 235: , 877–880. |
[6] | Tanzi RE , Bird ED , Latt SA , Neve RL ((1987) ) The amyloid beta protein gene is not duplicated in brains from patients with Alzheimer’s disease. Science 238: , 666–669. |
[7] | Robakis NK , Ramakrishna N , Wolfe G , Wisniewski HM ((1987) ) Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A 84: , 4190–4194. |
[8] | Olson MI , Shaw CM ((1969) ) Presenile dementia and Alzheimer’s disease in mongolism. Brain 92: , 147–156. |
[9] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[10] | Selkoe DJ ((1991) ) The molecular pathology of Alzheimer’s disease. Neuron 6: , 487–498. |
[11] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[12] | Beyreuther K , Masters CL ((1991) ) Amyloid precursor protein (APP) and beta A4 amyloid in the etiology of Alzheimer’s disease: Precursor-product relationships in the derangement of neuronal function. Brain Pathol 1: , 241–251. |
[13] | Vassar R , Bennett BD , Babu-Khan S , Kahn S , Mendiaz EA , Denis P , Teplow DB , Ross S , Amarante P , Loeloff R , Luo Y , Fisher S , Fuller J , Edenson S , Lile J , Jarosinski MA , Biere AL , Curran E , Burgess T , Louis JC , Collins F , Treanor J , Rogers G , Citron M ((1999) ) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: , 735–741. |
[14] | Sinha S , Anderson JP , Barbour R , Basi GS , Caccavello R , Davis D , Doan M , Dovey HF , Frigon N , Hong J , Jacobson-Croak K , Jewett N , Keim P , Knops J , Lieberburg I , Power M , Tan H , Tatsuno G , Tung J , Schenk D , Seubert P , Suomensaari SM , Wang S , Walker D , Zhao J , McConlogue L , John V ((1999) ) Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402: , 537–540. |
[15] | Hussain I , Powell D , Howlett DR , Tew DG , Meek TD , Chapman C , Gloger IS , Murphy KE , Southan CD , Ryan DM , Smith TS , Simmons DL , Walsh FS , Dingwall C , Christie G ((1999) ) Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci 14: , 419–427. |
[16] | Yan R , Bienkowski MJ , Shuck ME , Miao H , Tory MC , Pauley AM , Brashier JR , Stratman NC , Mathews WR , Buhl AE , Carter DB , Tomasselli AG , Parodi LA , Heinrikson RL , Gurney ME ((1999) ) Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402: , 533–537. |
[17] | Lin X , Koelsch G , Wu S , Downs D , Dashti A , Tang J ((2000) ) Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A 97: , 1456–1460. |
[18] | Wimo A , Winblad B , Jonsson L ((2010) ) The worldwide societal costs of dementia: Estimates for 2009. Alzheimers Dement 6: , 98–103. |
[19] | Karran E , Mercken M , De Strooper B ((2011) ) The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov 10: , 698–712. |
[20] | Karran E , De Strooper B ((2016) ) The amyloid cascade hypothesis: Are we poised for success or failure? J Neurochem 139 Suppl 2: , 237–252. |
[21] | Hardy J , De Strooper B ((2017) ) Alzheimer’s disease: Where next for anti-amyloid therapies? Brain 140: , 853–855. |
[22] | Behl C , Ziegler C ((2017) ) Beyond amyloid - widening the view on Alzheimer’s disease. J Neurochem 143: , 394–395. |
[23] | Katzman R , Terry R , DeTeresa R , Brown T , Davies P , Fuld P , Renbing X , Peck A ((1988) ) Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol 23: , 138–144. |
[24] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[25] | Mochizuki A , Peterson JW , Mufson EJ , Trapp BD ((1996) ) Amyloid load and neural elements in Alzheimer’s disease and nondemented individuals with high amyloid plaque density. Exp Neurol 142: , 89–102. |
[26] | Rowe CC , Ellis KA , Rimajova M , Bourgeat P , Pike KE , Jones G , Fripp J , Tochon-Danguy H , Morandeau L , O’Keefe G , Price R , Raniga P , Robins P , Acosta O , Lenzo N , Szoeke C , Salvado O , Head R , Martins R , Masters CL , Ames D , Villemagne VL ((2010) ) Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31: , 1275–1283. |
[27] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2: , 135. |
[28] | Willem M , Garratt AN , Novak B , Citron M , Kaufmann S , Rittger A , DeStrooper B , Saftig P , Birchmeier C , Haass C ((2006) ) Control of peripheral nerve myelination by the beta-secretase BACE1. Science 314: , 664–666. |
[29] | Hu X , Hicks CW , He W , Wong P , Macklin WB , Trapp BD , Yan R ((2006) ) Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci 9: , 1520–1525. |
[30] | Kim DY , Carey BW , Wang H , Ingano LA , Binshtok AM , Wertz MH , Pettingell WH , He P , Lee VM , Woolf CJ , Kovacs DM ((2007) ) BACE1 regulates voltage-gated sodium channels and neuronal activity. Nat Cell Biol 9: , 755–764. |
[31] | Miyazaki H , Oyama F , Wong HK , Kaneko K , Sakurai T , Tamaoka A , Nukina N ((2007) ) BACE1 modulates filopodia-like protrusions induced by sodium channel beta4 subunit. Biochem Biophys Res Commun 361: , 43–48. |
[32] | Hemming ML , Elias JE , Gygi SP , Selkoe DJ ((2009) ) Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PLoS One 4: , e8477. |
[33] | Kuhn PH , Koroniak K , Hogl S , Colombo A , Zeitschel U , Willem M , Volbracht C , Schepers U , Imhof A , Hoffmeister A , Haass C , Rossner S , Brase S , Lichtenthaler SF ((2012) ) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 31: , 3157–3168. |
[34] | Zhou L , Barao S , Laga M , Bockstael K , Borgers M , Gijsen H , Annaert W , Moechars D , Mercken M , Gevaert K , De Strooper B ((2012) ) The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem 287: , 25927–25940. |
[35] | Hogl S , van Bebber F , Dislich B , Kuhn PH , Haass C , Schmid B , Lichtenthaler SF ((2013) ) Label-free quantitative analysis of the membrane proteome of Bace1 protease knock-out zebrafish brains. Proteomics 13: , 1519–1527. |
[36] | Dislich B , Wohlrab F , Bachhuber T , Muller SA , Kuhn PH , Hogl S , Meyer-Luehmann M , Lichtenthaler SF ((2015) ) Label-free quantitative proteomics of mouse cerebrospinal fluid detects beta-site APP cleaving enzyme (BACE1) protease substrates in vivo. Mol Cell Proteomics 14: , 2550–2563. |
[37] | Laird FM , Cai H , Savonenko AV , Farah MH , He K , Melnikova T , Wen H , Chiang HC , Xu G , Koliatsos VE , Borchelt DR , Price DL , Lee HK , Wong PC ((2005) ) BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25: , 11693–11709. |
[38] | Ma H , Lesne S , Kotilinek L , Steidl-Nichols JV , Sherman M , Younkin L , Younkin S , Forster C , Sergeant N , Delacourte A , Vassar R , Citron M , Kofuji P , Boland LM , Ashe KH ((2007) ) Involvement of beta-site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein-mediated enhancement of memory and activity-dependent synaptic plasticity. Proc Natl Acad Sci U S A 104: , 8167–8172. |
[39] | Feig S , Lipton P ((1993) ) Pairing the cholinergic agonist carbachol with patterned Schaffer collateral stimulation initiates protein synthesis in hippocampal CA1 pyramidal cell dendrites via a muscarinic, NMDA-dependent mechanism. J Neurosci 13: , 1010–1021. |
[40] | Kang H , Schuman EM ((1996) ) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273: , 1402–1406. |
[41] | Martin KC , Casadio A , Zhu H , Yaping E , Rose JC , Chen M , Bailey CH , Kandel ER ((1997) ) Synapse-specific, long-term facilitation of aplysia sensory to motor synapses: A function for local protein synthesis in memory storage. Cell 91: , 927–938. |
[42] | Huber KM , Kayser MS , Bear MF ((2000) ) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: , 1254–1257. |
[43] | Zhang X , Poo MM ((2002) ) Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron 36: , 675–688. |
[44] | Flexner JB , Flexner LB , Stellar E ((1963) ) Memory in mice as affected by intracerebral puromycin. Science 141: , 57–59. |
[45] | Agranoff BW , Klinger PD ((1964) ) Puromycin effect on memory fixation in the goldfish. Science 146: , 952–953. |
[46] | Flexner LB , Flexner JB , De La HG , Roberts RB ((1965) ) Loss of memory as related to inhibition of cerebral protein synthesis. J Neurochem 12: , 535–541. |
[47] | Barondes SH , Cohen HD ((1967) ) Delayed and sustained effect of acetoxycycloheximide on memory in mice. Proc Natl Acad Sci U S A 58: , 157–164. |
[48] | Squire LR , Barondes SH ((1972) ) Variable decay of memory and its recovery in cycloheximide-treated mice. Proc Natl Acad Sci U S A 69: , 1416–1420. |
[49] | Flood JF , Bennett EL , Orme E , Rosenzweig MR ((1975) ) Relation of memory formation to controlled amounts of brain protein synthesis. Physiol Behav 15: , 97–102. |
[50] | Dunn AJ , Leibmann S ((1977) ) The amnestic effect of protein synthesis inhibitors is not due to the inhibition of adrenal corticosteroidogenesis. Behav Biol 19: , 411–416. |
[51] | Cammarota M , Bevilaqua LR , Kerr D , Medina JH , Izquierdo I ((2003) ) Inhibition of mRNA and protein synthesis in the CA1 region of the dorsal hippocampus blocks reinstallment of an extinguished conditioned fear response. J Neurosci 23: , 737–741. |
[52] | Luft AR , Buitrago MM , Ringer T , Dichgans J , Schulz JB ((2004) ) Motor skill learning depends on protein synthesis in motor cortex after training. J Neurosci 24: , 6515–6520. |
[53] | Kelleher RJ III , Govindarajan A , Jung HY , Kang H , Tonegawa S ((2004) ) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116: , 467–479. |
[54] | Lima RH , Rossato JI , Furini CR , Bevilaqua LR , Izquierdo I , Cammarota M ((2009) ) Infusion of protein synthesis inhibitors in the entorhinal cortex blocks consolidation but not reconsolidation of object recognition memory. Neurobiol Learn Mem 91: , 466–472. |
[55] | Rossato JI , Bevilaqua LR , Izquierdo I , Medina JH , Cammarota M ((2010) ) Retrieval induces reconsolidation of fear extinction memory. Proc Natl Acad Sci U S A 107: , 21801–21805. |
[56] | Glassman E ((1969) ) The biochemistry of learning: An evaluation of the role of RNA and protein. Annu Rev Biochem 38: , 605–646. |
[57] | Matthies H ((1974) ) The biochemical basis of learning and memory. Life Sci 15: , 2017–2031. |
[58] | Davis HP , Squire LR ((1984) ) Protein synthesis and memory: A review. Psychol Bull 96: , 518–559. |
[59] | Schuman EM ((1999) ) mRNA trafficking and local protein synthesis at the synapse. Neuron 23: , 645–648. |
[60] | Klann E , Dever TE ((2004) ) Biochemical mechanisms for translational regulation in synaptic plasticity. Nat Rev Neurosci 5: , 931–942. |
[61] | Klann E , Antion MD , Banko JL , Hou L ((2004) ) Synaptic plasticity and translation initiation. Learn Mem 11: , 365–372. |
[62] | Kindler S , Wang H , Richter D , Tiedge H ((2005) ) RNA transport and local control of translation. Annu Rev Cell Dev Biol 21: , 223–245. |
[63] | Sutton MA , Schuman EM ((2005) ) Local translational control in dendrites and its role in long-term synaptic plasticity. J Neurobiol 64: , 116–131. |
[64] | Sutton MA , Schuman EM ((2006) ) Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127: , 49–58. |
[65] | Bramham CR , Wells DG ((2007) ) Dendritic mRNA: Transport, translation and function. Nat Rev Neurosci 8: , 776–789. |
[66] | Gold PE ((2008) ) Protein synthesis inhibition and memory: Formation vs amnesia. Neurobiol Learn Mem 89: , 201–211. |
[67] | Costa-Mattioli M , Sossin WS , Klann E , Sonenberg N ((2009) ) Translational control of long-lasting synaptic plasticity and memory. Neuron 61: , 10–26. |
[68] | Bekinschtein P , Katche C , Slipczuk L , Gonzalez C , Dorman G , Cammarota M , Izquierdo I , Medina JH ((2010) ) Persistence of long-term memory storage: New insights into its molecular signatures in the hippocampus and related structures. Neurotox Res 18: , 377–385. |
[69] | O’Connor T , Sadleir KR , Maus E , Velliquette RA , Zhao J , Cole SL , Eimer WA , Hitt B , Bembinster LA , Lammich S , Lichtenthaler SF , Hebert SS , De SB , Haass C , Bennett DA , Vassar R ((2008) ) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60: , 988–1009. |
[70] | Mouton-Liger F , Paquet C , Dumurgier J , Bouras C , Pradier L , Gray F , Hugon J ((2012) ) Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim Biophys Acta 1822: , 885–896. |
[71] | ILL-Raga G , Palomer E , Wozniak MA , Ramos-Fernandez E , Bosch-Morato M , Tajes M , Guix FX , Galan JJ , Clarimon J , Antunez C , Real LM , Boada M , Itzhaki RF , Fandos C , Munoz FJ ((2011) ) Activation of PKR causes amyloid beta-peptide accumulation via de-repression of BACE1 expression. PLoS One 6: , e21456. |
[72] | Goate A , Chartier-Harlin MC , Mullan M , Brown J , Crawford F , Fidani L , Giuffra L , Haynes A , Irving N , James L , et al. ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: , 704–706. |
[73] | Hardy J ((1992) ) Framing beta-amyloid. Nat Genet 1: , 233–234. |
[74] | De Strooper B , Saftig P , Craessaerts K , Vanderstichele H , Guhde G , Annaert W , Von FK , Van LF ((1998) ) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: , 387–390. |
[75] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K , et al. ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[76] | Rogaev EI , Sherrington R , Rogaeva EA , Levesque G , Ikeda M , Liang Y , Chi H , Lin C , Holman K , Tsuda T , et al. ((1995) ) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376: , 775–778. |
[77] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G , Ikeda M , Chi H , Lin C , Li G , Holman K , Tsuda T , Mar L , Foncin JF , Bruni AC , Montesi MP , Sorbi S , Rainero I , Pinessi L , Nee L , Chumakov I , Pollen D , Brookes A , Sanseau P , Polinsky RJ , Wasco W , Da Silva HA , Haines JL , Perkicak-Vance MA , Tanzi RE , Roses AD , Fraser PE , Rommens JM , George-Hyslop PH ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[78] | Rovelet-Lecrux A , Hannequin D , Raux G , Le Meur N , Laquerriere A , Vital A , Dumanchin C , Feuillette S , Brice A , Vercelletto M , Dubas F , Frebourg T , Campion D ((2006) ) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet 38: , 24–26. |
[79] | Wisniewski T , Ghiso J , Frangione B ((1991) ) Peptides homologous to the amyloid protein of Alzheimer’s disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem Biophys Res Commun 180: , 1528. |
[80] | Wisniewski T , Frangione B ((1992) ) Molecular biology of Alzheimer’s amyloid–Dutch variant. Mol Neurobiol 6: , 75–86. |
[81] | Nilsberth C , Westlind-Danielsson A , Eckman CB , Condron MM , Axelman K , Forsell C , Stenh C , Luthman J , Teplow DB , Younkin SG , Naslund J , Lannfelt L ((2001) ) The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat Neurosci 4: , 887–893. |
[82] | Citron M , Oltersdorf T , Haass C , McConlogue L , Hung AY , Seubert P , Vigo-Pelfrey C , Lieberburg I , Selkoe DJ ((1992) ) Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 360: , 672–674. |
[83] | Scheuner D , Eckman C , Jensen M , Song X , Citron M , Suzuki N , Bird TD , Hardy J , Hutton M , Kukull W , Larson E , Levy-Lahad E , Viitanen M , Peskind E , Poorkaj P , Schellenberg G , Tanzi R , Wasco W , Lannfelt L , Selkoe D , Younkin S ((1996) ) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased} by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2: , 864–870. |
[84] | Hardy J ((2017) ) The discovery of Alzheimer-causing mutations in the APP gene and the formulation of the “amyloid cascade hypothesis”. FEBS J 284: , 1040–1044. |
[85] | Pike CJ , Ramezan-Arab N , Cotman CW ((1997) ) Beta-amyloid neurotoxicity in vitro: Evidence of oxidative stress but not protection by antioxidants. J Neurochem 69: , 1601–1611. |
[86] | Huang X , Atwood CS , Hartshorn MA , Multhaup G , Goldstein LE , Scarpa RC , Cuajungco MP , Gray DN , Lim J , Moir RD , Tanzi RE , Bush AI ((1999) ) The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38: , 7609–7616. |
[87] | Huang X , Cuajungco MP , Atwood CS , Hartshorn MA , Tyndall JD , Hanson GR , Stokes KC , Leopold M , Multhaup G , Goldstein LE , Scarpa RC , Saunders AJ , Lim J , Moir RD , Glabe C , Bowden EF , Masters CL , Fairlie DP , Tanzi RE , Bush AI ((1999) ) Cu(II) potentiation of AlzheimerAbeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274: , 37111–37116. |
[88] | Ali FE , Separovic F , Barrow CJ , Cherny RA , Fraser F , Bush AI , Masters CL , Barnham KJ ((2005) ) Methionine regulates copper/hydrogen peroxide oxidation products of Abeta. J Pept Sci 11: , 353–360. |
[89] | Behl C , Davis JB , Lesley R , Schubert D ((1994) ) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77: , 817–827. |
[90] | Lovell MA , Ehmann WD , Butler SM , Markesbery WR ((1995) ) Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45: , 1594–1601. |
[91] | Smith MA , Perry G , Richey PL , Sayre LM , Anderson VE , Beal MF , Kowall N ((1996) ) Oxidative damage in Alzheimer’s. Nature 382: , 120–121. |
[92] | Miranda S , Opazo C , Larrondo LF , Munoz FJ , Ruiz F , Leighton F , Inestrosa NC ((2000) ) The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s disease. Prog Neurobiol 62: , 633–648. |
[93] | Tamagno E , Bardini P , Obbili A , Vitali A , Borghi R , Zaccheo D , Pronzato MA , Danni O , Smith MA , Perry G , Tabaton M ((2002) ) Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 10: , 279–288. |
[94] | Lambert MP , Barlow AK , Chromy BA , Edwards C , Freed R , Liosatos M , Morgan TE , Rozovsky I , Trommer B , Viola KL , Wals P , Zhang C , Finch CE , Krafft GA , Klein WL ((1998) ) Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95: , 6448–6453. |
[95] | De Felice FG , Velasco PT , Lambert MP , Viola K , Fernandez SJ , Ferreira ST , Klein WL ((2007) ) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem 282: , 11590–11601. |
[96] | Savva GM , Wharton SB , Ince PG , Forster G , Matthews FE , Brayne C ((2009) ) Age, neuropathology, and dementia. N Engl J Med 360: , 2302–2309. |
[97] | Crews L , Masliah E ((2010) ) Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum Mol Genet 19: , R12–R20. |
[98] | Walsh DM , Selkoe DJ ((2004) ) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44: , 181–193. |
[99] | Willem M , Lammich S , Haass C ((2009) ) Function, regulation and therapeutic properties of beta-secretase (BACE1). Semin Cell Dev Biol 20: , 175–182. |
[100] | Dislich B , Lichtenthaler SF ((2012) ) The membrane-bound aspartyl protease BACE1: Molecular and functional properties in Alzheimer’s disease and beyond. Front Physiol 3: , 8. |
[101] | Vassar R , Kuhn PH , Haass C , Kennedy ME , Rajendran L , Wong PC , Lichtenthaler SF ((2014) ) Function, therapeutic potential and cell biology of BACE proteases: Current status and future prospects. J Neurochem 130: , 4–28. |
[102] | Barão S , Moechars D , Lichtenthaler SF , De Strooper B ((2016) ) BACE1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci 39: , 158–169. |
[103] | Munro KM , Nash A , Pigoni M , Lichtenthaler SF , Gunnersen JM ((2016) ) Functions of the Alzheimer’s disease protease BACE1 at the synapse in the central nervous system. J Mol Neurosci 60: , 305–315. |
[104] | Holsinger RM , McLean CA , Beyreuther K , Masters CL , Evin G ((2002) ) Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol 51: , 783–786. |
[105] | Preece P , Virley DJ , Costandi M , Coombes R , Moss SJ , Mudge AW , Jazin E , Cairns NJ ((2003) ) Beta-secretase (BACE) and GSK-3 mRNA levels in Alzheimer’s disease. Brain Res Mol Brain Res 116: , 155–158. |
[106] | Matsui T , Ingelsson M , Fukumoto H , Ramasamy K , Kowa H , Frosch MP , Irizarry MC , Hyman BT ((2007) ) Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res 1161: , 116–123. |
[107] | Borghi R , Patriarca S , Traverso N , Piccini A , Storace D , Garuti A , Gabriella C , Patrizio O , Massimo T ((2007) ) The increased activity of BACE1 correlates with oxidative stress in Alzheimer’s disease. Neurobiol Aging 28: , 1009–1014. |
[108] | Welsh GI , Proud CG ((1993) ) Glycogen synthase kinase-3 is rapidly inactivated in response to insulin and phosphorylates eukaryotic initiation factor eIF-2B. Biochem J 294 (Pt 3): , 625–629. |
[109] | Rowlands AG , Panniers R , Henshaw EC ((1988) ) The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J Biol Chem 263: , 5526–5533. |
[110] | Ishiguro K , Shiratsuchi A , Sato S , Omori A , Arioka M , Kobayashi S , Uchida T , Imahori K ((1993) ) Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett 325: , 167–172. |
[111] | Llorens-Martin M , Jurado J , Hernandez F , Avila J ((2014) ) GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci 7: , 46. |
[112] | De Pietri-Tonelli D , Mihailovich M , Di Cesare A , Codazzi F , Grohovaz F , Zacchetti D ((2004) ) Translational regulation of BACE-1 expression in neuronal and non-neuronal cells. Nucleic Acids Res 32: , 1808–1817. |
[113] | Lammich S , Schobel S , Zimmer AK , Lichtenthaler SF , Haass C ((2004) ) Expression of the Alzheimer protease BACE1 is suppressed via its 5’-untranslated region. EMBO Rep 5: , 620–625. |
[114] | Rogers GW Jr. , Edelman GM , Mauro VP ((2004) ) Differential utilization of upstream AUGs in the beta-secretase mRNA suggests that a shunting mechanism regulates translation. Proc Natl Acad Sci U S A 101: , 2794–2799. |
[115] | Zhou W , Song W ((2006) ) Leaky scanning and reinitiation regulate BACE1 gene expression. Mol Cell Biol 26: , 3353–3364. |
[116] | Mihailovich M , Thermann R , Grohovaz F , Hentze MW , Zacchetti D ((2007) ) Complex translational regulation of BACE1 involves upstream AUGs and stimulatory elements within the 5’ untranslated region. Nucleic Acids Res 35: , 2975–2985. |
[117] | Garthwaite J , Charles SL , Chess-Williams R ((1988) ) Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336: , 385–388. |
[118] | Bult H , Boeckxstaens GE , Pelckmans PA , Jordaens FH , Van Maercke YM , Herman AG ((1990) ) Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature 345: , 346–347. |
[119] | Boeckxstaens GE , Pelckmans PA , Bult H , De Man JG , Herman AG , Van Maercke YM ((1990) ) Non-adrenergic non-cholinergic relaxation mediated by nitric oxide in the canine ileocolonic junction. Eur J Pharmacol 190: , 239–246. |
[120] | Zakhary R , Poss KD , Jaffrey SR , Ferris CD , Tonegawa S , Snyder SH ((1997) ) Targeted gene deletion of heme oxygenase 2 reveals neural role for carbon monoxide. Proc Natl Acad Sci U S A 94: , 14848–14853. |
[121] | Xue L , Farrugia G , Miller SM , Ferris CD , Snyder SH , Szurszewski JH ((2000) ) Carbon monoxide and nitric oxide as coneurotransmitters in the enteric nervous system: Evidence from genomic deletion of biosynthetic enzymes. Proc Natl Acad Sci U S A 97: , 1851–1855. |
[122] | Son H , Hawkins RD , Martin K , Kiebler M , Huang PL , Fishman MC , Kandel ER ((1996) ) Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 87: , 1015–1023. |
[123] | Arancio O , Kiebler M , Lee CJ , Lev-Ram V , Tsien RY , Kandel ER , Hawkins RD ((1996) ) Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87: , 1025–1035. |
[124] | Bohme GA , Bon C , Lemaire M , Reibaud M , Piot O , Stutzmann JM , Doble A , Blanchard JC ((1993) ) Altered synaptic plasticity and memory formation in nitric oxide synthase inhibitor-treated rats. Proc Natl Acad Sci U S A 90: , 9191–9194. |
[125] | Susswein AJ , Katzoff A , Miller N , Hurwitz I ((2004) ) Nitric oxide and memory. Neuroscientist 10: , 153–162. |
[126] | Zinn CG , Bevilaqua LR , Rossato JI , Medina JH , Izquierdo I , Cammarota M ((2009) ) On the requirement of nitric oxide signaling in the amygdala for consolidation of inhibitory avoidance memory. Neurobiol Learn Mem 91: , 266–272. |
[127] | Furini CR , Rossato JI , Bitencourt LL , Medina JH , Izquierdo I , Cammarota M ((2010) ) Beta-adrenergic receptors link NO/sGC/PKG signaling to BDNF expression during the consolidation of object recognition long-term memory. Hippocampus 20: , 672–683. |
[128] | Garthwaite J ((2008) ) Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci 27: , 2783–2802. |
[129] | Uma S , Matts RL , Guo Y , White S , Chen JJ ((2000) ) The N-terminal region of the heme-regulated eIF2alpha kinase is an autonomous heme binding domain. Eur J Biochem 267: , 498–506. |
[130] | Uma S , Yun BG , Matts RL ((2001) ) The heme-regulated eukaryotic initiation factor 2alpha kinase. A potential regulatory target for control of protein synthesis by diffusible gases. J Biol Chem 276: , 14875–14883. |
[131] | Ishikawa H , Yun BG , Takahashi S , Hori H , Matts RL , Ishimori K , Morishima I ((2002) ) NO-induced activation mechanism of the heme-regulated eIF2alpha kinase. J Am Chem Soc 124: , 13696–13697. |
[132] | Igarashi J , Sato A , Kitagawa T , Yoshimura T , Yamauchi S , Sagami I , Shimizu T ((2004) ) Activation of heme-regulated eukaryotic initiation factor 2alpha kinase by nitric oxide is induced by the formation of a five-coordinate NO-heme complex: Optical absorption, electron spin resonance, and resonance raman spectral studies. J Biol Chem 279: , 15752–15762. |
[133] | Girvan HM , Munro AW ((2013) ) Heme sensor proteins. J Biol Chem 288: , 13194–13203. |
[134] | Guix FX , Uribesalgo I , Coma M , Munoz FJ ((2005) ) The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol 76: , 126–152. |
[135] | Hall CN , Garthwaite J ((2009) ) What is the real physiological NO concentration in vivo? Nitric Oxide 21: , 92–103. |
[136] | Wek RC , Jiang HY , Anthony TG ((2006) ) Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans 34: , 7–11. |
[137] | Costa-Mattioli M , Gobert D , Harding H , Herdy B , Azzi M , Bruno M , Bidinosti M , Ben MC , Marcinkiewicz E , Yoshida M , Imataka H , Cuello AC , Seidah N , Sossin W , Lacaille JC , Ron D , Nader K , Sonenberg N ((2005) ) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature 436: , 1166–1173. |
[138] | Costa-Mattioli M , Gobert D , Stern E , Gamache K , Colina R , Cuello C , Sossin W , Kaufman R , Pelletier J , Rosenblum K , Krnjevic K , Lacaille JC , Nader K , Sonenberg N ((2007) ) eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell 129: , 195–206. |
[139] | Costa-Mattioli M , Sonenberg N ((2006) ) Translational control of long-term synaptic plasticity and memory storage by eIF2alpha. Crit Rev Neurobiol 18: , 187–195. |
[140] | Costa-Mattioli M , Sonenberg N ((2008) ) Translational control of gene expression: A molecular switch for memory storage. Prog Brain Res 169: , 81–95. |
[141] | Gkogkas C , Sonenberg N , Costa-Mattioli M ((2010) ) Translational control mechanisms in long-lasting synaptic plasticity and memory. J Biol Chem 285: , 31913–31917. |
[142] | ILL-Raga G , Kohler C , Radiske A , Lima RH , Rosen MD , Munoz FJ , Cammarota M ((2013) ) Consolidation of object recognition memory requires HRI kinase-dependent phosphorylation of eIF2alpha in the hippocampus. Hippocampus 23: , 431–436. |
[143] | Mellor H , Flowers KM , Kimball SR , Jefferson LS ((1994) ) Cloning and characterization of cDNA encoding rat hemin-sensitive initiation factor-2 alpha (eIF-2 alpha) kinase. Evidence for multitissue expression. J Biol Chem 269: , 10201–10204. |
[144] | Chen JJ , London IM ((1995) ) Regulation of protein synthesis by heme-regulated eIF-2 alpha kinase. Trends Biochem Sci 20: , 105–108. |
[145] | ILL-Raga G , Tajes M , Busquets-Garcia A , Ramos-Fernandez E , Vargas LM , Bosch-Morato M , Guivernau B , Valls-Comamala V , Eraso-Pichot A , Guix FX , Fandos C , Rosen MD , Rabinowitz MH , Maldonado R , Alvarez AR , Ozaita A , Munoz FJ ((2015) ) Physiological control of nitric oxide in neuronal BACE1 translation by heme-regulated eIF2alpha kinase HRI induces synaptogenesis. Antioxid Redox Signal 22: , 1295–1307. |
[146] | Bellato HM , Hajj GN ((2016) ) Translational control by eIF2alpha in neurons: Beyond the stress response. Cytoskeleton (Hoboken) 73: , 551–565. |
[147] | Chesnokova E , Bal N , Kolosov P ((2017) ) Kinases of eIF2a switch translation of mRNA subset during neuronal plasticity. Int J Mol Sci 18: , E2213. |
[148] | Trinh MA , Klann E ((2013) ) Translational control by eIF2alpha kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol Learn Mem 105: , 93–99. |
[149] | Ramos-Fernandez E , Tajes M , ILL-Raga G , Vargas L , Busquets-Garcia A , Bosch-Morato M , Guivernau B , Valls-Comamala V , Gomis M , Grau C , Fandos C , Rosen MD , Rabinowitz MH , Inestrosa N , Maldonado R , Altafaj X , Ozaita A , Alvarez A , Vicente R , Valverde MA , Munoz FJ ((2016) ) Glutamatergic stimulation induces GluN2B translation by the nitric oxide-Heme-Regulated eIF2alpha kinase in cortical neurons. Oncotarget 7: , 58876–58892. |
[150] | Bailey CH , Kandel ER ((1993) ) Structural changes accompanying memory storage. Annu Rev Physiol 55: , 397–426. |
[151] | Bailey CH , Kandel ER , Si K ((2004) ) The persistence of long-term memory: A molecular approach to self-sustaining changes in learning-induced synaptic growth. Neuron 44: , 49–57. |
[152] | Saftig P , Bovolenta P ((2015) ) Proteases at work: Cues for understanding neural development and degeneration. Front Mol Neurosci 8: , 13. |
[153] | Park S , Lee C , Sabharwal P , Zhang M , Meyers CL , Sockanathan S ((2013) ) GDE2 promotes neurogenesis by glycosylphosphatidylinositol-anchor cleavage of RECK. Science 339: , 324–328. |
[154] | Jorissen E , Prox J , Bernreuther C , Weber S , Schwanbeck R , Serneels L , Snellinx A , Craessaerts K , Thathiah A , Tesseur I , Bartsch U , Weskamp G , Blobel CP , Glatzel M , De Strooper B , Saftig P ((2010) ) The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci 30: , 4833–4844. |
[155] | Santiago-Medina M , Gregus KA , Nichol RH , O’Toole SM , Gomez TM ((2015) ) Regulation of ECM degradation and axon guidance by growth cone invadosomes. Development 142: , 486–496. |
[156] | Bai G , Chivatakarn O , Bonanomi D , Lettieri K , Franco L , Xia C , Stein E , Ma L , Lewcock JW , Pfaff SL ((2011) ) Presenilin-dependent receptor processing is required for axon guidance. Cell 144: , 106–118. |
[157] | Bai G , Pfaff SL ((2011) ) Protease regulation: The Yin and Yang of neural development and disease. Neuron 72: , 9–21. |
[158] | Tsilibary E , Tzinia A , Radenovic L , Stamenkovic V , Lebitko T , Mucha M , Pawlak R , Frischknecht R , Kaczmarek L ((2014) ) Neural ECM proteases in learning and synaptic plasticity. Prog Brain Res 214: , 135–157. |
[159] | Mozrzymas JW , Kaczmarek L ((2016) ) Editorial: Neuroplasticity and extracellular proteolysis. Front Cell Neurosci 10: , 59. |
[160] | Salazar IL , Caldeira MV , Curcio M , Duarte CB ((2016) ) The role of proteases in hippocampal synaptic plasticity: Putting together small pieces of a complex puzzle. Neurochem Res 41: , 156–182. |
[161] | Prox J , Bernreuther C , Altmeppen H , Grendel J , Glatzel M , D’Hooge R , Stroobants S , Ahmed T , Balschun D , Willem M , Lammich S , Isbrandt D , Schweizer M , Horre K , De Strooper B , Saftig P ((2013) ) Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J Neurosci 33: , 12915–12928, 12928a. |
[162] | Dziembowska M , Milek J , Janusz A , Rejmak E , Romanowska E , Gorkiewicz T , Tiron A , Bramham CR , Kaczmarek L ((2012) ) Activity-dependent local translation of matrix metalloproteinase-9. J Neurosci 32: , 14538–14547. |
[163] | de Haro C , Mendez R , Santoyo J ((1996) ) The eIF-2alpha kinases and the control of protein synthesis. FASEB J 10: , 1378–1387. |
[164] | Ohno M ((2014) ) Roles of eIF2alpha kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 7: , 22. |
[165] | Devi L , Ohno M ((2010) ) Phospho-eIF2alpha level is important for determining abilities of BACE1 reduction to rescue cholinergic neurodegeneration and memory defects in 5XFAD mice. PLoS One 5: , e12974. |
[166] | Itzhaki RF , Wozniak MA ((2008) ) Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J Alzheimers Dis 13: , 393–405. |
[167] | Itzhaki RF ((2014) ) Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front Aging Neurosci 6: , 202. |
[168] | Itzhaki RF ((2016) ) Herpes and Alzheimer’s disease: Subversion in the central nervous system and how it might be halted. J Alzheimers Dis 54: , 1273–1281. |
[169] | Jamieson GA , Maitland NJ , Wilcock GK , Craske J , Itzhaki RF ((1991) ) Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol 33: , 224–227. |
[170] | Itzhaki RF , Lin WR , Wilcock GK , Faragher B ((1998) ) HSV-1 and risk of Alzheimer’s disease. Lancet 352: , 238. |
[171] | Wozniak MA , Itzhaki RF , Shipley SJ , Dobson CB ((2007) ) Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett 429: , 95–100. |
[172] | Wozniak MA , Mee AP , Itzhaki RF ((2009) ) Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol 217: , 131–138. |
[173] | Zhang F , Romano PR , Nagamura-Inoue T , Tian B , Dever TE , Mathews MB , Ozato K , Hinnebusch AG ((2001) ) Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J Biol Chem 276: , 24946–24958. |
[174] | Lindholm D , Wootz H , Korhonen L ((2006) ) ER stress and neurodegenerative diseases. Cell Death Differ 13: , 385–392. |
[175] | Jang H , Zheng J , Nussinov R ((2007) ) Models of beta-amyloid ion channels in the membrane suggest that channel formation in the bilayer is a dynamic process. Biophys J 93: , 1938–1949. |
[176] | Shankar GM , Bloodgood BL , Townsend M , Walsh DM , Selkoe DJ , Sabatini BL ((2007) ) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27: , 2866–2875. |
[177] | Zhao J , Fu Y , Yasvoina M , Shao P , Hitt B , O’Connor T , Logan S , Maus E , Citron M , Berry R , Binder L , Vassar R ((2007) ) Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: Implications for Alzheimer’s disease pathogenesis. J Neurosci 27: , 3639–3649. |
[178] | Zhang XM , Cai Y , Xiong K , Cai H , Luo XG , Feng JC , Clough RW , Struble RG , Patrylo PR , Yan XX ((2009) ) Beta-secretase-1 elevation in transgenic mouse models of Alzheimer’s disease is associated with synaptic/axonal pathology and amyloidogenesis: Implications for neuritic plaque development. Eur J Neurosci 30: , 2271–2283. |
[179] | Kandalepas PC , Sadleir KR , Eimer WA , Zhao J , Nicholson DA , Vassar R ((2013) ) The Alzheimer’s beta-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol 126: , 329–352. |
[180] | Sadleir KR , Eimer WA , Kaufman RJ , Osten P , Vassar R ((2014) ) Genetic inhibition of phosphorylation of the translation initiation factor eIF2alpha does not block Abeta-dependent elevation of BACE1 and APP levels or reduce amyloid pathology in a mouse model of Alzheimer’s disease. PLoS One 9: , e101643. |
[181] | Mouton-Liger F , Rebillat AS , Gourmaud S , Paquet C , Leguen A , Dumurgier J , Bernadelli P , Taupin V , Pradier L , Rooney T , Hugon J ((2015) ) PKR downregulation prevents neurodegeneration and beta-amyloid production in a thiamine-deficient model. Cell Death Dis 6: , e1594. |
[182] | Devi L , Ohno M ((2014) ) PERK mediates eIF2alpha phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol Aging 35: , 2272–2281. |
[183] | Carret-Rebillat AS , Pace C , Gourmaud S , Ravasi L , Montagne-Stora S , Longueville S , Tible M , Sudol E , Chang RC , Paquet C , Mouton-Liger F , Hugon J ((2015) ) Neuroinflammation and Abeta accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci Rep 5: , 8489. |
[184] | Yan L , Deng Y , Gao J , Liu Y , Li F , Shi J , Gong Q ((2017) ) Icariside II Effectively Reduces Spatial Learning and memory impairments in Alzheimer’s disease model mice targeting beta-amyloid production. Front Pharmacol 8: , 106. |
[185] | Li C , Guo XD , Lei M , Wu JY , Jin JZ , Shi XF , Zhu ZY , Rukachaisirikul V , Hu LH , Wen TQ , Shen X ((2017) ) Thamnolia vermicularis extract improves learning ability in APP/PS1 transgenic mice by ameliorating both Abeta and Tau pathologies. Acta Pharmacol Sin 38: , 9–28. |
[186] | Zhang JS , Zhou SF , Wang Q , Guo JN , Liang HM , Deng JB , He WY ((2016) ) Gastrodin suppresses BACE1 expression under oxidative stress condition via inhibition of the PKR/eIF2alpha pathway in Alzheimer’s disease. Neuroscience 325: , 1–9. |
[187] | Sze SC , Tong Y , Ng TB , Cheng CL , Cheung HP ((2010) ) Herba Epimedii: Anti-oxidative properties and its medical implications. Molecules 15: , 7861–7870. |
[188] | Luo H , Ren M , Lim KM , Koh YJ , Wang LS , Hur JS ((2006) ) Antioxidative activity of lichen Thamnolia vermicularis in vitro. Mycobiology 34: , 124–127. |
[189] | Gannon PJ , Akay-Espinoza C , Yee AC , Briand LA , Erickson MA , Gelman BB , Gao Y , Haughey NJ , Zink MC , Clements JE , Kim NS , Van De WG , Jensen BK , Vassar R , Pierce RC , Gill AJ , Kolson DL , Diehl JA , Mankowski JL , Jordan-Sciutto KL ((2017) ) HIV protease inhibitors alter amyloid precursor protein processing via beta-site amyloid precursor protein cleaving enzyme-1 translational up-regulation. Am J Pathol 187: , 91–109. |
[190] | Sanchez AB , Kaul M ((2017) ) Neuronal stress and injury caused by HIV-1, cART and drug abuse: Converging contributions to HAND. Brain Sci 7: , E25. |
[191] | Mori K ((2000) ) Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 101: , 451–454. |
[192] | Kaufman RJ ((2002) ) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: , 1389–1398. |
[193] | Schroder M , Kaufman RJ ((2005) ) ER stress and the unfolded protein response. Mutat Res 569: , 29–63. |
[194] | Rozpedek W , Markiewicz L , Diehl JA , Pytel D , Majsterek I ((2015) ) Unfolded protein response and PERK kinase as a new therapeutic target in the pathogenesis of Alzheimer’s disease. Curr Med Chem 22: , 3169–3184. |
[195] | Dreses-Werringloer U , Lambert JC , Vingtdeux V , Zhao H , Vais H , Siebert A , Jain A , Koppel J , Rovelet-Lecrux A , Hannequin D , Pasquier F , Galimberti D , Scarpini E , Mann D , Lendon C , Campion D , Amouyel P , Davies P , Foskett JK , Campagne F , Marambaud P ((2008) ) A polymorphism in CALHM1 influences Ca2+homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 133: , 1149–1161. |
[196] | Suzuki N , Cheung TT , Cai XD , Odaka A , Otvos L Jr. , Eckman C , Golde TE , Younkin SG ((1994) ) An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264: , 1336–1340. |
[197] | LaFerla FM ((2002) ) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3: , 862–872. |
[198] | Liang SH , Zhang W , McGrath BC , Zhang P , Cavener DR ((2006) ) PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem J 393: , 201–209. |
[199] | Querfurth HW , Jiang J , Geiger JD , Selkoe DJ ((1997) ) Caffeine stimulates amyloid beta-peptide release from beta-amyloid precursor protein-transfected HEK293 cells. J Neurochem 69: , 1580–1591. |
[200] | Cheung KH , Shineman D , Muller M , Cardenas C , Mei L , Yang J , Tomita T , Iwatsubo T , Lee VM , Foskett JK ((2008) ) Mechanism of Ca2+disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58: , 871–883. |
[201] | Querfurth HW , Selkoe DJ ((1994) ) Calcium ionophore increases amyloid beta peptide production by cultured cells. Biochemistry 33: , 4550–4561. |
[202] | Alzheimer’s Association Calcium Hypothesis Workgroup ((2017) ) Calcium hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 13: , 178–182 e117. |
[203] | Lu L , Han AP , Chen JJ ((2001) ) Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol 21: , 7971–7980. |
[204] | Ochoa S ((1983) ) Regulation of protein synthesis initiation in eucaryotes. Arch Biochem Biophys 223: , 325–349. |
[205] | Atamna H , Frey WH ((2004) ) A role for heme in Alzheimer’s disease: Heme binds amyloid beta and has altered metabolism. Proc Natl Acad Sci U S A 101: , 11153–11158. |
[206] | Dwyer BE , Smith MA , Richardson SL , Perry G , Zhu X ((2009) ) Down-regulation of aminolevulinate synthase, the rate-limiting enzyme for heme biosynthesis in Alzheimer’s disease. Neurosci Lett 460: , 180–184. |
[207] | Perry G , Nunomura A , Raina AK , Aliev G , Siedlak SL , Harris PL , Casadesus G , Petersen RB , Bligh-Glover W , Balraj E , Petot GJ , Smith MA ((2003) ) A metabolic basis for Alzheimer disease. Neurochem Res 28: , 1549–1552. |
[208] | Tyrrell RM , Applegate LA , Tromvoukis Y ((1993) ) The proximal promoter region of the human heme oxygenase gene contains elements involved in stimulation of transcriptional activity by a variety of agents including oxidants. Carcinogenesis 14: , 761–765. |
[209] | Keyse SM , Tyrrell RM ((1989) ) Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc Natl Acad Sci U S A 86: , 99–103. |
[210] | Keyse SM , Applegate LA , Tromvoukis Y , Tyrrell RM ((1990) ) Oxidant stress leads to transcriptional activation of the human heme oxygenase gene in cultured skin fibroblasts. Mol Cell Biol 10: , 4967–4969. |
[211] | Smith MA , Kutty RK , Richey PL , Yan SD , Stern D , Chader GJ , Wiggert B , Petersen RB , Perry G ((1994) ) Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol 145: , 42–47. |
[212] | Kimpara T , Takeda A , Yamaguchi T , Arai H , Okita N , Takase S , Sasaki H , Itoyama Y ((2000) ) Increased bilirubins and their derivatives in cerebrospinal fluid in Alzheimer’s disease. Neurobiol Aging 21: , 551–554. |
[213] | Nixon RA , Yang DS ((2011) ) Autophagy failure in Alzheimer’s disease–locating the primary defect. Neurobiol Dis 43: , 38–45. |
[214] | Raposo G , Stoorvogel W ((2013) ) Extracellular vesicles: Exosomes, microvesicles, and friends. J Cell Biol 200: , 373–383. |
[215] | Dobrowolski R , Vick P , Ploper D , Gumper I , Snitkin H , Sabatini DD , De Robertis EM ((2012) ) Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep 2: , 1316–1328. |
[216] | Esselens C , Oorschot V , Baert V , Raemaekers T , Spittaels K , Serneels L , Zheng H , Saftig P , De Strooper B , Klumperman J , Annaert W ((2004) ) Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol 166: , 1041–1054. |
[217] | Lee JH , Yu WH , Kumar A , Lee S , Mohan PS , Peterhoff CM , Wolfe DM , Martinez-Vicente M , Massey AC , Sovak G , Uchiyama Y , Westaway D , Cuervo AM , Nixon RA ((2010) ) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141: , 1146–1158. |
[218] | Coffey EE , Beckel JM , Laties AM , Mitchell CH ((2014) ) Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 263: , 111–124. |
[219] | Dolzhanskaya N , Gonzalez MA , Sperziani F , Stefl S , Messing J , Wen GY , Alexov E , Zuchner S , Velinov M ((2014) ) A novel p.Leu(381)Phe mutation in presenilin 1 is associated with very early onset and unusually fast progressing dementia as well as lysosomal inclusions typically seen in Kufs disease. J Alzheimers Dis 39: , 23–27. |
[220] | Willnow TE , Andersen OM ((2013) ) Sorting receptor SORLA–a trafficking path to avoid Alzheimer disease. J Cell Sci 126: , 2751–2760. |
[221] | Scherzer CR , Offe K , Gearing M , Rees HD , Fang G , Heilman CJ , Schaller C , Bujo H , Levey AI , Lah JJ ((2004) ) Loss of apolipoprotein E receptor LR11 in Alzheimer disease. Arch Neurol 61: , 1200–1205. |
[222] | Grear KE , Ling IF , Simpson JF , Furman JL , Simmons CR , Peterson SL , Schmitt FA , Markesbery WR , Liu Q , Crook JE , Younkin SG , Bu G , Estus S ((2009) ) Expression of SORL1 and a novel SORL1 splice variant in normal and Alzheimer’s disease brain. Mol Neurodegener 4: , 46. |
[223] | La Rosa LR , Perrone L , Nielsen MS , Calissano P , Andersen OM , Matrone C ((2015) ) Y682G mutation of amyloid precursor protein promotes endo-lysosomal dysfunction by disrupting APP-SorLA interaction. Front Cell Neurosci 9: , 109. |
[224] | Xu W , Weissmiller AM , White JA , Fang F , Wang X , Wu Y , Pearn ML , Zhao X , Sawa M , Chen S , Gunawardena S , Ding J , Mobley WC , Wu C ((2016) ) Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest 126: , 1815–1833. |
[225] | Israel MA , Yuan SH , Bardy C , Reyna SM , Mu Y , Herrera C , Hefferan MP , Van Gorp S , Nazor KL , Boscolo FS , Carson CT , Laurent LC , Marsala M , Gage FH , Remes AM , Koo EH , Goldstein LS ((2012) ) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482: , 216–220. |
[226] | Cataldo AM , Petanceska S , Peterhoff CM , Terio NB , Epstein CJ , Villar A , Carlson EJ , Staufenbiel M , Nixon RA ((2003) ) App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. J Neurosci 23: , 6788–6792. |
[227] | Salehi A , Delcroix JD , Belichenko PV , Zhan K , Wu C , Valletta JS , Takimoto-Kimura R , Kleschevnikov AM , Sambamurti K , Chung PP , Xia W , Villar A , Campbell WA , Kulnane LS , Nixon RA , Lamb BT , Epstein CJ , Stokin GB , Goldstein LS , Mobley WC ((2006) ) Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51: , 29–42. |
[228] | Jiang Y , Mullaney KA , Peterhoff CM , Che S , Schmidt SD , Boyer-Boiteau A , Ginsberg SD , Cataldo AM , Mathews PM , Nixon RA ((2010) ) Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 107: , 1630–1635. |
[229] | Jiang Y , Rigoglioso A , Peterhoff CM , Pawlik M , Sato Y , Bleiwas C , Stavrides P , Smiley JF , Ginsberg SD , Mathews PM , Levy E , Nixon RA ((2016) ) Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: Role of APP-CTF. Neurobiol Aging 39: , 90–98. |
[230] | Das U , Wang L , Ganguly A , Saikia JM , Wagner SL , Koo EH , Roy S ((2016) ) Visualizing APP and BACE-1 approximation in neurons yields insight into the amyloidogenic pathway. Nat Neurosci 19: , 55–64. |
[231] | Sannerud R , Declerck I , Peric A , Raemaekers T , Menendez G , Zhou L , Veerle B , Coen K , Munck S , De Strooper B , Schiavo G , Annaert W ((2011) ) ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A 108: , E559–E568. |
[232] | Morel E , Chamoun Z , Lasiecka ZM , Chan RB , Williamson RL , Vetanovetz C , Dall’Armi C , Simoes S , Point Du Jour KS , McCabe BD , Small SA , Di Paolo G ((2013) ) Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun 4: , 2250. |
[233] | Guix FX , Sannerud R , Berditchevski F , Arranz AM , Horre K , Snellinx A , Thathiah A , Saido T , Saito T , Rajesh S , Overduin M , Kumar-Singh S , Radaelli E , Corthout N , Colombelli J , Tosi S , Munck S , Salas IH , Annaert W , De Strooper B ((2017) ) Tetraspanin 6: A pivotal protein of the multiple vesicular body determining exosome release and lysosomal degradation of amyloid precursor protein fragments. Mol Neurodegener 12: , 25. |
[234] | Kim S , Sato Y , Mohan PS , Peterhoff C , Pensalfini A , Rigoglioso A , Jiang Y , Nixon RA ((2016) ) Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol Psychiatry 21: , 707–716. |
[235] | Stefanova NA , Muraleva NA , Korbolina EE , Kiseleva E , Maksimova KY , Kolosova NG ((2015) ) Amyloid accumulation is a late event in sporadic Alzheimer’s disease-like pathology in nontransgenic rats. Oncotarget 6: , 1396–1413. |
[236] | Bellingham SA , Guo B , Hill AF ((2015) ) The secret life of extracellular vesicles in metal homeostasis and neurodegeneration. Biol Cell 107: , 389–418. |
[237] | Johnstone RM , Mathew A , Mason AB , Teng K ((1991) ) Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalization of obsolete membrane proteins. J Cell Physiol 147: , 27–36. |
[238] | Eitan E , Suire C , Zhang S , Mattson MP ((2016) ) Impact of lysosome status on extracellular vesicle content and release. Ageing Res Rev 32: , 65–74. |
[239] | Alvarez-Erviti L , Seow Y , Schapira AH , Gardiner C , Sargent IL , Wood MJ , Cooper JM ((2011) ) Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 42: , 360–367. |
[240] | Hasegawa T , Konno M , Baba T , Sugeno N , Kikuchi A , Kobayashi M , Miura E , Tanaka N , Tamai K , Furukawa K , Arai H , Mori F , Wakabayashi K , Aoki M , Itoyama Y , Takeda A ((2011) ) The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of alpha-synuclein. PLoS One 6: , e29460. |
[241] | Rajendran L , Honsho M , Zahn TR , Keller P , Geiger KD , Verkade P , Simons K ((2006) ) Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A 103: , 11172–11177. |
[242] | Perez-Gonzalez R , Gauthier SA , Kumar A , Levy E ((2012) ) The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem 287: , 43108–43115. |
[243] | Fiandaca MS , Kapogiannis D , Mapstone M , Boxer A , Eitan E , Schwartz JB , Abner EL , Petersen RC , Federoff HJ , Miller BL , Goetzl EJ ((2015) ) Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement 11: , 600–607. |
[244] | Goetzl EJ , Boxer A , Schwartz JB , Abner EL , Petersen RC , Miller BL , Kapogiannis D ((2015) ) Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85: , 40–47. |
[245] | Miao Y , Li G , Zhang X , Xu H , Abraham SN ((2015) ) A TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell 161: , 1306–1319. |
[246] | Eitan E , Hutchison ER , Marosi K , Comotto J , Mustapic M , Nigam SM , Suire C , Maharana C , Jicha GA , Liu D , Machairaki V , Witwer KW , Kapogiannis D , Mattson MP ((2016) ) Extracellular vesicle-associated Abeta mediates trans-neuronal bioenergetic and Ca2+-handling deficits in Alzheimer’s disease models. NPJ Aging Mech Dis 2: , 16019. |
[247] | Dinkins MB , Enasko J , Hernandez C , Wang G , Kong J , Helwa I , Liu Y , Terry AV Jr. , Bieberich E ((2016) ) Neutral sphingomyelinase-2 deficiency ameliorates Alzheimer’s disease pathology and improves cognition in the 5XFAD mouse. J Neurosci 36: , 8653–8667. |
[248] | Jin S , Kedia N , Illes-Toth E , Haralampiev I , Prisner S , Herrmann A , Wanker EE , Bieschke J ((2016) ) Amyloid-beta(1-42) aggregation initiates its cellular uptake and cytotoxicity. J Biol Chem 291: , 19590–19606. |
[249] | Ubelmann F , Burrinha T , Salavessa L , Gomes R , Ferreira C , Moreno N , Guimas AC ((2017) ) Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep 18: , 102–122. |
[250] | Tian Y , Chang JC , Fan EY , Flajolet M , Greengard P ((2013) ) Adaptor complex AP2/PICALM, through interaction with LC 3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A 110: , 17071–17076. |
[251] | Pickford F , Masliah E , Britschgi M , Lucin K , Narasimhan R , Jaeger PA , Small S , Spencer B , Rockenstein E , Levine B , Wyss-Coray T ((2008) ) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118: , 2190–2199. |
[252] | Fazzari P , Horre K , Arranz AM , Frigerio CS , Saito T , Saido TC , De Strooper B ((2017) ) PLD3 gene and processing of APP. Nature 541: , E1–E2. |
[253] | Doody RS , Raman R , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , He F , Sun X , Thomas RG , Aisen PS , Siemers E , Sethuraman G , Mohs R ((2013) ) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369: , 341–350. |
[254] | De Strooper B ((2014) ) Lessons from a failed gamma-secretase Alzheimer trial. Cell 159: , 721–726. |
[255] | Penninkilampi R , Brothers HM , Eslick GD ((2016) ) Pharmacological agents targeting gamma-secretase increase risk of cancer and cognitive decline in Alzheimer’s disease patients: A systematic review and meta-analysis. J Alzheimers Dis 53: , 1395–1404. |
[256] | Cirrito JR , Yamada KA , Finn MB , Sloviter RS , Bales KR , May PC , Schoepp DD , Paul SM , Mennerick S , Holtzman DM ((2005) ) Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48: , 913–922. |
[257] | Garcia-Osta A , Alberini CM ((2009) ) Amyloid beta mediates memory formation. Learn Mem 16: , 267–272. |
[258] | Filser S , Ovsepian SV , Masana M , Blazquez-Llorca L , Brandt EA , Volbracht C , Muller MB , Jung CK , Herms J ((2015) ) Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry 77: , 729–739. |
[259] | Chen Q , Kimura H , Schubert D ((2002) ) A novel mechanism for the regulation of amyloid precursor protein metabolism. J Cell Biol 158: , 79–89. |
[260] | Coulson EJ , Paliga K , Beyreuther K , Masters CL ((2000) ) What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int 36: , 175–184. |
[261] | Pigoni M , Wanngren J , Kuhn PH , Munro KM , Gunnersen JM , Takeshima H , Feederle R , Voytyuk I , De Strooper B , Levasseur MD , Hrupka BJ , Muller SA , Lichtenthaler SF ((2016) ) Seizure protein 6 and its homolog seizure 6-like protein are physiological substrates of BACE1 in neurons. Mol Neurodegener 11: , 67. |
[262] | Pigoni M , Gunnersen JM , Lichtenthaler SF ((2017) ) Seizure-6 proteins highlight BACE1 functions in neurobiology. Oncotarget 8: , 7214–7215. |
[263] | Suzuki K , Hayashi Y , Nakahara S , Kumazaki H , Prox J , Horiuchi K , Zeng M , Tanimura S , Nishiyama Y , Osawa S , Sehara-Fujisawa A , Saftig P , Yokoshima S , Fukuyama T , Matsuki N , Koyama R , Tomita T , Iwatsubo T ((2012) ) Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 76: , 410–422. |
[264] | Wang XB , Bozdagi O , Nikitczuk JS , Zhai ZW , Zhou Q , Huntley GW ((2008) ) Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc Natl Acad Sci U S A 105: , 19520–19525. |
[265] | Michaluk P , Wawrzyniak M , Alot P , Szczot M , Wyrembek P , Mercik K , Medvedev N , Wilczek E , De Roo M , Zuschratter W , Muller D , Wilczynski GM , Mozrzymas JW , Stewart MG , Kaczmarek L , Wlodarczyk J ((2011) ) Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J Cell Sci 124: , 3369–3380. |
[266] | Szepesi Z , Hosy E , Ruszczycki B , Bijata M , Pyskaty M , Bikbaev A , Heine M , Choquet D , Kaczmarek L , Wlodarczyk J ((2014) ) Synaptically released matrix metalloproteinase activity in control of structural plasticity and the cell surface distribution of GluA1-AMPA receptors. PLoS One 9: , e98274. |
[267] | Matsumoto-Miyai K , Ninomiya A , Yamasaki H , Tamura H , Nakamura Y , Shiosaka S ((2003) ) NMDA-dependent proteolysis of presynaptic adhesion molecule L1 in the hippocampus by neuropsin. J Neurosci 23: , 7727–7736. |
[268] | Bot N , Schweizer C , Ben Halima S , Fraering PC ((2011) ) Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J Biol Chem 286: , 2762–2773. |
[269] | Bayes A , van de Lagemaat LN , Collins MO , Croning MD , Whittle IR , Choudhary JS , Grant SG ((2011) ) Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci 14: , 19–21. |
[270] | Liu G , Jiang Y , Wang P , Feng R , Jiang N , Chen X , Song H , Chen Z ((2012) ) Cell adhesion molecules contribute to Alzheimer’s disease: Multiple pathway analyses of two genome-wide association studies. J Neurochem 120: , 190–198. |
[271] | Viader A , Sasaki Y , Kim S , Strickland A , Workman CS , Yang K , Gross RW , Milbrandt J ((2013) ) Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron 77: , 886–898. |
[272] | Bicker G ((2005) ) STOP and GO with NO: Nitric oxide as a regulator of cell motility in simple brains. Bioessays 27: , 495–505. |
[273] | Munoz FJ , Godoy JA , Cerpa W , Poblete IM , Huidobro-Toro JP , Inestrosa NC ((2014) ) Wnt-5a increases NO and modulates NMDA receptor in rat hippocampal neurons. Biochem Biophys Res Commun 444: , 189–194. |
[274] | Lee HG , Kim SY , Kim dS , Seo SR , Lee SI , Shin DM , De Smet P , Seo JT ((2009) ) 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one inhibits neurite outgrowth and causes neurite retraction in PC12 cells independently of soluble guanylyl cyclase. J Neurosci Res 87: , 269–277. |
[275] | Zhao Y , Brandish PE , Di Valentin M , Schelvis JP , Babcock GT , Marletta MA ((2000) ) Inhibition of soluble guanylate cyclase by ODQ. Biochemistry 39: , 10848–10854. |
[276] | Furchgott RF , Zawadzki JV ((1980) ) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: , 373–376. |
[277] | Ignarro LJ , Lippton H , Edwards JC , Baricos WH , Hyman AL , Kadowitz PJ , Gruetter CA ((1981) ) Mechanism of vascular smooth muscle relaxation by organic nitrates, nitrites, nitroprusside and nitric oxide: Evidence for the involvement of S-nitrosothiols as active intermediates. J Pharmacol Exp Ther 218: , 739–749. |
[278] | Palmer RM , Ferrige AG , Moncada S ((1987) ) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: , 524–526. |
[279] | Tare M , Parkington HC , Coleman HA , Neild TO , Dusting GJ ((1990) ) Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature 346: , 69–71. |
[280] | Cornwell TL , Pryzwansky KB , Wyatt TA , Lincoln TM ((1991) ) Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Mol Pharmacol 40: , 923–931. |
[281] | Furukawa K , Ohshima N , Tawada-Iwata Y , Shigekawa M ((1991) ) Cyclic GMP stimulates Na+/Ca2+exchange in vascular smooth muscle cells in primary culture. J Biol Chem 266: , 12337–12341. |
[282] | Mery PF , Lohmann SM , Walter U , Fischmeister R ((1991) ) Ca2+current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci U S A 88: , 1197–1201. |
[283] | Bolotina VM , Najibi S , Palacino JJ , Pagano PJ , Cohen RA ((1994) ) Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 368: , 850–853. |
[284] | Kawada T , Toyosato A , Islam MO , Yoshida Y , Imai S ((1997) ) cGMP-kinase mediates cGMP- and cAMP-induced Ca2+desensitization of skinned rat artery. Eur J Pharmacol 323: , 75–82. |
[285] | Rapoport RM , Murad F ((1983) ) Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res 52: , 352–357. |
[286] | Rembold CM ((1992) ) Regulation of contraction and relaxation in arterial smooth muscle. Hypertension 20: , 129–137. |
[287] | Feletou M , Vanhoutte PM ((2009) ) EDHF: An update. Clin Sci (Lond) 117: , 139–155. |
[288] | Ledoux J , Werner ME , Brayden JE , Nelson MT ((2006) ) Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 21: , 69–78. |
[289] | Marchenko SM , Sage SO ((1996) ) Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol 492 (Pt 1): , 53–60. |
[290] | Bychkov R , Burnham MP , Richards GR , Edwards G , Weston AH , Feletou M , Vanhoutte PM ((2002) ) Characterization of a charybdotoxin-sensitive intermediate conductance Ca2+-activated+channel in porcine coronary endothelium: Relevance to EDHF. Br J Pharmacol 137: , 1346–1354. |
[291] | Gauthier KM , Liu C , Popovic A , Albarwani S , Rusch NJ ((2002) ) Freshly isolated bovine coronary endothelial cells do not express the BK Ca channel gene. J Physiol 545: , 829–836. |
[292] | Knaus HG , Eberhart A , Glossmann H , Munujos P , Kaczorowski GJ , Garcia ML ((1994) ) Pharmacology and structure of high conductance calcium-activated potassium channels. Cell Signal 6: , 861–870. |
[293] | Tanaka Y , Meera P , Song M , Knaus HG , Toro L ((1997) ) Molecular constituents of maxi KCa channels in human coronary smooth muscle: Predominant alpha+beta subunit complexes. J Physiol 502 (Pt 3): , 545–557. |
[294] | Brenner R , Perez GJ , Bonev AD , Eckman DM , Kosek JC , Wiler SW , Patterson AJ , Nelson MT , Aldrich RW ((2000) ) Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: , 870–876. |
[295] | Pluger S , Faulhaber J , Furstenau M , Lohn M , Waldschutz R , Gollasch M , Haller H , Luft FC , Ehmke H , Pongs O ((2000) ) Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res 87: , E53–E60. |
[296] | Lohn M , Lauterbach B , Haller H , Pongs O , Luft FC , Gollasch M ((2001) ) beta(1)-Subunit of BK channels regulates arterial wall[Ca(2+)] and diameter in mouse cerebral arteries. J Appl Physiol (1985) 91: , 1350–1354. |
[297] | Brayden JE , Nelson MT ((1992) ) Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 256: , 532–535. |
[298] | Knot HJ , Nelson MT ((1998) ) Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508 (Pt 1): , 199–209. |
[299] | Gebremedhin D , Kaldunski M , Jacobs ER , Harder DR , Roman RJ ((1996) ) Coexistence of two types of Ca(2+)-activated+channels in rat renal arterioles. Am J Physiol 270: , F69–F81. |
[300] | Quignard JF , Feletou M , Edwards G , Duhault J , Weston AH , Vanhoutte PM ((2000) ) Role of endothelial cell hyperpolarization in EDHF-mediated responses in the guinea-pig carotid artery. Br J Pharmacol 129: , 1103–1112. |
[301] | Gauthier KM , Spitzbarth N , Edwards EM , Campbell WB ((2004) ) Apamin-sensitive+currents mediate arachidonic acid-induced relaxations of rabbit aorta. Hypertension 43: , 413–419. |
[302] | Taylor MS , Bonev AD , Gross TP , Eckman DM , Brayden JE , Bond CT , Adelman JP , Nelson MT ((2003) ) Altered expression of small-conductance Ca2+-activated+(SK3) channels modulates arterial tone and blood pressure. Circ Res 93: , 124–131. |
[303] | Archer SL , Huang JM , Hampl V , Nelson DP , Shultz PJ , Weir EK ((1994) ) Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci U S A 91: , 7583–7587. |
[304] | Kadekaro M , Su G , Chu R , Lei Y , Li J , Fang L ((2006) ) Nitric oxide up-regulates the expression of calcium-dependent potassium channels in the supraoptic nuclei and neural lobe of rats following dehydration. Neurosci Lett 404: , 50–55. |
[305] | Kadekaro M , Su G , Chu R , Lei Y , Li J , Fang L ((2007) ) Effects of nitric oxide on expressions of nitrosocysteine and calcium-activated potassium channels in the supraoptic nuclei and neural lobe of dehydrated rats. Neurosci Lett 411: , 117–122. |
[306] | Ramón y Cajal S ((1894) ) The Croonian Lecture: La Fine Structure des Centres Nerveux. Proc R Soc Lond 55: , 444–468. |
[307] | Huang YS , Carson JH , Barbarese E , Richter JD ((2003) ) Facilitation of dendritic mRNA transport by CPEB. Genes Dev 17: , 638–653. |


