
Identification of candidate biomarkers and prognostic analysis of recurrence in colorectal cancer
Abstract
Colorectal cancer (CRC) is one of the most common digestive tract malignant tumors, which has a high mortality rate especially for patients with CRC recurrence. However, the pathological mechanism of recurrence of CRC is unclear. In this study, we integrated multiple cohort datasets and databases to clarify and verify potential key candidate biomarkers and signal transduction pathways in recurrence of CRC. As results, 628 DEGs were identified from GSE33113 and GSE2630 datasets and their function and pathway were analyzed. 14 hub genes related to CRC recurrence were screened from and their influence on survival were analyzed. Two key genes (IL1B and DDAH1) regarded as prognostic factors were further screened. Relapse-free survival results indicated the interaction between IL1B and DDAH1 genes and B cells was the most obvious and correlated with survival, with statistical significance (
1.Introduction
Table 1
Information about the GEO data and series
| Datasets | Case ( | Primary CRC ( | Recurrent CRC ( | Platforms |
|---|---|---|---|---|
| GSE2630 | 16 | 10 | 6 | GPL2006 Human 19K oligo array |
| GSE33113 | 90 | 72 | 18 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array |
Colorectal cancer (CRC) a common malignant tumor in the gastrointestinal tract has become one of the most common disease with the highest incidence and mortality rate in the world [1, 2, 3]. With the development of various therapeutic methods such as surgery, chemoradiotherapy, targeted therapy and local therapy, the survival of CRC patients has been significantly extended [4, 5]. However, due to the limitations of traditional therapy and tumor heterogeneity, the clinical efficacy of CRC is still unsatisfactory. Although the tumor cannot be detected by conventional imaging (including PET/CT) or laboratory methods after CRC radical resection surgery, those tumor cells may not be completely eliminated, noting as minimal residual disease (MRD) [6]. Those MRD may reappear in the primary organ or the cancer will invade the lymph, blood vessels or body cavity, and migrate to other sites and continue growing, forming a tumor with the same pathological type and biological behavior as the primary tumor. Studies shows that colorectal cancer patients who test positive for MRD 2–8 weeks after surgery have a 13-fold higher risk of recurrence than patients who test negative [7]. For the patients who undergo radical surgery alone, the overall recurrence rate can reach 30–40% [2, 8]. Therefore, postoperative recurrence of CRC has become a big challenge for the therapy of CRC patients. To prevent the CRC recurrence, it is important to understand the mechanism of CRC recurrence, search for related molecular markers, and conduct translational research on related targets.
The identification of molecular markers is important for the treatment of CRC. It is recommended that routine molecular marker detection should include RAS (including KRAS and NRAS), BRAF, and MMR/MSI, which are of great significance in guiding treatment and prognosis [9]. However, the key candidate biomarkers and signal transduction pathways in CRC recurrence are still unclear. With the development of medical science and technology, Next-Generation Sequencing (NGS) is gradually being applied in all aspects of scientific research and clinical practice [10, 11, 12]. It can significantly improve sequencing throughput, sequencing hundreds of thousands to millions of DNA molecules simultaneously. In oncology, databases such as Gene Expression Profiling Interactive Analysis (GEPIA) [13] and The Cancer Genome Atlas (TCGA) [14] provided new approach for the study of markers related to colorectal cancer. By using microarray technology to analyze the differentially expressed genes (DEGs), the pathological process of CRC recurrence can be revealed, which is helpful for the prevent of CRC recurrence [15, 16, 17].
In this study, we took GEO, TCGA and other public databases to identify the key genes related to colorectal cancer recurrence, study the relationship between genes expression and clinical prognosis, and analyze the mechanism of key genes on CRC recurrence.
2.Materials and methods
2.1Microarray data
The microarray data of CRC were obtained from GEO database. All included datasets should meet the following criteria: (1) the data sets using surgically resected samples for RNA detection, (2) total sample size should
2.2Bioinformatics related analysis
Gene function and pathway analysis: ClueGO of Cytoscape3.9.1 software was used to analyse the DEGs gene ontology (Gene Ontology, GO) and Kyoto encyclopedia (Kyoto Encyclopedia of Genes and Genomes, KEGG) GO analysis was consisted by three parts: biologicalprocess, cellularcomponent, and molecularfunction. STRING tools were used to construct protein-proteininteraction (PPI) networks to search for the interactions among proteins. Screening of hub genes was generated via PPI networks using cytoHubba calculations in Cytoscape. Survival analysis and expression analysis were performed using GEPIA online Survival analysis tool. The correlation between gene expression and the abundance of 6 kinds of infiltrating immune cells and the effect on patient survival were evaluated by the TIMER database. The web addresses of all databases and analysis tools are shown in Table 2.
Table 2
Databases related to bioinformatics analysis
| Databases | Website |
|---|---|
| GEO | |
| GEO2R | |
| STRING | |
| GEPIA | |
| TIMER2.0 | |
| Venn map tool | |
| GEPIA Survival analysis | |
| CBIOPORTAL |
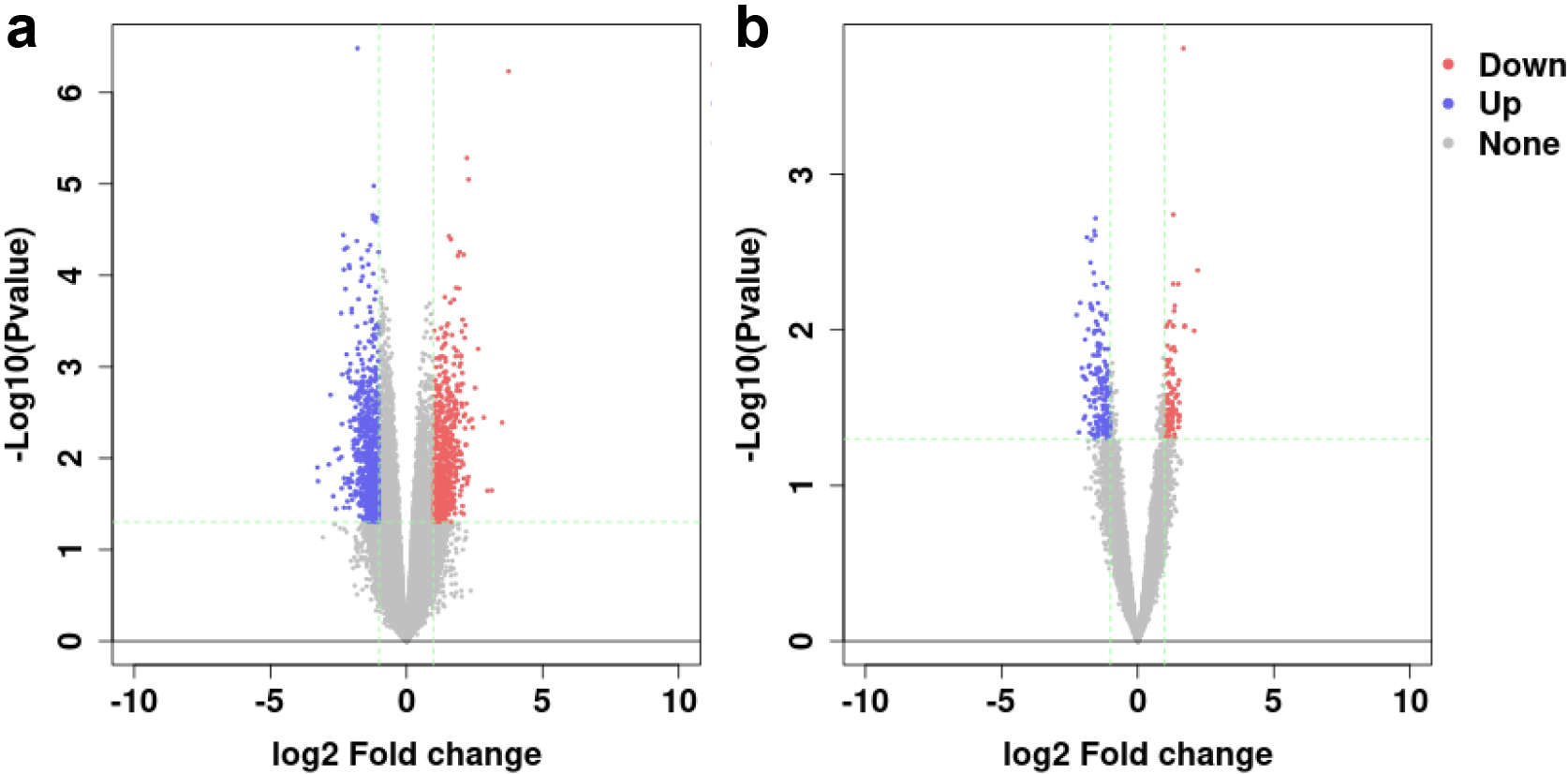
Figure 1.
Volcano plot of (a) GSE33113 and (b) GSE2630. Red spots represent the up-regulated genes, blue spots represent the down-regulated genes.
2.3Statistical processing
The statistical analyses of experimental data were performed using SPSS version 22.0. All experiments were performed with independent samples. GEO2R was used to identify DEGs associated with the recurrence of CRC. GEO2R is an interactive online analysis tool based on limma algorithm, which can be used to compare two or more datasets in the GEO series to determine DEGs. Previously, the cases in datasets were divided into primary CRC group and recurrent CRC group. Via GEO2R tool, all differentially expressed genes of two groups were identified. With
3.Results
3.1Screening and identification of DEGs
The DEGs between primary CRC and recurrent CRC were identified from GSE33113 and GSE2630 datasets by GEO2R. The GSE33113 dataset included 72 cases of primary CRC and 18 cases of recurrent CRC. The GSE2630 dataset included 10 cases of primary CRC and 6 cases of recurrent CRC. The screening criteria for DEGs were
Figure 2.
GO annotation and KEGG pathway enrichment of DEGs. ac: GSE33113 df: GSE2630.
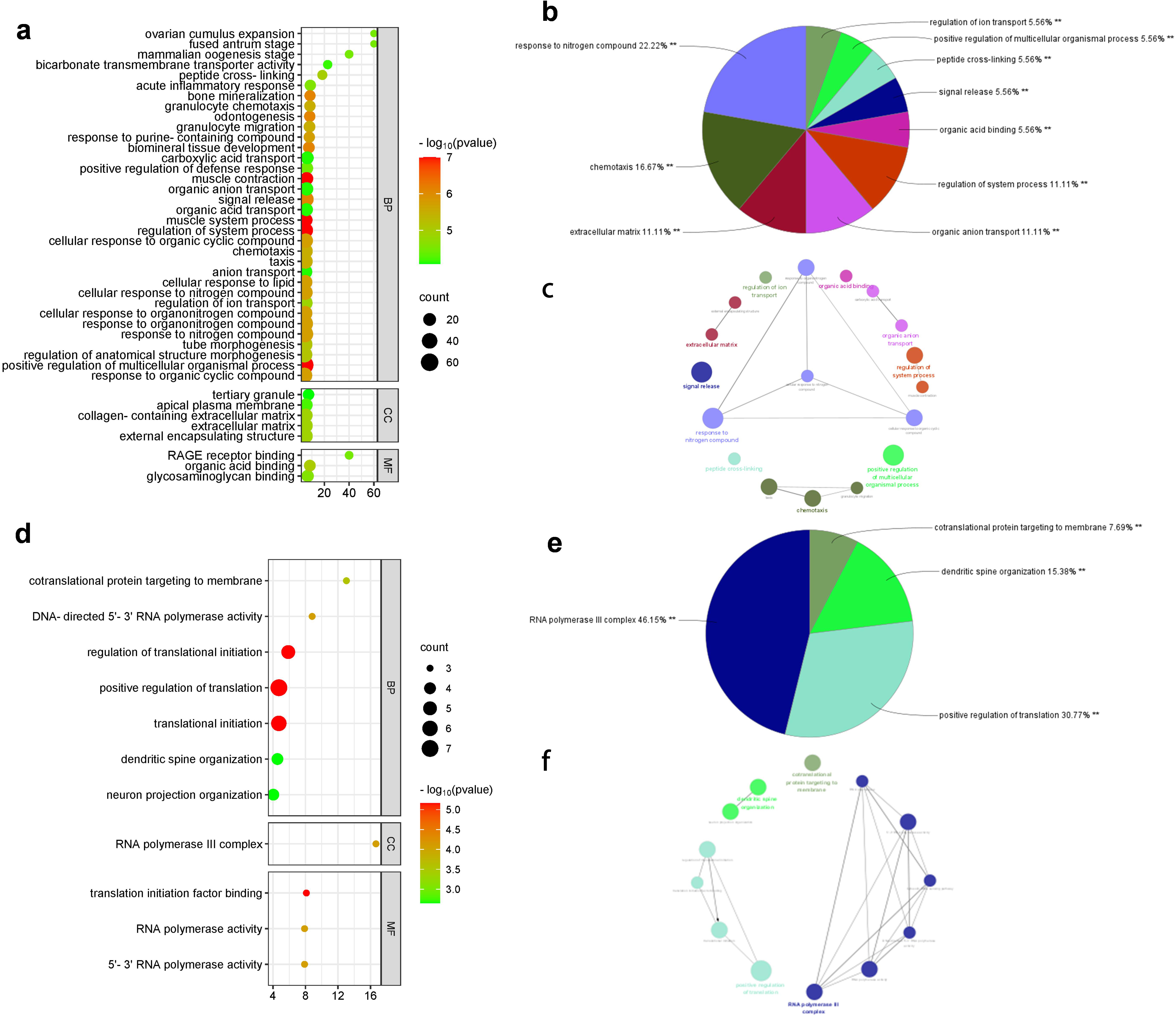
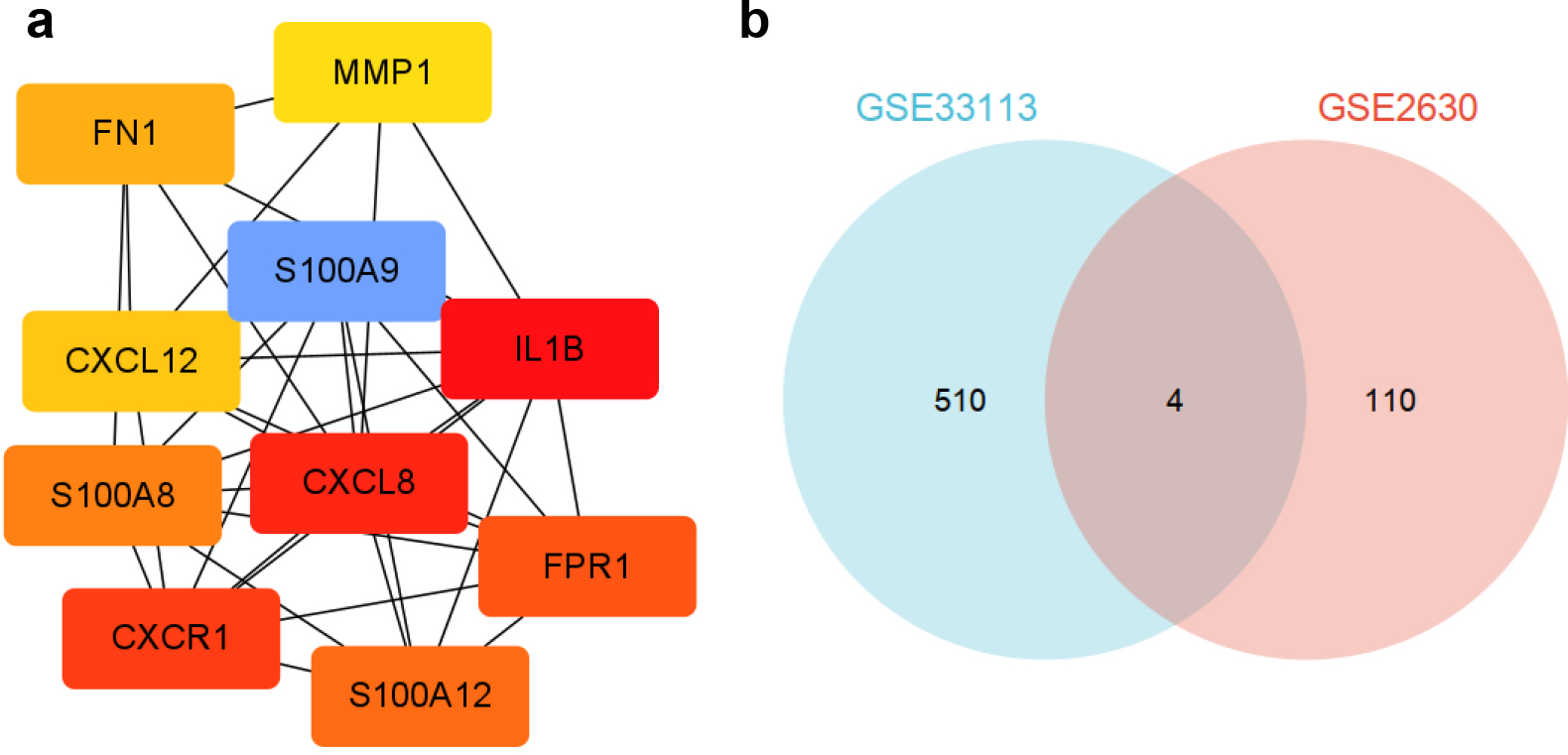
Figure 3.
(a) 10 hub genes selected from PPI network. (b) The intersected DEGs of GSE33113 and GSE2690.
3.2DEGs functional enrichment analysis
The functional and pathway enrichment analysis of identified DEGs were performed using ClueGO and KEGG of Cytoscape software. The GO analysis indicated that DEGs of GSE33113 were mainly enriched in peptide cross-linking, organic acid binding, granulocyte migration, chemotactic reaction, positive regulation of various biological processes and other physiological processes (Fig. 2a). The DEGs of GSE2630 was mainly involved in the translation of targeted membrane proteins, the synthesis of RNA polymerase III complex, the DNA sensing pathway in cytoplasm, and the positive regulation of translation (Fig. 2d).
Figure 4.
Survival analysis of TCGA database. (a) CXCL8, (b) IL1B, (c) CXCR1, (d) S100A12, (e) FPR1, (f) S100A8, (g) FN1, (h) CXCL12, (i) MMP1, (j) S100A9, (k) KCNE5, (l) DDAH1, (m) TM4SF4 and (n) DKK2.
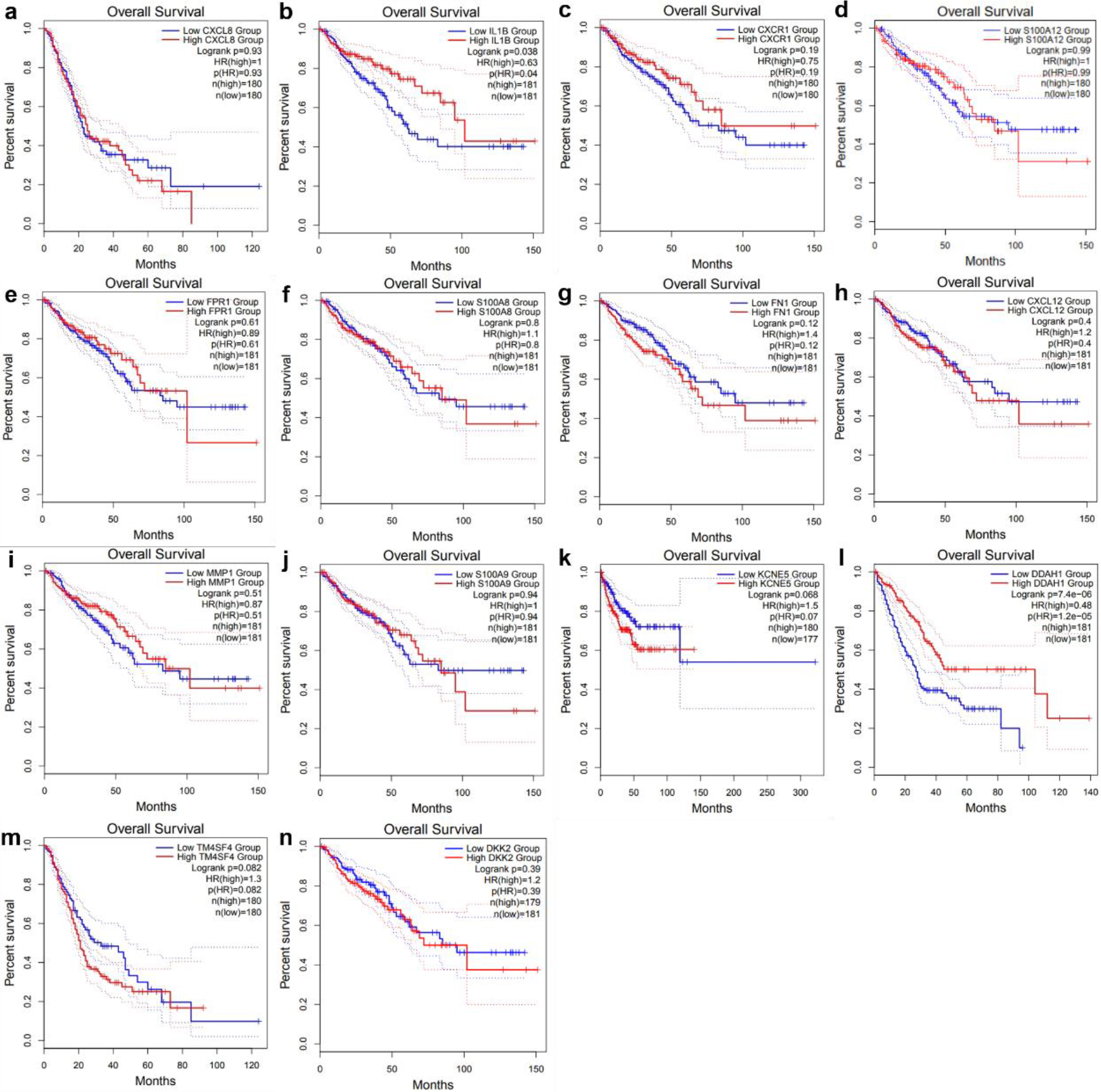
Figure 5.
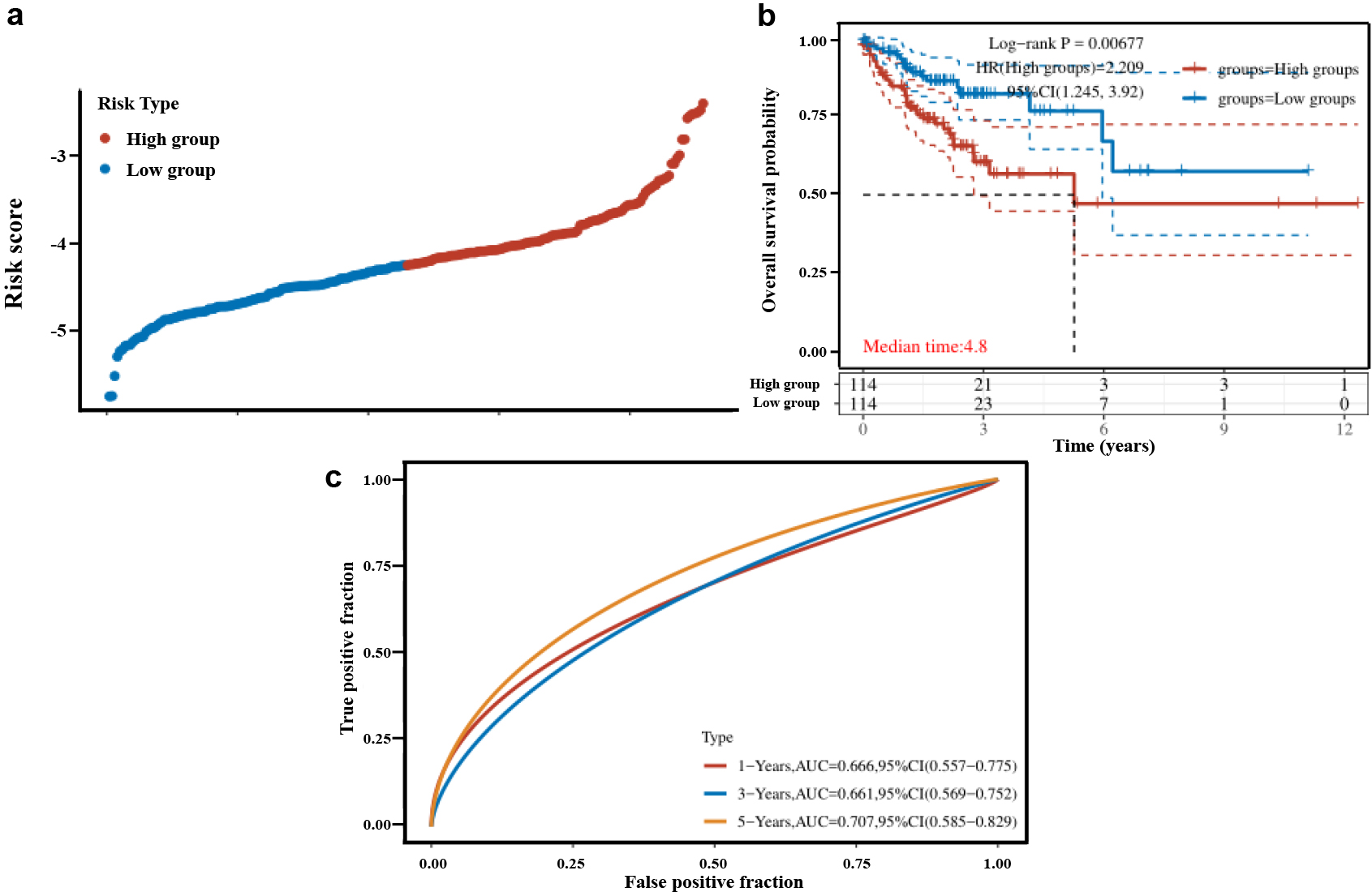
(a) Distribution of risk scores, (b) Recurrence-free survival curves for high and low risk patients, (c) ROC curve versus AUC at different times in this risk model.
The KEGG channel enrichment analysis results showed that the DEGs of GSE33113 was mainly involved in the AGE-RAGE signal path IL-17 signaling pathway, and the metabolic pathway of glycerophospholipids (Fig. 2b, 2c). The DEGs of GSE2630 was mainly involved in the cytoplasmic DNA sensing pathway and RNA polymerization pathway (Fig. 2e, 2f).
3.3PPI network construction and hub gene recognition
To select the key genes of recurrent CRC, a PPI network was built using the STRING database and the intersected DEGs of GSE33113 and GSE2690 was analyzed [19, 20]. The key subnetworks were extracted by cytoHubba calculations in Cytoscape software using MCC algorithm and Molecular Complex Detection plugin. It can be found that in the PPI network, some regions were closely linked to each other, which may represent the molecules complex. Based on the PPI network results and connection data function, the top10 genes were filtered. Including CXCL8, IL1B, CXCR1, S100A12, FPR1, S100A8, FN1, CXCL12, MMP1 and S100A9 genes (Fig. 3a). Furthermore, the intersected DEGs of GSE33113 and GSE2690 chips was taken out by Venn map tool, and 4 intersected genes such as KCNE5, DDAH1, TM4SF4 and DKK2 were observed (Fig. 3b). The 14 genes selected by PPI and Venn map were identified as candidate biomarkers.
3.4Prognostic analysis of DEGs
The 10 hub genes and 4 shared genes were divided into two groups according to the expression level of each gene. To identify the prognostic factors of the 14 candidate genes to the survival time of the patients survival analysis was performed by GEPIA online Survival analysis tool. The overall survival rate was compared by
3.5Establishment of prognostic model
Multivariate COX regression analysis was used to construct a molecular prognostic model for CRC recurrence based on the IL1B and DDAH1. The risk score of each colon cancer patient was calculated by summing the multiplication of normalized expression level of the gene by its corresponding coefficient obtained by GEO2R. The risk scores
Figure 6.
mRNA expression of IL1Band DDAH1 genes in control tissues and CRC (*P< 0.05). Adenocarcinoma is the most common type of colorectal cancer, accounting for more than 95% of colorectal cancer. COAD refers to colon adenocarcinoma, READ refers to rectum adenocarcinoma.
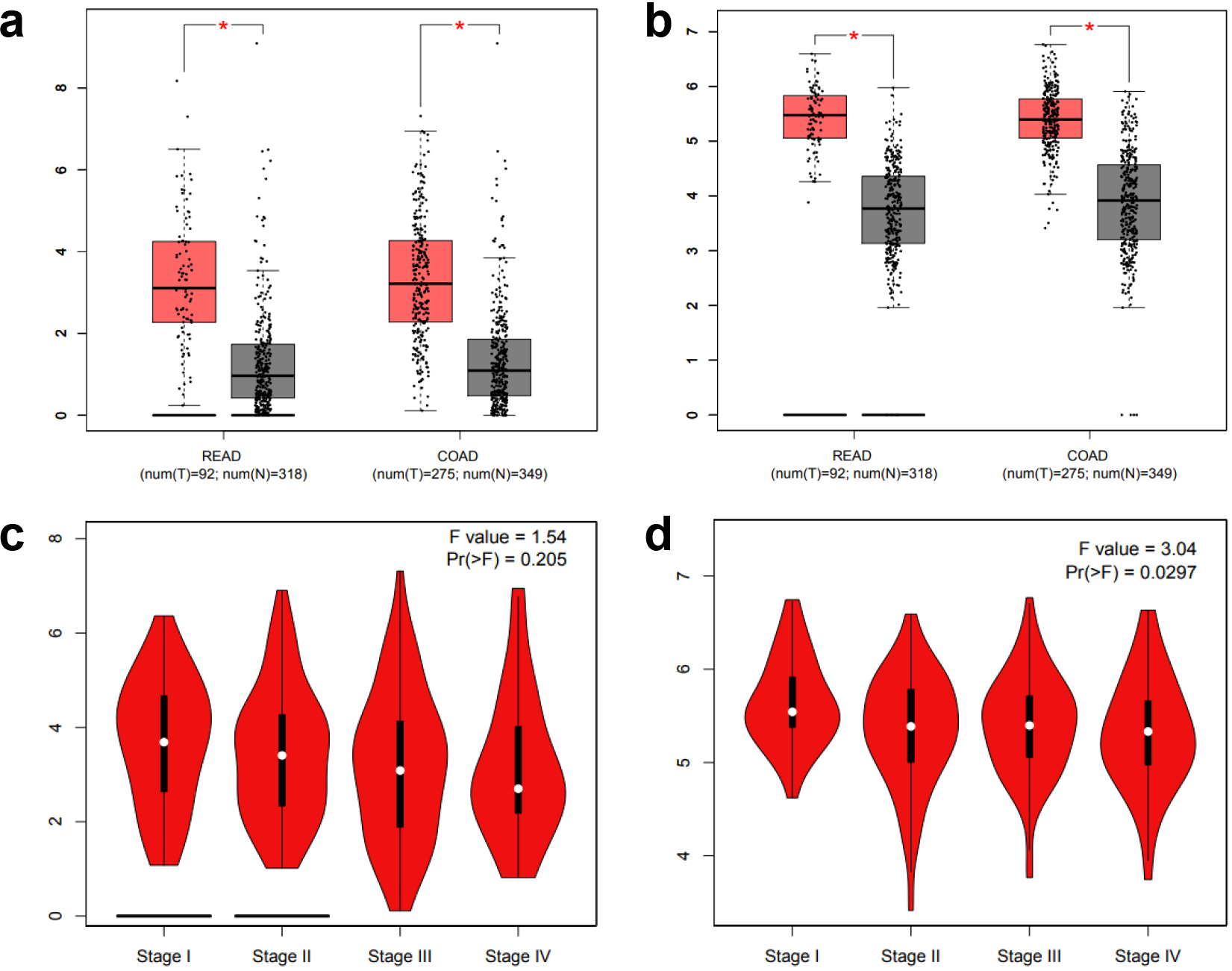
3.6Expression of IL1B and DDAH1 mRNA in CRC
The expression of IL1B and DDAH1 in CRC were further analyzed by GEPIA. The expression levels of IL1B and DDAH1 genes in colorectal adenocarcinoma tissues were significantly higher than those in control tissues (Fig. 6a–b). The expression of IL1B and DDAH1 genes among tumor tissues at different stages (stage I, II, III, and IV) were further analyzed. Figure 6c–d show that the expression of DDAH1 was significantly different among the four stages (
Figure 7.
(a) The correlation analysis of IL1B and B lymphocyte expression level in CRC. (b) The prognostic value of IL1B and B lymphocyte patients in the survival curve. (c) The correlation analysis of DDAH1 and B lymphocyte expression level in CRC. (d) The prognostic value of DDAH1 and B lymphocyte patients in the survival curve.
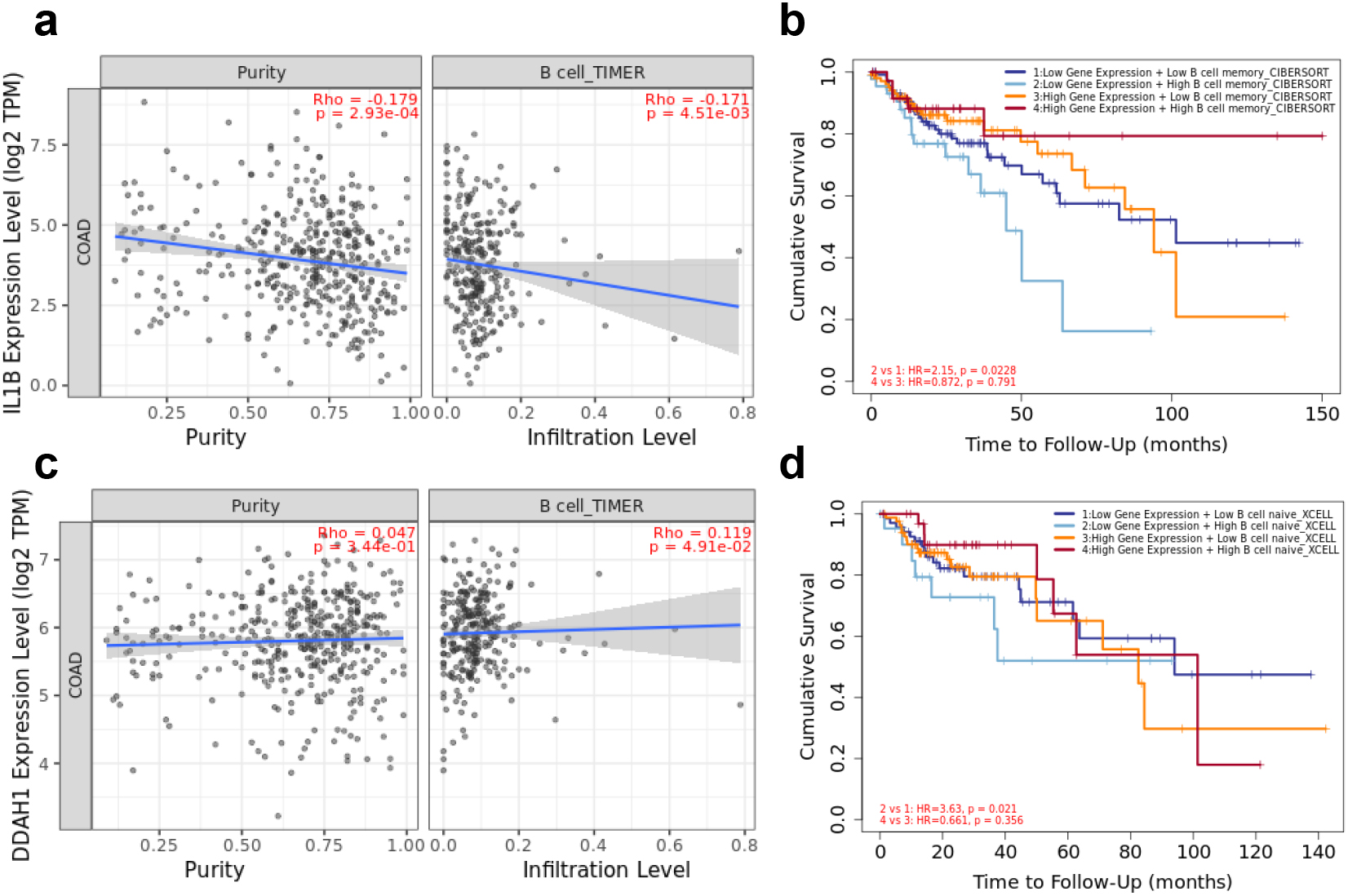
Figure 8.
(a) The genetic alteration of IL1B and DDAH1. (b) The potential co-expression of IL1B and DDAH1. (c–f) Promoter methylation level of IL1B and DDAH1.
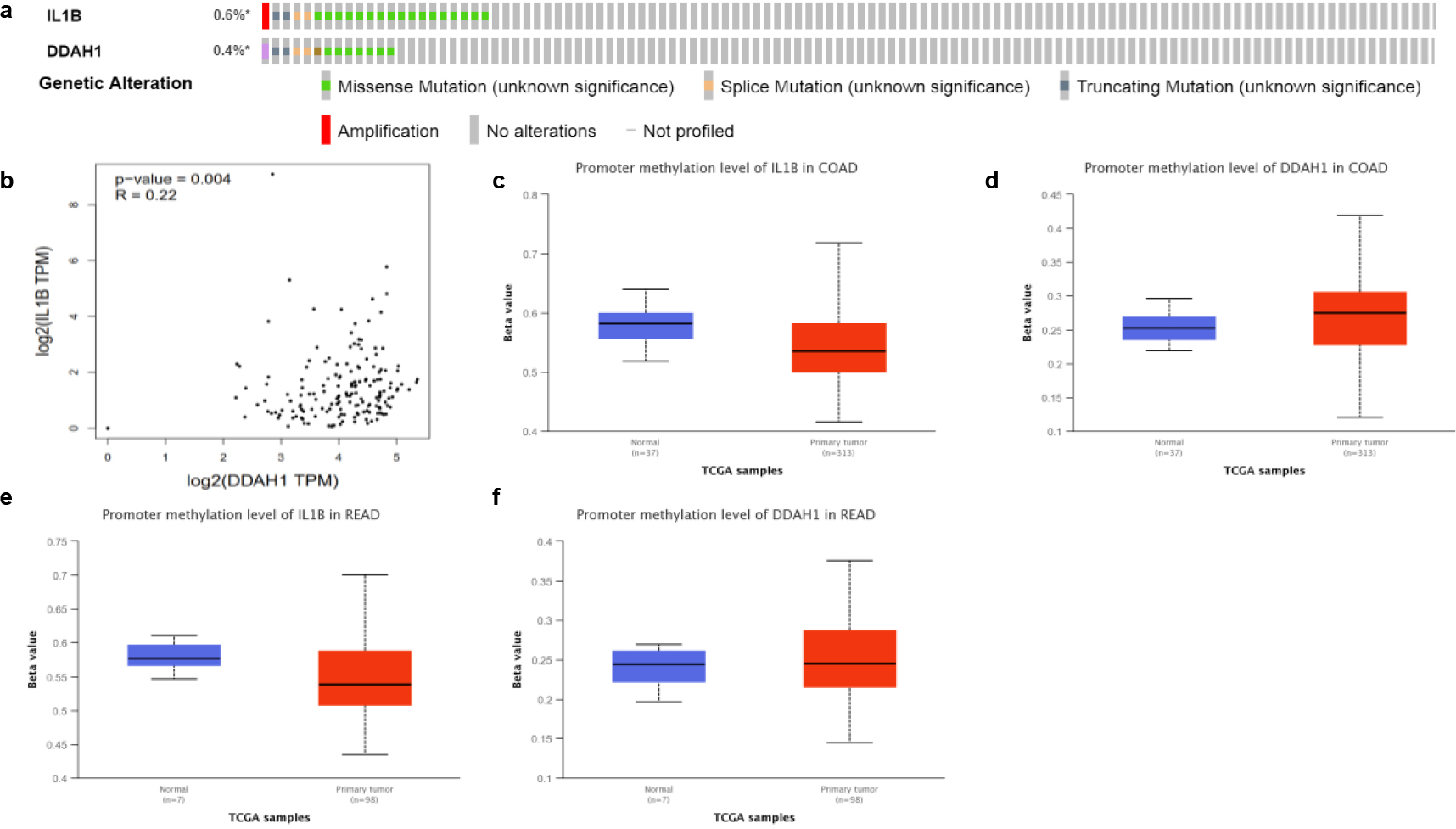
Figure 9.
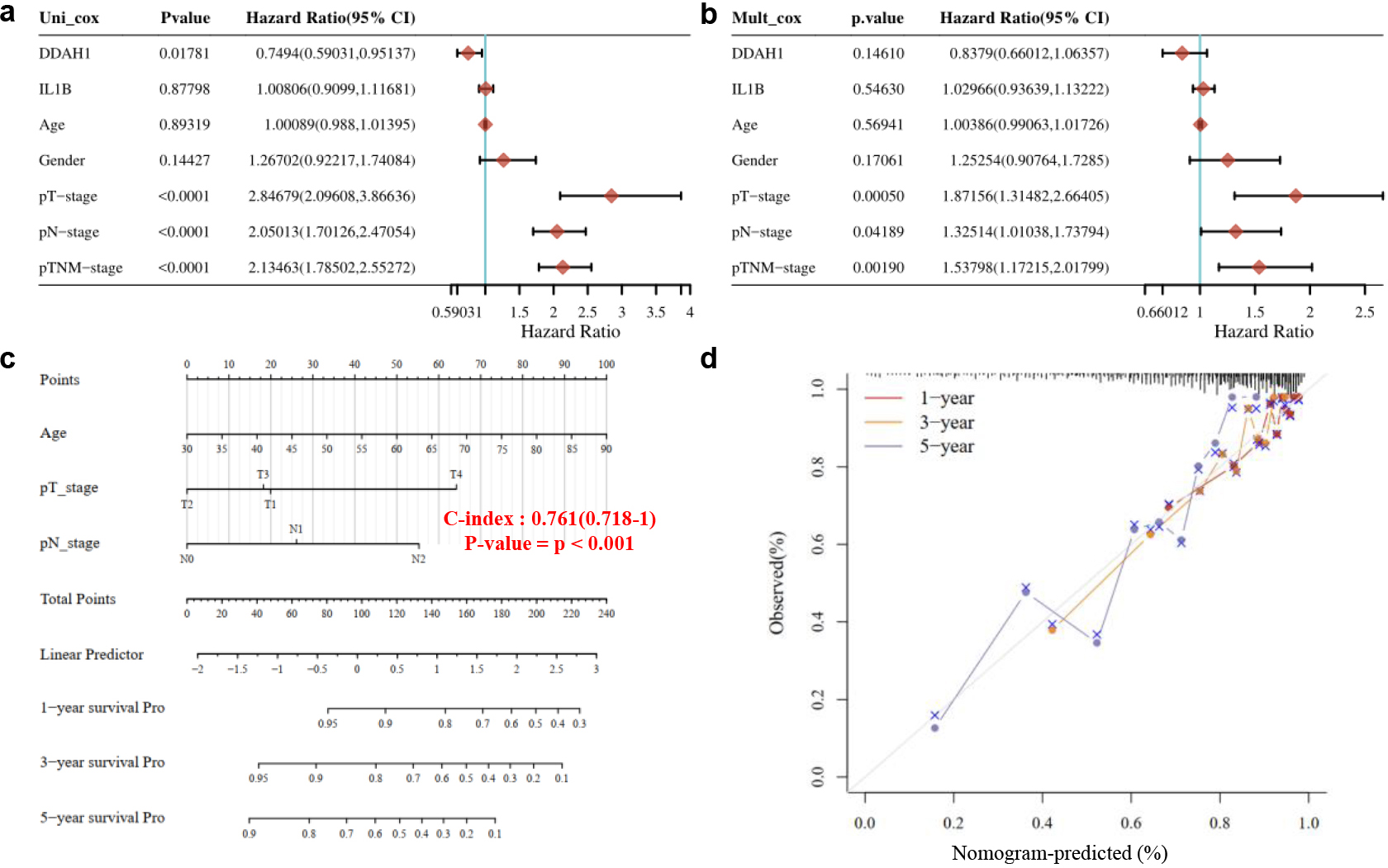
(a) Univariate cox regression analysis. (b) Multivariate cox regression analysis. (c) The predictive nomogram for RFS of CRC patients. (d) The 1,3,5-year calibration curves for CRC patients.
3.7Immune cell infiltration analysis
To reveal the function of IL1B and DDAH1 for the prediction performance of CRC patients, the expression levels of IL1B and DDAH1 in association with 6 types of infiltrating immune cells (B lymphocyte, CD4
3.8Genetic alteration co-expression and promotor methylation level analysis
Further, the molecular characteristics of IL1B and DDAH1 were analyzed using the Pan Cancer Atlas of TCGA to evaluate their impact on the genetic expression by CBIOPORTAL database. As shown in Fig. 8a, 0.6% and 0.4% of the CRC samples were altered by IL1B and DDAH1 respectively. Next the potential co-expression of the two DEGs was analyzed. Both the expression of IL1B and DDAH1 exhibited significant correlations as shown in Fig. 8b. Then we explored the promotor methylation level of IL1B and DDAH1 and found that the promotor for IL1B was hyper-methylation, while for DDAH1 was hypo- methylation (Fig. 8c–f).
3.9Cox regression analysis and clinical information
Univariate and multivariate cox regression analysis were performed and a nomogram was developed to predict the 1,3,5-year overall recurrence [21]. The forest was used to show the
4.Discussion
In recent years, many basic and clinical studies have revealed the causes and potential mechanisms mediating the formation and development of CRC [2]. However, the high recurrence rate of the disease is the main factor for the death of CRC patients [22]. With the wide application of gene chip generation technology, a large amount of core slice data was generated, and most of the data were stored in the public database. Therefore, integrating and reanalyzing these datasets can provide valuable clues for new research. Recently, researchers have conducted a large number of microarray data analysis studies on CRC and obtained hundreds of DEGs [23], but reliable biomarkers related to CRC recurrence have not been identified.
In this study, we integrated datasets from two coincidences of CRC recurrence from different sources and analyzed these data using a variety of bioinformatics methods. 514 DEGs between primary CRC and recurrent CRC patients were screened from the GSE33113 and GSE2630 datasets. The function and pathway of the 514 DEGs were analyzed by GO functional and KEGG genome pathway enrichment analysis. Results showed that the GSE33113 DEGs were mainly involved in the AGE-RAGE signaling pathways, IL-17 signaling pathways, and glycerol phospholipid metabolic pathways [24]. And the DEGS of GSE2630 were mainly involved in plasma DNA sensing pathway and RNA polymerization.
Next, 10 hub genes and 4 intersected genes were screened form the 514 DEGs through PPI and Venn map [17]. GEPIA survival analysis indicated only the IL1B and DDAH1 genes can be regarded as prognostic factors [11, 12]. The expression levels analyze of IL1B and DDAH1 genes in colorectal adenocarcinoma tissues were high, suggested that the two genes significantly affected the survival of CRC patients. Finally, we studied the mechanism of the expression of IL1B and DDAH1 genes on the CRC patients. Results suggested that the expression IL1B and DDAH1 genes affected the immune system of CRC patients, which caused the alter of prognosis of CRC patients Univariate analysis suggests DDAH1 and TNM clinical stage were independent risk factors for relapse-free survival. In multivariate analysis, TNM clinical stage was an independent risk factor for relapse-free survival in patients with CRC recurrence. Surprisingly, the patients at T1and N0 stages had a higher risk of recurrence than patients at T2 and N1, Therefore, for these patients, it is of great significance to assess the risk of recurrence as early as possible after surgery and take measures as early as possible.
Using biomarkers to predict the possibility of CRC recurrence could help to identify MRD, which benefits the early screening and diagnosis. Based on biomarkers results, a better early treatment can be realized, the rate of early detection can be increased, which possess significant influence on the prevention of CRC recurrence.
Although the two datasets were accessed from a same platform, batch effects may have an impact on sample expression differences. In microarray experiments, the absolute optical density values of each chip were not the same. After background processing and data cleaning, the corrected values reflect the gene expression level. Before comparing the results of each test, they must be normalized to adjust for errors caused by gene chip technology. Both datasets were normalized by GEO2R analysis.
The limitations of this study were that the data used in this study were obtained from the open database, so the time of data collection were not consistent. Furthermore, the results of this study were not validated by large clinical samples.
5.Conclusion
In conclusion, 14 pivotal genes related to CRC recurrence were screened by bioinformatics analysis tools Among them, two genes (IL1B and DDAH1) were regarded as prognostic factors which impacted the survival of CRC patients. Mechanism study suggested IL1B and DDAH1 significantly related to the prognosis by affecting the invasion of immune cells through IL-17 signaling pathway. Our study also found that patients at T1 and N0 stages had a higher risk of recurrence than patients at T2 and N1, Therefore, for these patients, it is of great significance to assess the risk of recurrence as early as possible.
Ethics approval and consent to participate
The study has been approved by the Ethics Committee of Shanxi People’s Hospital (Shanxi provincial people’s Hospital, Yingze District, Taiyuan, Shanxi, China; Dr. Huazhong Sun, Chair; Approval No. 2023-210) on 18 April, 2023.
Consent for publication
All authors have read and approved the submission of the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Funding
This work was supported by the Fundamental Research Program of Shanxi Province (202303021222360 202103021223086).
Author contributions
Preparation of the manuscript: R. Xu and H. Liang. Interpretation or analysis of data: R. Xu, H. Liang and H. Feng. Supervision: H. Liang and Y. Li.
Acknowledgments
Not applicable.
Conflict of interest
The authors declare that they have no conflict of interest.
References
[1] | J. Guinney, R. Dienstmann, X. Wang, A. de Reynies, A. Schlicker, C. Soneson, L. Marisa, P. Roepman, G. Nyamundanda, P. Angelino, B.M. Bot, J.S. Morris, I.M. Simon, S. Gerster, E. Fessler, E.M.F. De Sousa, E. Missiaglia, H. Ramay, D. Barras, K. Homicsko, D. Maru, G.C. Manyam, B. Broom, V. Boige, B. Perez-Villamil, T. Laderas, R. Salazar, J.W. Gray, D. Hanahan, J. Tabernero, R. Bernards, S.H. Friend, P. Laurent-Puig, J.P. Medema, A. Sadanandam, L. Wessels, M. Delorenzi, S. Kopetz, L. Vermeulen and S. Tejpar, The consensus molecular subtypes of colorectal cancer, Nat Med 21: ((2015) ), 1350–1356. |
[2] | L.H. Biller and D. Schrag, Diagnosis and Treatment of metastatic colorectal cancer: A review, JAMA 325: ((2021) ), 669–685. |
[3] | E. Morgan, M. Arnold, A. Gini, V. Lorenzoni, C.J. Cabasag, M. Laversanne, J. Vignat, J. Ferlay, N. Murphy and F. Bray, Global burden of colorectal cancer in 2020 and 2040: incidence and mortality estimates from GLOBOCAN, Gut 72: ((2023) ), 338. |
[4] | J.R. Huyghe, S.A. Bien, T.A. Harrison, H.M. Kang, S. Chen, S.L. Schmit, D.V. Conti, C. Qu, J. Jeon, C.K. Edlund, P. Greenside, M. Wainberg, F.R. Schumacher, J.D. Smith, D.M. Levine, S.C. Nelson, N.A. Sinnott-Armstrong, D. Albanes, M.H. Alonso, K. Anderson, C. Arnau-Collell, V. Arndt, C. Bamia, B.L. Banbury, J.A. Baron, S.I. Berndt, S. Bézieau, D.T. Bishop, J. Boehm, H. Boeing, H. Brenner, S. Brezina, S. Buch, D.D. Buchanan, A. Burnett-Hartman, K. Butterbach, B.J. Caan, P.T. Campbell, C.S. Carlson, S. Castellví-Bel, A.T. Chan, J. Chang-Claude, S.J. Chanock, M.-D. Chirlaque, S.H. Cho, C.M. Connolly, A.J. Cross, K. Cuk, K.R. Curtis, A. de la Chapelle, K.F. Doheny, D. Duggan, D.F. Easton, S.G. Elias, F. Elliott, D.R. English, E.J.M. Feskens, J.C. Figueiredo, R. Fischer, L.M. FitzGerald, D. Forman, M. Gala, S. Gallinger, W.J. Gauderman, G.G. Giles, E. Gillanders, J. Gong, P.J. Goodman, W.M. Grady, J.S. Grove, A. Gsur, M.J. Gunter, R.W. Haile, J. Hampe, H. Hampel, S. Harlid, R.B. Hayes, P. Hofer, M. Hoffmeister, J.L. Hopper, W.-L. Hsu, W.-Y. Huang, T.J. Hudson, D.J. Hunter, G. Ibañez-Sanz, G.E. Idos, R. Ingersoll, R.D. Jackson, E.J. Jacobs, M.A. Jenkins, A.D. Joshi, C.E. Joshu, T.O. Keku, T.J. Key, H.R. Kim, E. Kobayashi, L.N. Kolonel, C. Kooperberg, T. Kühn, S. Küry, S.-S. Kweon, S.C. Larsson, C.A. Laurie, L. Le Marchand, S.M. Leal, S.C. Lee, F. Lejbkowicz, M. Lemire, C.I. Li, L. Li, W. Lieb, Y. Lin, A. Lindblom, N.M. Lindor, H. Ling, T.L. Louie, S. Männistö, S.D. Markowitz, V. Martín, G. Masala, C.E. McNeil, M. Melas, R.L. Milne, L. Moreno, N. Murphy, R. Myte, A. Naccarati, P.A. Newcomb, K. Offit, S. Ogino, N.C. Onland-Moret, B. Pardini, P.S. Parfrey, R. Pearlman, V. Perduca, P.D.P. Pharoah, M. Pinchev, E.A. Platz, R.L. Prentice, E. Pugh, L. Raskin, G. Rennert, H.S. Rennert, E. Riboli, M. Rodríguez-Barranco, J. Romm, L.C. Sakoda, C. Schafmayer, R.E. Schoen, D. Seminara, M. Shah, T. Shelford, M.-H. Shin, K. Shulman, S. Sieri, M.L. Slattery, M.C. Southey, Z.K. Stadler, C. Stegmaier, Y.-R. Su, C.M. Tangen, S.N. Thibodeau, D.C. Thomas, S.S. Thomas, A.E. Toland, A. Trichopoulou, C.M. Ulrich, D.J. Van Den Berg, F.J.B. van Duijnhoven, B. Van Guelpen, H. van Kranen, J. Vijai, K. Visvanathan, P. Vodicka, L. Vodickova, V. Vymetalkova, K. Weigl, S.J. Weinstein, E. White, A.K. Win, C.R. Wolf, A. Wolk, M.O. Woods, A.H. Wu, S.H. Zaidi, B.W. Zanke, Q. Zhang, W. Zheng, P.C. Scacheri, J.D. Potter, M.C. Bassik, A. Kundaje, G. Casey, V. Moreno, G.R. Abecasis, D.A. Nickerson, S.B. Gruber, L. Hsu and U. Peters, Discovery of common and rare genetic risk variants for colorectal cancer, Nat Genet 51: ((2019) ), 76–87. |
[5] | J.A. Meyerhardt and R.J. Mayer, Systemic therapy for colorectal cancer, New Engl J Med 352: ((2005) ), 476–487. |
[6] | A.R. Parikh, E.E. Van Seventer, G. Siravegna, A.V. Hartwig, A. Jaimovich, Y. He, K. Kanter, M.G. Fish, K.D. Fosbenner, B. Miao, S. Phillips, J.H. Carmichael, N. Sharma, J. Jarnagin, I. Baiev, Y.S. Shah, I.J. Fetter, H.A. Shahzade, J.N. Allen, L.S. Blaszkowsky, J.W. Clark, J.S. Dubois, J.W. Franses, B.J. Giantonio, L. Goyal, S.J. Klempner, R.D. Nipp, E.J. Roeland, D.P. Ryan, C.D. Weekes, J.Y. Wo, T.S. Hong, L. Bordeianou, C.R. Ferrone, M. Qadan, H. Kunitake, D. Berger, R. Ricciardi, J.C. Cusack, V.M. Raymond, A. Talasaz, G.M. Boland and R.B. Corcoran, Minimal residual disease detection using a plasma-only circulating tumor DNA assay in patients with colorectal cancer, Clin Cancer Res 27: ((2021) ), 5586–5594. |
[7] | S.A. Cohen, P.M. Kasi, V.N. Aushev, D.L. Hanna, G.P. Botta, S. Sharif, G. Laliotis, S. Sharma, V. Sharma, A. Alqahtani, S.R. Chandana, S. Kang, S. Chakrabarti, B.G. Somer, A. Kasi, F. Dayyani, M. Malla, A. Jurdi, A. Aleshin and S. Kopetz, Real-world monitoring of circulating tumor DNA reliably predicts cancer recurrence in patients with resected stages I–III colorectal cancer, Ann Oncol 33: ((2022) ), S683–S684. |
[8] | P.E. Teloken, D. Ransom, I. Faragher, I. Jones, P. Gibbs and C. Platell, Recurrence in patients with stage I colorectal cancer, ANZ J Surg 86: ((2016) ), 49–53. |
[9] | M. Gutierrez, K.S. Price, R.B. Lanman, R. Nagy, I. Shah, S. Mathura, A.D. Norden and S.L. Goldberg, Rates of genotyping for KRAS, NRAS, BRAF, microsatellite instability (MSI), and mismatch repair (MMR) in metastatic colon cancer patients: Gaps and implications, J Clin Oncol 37: ((2019) ), e15123–e15123. |
[10] | J. Shendure and H. Ji, Next-generation DNA sequencing, Nat Biotechnol 26: ((2008) ), 1135–1145. |
[11] | M. Ni, X. Liu, J. Wu, D. Zhang, J. Tian, T. Wang, S. Liu, Z. Meng, K. Wang, X. Duan, W. Zhou and X. Zhang, Identification of candidate biomarkers correlated with the pathogenesis and prognosis of non-small cell lung cancer via integrated bioinformatics analysis, Front Genet 9: ((2018) ), 469. |
[12] | L. Yu, X. Yang, W. Guan, D. Zhang, S. Ren, Y. Xing, D. An, J. Zhang, Y. Zhu and A. Zhu, Analysis of key genes for slow transit constipation based on RNA sequencing, Int J Gen Med 15: ((2022) ), 7569–7579. |
[13] | Z. Tang, C. Li, B. Kang, G. Gao, C. Li and Z. Zhang, GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses, Nucleic Acids Res 45: ((2017) ), W98–W102. |
[14] | C. Hutter and J.C. Zenklusen, The cancer genome atlas: Creating lasting value beyond its data, Cell 173: ((2018) ), 283–285. |
[15] | H.Y. Liu and C.J. Zhang, Identification of differentially expressed genes and their upstream regulators in colorectal cancer, Cancer Gene Ther 24: ((2017) ), 244–250. |
[16] | B. Zhao, Z. Baloch, Y. Ma, Z. Wan, Y. Huo, F. Li and Y. Zhao, Identification of potential key genes and pathways in early-onset colorectal cancer through bioinformatics analysis, Cancer Control 26: ((2019) ), 1073274819831260. |
[17] | X. Ding, H. Duan and H. Luo, Identification of core gene expression signature and key pathways in colorectal cancer, Front Genet 11: ((2020) ), 45. |
[18] | T. Barrett, S.E. Wilhite, P. Ledoux, C. Evangelista, I.F. Kim, M. Tomashevsky, K.A. Marshall, K.H. Phillippy, P.M. Sherman, M. Holko, A. Yefanov, H. Lee, N. Zhang, C.L. Robertson, N. Serova, S. Davis and A. Soboleva, NCBI GEO: archive for functional genomics data sets – update, Nucleic Acids Research 41: ((2012) ), D991–D995. |
[19] | S. Wu, F. Wu and Z. Jiang, Identification of hub genes, key miRNAs and potential molecular mechanisms of colorectal cancer, Oncol Rep 38: ((2017) ), 2043–2050. |
[20] | T. Zhang, J. Guo, J. Gu, Z. Wang, G. Wang, H. Li and J. Wang, Identifying the key genes and microRNAs in colorectal cancer liver metastasis by bioinformatics analysis and in vitro experiments, Oncol Rep 41: ((2019) ), 279–291. |
[21] | X. Zhai, Q. Xue, Q. Liu, Y. Guo and Z. Chen, Colon cancer recurrence-associated genes revealed by WGCNA co-expression network analysis, Mol Med Rep 16: ((2017) ), 6499–6505. |
[22] | M. Kruschewski, M. Ciurea, S. Lipka, S. Daum, L. Moser, B. Meyer, J. Gröne, J. Budczies and H.J. Buhr, Locally recurrent colorectal cancer: results of surgical therapy, Langenbeck Arch Surg 397: ((2012) ), 1059–1067. |
[23] | J. Hu, X. Yue, J. Liu and D. Kong, Construction of an miRNA-gene regulatory network in colorectal cancer through integrated analysis of mRNA and miRNA microarrays, Mol Med Rep 18: ((2018) ), 5109–5116. |
[24] | M. Kanehisa, M. Furumichi, M. Tanabe, Y. Sato and K. Morishima, KEGG: new perspectives on genomes, pathways, diseases and drugs, Nucleic Acids Res 45: ((2017) ), D353–D361. |


