
The paradoxical role of transforming growth factor-β in controlling oral squamous cell carcinoma development
Abstract
Transforming growth factor-
1.Introduction
Oral cancer is the most common malignant tumor in the head and neck region, and its incidence has been on the rise. According to the latest data from the International Agency for Research on Cancer, there were approximately 377,713 new cases and 177,757 deaths worldwide [1]. Oral squamous cell carcinoma (OSCC) accounts for 90% of all oral cancer cases. Despite the effectiveness of surgical treatment combined with postoperative radiotherapy and chemotherapy, the 5-year survival rate of oral cancer patients is still around 50
The transforming growth factor-
TGF-
2.TGF-𝜷
Figure 1.
The double role of TGF-
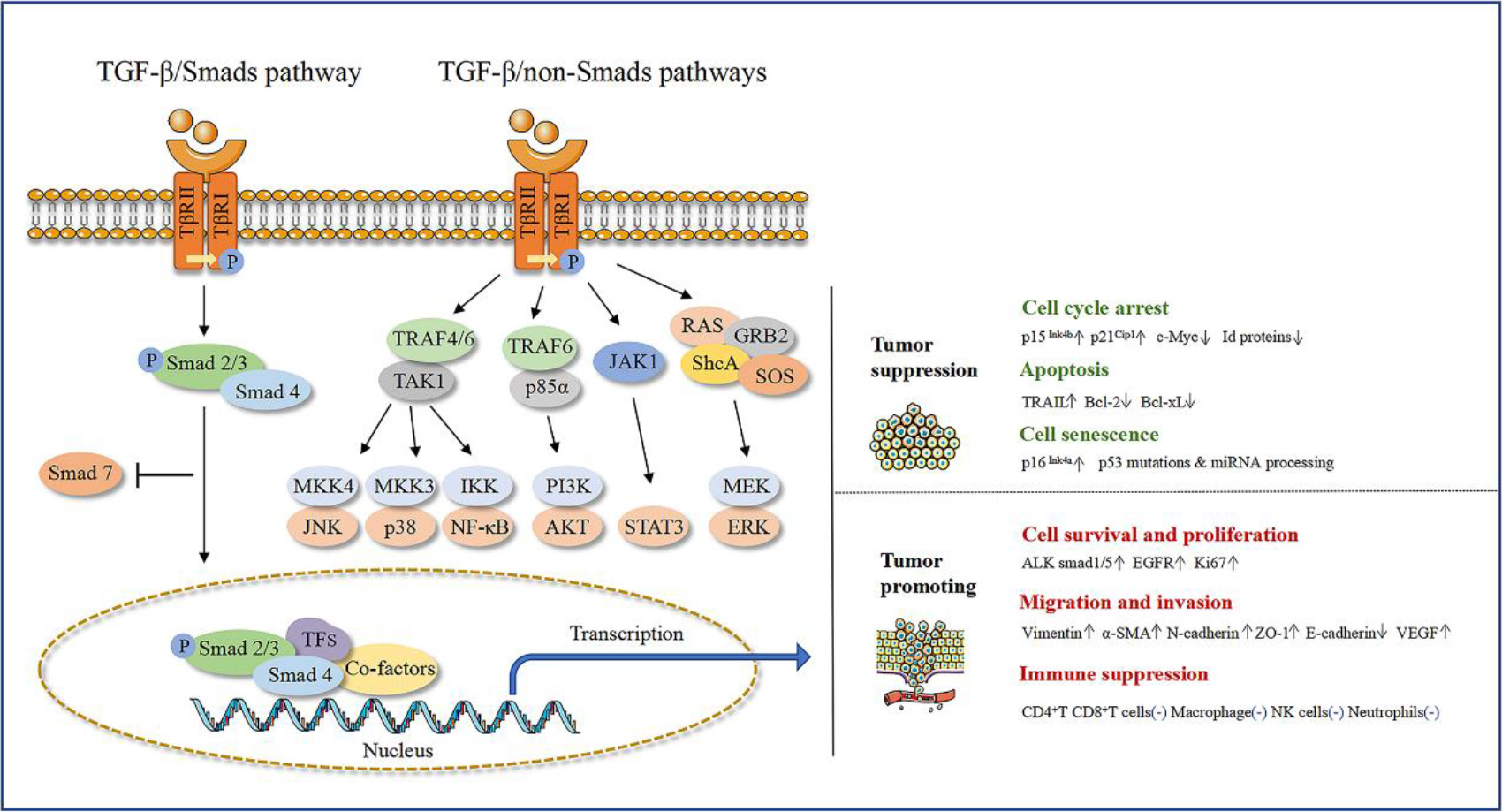
TGF-
2.1TGF-β
Firstly, TGF-
TGF-
In summary, the inhibition of oral squamous cell carcinoma cell proliferation by TGF-
2.2TGF-β
Apoptosis is a genetically controlled autonomous and orderly death process that eliminates excessive or damaged cells during embryonic organogenesis and adult organ homeostasis, maintaining stable internal environment [35]. TGF-
Cellular senescence is a stress response program that limits cell proliferation by inducing persistent and irreversible cell cycle arrest [39]. Cellular senescence can be triggered by telomere shortening (referred to as replicative senescence) or various cell stresses, such as activation of oncogenes, mitochondrial dysfunction, inactivation of tumor suppressor genes, endoplasmic reticulum stress, DNA damage, etc [40]. The cell cycle arrest associated with cellular senescence is one of the key mechanisms that limit the proliferation of epithelial tumor cells. Loss of genes such as p53 and p16Ink4a in late-stage malignant tumors leads to the occurrence of genetically unstable OSCC (GU-OSCC) [41]. Hassona et al. found that fibroblasts extracted from GU-OSCC showed high levels of cellular senescence, with an increased activity of senescence-associated
3.TGF-𝜷
TGF-
3.1TGF-β
TGF-
TGF-
In summary, researchers have found that TGF-
3.2TGF-β
Epithelial to mesenchymal transition (EMT) plays a key driving role in tumor cell migration and invasion. When epithelial-mesenchymal-like cells lose adhesion to the extracellular matrix, they undergo mesenchymal-to-amoeboid transition (MAT), which enhances the contractile ability of actin and forms cell membrane protrusions, thereby enhancing the movement of deformed cells [54]. During the process of EMT, the expression of epithelial cell-cell adhesion proteins such as E-cadherin and zonula occludens-1(ZO-1) is downregulated, while the expression of mesenchymal markers such as N-cadherin, vimentin, and
Increasing evidence suggests that EMT is not a single, rigid process, but a multi-step process that passes through a partial EMT state (P-EMT), and the TGF-
Angiogenesis provides the necessary nutrients and oxygen for tumors, and TGF-
In summary, recent studies pointed out that TGF-
3.3TGF-β
The tumor microenvironment (TME) is a complex system composed of various cell components, such as fibroblasts, endothelial cells, and various immune cells, including T cells and natural killer (NK) cells, which have natural cytotoxic effects on tumor cells [68]. Dendritic cells present tumor antigens to T cells, while macrophages and neutrophils clear cell debris through phagocytosis. TGF-
TGF-
TGF-
In summary, recent studies have found that TGF-
4.Conclusions and outlooks
Indeed, the dual role of TGF-
The dual role of TGF-
Funding
This study was supported by Science Foundation of Sichuan (2023NSFSC0558).
Author contributions
PENG Ruiting: Contributed to conception, interpretation of data, preparation of the manuscript.
HUANG Yun & HUANG Ping & LIU Linyi: Contributed to conception, preparation of the manuscript.
CHENG Lei: Contributed to conception, revision for important intellectual content.
PENG Xian: Contributed to revision for important intellectual content, supervision.
All authors gave their final approval and agree to be accountable for all aspects of the work.
Conflict of interest
The authors declare no conflict of interest.
References
[1] | H. Sung, J. Ferlay, R.L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J Clin 71: ((2021) ), 209–249. doi: 10.3322/caac.21660. |
[2] | J.P. Machiels, C. Rene Leemans, W. Golusinski, C. Grau, L. Licitra and V. Gregoire, E.E.B.E. a. [email protected], E.G.C.E. a. [email protected], E.E.B.E. a. [email protected], Squamous cell carcinoma of the oral cavity, larynx, oropharynx and hypopharynx: EHNS-ESMO-ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up, Annals of Oncology: Official Journal of the European Society for Medical Oncology 31: ((2020) ), 1462–1475. doi: 10.1016/j.annonc.2020.07.011. |
[3] | Z.H. Ren, C.Y. Hu, H.R. He, Y.J. Li and J. Lyu, Global and regional burdens of oral cancer from 1990 to 2017: Results from the global burden of disease study, Cancer Commun (Lond) 40: ((2020) ), 81–92. doi: 10.1002/cac2.12009. |
[4] | F.W. Mello, G. Melo, J.J. Pasetto, C.A.B. Silva, S. Warnakulasuriya and E.R.C. Rivero, The synergistic effect of tobacco and alcohol consumption on oral squamous cell carcinoma: a systematic review and meta-analysis, Clinical Oral Investigations 23: ((2019) ), 2849–2859. doi: 10.1007/s00784-019-02958-1. |
[5] | I. Ganly, L. Yang, R.A. Giese, Y. Hao, C.W. Nossa, L.G.T. Morris, M. Rosenthal, J. Migliacci, D. Kelly, W. Tseng, J. Hu, H. Li, S. Brown and Z. Pei, Periodontal pathogens are a risk factor of oral cavity squamous cell carcinoma, independent of tobacco and alcohol and human papillomavirus, Int J Cancer 145: ((2019) ), 775–784. doi: 10.1002/ijc.32152. |
[6] | X. Hu, J. Wu, H. Xiong, L. Zeng, Z. Wang, C. Wang, D. Huang, T. Zhang, Y. Peng, W. Chen, K. Xia and T. Su, Type 2 diabetes mellitus promotes the proliferation, metastasis, and suppresses the apoptosis in oral squamous cell carcinoma, Journal of Oral Pathology and Medicine: Official Publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology 51: ((2022) ), 483–492. doi: 10.1111/jop.13244. |
[7] | Y. Yu and X.H. Feng, TGF-beta signaling in cell fate control and cancer, Curr Opin Cell Biol 61: ((2019) ), 56–63. doi: 10.1016/j.ceb.2019.07.007. |
[8] | Z. Lu, L. Ding, H. Ding, F. Hao, Y. Pu, Y. Wang, S. Chen, Y. Yang, X. Zhao, X. Huang, L. Zhang, Z. Wang, Q. Hu and Y. Ni, Tumor cell-derived TGF-beta at tumor center independently predicts recurrence and poor survival in oral squamous cell carcinoma, Journal of Oral Pathology and Medicine: Official Publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology 48: ((2019) ), 696–704. doi: 10.1111/jop.12888. |
[9] | I.T.S. Santana, J.N.A. Dos Santos, V.L. de Almeida, W.N.S. Ferreira, E.M. Santos, R. de Almeida Freitas, C.C.K. Pinto, I.D. de Carvalho Barreto and F.R. de Matos, Association of PON1, TNF-alpha and TGF-beta gene polymorphisms with prognosis in oral and oropharyngeal squamous cell carcinoma, Acta Odontol Scand 79: ((2021) ), 327–334. doi: 10.1080/00016357.2020.1850856. |
[10] | D. Peng, M. Fu, M. Wang, Y. Wei and X. Wei, Targeting TGF-beta signal transduction for fibrosis and cancer therapy, Mol Cancer 21: ((2022) ), 104. doi: 10.1186/s12943-022-01569-x. |
[11] | A. Vander Ark, J. Cao and X. Li, TGF-beta receptors: In and beyond TGF-beta signaling, Cell Signal 52: ((2018) ), 112–120. doi: 10.1016/j.cellsig.2018.09.002. |
[12] | Y.E. Zhang, Non-smad signaling pathways of the TGF-beta family, Cold Spring Harb Perspect Biol 9: ((2017) ). doi: 10.1101/cshperspect.a022129. |
[13] | A. Sorrentino, N. Thakur, S. Grimsby, A. Marcusson, V. von Bulow, N. Schuster, S. Zhang, C.H. Heldin and M. Landstrom, The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner, Nat Cell Biol 10: ((2008) ), 1199–1207. doi: 10.1038/ncb1780. |
[14] | M. Yamashita, K. Fatyol, C. Jin, X. Wang, Z. Liu and Y.E. Zhang, TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta., Mol Cell 31: ((2008) ), 918–924. doi: 10.1016/j.molcel.2008.09.002. |
[15] | A. Hamidi, J. Song, N. Thakur, S. Itoh, A. Marcusson, A. Bergh, C.H. Heldin and M. Landstrom, TGF-beta promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85alpha, Sci Signal 10: ((2017) ). doi: 10.1126/scisignal.aal4186. |
[16] | L.Y. Tang, M. Heller, Z. Meng, L.R. Yu, Y. Tang, M. Zhou and Y.E. Zhang, Transforming growth factor-beta (TGF-beta) directly activates the JAK1-STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway, J Biol Chem 292: ((2017) ), 4302–4312. doi: 10.1074/jbc.M116.773085. |
[17] | M.K. Lee, C. Pardoux, M.C. Hall, P.S. Lee, D. Warburton, J. Qing, S.M. Smith and R. Derynck, TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA, EMBO J 26: ((2007) ), 3957–3967. doi: 10.1038/sj.emboj.7601818. |
[18] | Y.E. Zhang, Mechanistic insight into contextual TGF-beta signaling, Curr Opin Cell Biol 51: ((2018) ), 1–7. doi: 10.1016/j.ceb.2017.10.001. |
[19] | K. Luo, Signaling cross talk between TGF-beta/Smad and other signaling pathways, Cold Spring Harb Perspect Biol 9: ((2017) ). doi: 10.1101/cshperspect.a022137. |
[20] | H.L. Moses, A.B. Roberts and R. Derynck, The discovery and early days of TGF-beta: A historical perspective, Cold Spring Harb Perspect Biol 8: ((2016) ). doi: 10.1101/cshperspect.a021865. |
[21] | Y. Zhang, P.B. Alexander and X.F. Wang, TGF-beta family signaling in the control of cell proliferation and survival, Cold Spring Harb Perspect Biol 9: ((2017) ). doi: 10.1101/cshperspect.a022145. |
[22] | H. Zhang, Y. Zhan, Y. Zhang, G. Yuan and G. Yang, Dual roles of TGF-beta signaling in the regulation of dental epithelial cell proliferation, J Mol Histol ((2020) ). doi: 10.1007/s10735-020-09925-1. |
[23] | F. Wu, K.J. Weigel, H. Zhou and X.J. Wang, Paradoxical roles of TGF-beta signaling in suppressing and promoting squamous cell carcinoma, Acta Biochim Biophys Sin (Shanghai) 50: ((2018) ), 98–105. doi: 10.1093/abbs/gmx127. |
[24] | J. Seoane and R.R. Gomis, TGF-beta family signaling in tumor suppression and cancer progression, Cold Spring Harb Perspect Biol 9: ((2017) ). doi: 10.1101/cshperspect.a022277. |
[25] | W. Sun, D. Yi, L. Zhu, J. Zeng, Y. Liu, J. Chang, J. Teng, Y. Zhang, Y. Dong, X. Pan, Y. Chen, Y. Zhou, M. Lai, Q. Zhou, J. Liu, B. Chen and F. Ma, RUNX1 overexpression triggers TGF-beta signaling to upregulate p15 and thereby blocks early hematopoiesis by inducing cell cycle arrest, Stem Cell Res 60: ((2022) ), 102694. doi: 10.1016/j.scr.2022.102694. |
[26] | K. Engeland, Cell cycle regulation: p53-p21-RB signaling, Cell Death and Differentiation 29: ((2022) ), 946–960. doi: 10.1038/s41418-022-00988-z. |
[27] | S. Nakamura, T. Kamakura and T. Ookura, Tongue epithelial KT-1 cell-cycle arrest by TGF-beta associated with induction of p21(Cip1) and p15 (Ink4b), Cytotechnology 61: ((2009) ), 109–116. doi: 10.1007/s10616-010-9251-7. |
[28] | H. Niimi, K. Pardali, M. Vanlandewijck, C.H. Heldin and A. Moustakas, Notch signaling is necessary for epithelial growth arrest by TGF-beta, J Cell Biol 176: ((2007) ), 695–707. doi: 10.1083/jcb.200612129. |
[29] | Z. Liu, T. Chen, D. Bai, W. Tian and Y. Chen, Smad7 regulates dental epithelial proliferation during tooth development, Journal of Dental Research 98: ((2019) ), 1376–1385. doi: 10.1177/0022034519872487. |
[30] | X. Wang, W. Sun, C. Zhang, G. Ji, Y. Ge, Y. Xu and Y. Zhao, TGF-beta1 inhibits the growth and metastasis of tongue squamous carcinoma cells through Smad4, Gene 485: ((2011) ), 160–166. doi: 10.1016/j.gene.2011.06.023. |
[31] | C.R. Chen, Y. Kang, P.M. Siegel and J. Massague, E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression, Cell 110: ((2002) ), 19–32. doi: 10.1016/s0092-8674(02)00801-2. |
[32] | X.H. Feng, Y.Y. Liang, M. Liang, W. Zhai and X. Lin, Direct interaction of c-Myc with Smad2 and Smad3 to inhibit TGF-beta-mediated induction of the CDK inhibitor p15(Ink4B), Mol Cell 63: ((2016) ), 1089. doi: 10.1016/j.molcel.2016.08.027. |
[33] | Y. Kang, C.R. Chen and J. Massague, A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells, Mol Cell 11: ((2003) ), 915–926. doi: 10.1016/s1097-2765(03)00109-6. |
[34] | M. Kowanetz, U. Valcourt, R. Bergstrom, C.H. Heldin and A. Moustakas, Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein, Mol Cell Biol 24: ((2004) ), 4241–4254. doi: 10.1128/MCB.24.10.4241-4254.2004. |
[35] | N. Ketelut-Carneiro and K.A. Fitzgerald, Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die, J Mol Biol 434: ((2022) ), 167378. doi: 10.1016/j.jmb.2021.167378. |
[36] | K. Zhang, M. Zhang, Z. Luo, Z. Wen and X. Yan, The dichotomous role of TGF-β in controlling liver cancer cell survival and proliferation, J Genet Genomics ((2020) ). doi: 10.1016/j.jgg.2020.09.005. |
[37] | A. Sanchez-Capelo, Dual role for TGF-beta1 in apoptosis, Cytokine Growth Factor Rev 16: ((2005) ), 15–34. doi: 10.1016/j.cytogfr.2004.11.002. |
[38] | M. Bakhshayesh, F. Zaker, M. Hashemi, M. Katebi and M. Solaimani, TGF- beta1-mediated apoptosis associated with SMAD-dependent mitochondrial Bcl-2 expression, Clin Lymphoma Myeloma Leuk 12: ((2012) ), 138–143. doi: 10.1016/j.clml.2011.12.001. |
[39] | M. Ogrodnik, H. Salmonowicz, D. Jurk and J.F. Passos, Expansion and cell-cycle arrest: Common denominators of cellular senescence, Trends Biochem Sci 44: ((2019) ), 996–1008. doi: 10.1016/j.tibs.2019.06.011. |
[40] | D. Munoz-Espin and M. Serrano, Cellular senescence: from physiology to pathology, Nat Rev Mol Cell Biol 15: ((2014) ), 482–496. doi: 10.1038/nrm3823. |
[41] | E. Natarajan, M. Saeb, C.P. Crum, S.B. Woo, P.H. McKee and J.G. Rheinwald, Co-expression of p16(INK4A) and laminin 5 gamma2 by microinvasive and superficial squamous cell carcinomas in vivo and by migrating wound and senescent keratinocytes in culture, Am J Pathol 163: ((2003) ), 477–491. doi: 10.1016/s0002-9440(10)63677-2. |
[42] | Y. Hassona, N. Cirillo, K.P. Lim, A. Herman, M. Mellone, G.J. Thomas, G.N. Pitiyage, E.K. Parkinson and S.S. Prime, Progression of genotype-specific oral cancer leads to senescence of cancer-associated fibroblasts and is mediated by oxidative stress and TGF-beta, Carcinogenesis 34: ((2013) ), 1286–1295. doi: 10.1093/carcin/bgt035. |
[43] | R. Elston and G.J. Inman, Crosstalk between p53 and TGF-beta Signalling, J Signal Transduct 2012: ((2012) ), 294097. doi: 10.1155/2012/294097. |
[44] | C.J. Braun, X. Zhang, I. Savelyeva, S. Wolff, U.M. Moll, T. Schepeler, T.F. Orntoft, C.L. Andersen and M. Dobbelstein, p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest, Cancer Research 68: ((2008) ), 10094–10104. doi: 10.1158/0008-5472.CAN-08-1569. |
[45] | A. Korkut, S. Zaidi, R.S. Kanchi, S. Rao, N.R. Gough, A. Schultz, X. Li, P.L. Lorenzi, A.C. Berger, G. Robertson, L.N. Kwong, M. Datto, J. Roszik, S. Ling, V. Ravikumar, G. Manyam, A. Rao, S. Shelley, Y. Liu, Z. Ju, D. Hansel, G. de Velasco, A. Pennathur, J.B. Andersen, C.J. O’Rourke, K. Ohshiro, W. Jogunoori, B.N. Nguyen, S. Li, H.U. Osmanbeyoglu, J.A. Ajani, S.A. Mani, A. Houseman, M. Wiznerowicz, J. Chen, S. Gu, W. Ma, J. Zhang, P. Tong, A.D. Cherniack, C. Deng, L. Resar, N. Cancer Genome Atlas Research, J.N. Weinstein, L. Mishra and R. Akbani, A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-beta superfamily, Cell Syst 7: ((2018) ), 422–437 e427. doi: 10.1016/j.cels.2018.08.010. |
[46] | R.T. Peng, Y. Sun, X.D. Zhou, S.Y. Liu, Q. Han, L. Cheng and X. Peng, Treponema denticola promotes OSCC development via the TGF-beta signaling pathway, Journal of Dental Research 101: ((2022) ), 704–713. doi: 10.1177/00220345211067401. |
[47] | H.K. Son, D. Kim, Y. Lim, J. Kim and I. Park, A novel TGF-beta receptor II mutation (I227T/N236D) promotes aggressive phenotype of oral squamous cell carcinoma via enhanced EGFR signaling, BMC Cancer 20: ((2020) ), 1163. doi: 10.1186/s12885-020-07669-5. |
[48] | D. Pena-Oyarzun, M. Reyes, M.P. Hernandez-Caceres, C. Kretschmar, E. Morselli, C.A. Ramirez-Sarmiento, S. Lavandero, V.A. Torres and A. Criollo, Role of Autophagy in the Microenvironment of Oral Squamous Cell Carcinoma, Front Oncol 10: ((2020) ), 602661. doi: 10.3389/fonc.2020.602661. |
[49] | L. Yu, Y. Chen and S.A. Tooze, Autophagy pathway: Cellular and molecular mechanisms, Autophagy 14: ((2018) ), 207–215. doi: 10.1080/15548627.2017.1378838. |
[50] | Y.S. Abd El-Aziz, L.Y.W. Leck, P.J. Jansson and S. Sahni, Emerging role of autophagy in the development and progression of oral squamous cell carcinoma, Cancers (Basel) 13: ((2021) ). doi: 10.3390/cancers13246152. |
[51] | M.L. Tan, E.K. Parkinson, L.F. Yap and I.C. Paterson, Autophagy is deregulated in cancer-associated fibroblasts from oral cancer and is stimulated during the induction of fibroblast senescence by TGF-beta1, Scientific Reports 11: ((2021) ), 584. doi: 10.1038/s41598-020-79789-8. |
[52] | M.G. Kellermann, L.M. Sobral, S.D. da Silva, K.G. Zecchin, E. Graner, M.A. Lopes, L.P. Kowalski and R.D. Coletta, Mutual paracrine effects of oral squamous cell carcinoma cells and normal oral fibroblasts: induction of fibroblast to myofibroblast transdifferentiation and modulation of tumor cell proliferation, Oral Oncology 44: ((2008) ), 509–517. doi: 10.1016/j.oraloncology.2007.07.001. |
[53] | K. Haga, M. Yamazaki, S. Maruyama, M. Kawaharada, A. Suzuki, E. Hoshikawa, N.N. Chan, A. Funayama, T. Mikami, T. Kobayashi, K. Izumi and J.I. Tanuma, Crosstalk between oral squamous cell carcinoma cells and cancer-associated fibroblasts via the TGF-beta/SOX9 axis in cancer progression, Transl Oncol 14: ((2021) ), 101236. doi: 10.1016/j.tranon.2021.101236. |
[54] | A. Dongre and R.A. Weinberg, New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer, Nat Rev Mol Cell Biol 20: ((2019) ), 69–84. doi: 10.1038/s41580-018-0080-4. |
[55] | S. Lamouille, J. Xu and R. Derynck, Molecular mechanisms of epithelial-mesenchymal transition, Nat Rev Mol Cell Biol 15: ((2014) ), 178–196. doi: 10.1038/nrm3758. |
[56] | F. Xie, L. Ling, H. van Dam, F. Zhou and L. Zhang, TGF-beta signaling in cancer metastasis, Acta Biochim Biophys Sin (Shanghai) 50: ((2018) ), 121–132. doi: 10.1093/abbs/gmx123. |
[57] | Y. Wang, C. Wu, C. Zhang, Z. Li, T. Zhu, J. Chen, Y. Ren, X. Wang, L. Zhang and X. Zhou, TGF-beta-induced STAT3 overexpression promotes human head and neck squamous cell carcinoma invasion and metastasis through malat1/miR-30a interactions, Cancer Lett 436: ((2018) ), 52–62. doi: 10.1016/j.canlet.2018.08.009. |
[58] | J.Q. Bu and F. Chen, TGF-beta1 promotes cells invasion and migration by inducing epithelial mesenchymal transformation in oral squamous cell carcinoma, Eur Rev Med Pharmacol Sci 21: ((2017) ), 2137–2144. |
[59] | L. Meng, Y. Zhao, W. Bu, X. Li, X. Liu, D. Zhou, Y. Chen, S. Zheng, Q. Lin, Q. Liu and H. Sun, Bone mesenchymal stem cells are recruited via CXCL8-CXCR2 and promote EMT through TGF-beta signal pathways in oral squamous carcinoma, Cell Prolif 53: ((2020) ), e12859. doi: 10.1111/cpr.12859. |
[60] | N. Cirillo, Y. Hassona, A. Celentano, K.P. Lim, S. Manchella, E.K. Parkinson and S.S. Prime, Cancer-associated fibroblasts regulate keratinocyte cell-cell adhesion via TGF-beta-dependent pathways in genotype-specific oral cancer, Carcinogenesis 38: ((2017) ), 76–85. doi: 10.1093/carcin/bgw113. |
[61] | Y. Hao, D. Baker and P. Ten Dijke, TGF-beta-mediated epithelial-mesenchymal transition and cancer metastasis, Int J Mol Sci 20: ((2019) ). doi: 10.3390/ijms20112767. |
[62] | S.V. Puram, A.S. Parikh and I. Tirosh, Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer, Mol Cell Oncol 5: ((2018) ), e1448244. doi: 10.1080/23723556.2018.1448244. |
[63] | S. Yokoyama, H. Shigeishi, H. Murodumi, M. Sakuma, H. Kato, K. Higashikawa, K. Ohta, M. Sugiyama, M. Takechi, TGF-beta1 induces amoeboid-to-mesenchymal transition of CD44(high) oral squamous cell carcinoma cells via miR-422a downregulation through ERK activation and Cofilin-1 phosphorylation, Journal of oral pathology and medicine: official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology 50: ((2021) ), 155–164. doi: 10.1111/jop.13113. |
[64] | T. Courau, D. Nehar-Belaid, L. Florez, B. Levacher, T. Vazquez, F. Brimaud, B. Bellier and D. Klatzmann, TGF-beta and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies, JCI Insight 1: ((2016) ), e85974. doi: 10.1172/jci.insight.85974. |
[65] | K.H. Pak, K.C. Park and J.H. Cheong, VEGF-C induced by TGF- beta1 signaling in gastric cancer enhances tumor-induced lymphangiogenesis, BMC Cancer 19: ((2019) ), 799. doi: 10.1186/s12885-019-5972-y. |
[66] | D. Liu, L. Li, X.X. Zhang, D.Y. Wan, B.X. Xi, Z. Hu, W.C. Ding, D. Zhu, X.L. Wang, W. Wang, Z.H. Feng, H. Wang, D. Ma and Q.L. Gao, SIX1 promotes tumor lymphangiogenesis by coordinating TGFbeta signals that increase expression of VEGF-C, Cancer Research 74: ((2014) ), 5597–5607. doi: 10.1158/0008-5472.CAN-13-3598. |
[67] | H. Sun, C. Miao, W. Liu, X. Qiao, W. Yang, L. Li and C. Li, TGF-beta1/TbetaRII/Smad3 signaling pathway promotes VEGF expression in oral squamous cell carcinoma tumor-associated macrophages, Biochem Biophys Res Commun 497: ((2018) ), 583–590. doi: 10.1016/j.bbrc.2018.02.104. |
[68] | E. Batlle and J. Massague, Transforming Growth Factor-beta Signaling in Immunity and Cancer, Immunity 50: ((2019) ), 924–940. doi: 10.1016/j.immuni.2019.03.024. |
[69] | S. Sanjabi, S.A. Oh and M.O. Li, Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection, Cold Spring Harb Perspect Biol 9: ((2017) ). doi: 10.1101/cshperspect.a022236. |
[70] | A. Dahmani and J.S. Delisle, TGF-beta in T Cell Biology: Implications for Cancer Immunotherapy, Cancers (Basel) 10: ((2018) ). doi: 10.3390/cancers10060194. |
[71] | D.A. Thomas and J. Massague, TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance, Cancer Cell 8: ((2005) ), 369–380. doi: 10.1016/j.ccr.2005.10.012. |
[72] | L. Gorelik, S. Constant and R.A. Flavell, Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation, J Exp Med 195: ((2002) ), 1499–1505. doi: 10.1084/jem.20012076. |
[73] | S.C. McKarns, R.H. Schwartz and N.E. Kaminski, Smad3 is essential for TGF-beta 1 to suppress IL-2 production and TCR-induced proliferation, but not IL-2-induced proliferation, J Immunol 172: ((2004) ), 4275–4284. doi: 10.4049/jimmunol.172.7.4275. |
[74] | J.S. Delisle, M. Giroux, G. Boucher, J.R. Landry, M.P. Hardy, S. Lemieux, R.G. Jones, B.T. Wilhelm and C. Perreault, The TGF-beta-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells, Genes Immun 14: ((2013) ), 115–126. doi: 10.1038/gene.2012.63. |
[75] | S.C. McKarns and R.H. Schwartz, Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: a role for T cell intrinsic Smad3, J Immunol 174: ((2005) ), 2071–2083. doi: 10.4049/jimmunol.174.4.2071. |
[76] | Y. Kondo, S. Suzuki, T. Takahara, S. Ono, M. Goto, S. Miyabe, Y. Sugita, T. Ogawa, H. Ito, A. Satou, T. Tsuzuki, K. Yoshikawa, R. Ueda and T. Nagao, Improving function of cytotoxic T-lymphocytes by transforming growth factor-beta inhibitor in oral squamous cell carcinoma, Cancer Science 112: ((2021) ), 4037–4049. doi: 10.1111/cas.15081. |
[77] | L.A.G. Maldonado, C.R. Nascimento, N.A. Rodrigues Fernandes, A.L.P. Silva, N.J. D’Silva, Jr., Influence of tumor cell-derived TGF-beta on macrophage phenotype and macrophage-mediated tumor cell invasion, Int J Biochem Cell Biol 153: ((2022) ), 106330. doi: 10.1016/j.biocel.2022.106330. |
[78] | J.C. Lee, K.M. Lee, D.W. Kim, D.S. Heo, Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients, J Immunol 172: ((2004) ), 7335–7340. doi: 10.4049/jimmunol.172.12.7335. |
[79] | S. Viel, A. Marcais, F.S. Guimaraes, R. Loftus, J. Rabilloud, M. Grau, S. Degouve, S. Djebali, A. Sanlaville, E. Charrier, J. Bienvenu, J.C. Marie, C. Caux, J. Marvel, L. Town, N.D. Huntington, L. Bartholin, D. Finlay, M.J. Smyth and T. Walzer, TGF-beta inhibits the activation and functions of NK cells by repressing the mTOR pathway, Sci Signal 9: ((2016) ), ra19. doi: 10.1126/scisignal.aad1884. |
[80] | Y. Gao, F. Souza-Fonseca-Guimaraes, T. Bald, S.S. Ng, A. Young, S.F. Ngiow, J. Rautela, J. Straube, N. Waddell, S.J. Blake, J. Yan, L. Bartholin, J.S. Lee, E. Vivier, K. Takeda, M. Messaoudene, L. Zitvogel, M.W.L. Teng, G.T. Belz, C.R. Engwerda, N.D. Huntington, K. Nakamura, M. Holzel and M.J. Smyth, Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells, Nat Immunol 18: ((2017) ), 1004–1015. doi: 10.1038/ni.3800. |
[81] | Y. Tao, T. Qinchao, C. Xueru, F. Wan, Z. Zhuoqian, H. Wanqian and L. Feixin, TGF-beta1 and IL-17A comediate the protumor phenotype of neutrophils to regulate the epithelial-mesenchymal transition in oral squamous cell carcinoma, Journal of Oral Pathology and Medicine: Official Publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology ((2020) ). doi: 10.1111/jop.13122. |


