
Droplet digital polymerase chain reaction for detection and quantification of cell-free DNA TP53 target somatic mutations in oral cancer
Abstract
BACKGROUND:
TP53 mutation is a driver mutation of oral carcinogenesis. This study investigated cancerous and cell-free DNA (cfDNA) in patients with oral squamous cell carcinoma (OSCC) to detect the target hotspot somatic mutation of TP53.
OBJECTIVE:
TP53 target hotspot mutations were determined in surgically resected primary tumor samples from 107 OSCC patients.
METHODS:
Cancerous and cfDNA samples were examined for mutations through droplet digital polymerase chain reaction (ddPCR) by using mutation-specific assays. The ddPCR results were evaluated alongside clinicopathological data.
RESULTS:
In total, 23 cases had target TP53 mutations in varying degrees. We found that OSCC had relatively low cfDNA shedding, and mutations were at low allele frequencies. Of these 23 cases, 13 had target TP53 mutations in their corresponding cfDNA. Target somatic mutations in cancerous DNA and cfDNA are related to cervical lymph node metastasis. The cfDNA concentration is related to primary tumor size, lymph node metastasis, and OSCC stage.
CONCLUSIONS:
Our results show that the detection of TP53 target somatic mutations in OSCC patients by using ddPCR is technically feasible. Low levels of cfDNA may produce different results between cancerous tissue and cfDNA analyses. Future research on cfDNA may quantify diagnostic biomarkers in the surveillance of OSCC patients.
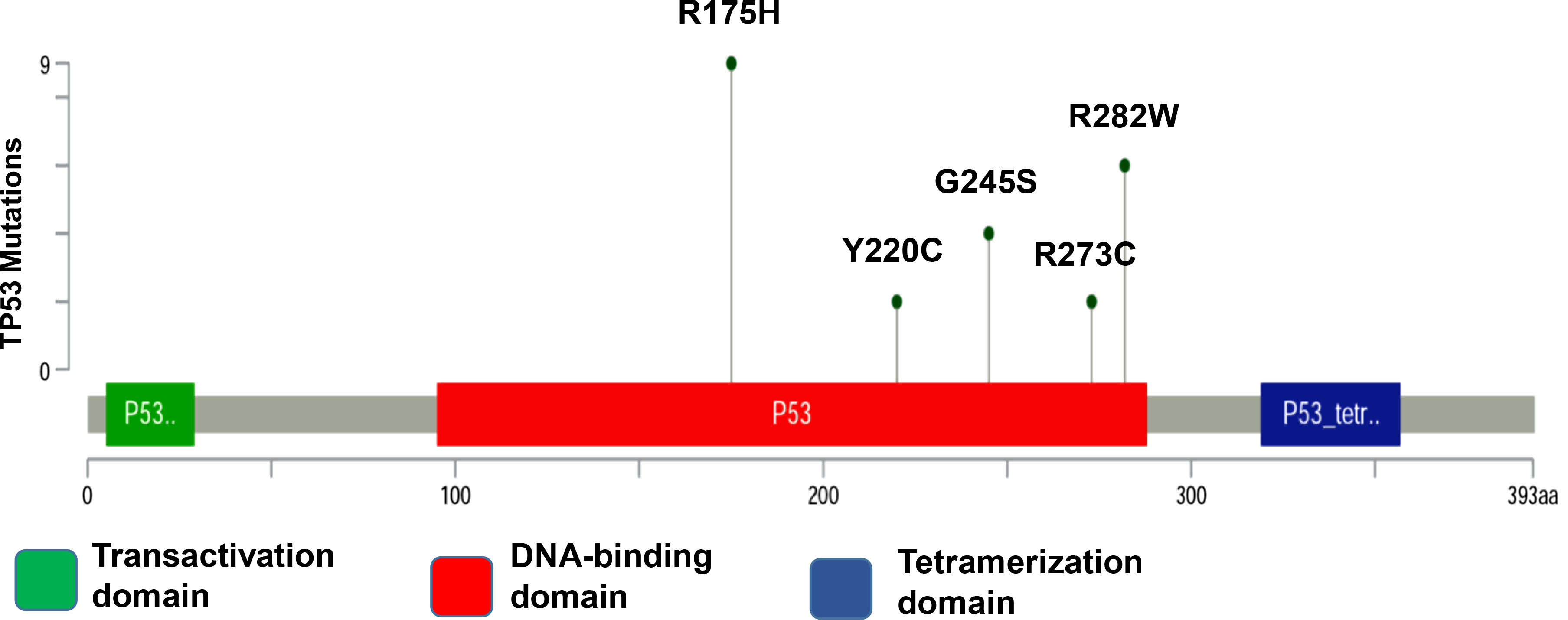
Figure 1.
TP53 mutations in patients with OSCC. Distribution of hotspot target mutations in the TP53 coding sequence. Mutation types are indicated by colored ovals, colored boxes are indicating domains.
1.Introduction
Head and neck squamous cell carcinoma (HNSCC) includes oral squamous cell carcinoma (OSCC) and is the sixth most prevalent malignancy worldwide [1]. Despite in the past 40 years ample knowledge has been gained regarding the carcinogenesis in HNSCC and OSCC, and a lot of innovative developments have been made in surgery, chemotherapy, and radiotherapy, the prognosis for many HNSCC types has not considerably improved [2, 3]. The Taiwan Ministry of Health and Welfare (MoHW), is reporting an average survival rate of 5 years after oral cancer has been diagnosed. This has not much improved in the past 20 years [4]. OSCC usually involves multistep carcinogenesis. Due to the large area that has been exposed to carcinogens, multiple lesions, also known as field cancerization, may develop at the same time at different neoplastic stages. This might be the reason for the high recurrence of OSCC after treatment [5]. Therefore, specific and sensitive biomarkers are highly desired for patients with OSCC and for individuals which have a high risk for developing OSCC [6]. Still, molecular biomarkers for guiding adequate diagnosis and management decisions have not been identified. In OSCC, TP53 is the gene with the highest mutation rate (65%–85%) [7], and the mutations in TP53 have predictive significance for the response to chemotherapy based on platinum complexes [8]. TP53 is a transcription factor that works as a tumor suppressor. TP53 is the most commonly mutated gene in various human cancers. In many types of cancer, the functions of the wild-type (WT) p53 protein are disturbed by these mutations [9]. The p53 pathway plays a critical role in the tumorigenesis and progression of HNSCC [10, 11, 12].
In cancerous cells, mutations in the TP53 gene frequently are found in its DNA-binding domain (DBD) between amino-acid residues 102 and 292 (out of 393 in total) [13]. Approximately 10% of these mutations result in the loss of function of p53 when no protein is synthesized due to nonsense or frameshift mutations or due to deletions. The remainder includes missense mutations, which result in a dysfunctional protein. Loss of function mutations include mutations that impair the ability of p53 to recognize its specific DNA sequence motifs [13, 14, 15, 16].
Table 1
TP53 ddPCR-specific PrimePCR ddPCR mutation assays
| TP53 primer/probe | Unique assay ID | Fluorophore | Amplicon length | Restriction enzyme |
|---|---|---|---|---|
| p.R175H | dHsaMDV2010105 | FAM | 65 | HaeIII |
| p.Y220C | dHsaMDV2510536 | FAM | 64 | HaeIII |
| p.G245S | dHsaMDV2516746 | FAM | 65 | MseI |
| p.R273C | dHsaMDV2510538 | FAM | 65 | HaeIII |
| p.R282W | dHsaMDV2516902 | FAM | 64 | HaeIII |
Of 190 mutation sites, the most common 10 that are located in the DBD are found in approximately 30% of all missense mutations in HNSCC. This apparent preference for some mutations may be the result of the selection process that favors alleles that are coding for proteins whose structures and possibly functions do maximally contribute to a cancerous phenotype [17]. Recent developments in molecular diagnostics foster the use of blood-based genetic biomarkers for the diagnosis of various cancer types [18]. Cell-free DNA (cfDNA) is found in healthy people and patients with benign or malign tumors. It may originate from normal cells, including healthy leukocytes that undergo apoptosis, and also may be shed from dead cells, from healthy or cancerous tissue [19, 20]. cfDNA has gained attention as a potential biomarker in oncology [21]. Cell-free circulating tumor DNA (ctDNA) may be released into the bloodstream by necrotic and apoptotic tumor cells and contains tumor-specific mutations [22]. These mutations may be detected in the blood of cancer patients by a simple blood sampling. This method has been termed “liquid biopsy” [23]. For head and neck cancer, so far studies have mainly focused on actionable oncogenic mutations, such as PIK3CA and HRAS, hotspot TP53 mutations, and also on human papillomavirus (HPV)-related biomarkers, which may serve as prognostic or predictive markers, in order to establish and to modify targeted therapy [20, 24, 25, 26, 27, 28]. However, the accurate detection of ctDNA in plasma is a challenge, as the concentration of ctDNA may be low. This may greatly impair the reliable and valid assessment of tumor dynamics. Droplet digital polymerase chain reaction (ddPCR) is a method for performing digital PCR that is based on water-oil emulsion droplet technology. The DNA sample is fractionated into about 20,000 water-in-oil droplets, and PCR amplification of the template molecules occurs in each individual droplet [29, 30]. High-copy templates and background are diluted, effectively enriching template concentration in target-positive partitions, allowing for the sensitive detection of rare targets especially in the cfDNA. In this study, we used ddPCR as a method to evaluate the five most frequent coding TP53 mutations, namely R175H, R282W, G245S, Y220C, and R273C, by using the OSCC patients cancerous tissue and cfDNA database (Fig. 1A) [17]. Clinicopathogenic variants were analyzed using the target mutation sites of OSCC patients.
2.Materials and methods
2.1Patients
In this study, we evaluated 107 patients with OSCC and 50 matched healthy controls. Tumor samples were obtained from OSCC patients during surgery. Before the surgery, none of the patients had received adjuvant chemotherapy or radiotherapy. Informed written consent was obtained from all participants. The Institutional Review Board (IRB) of Mackay Memorial Hospital has approved the study. IRB approval numbers are 12MMHIS178 and 15MMHIS104.
Cells were isolated from tissue sections by laser capture microdissection, following established protocols. DNA was extracted from cancerous tissue as previously reported [31]. 10 mL of whole blood were collected from the patients in the morning after fasting and before the surgery. Vacutainer blood collection tubes containing EDTA as anticoagulant (Becton Dickson, Franklin Lakes, NJ, USA) were used.
2.2cfDNA extraction
The procedure for cfDNA extraction has been previously described [32, 33]. In brief, plasma samples were spun at 1600
2.3Plasma DNA quantification
For sizing and quantifying the purified plasma DNA, a TapeStation 2200 (Agilent Technology, USA) with a high-sensitivity D1000 ScreenTape system (Agilent Technologies, USA) was used [33].
2.4Droplet digital polymerase chain reaction
The plasma and tissue samples from all 107 cancer patients were analyzed for TP53 point mutations which had been identified in the primary tumor tissue by next-generation sequencing (NGS). Mutant-type (MT) and wild-type (WT) TP53 sequences were used as DNA templates for designing the ddPCR experiments (Bio-Rad Laboratories, CA, USA) assays in accordance with the MIQE guidelines [34]. ddPCR reactions were carried out in a volume of 20
Table 2
TP53 mutation sites for Sanger sequencing
| TP53 mutation site | Sequence | TM ( | bp | |
|---|---|---|---|---|
| p.R175H | Forward | GTGCAGCTGTGGGTTGATT | 58 | 223 |
| Reverse | GGGCCAGACCTAAGAGCAAT | |||
| p.Y220C | Forward | GCCCCTCCTCAGCATCTTAT | 58 | 237 |
| Reverse | TTGCACATCTCATGGGGTTA | |||
| p.G245S | Forward | TGCTTGCCACAGGTCTCC | 58 | 236 |
| Reverse | GGTCAGAGGCAAGCAGAGG | |||
| p.R273C/ p.R282W | Forward | GGGAGTAGATGGAGCCTGGT | 58 | 248 |
| Reverse | GCTTCTTGTCCTGCTTGCTT |
2.5Validation of point mutations
We confirmed the identified somatic mutations by Sanger sequencing. For each point mutation, primer sets were designed in Primer3 version 0.4.0 (http://primer3. wi.mit.edu/), they are shown in Table 2. For Sanger sequencing, PCRs were performed using a standard hot start kit. Amplicons then were sequenced with the ABI BigDye Terminator Cycle Sequencing kit on an ABI 3730xl DNA analyzer instrument (Applied Biosystems, Foster City, CA, USA).
2.6Statistical analysis
For all statistics, the Prism 5 package (GraphPad, San Diego, CA, USA) and SPSS 18.0 (SPSS Inc, Chicago, IL) were used. Mann-Whitney, Fisher, and Kruskal-Wallis tests were used to compare differences among various variants. Their influence on survival was assessed with a Kaplan-Meier analysis. Differences were considered statistically significant for
3.Results
In this study, 107 patients with OSCC and 50 matched healthy controls were investigated. The distribution of cfDNA sizes was similar between OSCC patients and healthy donors, the average size of cfDNA was 150–200 bp (Fig. S1). Table 3 shows the clinical characteristics of the study participants. Preoperative cfDNA plasma concentrations were between 6.1 and 646 ng/mL (Fig. 2A). in OSCC, the mean concentration of cfDNA was 71.9
Table 3
Clinicopathological parameters with cfDNA concentration in patients with OSCC
| OSCC | C control | |||||
|---|---|---|---|---|---|---|
| Variables |
| Mean | |
| Mean |
|
| Gender | ||||||
| Male | 96 | 53.90 | 0.851 | 43 | 22.64 | 0.9 |
| Female | 11 | 41.25 | 7 | 32.04 | ||
| Tumor size | ||||||
| T1–T2 | 24 | 64.57 | 0.024* | |||
| T3–T4 | 83 | 106.9 | ||||
| Nodal stage | ||||||
| N0 | 67 | 46.45 | 0.009** | |||
| N | 40 | 127.4 | ||||
| Stage | ||||||
| I–II | 21 | 48.58 | 0.001** | |||
| III–IV | 86 | 96.51 | ||||
| TP53 mutation | ||||||
| No | 84 | 55.03 | 0.2 | |||
| Yes | 23 | 81.24 | ||||
Student’s
Figure 2.
Comparison of cfDNA plasma levels in (A) healthy controls versus patients with OSCC preoperatively; (B) patients with different tumor sizes preoperatively; (C) patients without metastasis in the neck lymph nodes; (D) patients with early- and late-stage carcinoma; and (E) patients with different statuses of lymphovascular invasion. (F) Kaplan-Meier analysis of patients with MT-TP53 and WT-TP53. Scatter plots displaying mean
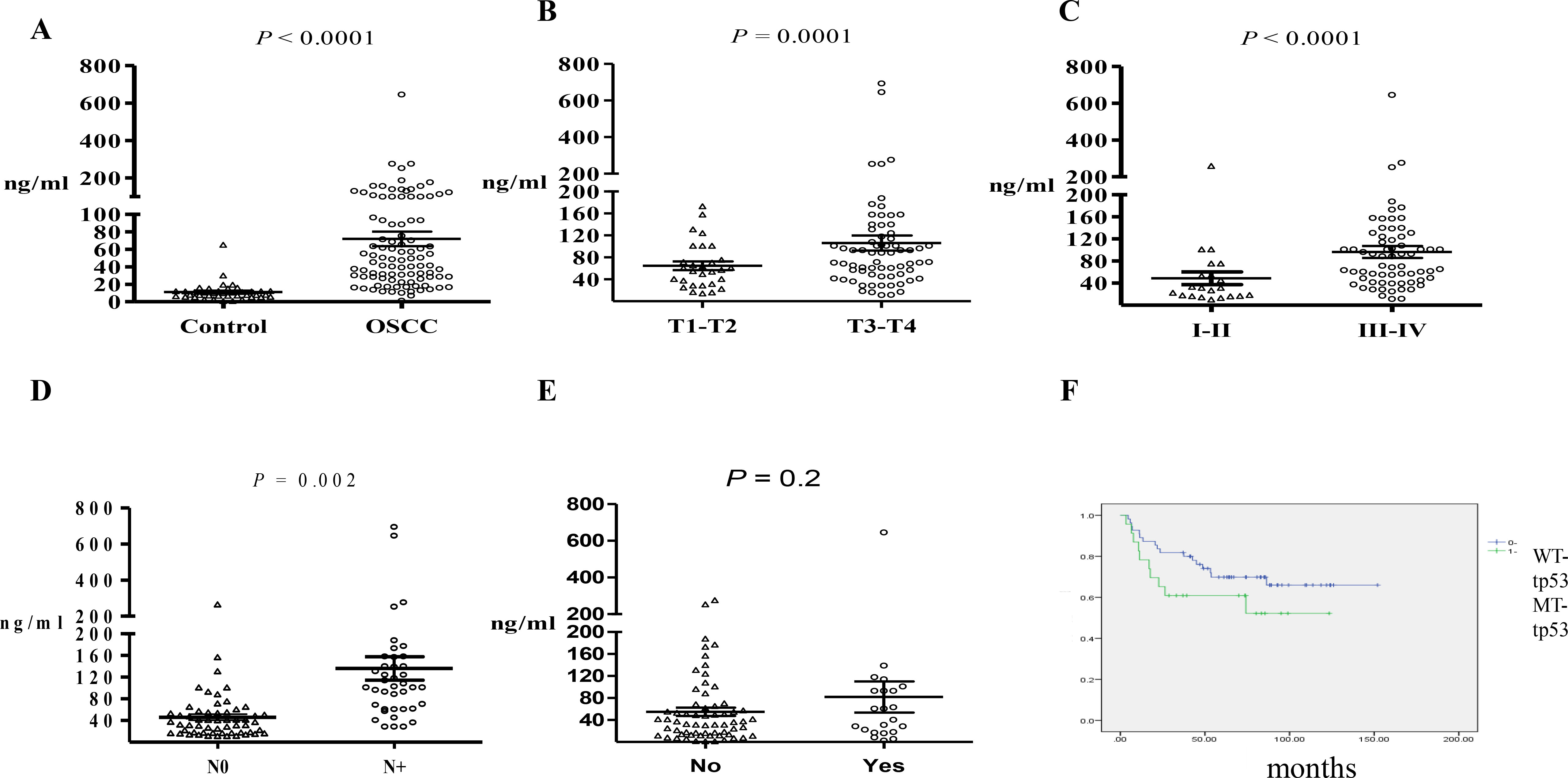
Figure 3.
Clinicopathological parameters of OSCC. Colored boxes indicate different parameters and patients with MT-TP53.
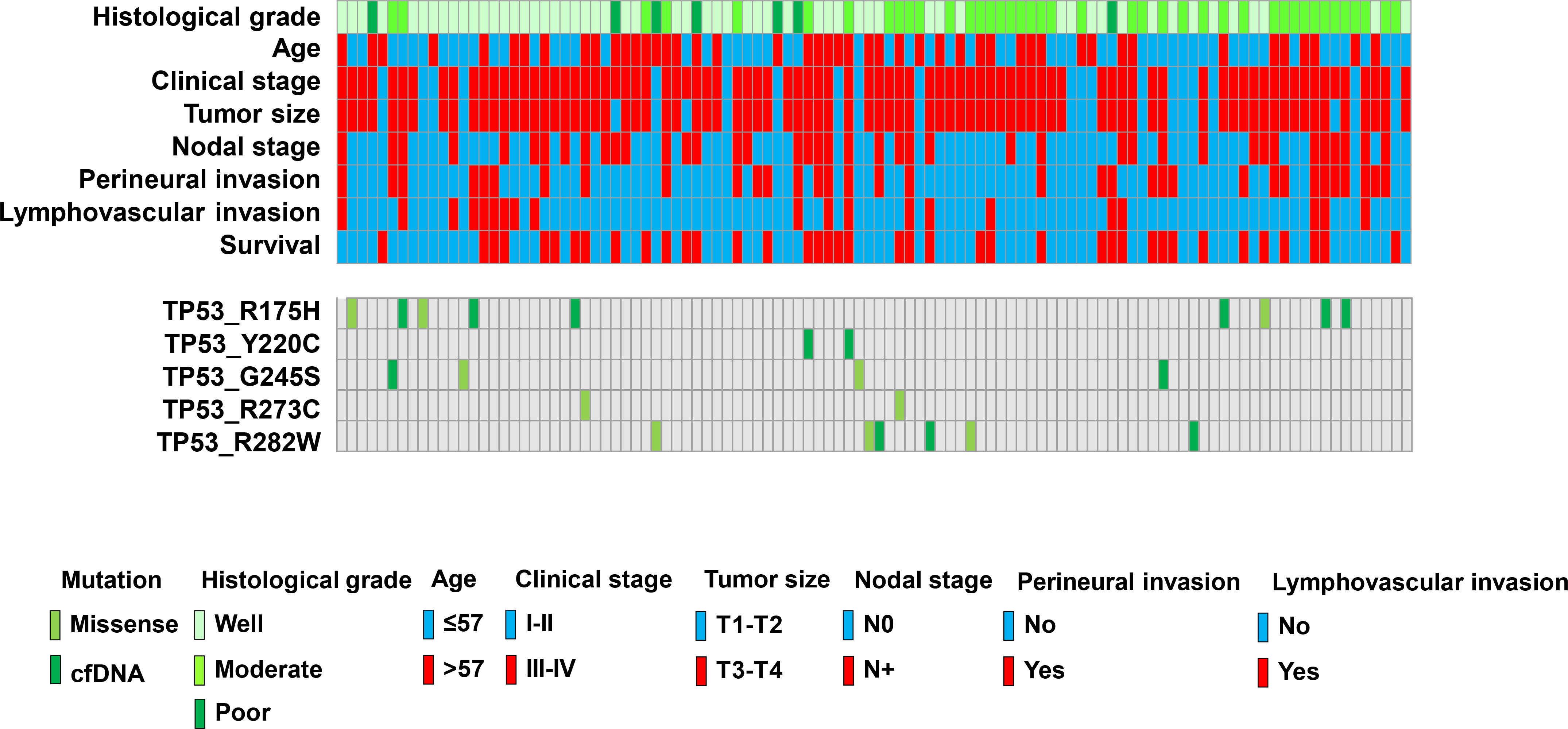
All 107 patients were assessed for TP53 mutations by performing ddPCR of primary cancerous tissue and preoperative cfDNA. Five hotspot target mutations were identified in this study. Five assays each were run to detect p.R282W, p.R175H, p.R273C, p.Y220C, and p.G245S mutations through ddPCR. PCR for wild type alleles was run in parallel, and samples were considered to have a mutated TP53 when mutations were detected at an FA of
Table 4
Clinicopathological parameters with TP53 hotspot target mutations
| TP53 mutation | Cancerous tissue | cfDNA | ||||
|---|---|---|---|---|---|---|
| MT-TP53 | WT-TP53 |
| MT-P53 | WT-TP53 |
| |
| Gender | ||||||
| Male | 21 | 75 | 12 | 84 | ||
| Female | 2 | 9 | 1 | 1 | 10 | 1 |
| Average age (y/o) site | 55.8 | 56.6 | 0.90 | 55.5 | 56.8 | 0.9 |
| Buccal | 6 | 30 | 4 | 33 | ||
| Tongue | 7 | 14 | 3 | 18 | ||
| Gingiva | 9 | 23 | 5 | 27 | ||
| Retromolar | 0 | 3 | 0 | 3 | ||
| Mouth floor | 0 | 3 | 0 | 3 | ||
| Lip | 1 | 2 | 1 | 2 | ||
| Other | 0 | 9 | 0.28 | 0 | 9 | 0.6 |
| Tumor size | ||||||
| T1–2 | 6 | 18 | 1 | 23 | ||
| T3–4 | 17 | 66 | 0.77 | 12 | 71 | 0.3 |
| Pathological stage | ||||||
| Stage 1–2 | 6 | 15 | 1 | 20 | ||
| Stage 3–4 | 17 | 69 | 0.38 | 12 | 74 | 0.4 |
| lymph node metastasis | ||||||
| N0 | 10 | 57 | 4 | 63 | ||
| N1–3 | 13 | 27 | 0.03 | 9 | 31 | 0.01* |
| ENE | ||||||
| Positive | 5 | 12 | 3 | 14 | ||
| Negative | 18 | 72 | 0.51 | 10 | 80 | 0.4 |
| Perineural invasion | ||||||
| Positive | 8 | 26 | 5 | 29 | ||
| Negative | 15 | 58 | 0.80 | 8 | 65 | 0.7 |
| Differentiation | ||||||
| Well | 13 | 46 | 7 | 52 | ||
| Moderate | 9 | 33 | 5 | 37 | ||
| Poor | 1 | 5 | 0.59 | 1 | 5 | 0.9 |
| Lymphovascular invasion | ||||||
| Positive | 5 | 15 | 3 | 17 | ||
| Negative | 18 | 69 | 0.76 | 10 | 77 | 0.7 |
| DM | ||||||
| Yes | 13 | 31 | 7 | 37 | ||
| No | 10 | 53 | 0.09 | 6 | 57 | 0.3 |
| Recurrence | ||||||
| Positive | 4 | 22 | 2 | 24 | ||
| Negative | 19 | 62 | 0.26 | 11 | 70 | 0.7 |
| Status | ||||||
| Expired | 12 | 28 | 6 | 34 | ||
| Alive | 11 | 56 | 0.09 | 7 | 60 | 0.5 |
Fisher’s exact test. *Statistically significant (
Figure 4.
Two-dimensional plot, showing the distribution of the droplet amounts of mutant MT-R175H in ddPCR of patient 969. (A) Merged ddPCR results in the duplicates of corresponding blood samples from patient 969. Blue dots represent MT-positive droplet clusters, dark gray dots represent negative droplet clusters, and orange dots represent MT/WT-positive droplets. WT-positive droplets are represented by green dots, demonstrating that cfDNA was present in the samples and that the ddPCR conditions were satisfactory. The purple lines are the thresholds that have been manually placed, in order to distinguish positive and negative droplets. They were set at fluorescence values based on the ddPCR data of (B) FFPE samples and (C) cfDNA samples. (D) Sanger sequencing indicated a G-to-A point mutation in the FFPE sample, but it was not detectable in the cfDNA sample.
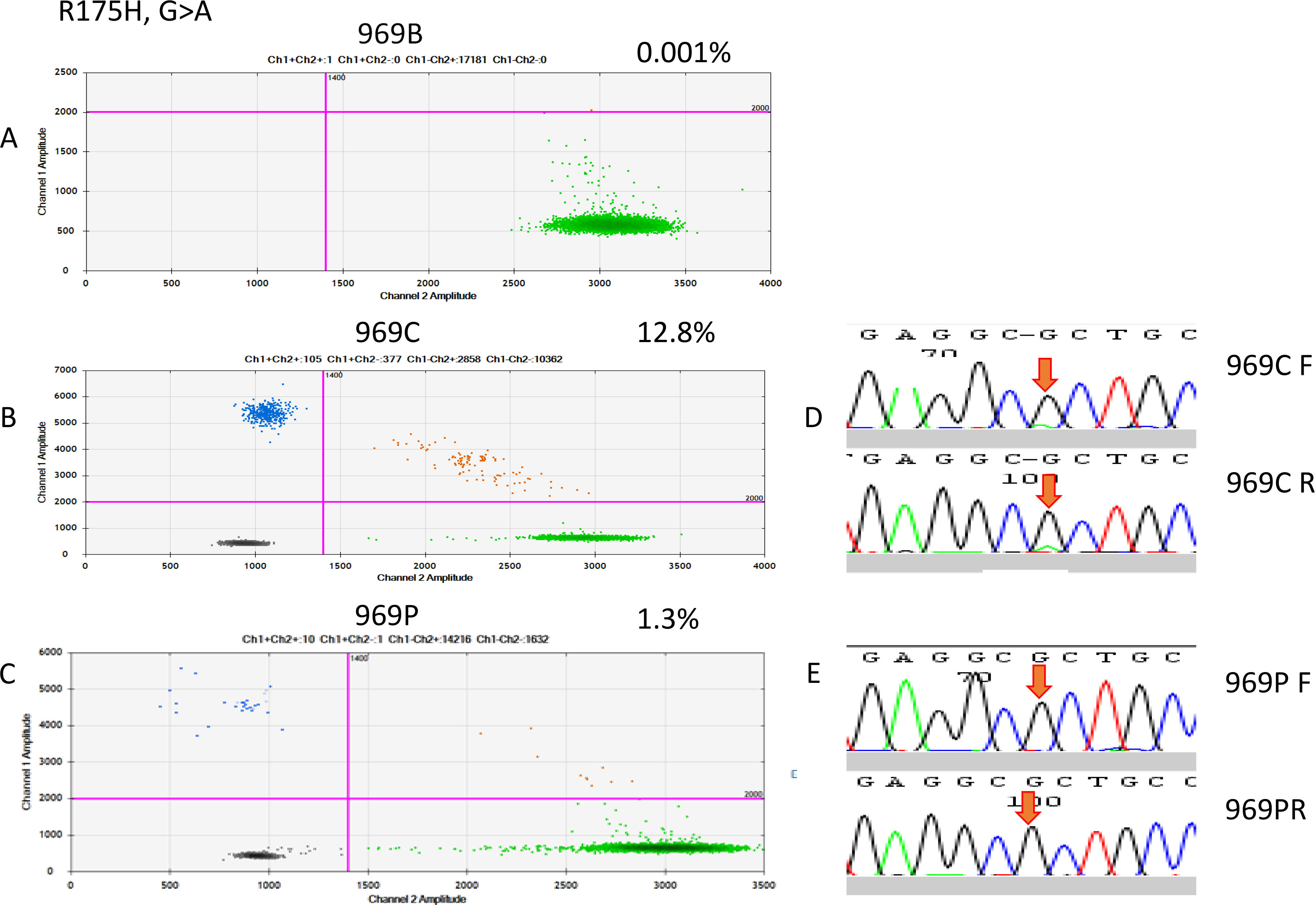
Figure 5.
ddRT-PCR results of patient 700. The two-dimensional plots show the amount of MT-R282W droplets in the analysis. (A) Blood sample, (B) FFPE sample, and (C) cfDNA sample. (D) Sanger sequencing indicated a C-to-T point mutation in the FFPE sample, but it was not detectable in the cfDNA sample.
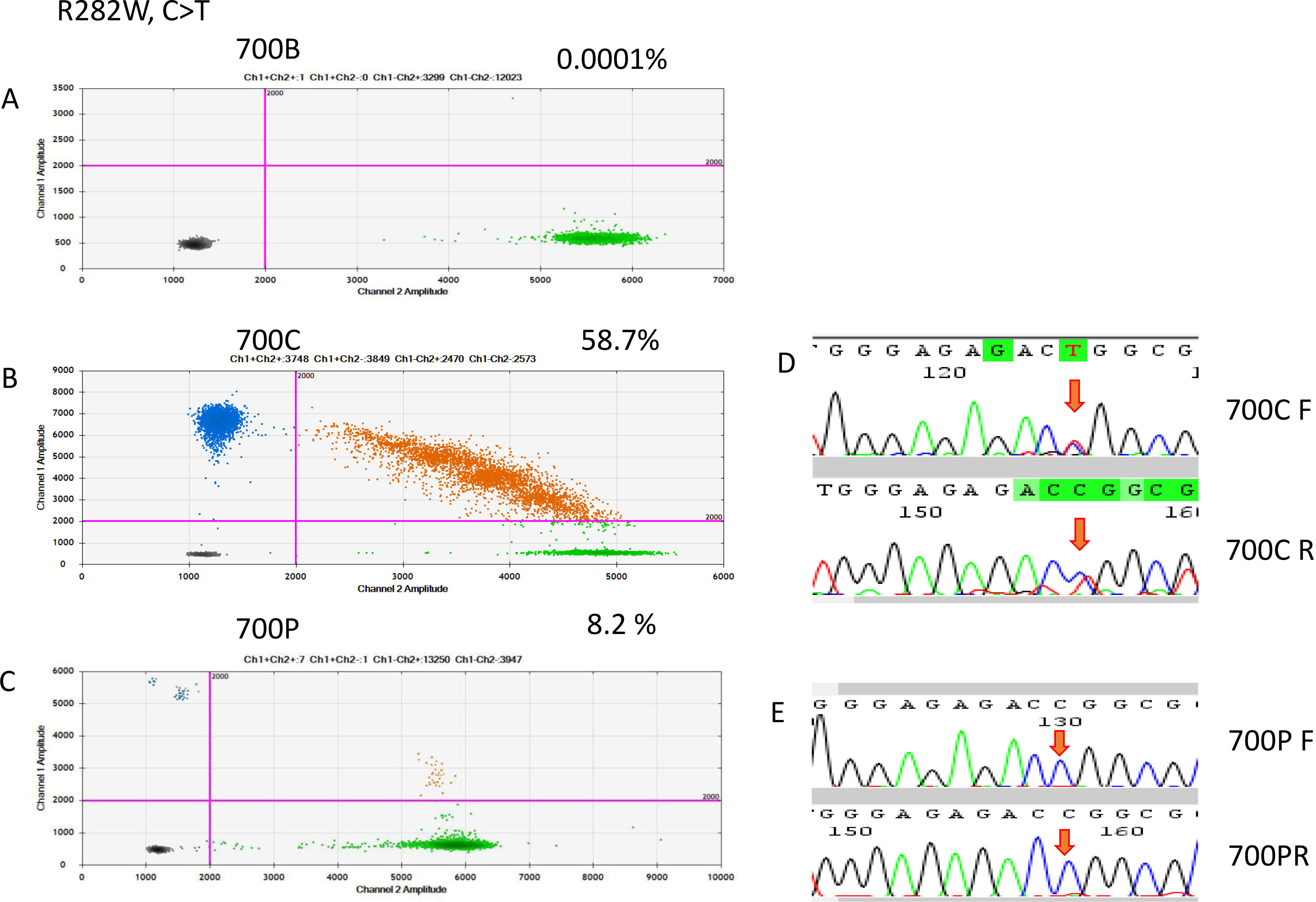
Figure 6.
ddRT-PCR results of patient 1044. The two-dimensional plots show the amount of MT-G245S droplets in the analysis. (A) Blood sample, (B) FFPE sample, and (C) cfDNA sample; (D) Sanger sequencing indicated a G-to-A point mutation in the FFPE sample, but it was not detectable in the cfDNA sample.
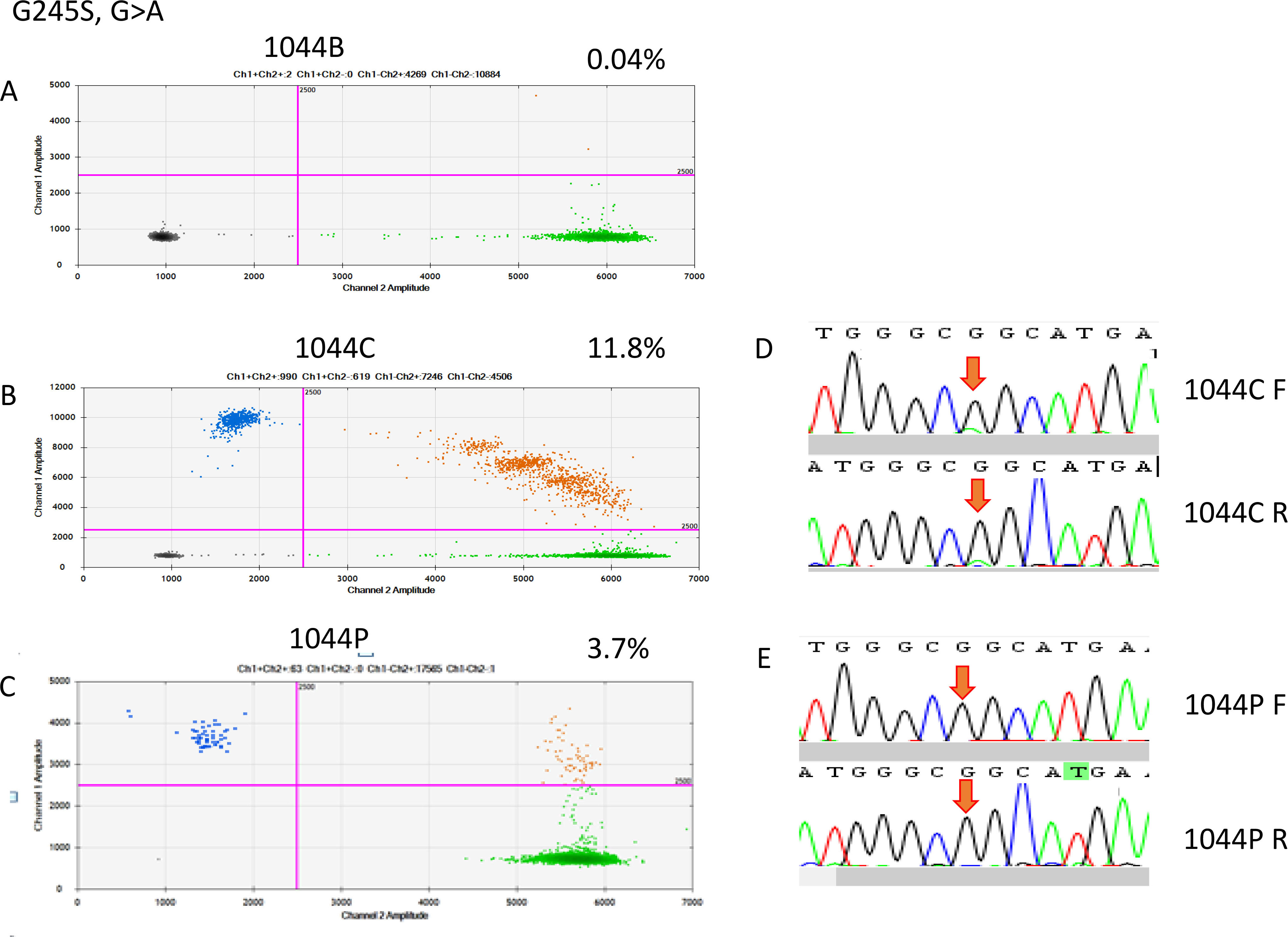
On the basis of the cancerous tissue, matched cfDNA samples were tested for the five TP53 target hotspot mutations by using ddPCR (Figs 4–6). In the circulating DNA however, identifying copies of mutant DNA with confidence was not possible in all analyzed cases (Fig. 3). A few samples with MT-TP53 had a limited number of droplets and fractional mutations below the minimum value which was predicted by the Poisson distribution analysis. In WT-TP53 background templates, we found mutations yielding FAs as low as 0.01%. In OSCC, cfDNA shedding was relatively low, and the allele frequencies of the mutations were low, too. Not in all patients where MT-TP53 was found in cancerous tissue had detectable amounts of cfDNA. Of these 23 patients, 13 (56.5%) had one of the target TP53 mutations. Six, three, two and one patients had R175H, R282W, G245S, and Y220C mutations, respectively. We could not confidently identify copies of mutant TP53-DNA with the R273C mutation in the circulating DNA (Table 5 and Fig. 3). In the comparison with clinicopathological parameters, cfDNA with TP53 hotspot target mutations was related to lymph node metastasis (Table 4). These targeted mutations were validated through Sanger sequencing in all cancerous tissue samples but could not be detected in cfDNA (Figs 4–6).
Table 5
MT-TP53 in cancerous tissue and cfDNA of patients with OSCC
| Mutation point | Cancerous tissue | cfDNA | Blood DNA | Correlation rate |
|---|---|---|---|---|
| R175H | 9 | 6 | 0 | 77.7% |
| R282W | 6 | 3 | 0 | 50.0% |
| G245S | 4 | 2 | 0 | 50.0% |
| Y220C | 2 | 1 | 0 | 50.0% |
| R273C | 2 | 0 | 0 | 0 |
| Total | 23 | 13 | 0 | 56.5% |
4.Discussion
Liquid biopsies may be used for the analysis of ctDNA, as recent research has shown. There is a wide range of potential applications, like early diagnosis, personalized treatment, and the prediction of disease progression [35]. Early cancer diagnosis and disease surveillance in precision medicine request to research the role that ctDNA is playing in OSCC. ctDNA is present in early cancers [36]. However, tumor fractions which are shedding detectable amounts of ctDNA are not yet well studied in respect to tumor type and stage [37]. In different malignancies, ctDNA has been widely reported, but it has been studied not much yet in OSCC. Tumor specific TP53 mutations in low levels of ctDNA from HNSCC have been reported by Ginkel et al. [38, 39]. However, only six cases of HNSCC were tested using ddPCR [39]. This study is, to the best of our knowledge, the first study that evaluates the presence of ctDNA in patients with OSCC through the detection of multiple target TP53 point mutations by ddPCR, requiring the use of very sensitive detection methods. Our data reveals that cancerous tissue and cfDNA are not associated. Only 56.5% of cases had matched target TP53 mutations between ctDNA and tumor DNA. However, the fraction of tumors that are shedding ctDNA at detectable levels, based on tumor type and stage, may be the reason for the discrepancy in MT-TP53 levels between tissue DNA and cfDNA [37]. In gastrointestinal stromal tumors, ctDNA shedding has been found to be relatively low, and also the allele frequencies of the mutations were low [40]. Only in 1 of the 13 cfDNA-matched samples ctDNA was detectable, the allele fraction was 12.3%. In a ctDNA screening experiment in hepatoma, negative results were observed in about 50% of the patients [41]. In our data, 56.5% of OSCC samples were found to have TP53 target mutations that are typical for cancerous tissues. This result might have been caused by the cancer type and by the tumor microenvironment, which are different in every patient, as well as effects of the immune system which are affecting the evolution and the lethality of a tumor. In contrast to infectious diseases, which are characterized by well-defined causative agents of highly conserved biology, cancer presents as a heterogeneous collection of many diseases, each of which is adding heterogeneity [37].
In particular, the low abundance of ctDNA and CTCs is limiting the usefulness of liquid biopsies in the early stages of cancer, and drawing large blood volumes in order to obtain more genetic material for the analysis is not always possible for clinical reasons. In cancer patients, the plasma concentration of cfDNA is higher than in healthy subjects, and it is increasing with the progression of the cancer. In healthy individuals, its blood concentration is approximately 13 ng/mL; in patients with metastatic cancer, up to 180 ng/mL have been reported [22, 42]. Most cfDNA consists of small linear fragments with a length of a few hundred base pairs. Its concentration is varying significantly between patients. In a previous study, the mean concentration of cfDNA in OSCC was found to be 53.1 ng/mL, in the control group it was less than half (24.0 ng/mL) [33]. For the efficient, non-selective amplification of small linear DNA fragments, T oligo-primed PCR was chosen. In accordance with modern molecular cloning strategies for NGS, which normally add an “A” to the 3
Although the sensitivity of ddPCR is high, one shortcoming is the limited number of mutations that may be detected in a single DNA sample of low abundance. The multiplexing of ddPCR assays may allow the simultaneous screening of several mutations [49]. This approach recently proved efficient for the genotyping of KRAS mutations in non-small-cell lung cancer [50]. and may further increase the usefulness of screening TP53 for mutations, because the mutations of interest are spanning the entire TP53 gene [51]. Therefore, hotspot mutations are a preferred target for the screening cfDNA by ddPCR. TP53 is the most commonly mutated gene in OSCC as reported by the Cancer Genome Atlas 2015 [12]. Most TP53 mutations are found in the DNA binding domain, they effectively block TP53 from binding to the transcription factor responsive elements and inhibit the transactivation of the target genes downstream of the binding site. R175H, R282W, G245S, Y220C, and R273C are the five most frequently mutated amino acids of p53 in HNSCC. They all are located in its DBD [12]. In this study, it was not clear whether our ddPCR strategy achieved comparable sensitivity in detecting ctDNA in OSCC patients. In addition, we were only able to select five mutations for the screening of cfDNA, and the results were negative in about half of the patients. Multiplex ddPCR therefore might be a feasible solution.
Regarding the association between TP53 mutations and metastasis in lymph nodes, the findings in this study have been inconsistent [8, 52, 53, 54, 55]. In HPV, DNA-negative HNSCC with TP53 mutations that were disrupting the protein function was independently associated with metastasis in lymph nodes [56]. In our study, target TP53 mutations were located on the DNA-binding site. Tumors with TP53 mutations at the surface of TP53 that is establishing the contact with the DNA (L2, L3
5.Conclusion
TP53 target somatic mutations can be detected with ddPCR in OSCC. Low levels of cfDNA and different cancer cell types may lead to diverse results between cancerous tissue and cfDNA analyses. Future research on cfDNA may quantify diagnostic biomarkers in the surveillance of these patients. The clinical significance of analyzing mutations in cfDNA samples as a diagnostic and predictive biomarker in patients with OSCC needs to be evaluated further. More studies should be performed with the goal to improve the sensitivity of the assays by targeting multiple mutations and determining the extraction of cfDNA from larger sample sizes and evaluating the usefulness of repeated, sequential, blood sampling.
Author contributions
Conception: CJ Liu.
Lab work: HW Cheng.
Interpretation or analysis of data: LH Lin, CJ Liu.
Preparation of the manuscript: LH Lin, CJ Liu.
Revision for important intellectual content: CJ Liu.
Supervision: CJ Liu.
All authors have approved the final version of the manuscript and agree to be accountable for it.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/CBM-210275.
Acknowledgments
This study was supported by grants from MacKay Memorial Hospital (MMH-E-105-12 and MMH-E-108-12) and the Ministry of Science and Technology, Taiwan (MOST 108-2314-B-195 -002 -MY2 and MOST 105-2314-B-195-005-MY3).
Conflict of interest
The authors declare no potential conflicts of interest.
References
[1] | J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D.M. Parkin, D. Forman and F. Bray, Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012, Int J Cancer 136: ((2015) ), E359–E386. |
[2] | S. Gupta, V.S. Kushwaha, S. Verma, H. Khan, M.L. Bhatt, N. Husain, M.P. Negi, V.V. Bhosale and A. Ghatak, Understanding molecular markers in recurrent oral squamous cell carcinoma treated with chemoradiation, Heliyon 2: ((2016) ), e00206. |
[3] | S. Gupta, W. Kong, Y. Peng, Q. Miao and W.J. Mackillop, Temporal trends in the incidence and survival of cancers of the upper aerodigestive tract in Ontario and the United States, Int J Cancer 125: ((2009) ), 2159–2165. |
[4] | M.o.H.a.W. Health Promotion Administration, Taiwan, Annual report on the causes of death statistics 2018, Available from: (accesse 21 Jun 2019), https://www.mohw.gov.tw/dl-54578-bd0c9197-80ae-4cae-b015-0cb3a055a069.html, (2019) . |
[5] | B.J. Braakhuis, M.P. Tabor, C.R. Leemans, I. van der Waal, G.B. Snow and R.H. Brakenhoff, Second primary tumors and field cancerization in oral and oropharyngeal cancer: Molecular techniques provide new insights and definitions, Head Neck 24: ((2002) ), 198–206. |
[6] | H. Schmidt, A. Kulasinghe, L. Kenny and C. Punyadeera, The development of a liquid biopsy for head and neck cancers, Oral Oncol 61: ((2016) ), 8–11. |
[7] | A. Lindemann, H. Takahashi, A.A. Patel, A.A. Osman and J.N. Myers, Targeting the DNA damage response in OSCC with TP53 mutations, J Dent Res 97: ((2018) ), 635–644. |
[8] | M.L. Poeta, J. Manola, M.A. Goldwasser, A. Forastiere, N. Benoit, J.A. Califano, J.A. Ridge, J. Goodwin, D. Kenady, J. Saunders, W. Westra, D. Sidransky and W.M. Koch, TP53 mutations and survival in squamous-cell carcinoma of the head and neck, N Engl J Med 357: ((2007) ), 2552–2561. |
[9] | E.H. Baugh, H. Ke, A.J. Levine, R.A. Bonneau and C.S. Chan, Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 25: ((2018) ), 154–160. |
[10] | N. Agrawal, M.J. Frederick, C.R. Pickering, C. Bettegowda, K. Chang, R.J. Li, C. Fakhry, T.X. Xie, J. Zhang, J. Wang, N. Zhang, A.K. El-Naggar, S.A. Jasser, J.N. Weinstein, L. Trevino, J.A. Drummond, D.M. Muzny, Y. Wu, L.D. Wood, R.H. Hruban, W.H. Westra, W.M. Koch, J.A. Califano, R.A. Gibbs, D. Sidransky, B. Vogelstein, V.E. Velculescu, N. Papadopoulos, D.A. Wheeler, K.W. Kinzler and J.N. Myers, Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1, Science 333: ((2011) ), 1154–1157. |
[11] | C.R. Pickering, J. Zhang, S.Y. Yoo, L. Bengtsson, S. Moorthy, D.M. Neskey, M. Zhao, M.V. Ortega Alves, K. Chang, J. Drummond, E. Cortez, T.X. Xie, D. Zhang, W. Chung, J.P. Issa, P.A. Zweidler-McKay, X. Wu, A.K. El-Naggar, J.N. Weinstein, J. Wang, D.M. Muzny, R.A. Gibbs, D.A. Wheeler, J.N. Myers and M.J. Frederick, Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers, Cancer Discov 3: ((2013) ), 770–781. |
[12] | N. Cancer Genome Atlas, Comprehensive genomic characterization of head and neck squamous cell carcinomas, Nature 517: ((2015) ), 576–582. |
[13] | P. Hainaut and G.P. Pfeifer, Somatic TP53 mutations in the era of genome sequencing, Cold Spring Harb Perspect Med 6: ((2016) ). |
[14] | R. Brosh and V. Rotter, When mutants gain new powers: News from the mutant p53 field, Nat Rev Cancer 9: ((2009) ), 701–713. |
[15] | P.A. Muller and K.H. Vousden, p53 mutations in cancer, Nat Cell Biol 15: ((2013) ), 2–8. |
[16] | T.X. Xie, G. Zhou, M. Zhao, D. Sano, S.A. Jasser, R.G. Brennan and J.N. Myers, Serine substitution of proline at codon 151 of TP53 confers gain of function activity leading to anoikis resistance and tumor progression of head and neck cancer cells, Laryngoscope 123: ((2013) ), 1416–1423. |
[17] | L. Bouaoun, D. Sonkin, M. Ardin, M. Hollstein, G. Byrnes, J. Zavadil and M. Olivier, TP53 Variations in human cancers: New lessons from the IARC TP53 database and genomics data, Hum Mutat 37: ((2016) ), 865–876. |
[18] | M. Kalia, Biomarkers for personalized oncology: Recent advances and future challenges, Metabolism 64: ((2015) ), S16–S21. |
[19] | E. Kidess and S.S. Jeffrey, Circulating tumor cells versus tumor-derived cell-free DNA: Rivals or partners in cancer care in the era of single-cell analysis? Genome Med 5: ((2013) ), 70. |
[20] | P.T. Spellman and J.W. Gray, Detecting cancer by monitoring circulating tumor DNA, Nat Med 20: ((2014) ), 474–475. |
[21] | P. Ulivi and R. Silvestrini, Role of quantitative and qualitative characteristics of free circulating DNA in the management of patients with non-small cell lung cancer, Cell Oncol (Dordr) 36: ((2013) ), 439–448. |
[22] | S. Jahr, H. Hentze, S. Englisch, D. Hardt, F.O. Fackelmayer, R.D. Hesch and R. Knippers, DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells, Cancer Res 61: ((2001) ), 1659–1665. |
[23] | L.A. Diaz and A. Bardelli, Liquid biopsies: Genotyping circulating tumor DNA, J Clin Oncol 32: ((2014) ), 579–586. |
[24] | J. Nemunaitis, G. Clayman, S.S. Agarwala, W. Hrushesky, J.R. Wells, C. Moore, J. Hamm, G. Yoo, J. Baselga, B.A. Murphy, K.A. Menander, L.L. Licato, S. Chada, R.D. Gibbons, M. Olivier, P. Hainaut, J.A. Roth, R.E. Sobol and W.J. Goodwin, Biomarkers predict p53 gene therapy efficacy in recurrent squamous cell carcinoma of the head and neck, Clin Cancer Res 15: ((2009) ), 7719–7725. |
[25] | V.W. Lui, M.L. Hedberg, H. Li, B.S. Vangara, K. Pendleton, Y. Zeng, Y. Lu, Q. Zhang, Y. Du, B.R. Gilbert, M. Freilino, S. Sauerwein, N.D. Peyser, D. Xiao, B. Diergaarde, L. Wang, S. Chiosea, R. Seethala, J.T. Johnson, S. Kim, U. Duvvuri, R.L. Ferris, M. Romkes, T. Nukui, P. Kwok-Shing Ng, L.A. Garraway, P.S. Hammerman, G.B. Mills and J.R. Grandis, Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers, Cancer Discov 3: ((2013) ), 761–769. |
[26] | C. Ndiaye, M. Mena, L. Alemany, M. Arbyn, X. Castellsague, L. Laporte, F.X. Bosch, S. de Sanjose and H. Trottier, HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis, Lancet Oncol 15: ((2014) ), 1319–1331. |
[27] | K. Koole, D. Brunen, P.M. van Kempen, R. Noorlag, R. de Bree, C. Lieftink, R.J. van Es, R. Bernards and S.M. Willems, FGFR1 is a potential prognostic biomarker and therapeutic target in head and neck squamous cell carcinoma, Clin Cancer Res 22: ((2016) ), 3884–3893. |
[28] | K.W. Chang, S. Sarraj, S.C. Lin, P.I. Tsai and D. Solt, P53 expression, p53 and Ha-ras mutation and telomerase activation during nitrosamine-mediated hamster pouch carcinogenesis, Carcinogenesis 21: ((2000) ), 1441–1451. |
[29] | S.E. Norton, J.M. Lechner, T. Williams and M.R. Fernando, A stabilizing reagent prevents cell-free DNA contamination by cellular DNA in plasma during blood sample storage and shipping as determined by digital PCR, Clin Biochem 46: ((2013) ), 1561–1565. |
[30] | C.M. Hindson, J.R. Chevillet, H.A. Briggs, E.N. Gallichotte, I.K. Ruf, B.J. Hindson, R.L. Vessella and M. Tewari, Absolute quantification by droplet digital PCR versus analog real-time PCR, Nat Methods 10: ((2013) ), 1003–1005. |
[31] | C.J. Liu, M.M. Tsai, P.S. Hung, S.Y. Kao, T.Y. Liu, K.J. Wu, S.H. Chiou, S.C. Lin and K.W. Chang, miR-31 ablates expression of the HIF regulatory factor FIH to activate the HIF pathway in head and neck carcinoma, Cancer Res 70: ((2010) ), 1635–1644. |
[32] | A.M. Newman, S.V. Bratman, J. To, J.F. Wynne, N.C. Eclov, L.A. Modlin, C.L. Liu, J.W. Neal, H.A. Wakelee, R.E. Merritt, J.B. Shrager, B.W. Loo, A.A. Alizadeh and M. Diehn, An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage, Nat Med 20: ((2014) ), 548–554. |
[33] | L.H. Lin, K.W. Chang, S.Y. Kao, H.W. Cheng and C.J. Liu, Increased plasma circulating cell-free DNA could be a potential marker for oral cancer, Int J Mol Sci 19: ((2018) ). |
[34] | J.F. Huggett, C.A. Foy, V. Benes, K. Emslie, J.A. Garson, R. Haynes, J. Hellemans, M. Kubista, R.D. Mueller, T. Nolan, M.W. Pfaffl, G.L. Shipley, J. Vandesompele, C.T. Wittwer and S.A. Bustin, The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments, Clin Chem 59: ((2013) ), 892–902. |
[35] | G. Rossi and M. Ignatiadis, Promises and pitfalls of using liquid biopsy for precision medicine, Cancer Res 79: ((2019) ), 2798–2804. |
[36] | C. Bettegowda, M. Sausen, R.J. Leary, I. Kinde, Y. Wang, N. Agrawal, B.R. Bartlett, H. Wang, B. Luber, R.M. Alani, E.S. Antonarakis, N.S. Azad, A. Bardelli, H. Brem, J.L. Cameron, C.C. Lee, L.A. Fecher, G.L. Gallia, P. Gibbs, D. Le, R.L. Giuntoli, M. Goggins, M.D. Hogarty, M. Holdhoff, S.M. Hong, Y. Jiao, H.H. Juhl, J.J. Kim, G. Siravegna, D.A. Laheru, C. Lauricella, M. Lim, E.J. Lipson, S.K. Marie, G.J. Netto, K.S. Oliner, A. Olivi, L. Olsson, G.J. Riggins, A. Sartore-Bianchi, K. Schmidt, M. Shih l, S.M. Oba-Shinjo, S. Siena, D. Theodorescu, J. Tie, T.T. Harkins, S. Veronese, T.L. Wang, J.D. Weingart, C.L. Wolfgang, L.D. Wood, D. Xing, R.H. Hruban, J. Wu, P.J. Allen, C.M. Schmidt, M.A. Choti, V.E. Velculescu, K.W. Kinzler, B. Vogelstein, N. Papadopoulos and L.A. Diaz, Detection of circulating tumor DNA in early- and late-stage human malignancies, Sci Transl Med 6: ((2014) ), 224ra24. |
[37] | A.M. Aravanis, M. Lee and R.D. Klausner, Next-generation sequencing of circulating tumor dna for early cancer detection, Cell 168: ((2017) ), 571–574. |
[38] | J.H. van Ginkel, W.W. de Leng, R. de Bree, R.J. van Es and S.M. Willems, Targeted sequencing reveals TP53 as a potential diagnostic biomarker in the post-treatment surveillance of head and neck cancer, Oncotarget 7: ((2016) ), 61575–61586. |
[39] | J.H. van Ginkel, M.M.H. Huibers, R.J.J. van Es, R. de Bree and S.M. Willems, Droplet digital PCR for detection and quantification of circulating tumor DNA in plasma of head and neck cancer patients, BMC Cancer 17: ((2017) ), 428. |
[40] | C. Serrano, A. Vivancos, A. Lopez-Pousa, J. Matito, F.M. Mancuso, C. Valverde, S. Quiroga, S. Landolfi, S. Castro, C. Dopazo, A. Sebio, A.C. Virgili, M.M. Menso, J. Martin-Broto, M. Sanso, A. Garcia-Valverde, J. Rosell, J.A. Fletcher, S. George, J. Carles and J. Arribas, Clinical value of next generation sequencing of plasma cell-free DNA in gastrointestinal stromal tumors, BMC Cancer 20: ((2020) ), 99. |
[41] | A. Huang, X. Zhang, S.L. Zhou, Y. Cao, X.W. Huang, J. Fan, X.R. Yang and J. Zhou, Detecting circulating tumor DNA in hepatocellular carcinoma patients using droplet digital PCR is feasible and reflects intratumoral heterogeneity, J Cancer 7: ((2016) ), 1907–1914. |
[42] | S.J. Dawson, D.W. Tsui, M. Murtaza, H. Biggs, O.M. Rueda, S.F. Chin, M.J. Dunning, D. Gale, T. Forshew, B. Mahler-Araujo, S. Rajan, S. Humphray, J. Becq, D. Halsall, M. Wallis, D. Bentley, C. Caldas and N. Rosenfeld, Analysis of circulating tumor DNA to monitor metastatic breast cancer, N Engl J Med 368: ((2013) ), 1199–1209. |
[43] | Y.S. Nai, T.H. Chen, Y.F. Huang, M.K. Midha, H.C. Shiau, C.Y. Shen, C.J. Chen, A.L. Yu and K.P. Chiu, T oligo-primed polymerase chain reaction (TOP-PCR): A robust method for the amplification of minute DNA fragments in body fluids, Sci Rep 7: ((2017) ), 40767. |
[44] | J.C.M. Wan, C. Massie, J. Garcia-Corbacho, F. Mouliere, J.D. Brenton, C. Caldas, S. Pacey, R. Baird and N. Rosenfeld, Liquid biopsies come of age: Towards implementation of circulating tumour DNA, Nat Rev Cancer 17: ((2017) ), 223–238. |
[45] | C. Abbosh, N.J. Birkbak, G.A. Wilson, M. Jamal-Hanjani, T. Constantin, R. Salari, J. Le Quesne, D.A. Moore, S. Veeriah, R. Rosenthal, T. Marafioti, E. Kirkizlar, T.B.K. Watkins, N. McGranahan, S. Ward, L. Martinson, J. Riley, F. Fraioli, M. Al Bakir, E. Gronroos, F. Zambrana, R. Endozo, W.L. Bi, F.M. Fennessy, N. Sponer, D. Johnson, J. Laycock, S. Shafi, J. Czyzewska-Khan, A. Rowan, T. Chambers, N. Matthews, S. Turajlic, C. Hiley, S.M. Lee, M.D. Forster, T. Ahmad, M. Falzon, E. Borg, D. Lawrence, M. Hayward, S. Kolvekar, N. Panagiotopoulos, S.M. Janes, R. Thakrar, A. Ahmed, F. Blackhall, Y. Summers, D. Hafez, A. Naik, A. Ganguly, S. Kareht, R. Shah, L. Joseph, A. Marie Quinn, P.A. Crosbie, B. Naidu, G. Middleton, G. Langman, S. Trotter, M. Nicolson, H. Remmen, K. Kerr, M. Chetty, L. Gomersall, D.A. Fennell, A. Nakas, S. Rathinam, G. Anand, S. Khan, P. Russell, V. Ezhil, B. Ismail, M. Irvin-Sellers, V. Prakash, J.F. Lester, M. Kornaszewska, R. Attanoos, H. Adams, H. Davies, D. Oukrif, A.U. Akarca, J.A. Hartley, H.L. Lowe, S. Lock, N. Iles, H. Bell, Y. Ngai, G. Elgar, Z. Szallasi, R.F. Schwarz, J. Herrero, A. Stewart, S.A. Quezada, K.S. Peggs, P. Van Loo, C. Dive, C.J. Lin, M. Rabinowitz, H. Aerts, et al., Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution, Nature 545: ((2017) ), 446–451. |
[46] | B.J. Hindson, K.D. Ness, D.A. Masquelier, P. Belgrader, N.J. Heredia, A.J. Makarewicz, I.J. Bright, M.Y. Lucero, A.L. Hiddessen, T.C. Legler, T.K. Kitano, M.R. Hodel, J.F. Petersen, P.W. Wyatt, E.R. Steenblock, P.H. Shah, L.J. Bousse, C.B. Troup, J.C. Mellen, D.K. Wittmann, N.G. Erndt, T.H. Cauley, R.T. Koehler, A.P. So, S. Dube, K.A. Rose, L. Montesclaros, S. Wang, D.P. Stumbo, S.P. Hodges, S. Romine, F.P. Milanovich, H.E. White, J.F. Regan, G.A. Karlin-Neumann, C.M. Hindson, S. Saxonov and B.W. Colston, High-throughput droplet digital PCR system for absolute quantitation of DNA copy number, Anal Chem 83: ((2011) ), 8604–8610. |
[47] | Y. Wang, L. Li, C. Douville, J.D. Cohen, T.T. Yen, I. Kinde, K. Sundfelt, S.K. Kjaer, R.H. Hruban, I.M. Shih, T.L. Wang, R.J. Kurman, S. Springer, J. Ptak, M. Popoli, J. Schaefer, N. Silliman, L. Dobbyn, E.J. Tanner, A. Angarita, M. Lycke, K. Jochumsen, B. Afsari, L. Danilova, D.A. Levine, K. Jardon, X. Zeng, J. Arseneau, L. Fu, L.A. Diaz, R. Karchin, C. Tomasetti, K.W. Kinzler, B. Vogelstein, A.N. Fader, L. Gilbert and N. Papadopoulos, Evaluation of liquid from the Papanicolaou test and other liquid biopsies for the detection of endometrial and ovarian cancers, Sci Transl Med 10: ((2018) ). |
[48] | N.S. Arildsen, L. Martin de la Fuente, A. Masback, S. Malander, O. Forslund, P. Kannisto and I. Hedenfalk, Detecting TP53 mutations in diagnostic and archival liquid-based Pap samples from ovarian cancer patients using an ultra-sensitive ddPCR method, Sci Rep 9: ((2019) ), 15506. |
[49] | D. Dobnik, D. Stebih, A. Blejec, D. Morisset and J. Zel, Multiplex quantification of four DNA targets in one reaction with Bio-Rad droplet digital PCR system for GMO detection, Sci Rep 6: ((2016) ), 35451. |
[50] | A. Pender, I. Garcia-Murillas, S. Rana, R.J. Cutts, G. Kelly, K. Fenwick, I. Kozarewa, D. Gonzalez de Castro, J. Bhosle, M. O’Brien, N.C. Turner, S. Popat and J. Downward, Efficient genotyping of KRAS mutant non-small cell lung cancer using a multiplexed droplet digital PCR approach, PLoS One 10: ((2015) ), e0139074. |
[51] | A.A. Ahmed, D. Etemadmoghadam, J. Temple, A.G. Lynch, M. Riad, R. Sharma, C. Stewart, S. Fereday, C. Caldas, A. Defazio, D. Bowtell and J.D. Brenton, Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary, J Pathol 221: ((2010) ), 49–56. |
[52] | M. Lindenbergh-van der Plas, R.H. Brakenhoff, D.J. Kuik, M. Buijze, E. Bloemena, P.J. Snijders, C.R. Leemans and B.J. Braakhuis, Prognostic significance of truncating TP53 mutations in head and neck squamous cell carcinoma, Clin Cancer Res 17: ((2011) ), 3733–3741. |
[53] | S.N. Zanaruddin, P.S. Yee, S.Y. Hor, Y.H. Kong, W.M. Ghani, W.M. Mustafa, R.B. Zain, S.S. Prime, Z.A. Rahman and S.C. Cheong, Common oncogenic mutations are infrequent in oral squamous cell carcinoma of Asian origin, PLoS One 8: ((2013) ), e80229. |
[54] | D. Sano, T.X. Xie, T.J. Ow, M. Zhao, C.R. Pickering, G. Zhou, V.C. Sandulache, D.A. Wheeler, R.A. Gibbs, C. Caulin and J.N. Myers, Disruptive TP53 mutation is associated with aggressive disease characteristics in an orthotopic murine model of oral tongue cancer, Clin Cancer Res 17: ((2011) ), 6658–6670. |
[55] | J.K. Peltonen, H.M. Helppi, P. Paakko, T. Turpeenniemi-Hujanen and K.H. Vahakangas, p53 in head and neck cancer: Functional consequences and environmental implications of TP53 mutations, Head Neck Oncol 2: ((2010) ), 36. |
[56] | G. Wichmann, M. Rosolowski, K. Krohn, M. Kreuz, A. Boehm, A. Reiche, U. Scharrer, D. Halama, J. Bertolini, U. Bauer, D. Holzinger, M. Pawlita, J. Hess, C. Engel, D. Hasenclever, M. Scholz, P. Ahnert, H. Kirsten, A. Hemprich, C. Wittekind, O. Herbarth, F. Horn, A. Dietz, M. Loeffler, H. Leipzig and G. Neck, The role of HPV RNA transcription, immune response-related gene expression and disruptive TP53 mutations in diagnostic and prognostic profiling of head and neck cancer, Int J Cancer 137: ((2015) ), 2846–2857. |
[57] | J.K. Peltonen, K.H. Vahakangas, H.M. Helppi, R. Bloigu, P. Paakko and T. Turpeenniemi-Hujanen, Specific TP53 mutations predict aggressive phenotype in head and neck squamous cell carcinoma: A retrospective archival study, Head Neck Oncol 3: ((2011) ), 20. |
[58] | M. Migliavacca, L. Ottini, V. Bazan, V. Agnese, S. Corsale, M. Macaluso, R. Lupi, G. Dardanoni, M.R. Valerio, G. Pantuso, G. Di Fede, R.M. Tomasino, N. Gebbia, R. Mariani-Costantini and A. Russo, TP53 in gastric cancer: Mutations in the l3 loop and LSH motif DNA-binding domains of TP53 predict poor outcome, J Cell Physiol 200: ((2004) ), 476–485. |
[59] | V. Bazan, V. Agnese, S. Corsale, V. Calo, M.R. Valerio, M.A. Latteri, S. Vieni, N. Grassi, G. Cicero, G. Dardanoni, R.M. Tomasino, G. Colucci, N. Gebbia, A. Russo and M. Gruppo Oncologico dell’Italia, Specific TP53 and/or Ki-ras mutations as independent predictors of clinical outcome in sporadic colorectal adenocarcinomas: Results of a 5-year Gruppo Oncologico dell’Italia Meridionale (GOIM) prospective study, Ann Oncol 16: Suppl 4 ((2005) ), iv 50–55. https://www.mohw.gov.tw/dl-54578-bd0c9197-80ae-4cae-b015-0cb3a055a069.html. |


