
In-silico prediction of anti-breast cancer activity of ginger (Zingiber officinale) using machine learning techniques
Abstract
INTRODUCTION:
Indonesian civilization extensively uses traditional medicine to cure illnesses and preserve health. The lack of knowledge on the security and efficacy of medicinal plants is still a significant concern. Although the precise chemicals responsible for this impact are unknown, ginger is a common medicinal plant in Southeast Asia that may have anticancer qualities.
METHOD:
Using data from Dudedocking, a machine-learning model was created to predict possible breast anticancer chemicals from ginger. The model was used to forecast substances that block KIT and MAPK2 proteins, essential elements in breast cancer.
RESULT:
Beta-carotene, 5-Hydroxy-74′-dimethoxyflavone, [12]-Shogaol, Isogingerenone B, curcumin, Trans-[10]-Shogaol, Gingerenone A, Dihydrocurcumin, and demethoxycurcumin were all superior to the reference ligand for MAPK2, according to molecular docking studies. Lycopene, [8]-Shogaol, [6]-Shogaol, and [1]-Paradol exhibited low toxicity and no Lipinski violations, but beta carotene had toxic predictions and Lipinski violations. It was anticipated that all three substances would have anticarcinogenic qualities.
CONCLUSION:
Overall, this study shows the value of machine learning in drug development and offers insightful information on possible anticancer chemicals from ginger.
1.Introduction
Human life has long been a concern for public health issues. Traditional medicine consumption is still common across several ASEAN nations and other countries, such as Japan, Korea, and China [1,2]. For generations, Indonesian society has relied on traditional herbal medicines to treat illness and maintain wellness. The generic name for this herbal remedy is jamu [3]. However, herbal medicine treatment still needs to be improved due to inadequate efficacy monitoring and a need for more information about the effectiveness of diverse medicinal plants [4].
Southeast Asian-born ginger is a traditional medicine frequently found in meals and beverages. Ginger is a traditional herbal antioxidant and anti-inflammatory medicine [5]. Ginger has anticancer effects and antioxidant and anti-inflammatory properties, as reported in several publications. These publications include those on gastrointestinal cancer [6], pancreatic cancer [7], and anti-cancer activity against human breast cancer cell lines when ginger extract is combined with turmeric and garlic [8]. Clinical research and animal models have shown that ginger and its contents effectively prevent and treat disease. However, the exact molecules that give ginger its anticancer properties remain unknown. One substance that may be anticancer from ginger is called valinoids [9]. Bioinformatic and pharmacoinformatic research is required to understand the ginger component that may serve as an anti-cancer.
The quantitative structure-activity relationship (QSAR) model of the chemicals dataset is currently being built using machine learning as a component of artificial intelligence to obtain crucial descriptors to predict a specific biological activity from unknown compounds [10]. To develop a model prediction, machine learning requires a dataset for training and testing [11]. Several protein inhibitor datasets have been gathered to produce drugs, such as Dudedocking (http://dude.docking.org/). This website offers information on chemicals that inhibit proteins and components that act as a decoy, which are used to create and test machine-learning models that can predict protein inhibitors [12].
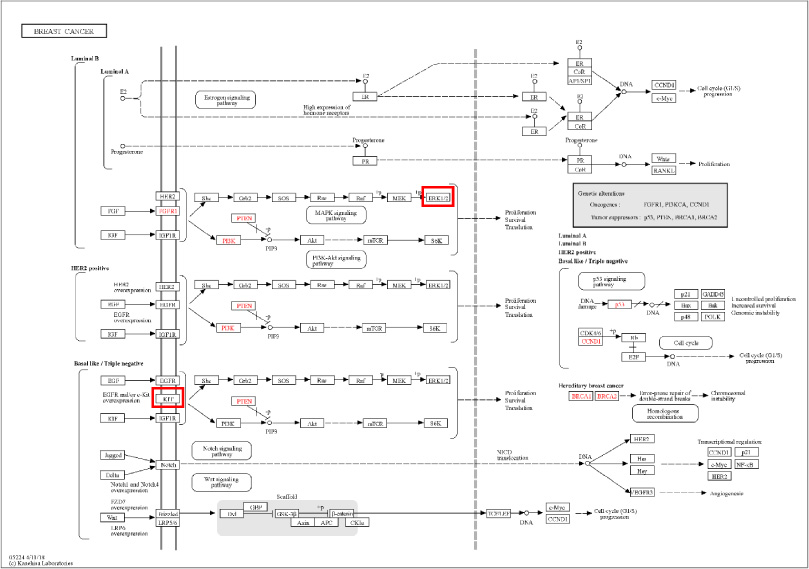
Cancer occurs when cells in a specific body area multiply and expand uncontrolled. Cancerous cells have the potential to penetrate and damage nearby healthy tissue, including organs. There are numerous pathways, and the cancer route includes several proteins. Over 30% of all human cancers include overactive MAPK1/2 (ERK1/2) proteins [13]. Mitogen-activated protein kinase (MAP kinase) cascades transmit and amplify signals relevant to cell growth and death. These signal transduction pathways allow us to assess the level of trafficking induced by various growth factors, steroid hormones, and G protein receptor-mediated ligands [13]. KIT protein is an additional crucial protein. KIT protein overexpression or mutations can accelerate the growth and spread of tumors in various human malignancies [14]. KIT signaling is linked to various physiological processes, including hematopoiesis, gastrointestinal motility, and pigmentation. It is an essential regulator of cell proliferation, survival, and migration [14]. This study’s objective is to identify possible breast anti-cancer chemicals in ginger, where both protein MAPK2 and KIT are mentioned as essential nodes in breast cancer in the KEGG pathway map (Fig. 1) utilizing machine learning approaches, molecular docking research, ADME analysis, and molecular pharmacophore analysis.
Fig. 1.
KIT and MAPK2 protein in breast cancer map pathway.
2.Material and methods
2.1.Data mining and fingerprint extraction
algorithm. A machine learning algorithm predicted protein KIT and MAPK2 inhibitors in ginger. Ginger compound data is gathered from the Knapsack database core system using the keyword Zingiber officinale [15]. Using the open-source RDkit in Python software base (https://www.rdkit.org/), smile structures from all compounds are extracted to create Klekota-Roth fingerprints. There are 4860 substructures in the Klekota-Roth fingerprint, each with a binary score of one or zero [16], through Dudedocking, a dataset of chemicals that inhibit KIT and MAPK2 proteins was gathered [12]. The website also provides a decoy compound that can be utilized as a non-active compound. Machine learning models would be created using active and inactive substances. Before developing a model, each substructure is obtained using the fingerprint extractor from RDKit.
2.2.Machine learning model development
Scikit-learn, a Python-based program, is used to create machine learning models [17]. Jupyter notebooks are used to write and implement the code [18]. The AUC/ROC score was used to select the most effective model. In this analysis, we use three algorithms that have been often referenced in other works. Random forest (RF), Support vector machine (SVM), and Logistic regression are three algorithms (RF). The three methods are compared, and the model with the highest Score is utilized to forecast the active chemicals in ginger.
2.3.Molecular docking from predicted active compound and interaction analysis
A machine learning-predicted active substance is subjected to molecular docking investigation. To determine the probable binding affinities of each component from ginger, we used the PLANTS 1.1 software [19]. The ligand’s minimal energy conformation in the protein’s binding region is determined using an artificial ant colony [20]. Two empirical scoring functions, PLANTSCHEMPLP and PLANTSPLP, were created for the docking method PLANTS (Protein-Ligand ANT System), which is based on ant colony optimization (ACO). The optimal parameter settings for the search algorithm have been found, and they can be used to Balance pose prediction accuracy with search speed [19]. PRODIGY was used to calculate binding affinity (ΔG) after molecular docking [21]. PRODIGY protein-ligand principles (https://wenmr.science.uu.nl/prodigy/lig) by appropriately modifying the small ligand prediction technique to use atomic interactions rather than residue contacts. PRODIGY-advantages LIGs include their simplicity, generality, and applicability to all protein-ligand complexes [22].
2.4.ADME (Absorption, Distribution, Metabolism, and Excretion) and toxicity analysis
The purpose of ADME analysis is to evaluate how much each chemical resembles a medicine expected to be active by machine learning. To ensure a drug’s pharmacokinetics, an examination called an ADME analysis (Absorption, Distribution, Metabolism, and Excretion) is performed. SwissADME is used for the ADME analysis (http://www.swissadme.ch/) [23]. Using Toxtree, each compound’s toxicity is examined [24]. The toxicological concern threshold is frequently called the toxicity level (TTC). TTC refers to determining an exposure threshold for all substances below which there is no appreciable danger to human health [24]. The Lipinski rule of five is a frequently used metric to measure drug-likeness, which includes (1). a maximum of five hydrogen bond donors (the sum of the bonds between hydrogen and oxygen and nitrogen), (2). ten or fewer (all nitrogen or oxygen atoms) hydrogen bond acceptors; (3). fewer than 500 daltons for the molecular weight (4). No more than 4.15 LogP [25].
Blood-brain barrier (BBB) and human intestinal absorption (HIA) analyses are frequently combined with toxicity analysis to predict the number of compounds that can be absorbed by the gastrointestinal tract (GI) [26]. Toxicity analysis indicates whether these compounds can pass through the blood-brain barrier.
2.5.Analysis of bioactivity prediction
The final technique we employed in this investigation was bioactivity prediction. The web-based PASS-SERVER allows for the prediction of bioactivity. This bioactivity prediction considers pharmacological effects, action mechanisms, toxic and adverse effects, interactions with metabolic transporters and enzymes, impact on gene expression, etc [27].
2.6.Tanimoto similarity for chemical structure and pharmacophore
Potential compounds already mentioned above are then converted to fingerprints. The fingerprint of each compound is used to calculate structure similarity structure with reference ligand control using RDKit in jupyter notebook [18]. The molecular docking result of each compound has to interact with several amino acids of the protein; the active residue that interacts with the ligand compound is also used to calculate the similarity interaction with the reference ligand as a control. This method tries to see if the ligand interacts similarly with the reference ligand using Pyplif-Hippos [28].
3.Results
3.1.Machine learning model development
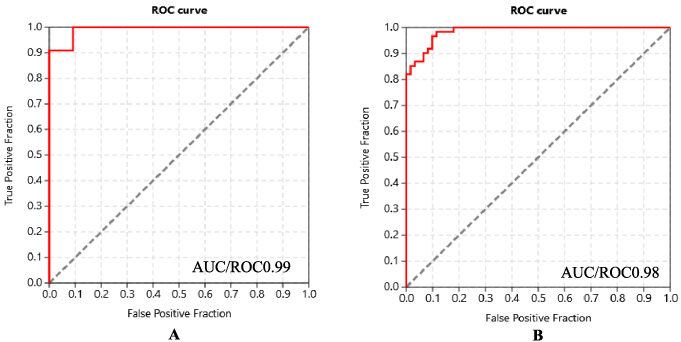
Table 1 displays the Score for the MAPK2 protein prediction model. Regarding the final result, the AUC/ROC score (0.997).
Table 1
The score of each of the three models for MAPK2 protein
| Model | Accuracy | Sensitivity | Specificity | AUC/ROC score |
| LR | 0.981 | 1.000 | 0.962 | 0.997 |
| SVM | 0.943 | 0.923 | 0.962 | 0.991 |
| RF | 0.905 | 0.961 | 0.962 | 0.981 |
It is also displayed by KIT protein, where logistic regression (LR) has the best Score regarding the AUC/ ROC score (0.984) (Table 2). The logistic regression (LR) model is then used to predict potential active compounds from ginger that possibly inhibit MAPK2 and KIT protein.
Table 2
The score of each of the three models for KIT protein
| Model | Accuracy | Sensitivity | Specificity | AUC/ROC score |
| LR | 0.954 | 0.909 | 1.000 | 0.984 |
| SVM | 0.954 | 0.909 | 1.000 | 0.983 |
| RF | 0.954 | 0.909 | 1.000 | 0.980 |
We discovered ten compounds that a machine learning model suggested could potentially be a KIT protein inhibitor (Table 3), and a total of 64 compounds are predicted as active chemicals because of the MAPK2 protein model (Table 4). Compounds predicted as inhibitors for KIT and MAPK2 protein are subjected to molecular docking to explore possible attachment in the binding pocket of KIT and MAPK2 protein.
3.2.Molecular docking from predicted active compound and interaction analysis
Software validation has been performed before using PLANTS 1.1 software for protein-ligand-docking. The PLANTS 1.1 software is used in a benchmarking analysis with data from Dudedocking [12]. Docking and binding affinity scores (ΔG) are calculated using active KIT and MAPK2 protein inhibitors and non-active (decoy) compounds. The docking and binding affinity scores (ΔG) are then projected to generate the AUC/ROC score for the PLANTS 1.1 software results using the predicted docking score and binding affinity of active and decoy compounds. This method was used to test whether the PLANTS 1.1 software could distinguish between active and decoy compounds based on the binding and affinity scores. The AUC/ROC score of retrospective analysis from PLANTS 1.1 software yields excellent results, with AUC ROC scores of 0.992 for KIT protein and 0.986 for MAPK2 protein (scale 0–1, the getting closer to 1 the better result) (Fig. 3).
Fig. 2.
AUC/ROC score for KIT protein (A) and AUC/ROC score for MAPK2 protein.
KIT (PDB: 1T46) and MAPK2 (PDB: 3M2W) were the proteins used in this study. Both proteins have a reference ligand that acts as an inhibitor. Imatinib (DrugBank code: DB00619) is an inhibitor of the KIT protein (PubChem code: 5291). The drug has already docked with the KIT protein (PDB: 1T46). While the reference ligand for the MAPK2 protein is 2′-(2-fluorophenyl)-1-methyl-6′,8′,9′,11′-tetrahydrospiro [azetidine-3,10′-pyrido [3′,4′:4,5] pyrrolo [2,3-f] isoquinolin], -7′(5′H) -one (PubChem code: 42646698). PLANTS 1.1 was used to perform 100 repetitions of re-docking reference ligand to validate the accuracy of the PLANTS 1.1 software (Fig. 3). The average docking score of a reference ligand to KIT protein is −142.904, while that of a MAPK2 reference ligand is −101.628.
Fig. 3.
PLANTS 1.1 docking Score between ref_ligand to KIT protein (A) and MAPK2 protein (B) for 100 repetitions using PLANTS 1.1.
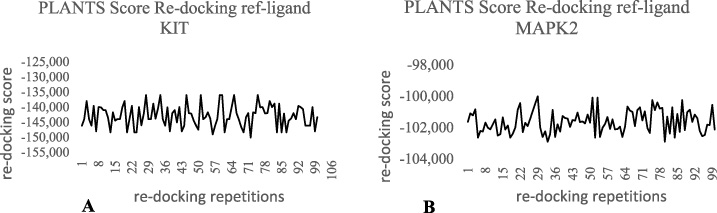
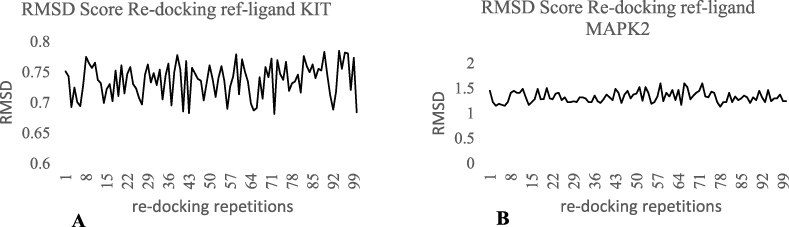
Figure 3 shows the binding affinity calculation result after 100 repetitions. The average Root mean square deviation (RMSD) of the reference ligand to KIT protein is 0.7365, while the RMSD of the reference ligand to MAPK2 protein is 1.3256. The RMSD after 100 repetitions of re-docking was stable. The figure shows that the RMSD of docking ligand to reference ligand after 100 repetitions to protein was stable below 2.0 Å [29].
Fig. 4.
Root means square deviation (RMSD) of re-docking between ref_ligand to KIT protein (A) and MAPK2 (B) protein for 100 repetitions using PLANTS 1.1.
Beta_caroten is the only compound predicted as active from a machine learning model for KIT protein with a lower binding affinity score (ΔG) than the reference ligand (ref_ligand). The binding energies score (ΔG) for the reference ligand is −10.4 Kcal/mol, while the Score for beta-carotene is −14.3 Kcal/mol. This finding indicates that beta-carotene may bind better to the KIT protein (Table 3). On the other side, 14 of the 64 compounds predicted as active by the machine learning model for the MAPK2 protein have lower binding energies than the reference ligand. Lycopene, 5-Hydroxy-7,4′-dimethoxyflavone, [12]-Shogaol, Isogingerenone B, Curcumin, trans-[10]-Shogaol, Gingerenone A, Dihydrocurcumin, Demethoxycurcumin, [8]-Shogaol, [6]-Shogaol, [1]-Paradol, Bisdemethoxycurcumin, and alpha-Zingiberene are among the 14 compounds (Table 4).
After calculating compounds classified as active and inactive KIT and MAPK2 protein inhibitors, we found that a machine-learning model revealed promising results. Compounds grouped as an active protein to inhibit KIT have a binding affinity (ΔG) ranging from −7.1 (Isovanilin) to −14.3 (beta-carotene). No compounds with a lower binding affinity score than −7.0 Kcal/mol are classified as active inhibitors of the KIT protein. A similar accurate prediction result was also shown in the MAPK2 protein. Binding affinity (ΔG) ranges from −7.4 Kcal/mol (3-Octen-2-one) to −10.6 Kcal/mol (Lycopene and 5-Hydroxy-7,4′-dimethoxyflavone) (Table 4). The compounds 5-Hydroxy-74′-dimethoxyflavone, lycopene [12] -Shogaol, Isogingerenone B, curcumin, Trans-[10]-Shogaol, Gingerenone A, Dihydrocurcumin, and Demethoxycurcumin were found to be more effective than the reference ligand, despite having a lower binding affinity. While [8]-Shogaol, [6]-Shogaol, [1]-Paradol, Bisdemethoxycurcumin, and alpha-Zingiberene appear to be similar but better than the reference ligand.
Table 3
Docking and binding affinity score (ΔG) for the predicted compound as an inhibitor for KIT protein
| Compound | Docking score | Binding affinity (ΔG) Kcal/mol |
| ref_ligand | −148 | −10.4 |
| Zingiberoside A | −75.4 | −9.1 |
| Vanillin | −65 | −7.2 |
| Vanilic acid | −65.4 | −7.2 |
| Trans 2 Octenal | −66.6 | −7.5 |
| Isovanilin | −66 | −7.1 |
| beta caroten | −121 | −14.3 |
| alpha,4-Dimethylstyrene | −68 | −8.2 |
| 4-Hydroxybenzaldehyde | −62 | −7.3 |
| 3-Octen-2-one | −66.6 | −7.4 |
Table 4
Docking and binding affinity score (ΔG) for the predicted compound as an inhibitor for MAPK2 protein
| Compounds | Docking score | Binding affinity (ΔG) Kcal/mol |
| ref_ligand | −101 | −9.2 |
| Lycopene | 182 | −10.6 |
| 5-Hydroxy-7,4′-dimethoxyflavone | −83 | −10.6 |
| [12]-Shogaol | −102 | −10.2 |
| Isogingerenone B | −88 | −10.2 |
| Curcumin | −90.6 | −10 |
| trans-[10]-Shogaol | −96 | −9.8 |
| Gingerenone A | −93.4 | −9.8 |
| Dihydrocurcumin | −91.2 | −9.7 |
| Demethoxycurcumin | −93.6 | −9.5 |
| [8]-Shogaol | −93 | −9.3 |
| [6]-Shogaol | −92 | −9.3 |
| [1]-Paradol | −90.4 | −9.3 |
| Bisdemethoxycurcumin | −93 | −9.3 |
| alpha-Zingiberene | −76 | −9.3 |
| 5,7-Dimethoxyflavone | −76 | −9.1 |
| Demethoxy[6]-shogaol | −93 | −9.1 |
| Zerumbone | −72 | −9 |
| alpha-calacorene | −76.4 | −9 |
| alpha-Caryophyllene | −72 | −9 |
| Dibutyl phthalate | −87 | −9 |
| Xanthorrhizol | −84.5 | −8.9 |
| [4]-Shogaol | −83.7 | −8.9 |
| Delphinidin | −92 | −8.9 |
| cis-Nuciferol | −83 | −8.9 |
| Diisobutyl phthalate | −84 | −8.9 |
| Farnesal | −85 | −8.9 |
| (E,E)-Farnesal | −86 | −8.8 |
| (R)-(-)-alpha-Curcumene | −81.4 | −8.8 |
| 3,7-Dimethyl-2,6-octadiene | −79 | −8.8 |
| alpha-Curcumene | −81 | −8.8 |
| alpha-Farnesene | −82 | −8.8 |
| cis-beta-Farnesene | −82 | −8.8 |
| Quercetin | −81 | −8.8 |
| Farnesol | −87 | −8.8 |
| Genistein | −82 | −8.8 |
| Homofarnesyl cyanide | −85 | −8.7 |
| Galangin | −78 | −8.7 |
| (E)-Nuciferol | −86 | −8.6 |
| (S)-(+)-Curcumene | −74 | −8.4 |
| Methyl [6]-Shogaol | −80 | −8.4 |
| Zingerone methyl ether | −72 | −8.1 |
| Zingerol | −73 | −8.1 |
| 4-(4-Hydroxy-3- methoxyphenyl)-2-butanone | −73 | −8.1 |
| alpha-Phellandrene | −63 | −7.9 |
| Myristicin | −69 | −7.9 |
| Dihydroferulic acid | −74 | −7.9 |
| Nerol | −71 | −7.8 |
Table 4 (Continued).
| Compounds | Docking score | Binding affinity (ΔG) Kcal/mol |
| 8-Hydroxygeraniol | −74 | −7.8 |
| cis-Citral | −70 | −7.8 |
| Geraniol | −71 | −7.8 |
| trans-Citral | −70 | −7.8 |
| Perillene | −66 | −7.8 |
| Safrole | −68 | −7.7 |
| cis-ocimene | −63 | −7.6 |
| cis-beta-Ocimene | −63 | −7.6 |
| (E)-beta-Ocimene | −65 | −7.6 |
| (E)-Ocimene | −65 | −7.6 |
| Isogeraniol | −67 | −7.5 |
| beta-Myrcene | −64 | −7.5 |
| 6-Methyl-5-hepten-2-one | −60 | −7.5 |
| 3-Octen-2-one | −62 | −7.4 |
Table 5
ADME and toxicity prediction of predicted ligand inhibitor for KIT protein
| Molecule | Canonical SMILES | PubChem ID | Lipinski rule | GI absorption | BBB permeant | Lipinski violations | Toxic prediction | ||||
| Molecular weight | H-bond donor | H-bond acceptors | Log P | Creamer rules | Cramer rules with extensions | ||||||
| Beta-Carotene | CC(=CC=CC=C(C=CC=C(C=CC1= C(C)CCCC1(C)C)C)C)C=CC=C(C=CC1=C(C)CCCC1(C)C)C | 10556789 | 536.87 | 0 | 0 | 11.11 | Low | No | 2 | Intermediate (Class II) | Intermediate (Class II) |
Table 6
ADME and toxicity prediction of predicted ligand inhibitor for MAPK2 protein
| Molecule | Canonical SMILES | PubChem ID | Lipinski rule | GI absorption | BBB permeant | Lipinski violations | Toxic prediction | ||||
| Molecular weight | H-bond donor | H-bond acceptors | Log P | Cramer rules | Cramer rules with extensions | ||||||
| Lycopene | CC(=CC=CC=C(C=CC=C(C=CC=C (CCC=C(C)C)C)C)C)C=CC=C(C=C C=C(CCC=C(C)C)C)C | 446925 | 536.87 | 0 | 0 | 9.21 | Low | No | 2 | Low (Class I) | Low (Class I) |
| 5-Hydroxy-7,4′-dimethoxyflavone | COC1=CC=C(C=C1)C2=CC(=O)C3 =C(C(=C(C(=C3O2)C4C(C(C(CO4) O)O)O)OC)C5C(C(C(CO5)O)O)O)O | 44258362 | 562.52 | 7 | 13 | −3.02 | Low | No | 3 | High (Class III) | High (Class III) |
| [12]-Shogaol | CCCCCCCCCCCC=CC(=O)CCC1 =CC(=C(C=C1)O)OC | 9975813 | 360.53 | 1 | 3 | 4.25 | High | No | 1 | Low (Class I) | Low (Class I) |
| Isogingerenone B | COC1=CC(=CC(=C1O)OC)CCC(=O) C=CCCC2=CC(=C(C=C2)O)OC | 5318568 | 386.44 | 2 | 6 | 2.11 | High | No | 0 | High (Class III) | High (Class III) |
| Curcumin | COC1=C(C=CC(=C1)C=CC(=O)CC (=O)C=CC2=CC(=C(C=C2)O)OC)O | 969516 | 368.38 | 2 | 6 | 1.47 | High | No | 0 | High (Class III) | High (Class III) |
| trans-[10]-Shogaol | CCCCCCCCCC=CC(=O)CCC1=CC (=C(C=C1)O)OC | 6442612 | 332.48 | 1 | 3 | 3.82 | High | Yes | 0 | Low (Class I) | Low (Class I) |
| Gingerenone A | COC1=C(C=CC(=C1)CCC=CC(=O) CCC2=CC(=C(C=C2)O)OC)O | 5281775 | 356.41 | 2 | 5 | 2.44 | High | Yes | 0 | High (Class III) | High (Class III) |
| Dihydrocurcumin | COC1=C(C=CC(=C1)CCC(=O)CC (=O)C=CC2=CC(=C(C=C2)O)OC)O | 10429233 | 370.40 | 2 | 6 | 1.55 | High | No | 0 | High (Class III) | High (Class III) |
| Demethoxycur cumin | COC1=C(C=CC(=C1)C=CC(=O)CC (=O)C=CC2=CC=C(C=C2)O)O | 5469424 | 338.35 | 2 | 5 | 1.80 | High | No | 0 | High (Class III) | High (Class III) |
| [8]-Shogaol | CCCCCCCC=CC(=O)CCC1=CC (=C(C=C1)O)OC | 6442560 | 304.42 | 1 | 3 | 3.37 | High | Yes | 0 | Low (Class I) | Low (Class I) |
| [6]-Shogaol | CCCCCC=CC(=O)CCC1=CC(= C(C=C1)O)OC | 5281794 | 276.37 | 1 | 3 | 2.90 | High | Yes | 0 | Low (Class I) | Low (Class I) |
| [1]-Paradol | CCC(=O)CCC1=CC(=C(C=C1)O) OC | 51352033 | 208.25 | 1 | 3 | 1.70 | High | Yes | 0 | Low (Class I) | Low (Class I) |
| Bisdemethoxy curcumin | C1=CC(=CC=C1C=CC(=O)CC (=O)C=CC2=CC=C(C=C2)O)O | 5315472 | 308.33 | 2 | 4 | 2.13 | High | Yes | 0 | High (Class III) | High (Class III) |
| alpha-Zingiberene | CC1=CCC(C=C1)C(C)CCC=C(C)C | 11127403 | 204.35 | 0 | 0 | 4.53 | Low | No | 1 | Low (Class I) | Low (Class I) |
3.3.ADME (Absorption, Distribution, Metabolism, and Excretion) and toxicity analysis
However, after ADME analysis, beta carotene is the only ligand with a lower binding affinity than the reference ligand of the KIT protein. Beta caroten has two Lipinski violations and is classified as intermediate or high due to toxic prediction using toxtree (Table 5). Lycopene, [8]-Shogaol, [6]-Shogaol, [1]-Paradol, and alpha-Zingiberene were identified as toxtree low toxicants. Due to Lipinski rules in ADME analysis, each compound also shows no Lipinski violation. The compounds [8]-Shogaol, [6]-Shogaol, and [1]-Paradol have docking scores identical to those of the reference ligand. The binding affinity score for reference ligands is −9.2 Kcal/mol, while the scores for [8]-Shogaol, [6]-Shogaol, and [1]-Paradol are all −9.3 Kcal/mol. Compounds classified as at least intermediate (Class 2) in toxic prediction using toxtree software [24] with no violations of the Lipinski rule [25] are subjected to bioactivity prediction analysis using pass-server (http://www.way2drug.com/passonline) [27]. Lycopene, [12]-Shogaol, [8]-Shogaol, [6]-Shogaol, [1]-Paradol, and alpha-Zingiberene are among the compounds filtered from Lipinski and toxtree (Table 6).
3.4.Analysis of bioactivity prediction
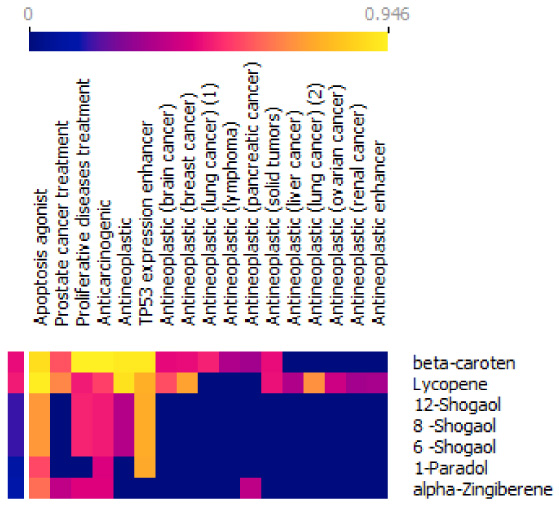
The outcome of bioactivity is then visualized using orange [30]. The heatmap analysis (Fig. 4) reveals a variety of potential activities for each compound. We discovered that each compound chosen has the potential to be an apoptosis agonist [31], which increases apoptosis in cancer cells. Except for [1]-Paradol, all compounds discovered have bioactivity as a proliferative disease treatment, which means that each of the compounds mentioned can both inhibit and aid in the proliferation of cancer cells. All chosen compounds have the potential to be anticarcinogenic. Only [1]-Paradol and alpha-Zingiberene lacked potential antineoplastic activity. Alpha-Zingiberene is also the only compound that did not exhibit TP53 expression enhancer activity, which is responsible for suppressing the cancer development process. The TP53 gene causes cell-cycle arrest, senescence, or apoptosis in response to cellular stressors such as DNA damage, hypoxia, nutritional deficiency, and oncogenic signaling [32]. Approximately half of all human cancers have lost or altered the tumor suppressor p53 [33].
Lycopene was discovered to be antineoplastic in certain cancers, including breast cancer. The anticancer activity is also shown in beta-carotene. Although beta-carotene has two Lipinski violations after ADME analysis, the antineoplastic properties of beta-carotene have been found in the brain, lung, lymphoma, pancreatic, and solid tumors. Beta-carotenes can increase apoptosis, stop the cell cycle at various stages, and prevent cell proliferation [34]. Lycopene is also one potential active compound that has antineoplastic potential in liver cancer, lung cancer, ovarian cancer, and renal cancer. Lycopene has also been identified as an antineoplastic enhancer. Lycopene has also been shown to have anti-cancer properties in the development of gastric cancer [35]. Some of the signal transduction pathways regulated by lycopene include the manipulation of the insulin-like growth factor system, the inhibition of the activity of sex steroid hormones, the modification of significant gene expression, and the alteration of mitochondrial function [36]. It is also found that alpha-zingiberene is the only compound with antineoplastic activity in pancreatic cancer cells (Fig. 5).
Fig. 5.
Bioactivity prediction from compounds selected from ADME and toxic prediction.
3.5.Tanimoto similarity for chemical structure and pharmacophore
Beta-carotene is shared about 6.55% similarity structure with the reference ligand for KIT protein inhibitor Imatinib (PubChem code: 5291), while MAPK2 protein reference ligand 2′-(2-fluorophenyl)-1-methyl-6′,8′,9′,11′-tetrahydrospiro [azetidine-3,10′-pyrido [3′,4′:4,5] pyrrolo [2,3-f] isoquinolin], -7′(5′H) -one (PubChem code: 42646698) has the highest similarity with 5-Hydroxy-7,4′-dimethoxyflavone (9%). None of the compounds selected have a similarity greater than 10%. The model can choose the possible active compound even with a smaller similarity fingerprint of compounds from ginger with an active compound of training and testing data (Table 7).
The Pharmacophore similarity of amino acids interacting with the compound selected as a ligand is also calculated. Beta-carotene only shares about 12.9% pharmacophore similarity with reference ligands for KIT protein. This means there are substantial differences between amino acids from proteins, which are amino acids from proteins that interact with beta-carotene compared to reference ligands. On the other hand, we found Gingerenone A share about 33.3% similarity with reference ligand for MAPK2 protein, Isogingerenone B, and [1]-Paradol also shows higher pharmacophore similarity with MAPK2 reference ligand with a similarity of 27.8% and 22.2% (Table 8). It is clear that even though 5-Hydroxy-7,4′-dimethoxyflavone ligands have the highest structure similarity (9%) with reference ligands, the compound does not always show the same result in pharmacophore similarity; the compound interaction only shares about 11.1% similarity.
Table 7
Structure similarity (%) between reference ligand and selected compound
| Molecule | PubChem ID | PubChem ID reference ligand | Structure similarity (%) | Protein target |
| Beta-carotene | 10556789 | 5291 | 6.5 | KIT |
| Lycopene | 446925 | 42646698 | 1.2 | MAPK2 |
| 5-Hydroxy-7,4′-dimethoxyflavone | 44258362 | 42646698 | 9.0 | MAPK2 |
| [12]-Shogaol | 9975813 | 42646698 | 6.5 | MAPK2 |
| Isogingerenone B | 5318568 | 42646698 | 6.6 | MAPK2 |
| Curcumin | 969516 | 42646698 | 8.6 | MAPK2 |
| trans-[10]-Shogaol | 6442612 | 42646698 | 6.5 | MAPK2 |
| Gingerenone A | 5281775 | 42646698 | 6.9 | MAPK2 |
| Dihydrocurcumin | 10429233 | 42646698 | 7.6 | MAPK2 |
| Demethoxycurcumin | 5469424 | 42646698 | 8.2 | MAPK2 |
| [8]-Shogaol | 6442560 | 42646698 | 6.5 | MAPK2 |
| [6]-Shogaol | 5281794 | 42646698 | 6.5 | MAPK2 |
| [1]-Paradol | 51352033 | 42646698 | 7.2 | MAPK2 |
| Bisdemethoxycurcumin | 5315472 | 42646698 | 5.3 | MAPK2 |
| alpha-Zingiberene | 11127403 | 42646698 | 4.6 | MAPK2 |
Table 8
Pharmacophore similarity (%) between reference ligand and selected compound
| Molecule | PubChem ID | PubChem ID reference ligand | Pharmacophore similarity (%) | Protein target |
| Beta-carotene | 10556789 | 5291 | 12.9 | KIT |
| Lycopene | 446925 | 42646698 | 10 | MAPK2 |
| 5-Hydroxy-7,4′-dimethoxyflavone | 44258362 | 42646698 | 11.1 | MAPK2 |
| [12]-Shogaol | 9975813 | 42646698 | 9.5 | MAPK2 |
| Isogingerenone B | 5318568 | 42646698 | 27.8 | MAPK2 |
| Curcumin | 969516 | 42646698 | 12.5 | MAPK2 |
| trans-[10]-Shogaol | 6442612 | 42646698 | 11.8 | MAPK2 |
| Gingerenone A | 5281775 | 42646698 | 33.3 | MAPK2 |
| Dihydrocurcumin | 10429233 | 42646698 | 10.5 | MAPK2 |
| Demethoxycurcumin | 5469424 | 42646698 | 11.8 | MAPK2 |
| [8]-Shogaol | 6442560 | 42646698 | 9.5 | MAPK2 |
| [6]-Shogaol | 5281794 | 42646698 | 25 | MAPK2 |
| [1]-Paradol | 51352033 | 42646698 | 22.2 | MAPK2 |
| Bisdemethoxycurcumin | 5315472 | 42646698 | 7.7 | MAPK2 |
| alpha-Zingiberene | 11127403 | 42646698 | 18.8 | MAPK2 |
4.Conclusion
Machine learning has been used to help classify anticancer compounds derived from ginger. Lycopene, [12]-shogaol, [8]-shogaol, [6]-shogaol, [1]-shogaol -Paradol and alpha-Zingiberene are two compounds chosen after ADME and toxicity testing. All of the compounds mentioned previously have the potential to be anticarcinogenic, with a different score assigned to each compound. Beta-carotene, although showing two violations of the Lipinski rule, and lycopene have shown potential activity in various cancers. Each compound also acts as an apoptosis agonist, apart from alpha-Zingiberene, which does function as a TP53 expression enhancer.
Conflict of interest
The authors declare no conflicts of interest.
Data availability statement
The data supporting the findings of this study are available on request from the corresponding author.
Author contribution
MEG and MRR wrote the original study design and protocol. MEG did the data collection. MRR participated in the data analysis and interpretation. The paper was written by MRR and was revised and edited by all authors who have approved the final version. We would like to acknowledge the equal contributions of MEG and MRR to this project.
References
[1] | Liu C-X, Overview on development of ASEAN traditional and herbal medicines, Chin Herb Med, 13: : 441–450, (2021) . |
[2] | Park H-L, , Lee H-S, , Shin B-C , Traditional medicine in China, Korea, and Japan: A brief introduction and comparison, Evid-Based Complement Alternat Med ECAM, 2012: : 429103, (2012) . |
[3] | Sumarni W, , Sudarmin S, , Sumarti SS, The scientification of jamu: A study of Indonesian’s traditional medicine, J Phys Conf Ser, 1321: : 032057, (2019) . |
[4] | Ekor M, The growing use of herbal medicines: Issues relating to adverse reactions and challenges in monitoring safety, Front Pharmacol, 4: : 177, (2014) . |
[5] | Mashhadi NS, , Ghiasvand R, , Askari G , Anti-oxidative and anti-inflammatory effects of ginger in health and physical activity: Review of current evidence, Int J Prev Med, 4: : S36–S42, (2013) . |
[6] | Prasad S, , Tyagi AK, Ginger and its constituents: Role in prevention and treatment of gastrointestinal cancer, Gastroenterol Res Pract, 2015: : e142979, (2015) . |
[7] | Akimoto M, , Iizuka M, , Kanematsu R , Anticancer effect of ginger extract against pancreatic cancer cells mainly through reactive oxygen species-mediated autotic cell death, PLoS One, 10: : e0126605, (2015) . |
[8] | Vemuri SK, , Banala RR, , Subbaiah GPV , Anti-cancer potential of a mix of natural extracts of turmeric, ginger and garlic: A cell-based study, Egypt J Basic Appl Sci, 4: : 332–344, (2017) . |
[9] | Rahmani AH, , shabrmi FMA, , Aly SM, Active ingredients of ginger as potential candidates in the prevention and treatment of diseases via modulation of biological activities, Int J Physiol Pathophysiol Pharmacol, 6: : 125–136, (2014) . |
[10] | Kumari M, , Tiwari N, , Chandra S , Comparative analysis of machine learning based QSAR models and molecular docking studies to screen potential anti-tubercular inhibitors against InhA of mycobacterium tuberculosis, Int J Comput Biol Drug Des, 11: : 209, (2018) . |
[11] | Genç B, , Tunc H, Optimal training and test sets design for machine learning, Turk J Electr Eng Comput Sci, 27: : 1534–1545, (2019) . |
[12] | Mysinger MM, , Carchia M, , Irwin John J , Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking, J Med Chem, 55: : 6582–6594, (2012) . |
[13] | Lotfaliansaremi S, , Sabio M, , Comwell S , The role of the Mitogen-Activated Protein Kinase (MAPK) signaling pathway in cancer, Med Res Arch, 8,: (2020) . Epub ahead of print 24 April 2020. doi:10.18103/mra.v8i4.2086. |
[14] | Sheikh E, , Tran T, , Vranic S , Role and significance of c-KIT receptor tyrosine kinase in cancer: A review, Bosn J Basic Med Sci, 22: : 683–698, (2022) . |
[15] | Shinbo Y, , Nakamura Y, , Altaf-Ul-Amin M KNApSAcK: A comprehensive species-metabolite relationship database, in: Saito K, Dixon RA, Willmitzer L (eds), Plant Metabolomics. 1 ed. Springer (Berlin, Heidelberg), 165–181, 2006. ISBN: 978-3-540-29782-6. |
[16] | Klekota J, , Roth FP, Chemical substructures that enrich for biological activity, Bioinforma Oxf Engl, 24: : 2518–2525, (2008) . |
[17] | Pedregosa F, , Varoquaux G, , Gramfort A , Scikit-learn: Machine learning in Python, J Mach Learn Res, 12: : 2825–2830, (2011) . |
[18] | Kluyver T, , Ragan-Kelley B, , Pérez Fernando Jupyter Note-books – A publishing format for reproducible computational workflows, in: Schmidt B, Loizides F (eds), Positioning and Power in Academic Publishing: Players, Agents and Agendas. IOS Press, 87–90, 2016. ISBN: 978-1-61499-649-1. |
[19] | Korb O, , Stützle T, , Exner TE, Empirical scoring functions for advanced protein–ligand docking with PLANTS, J Chem Inf Model, 49: : 84–96, (2009) . |
[20] | Korb O, , Stützle T, , Exner TE PLANTS: Application of ant colony optimization to structure-based drug design, in: Dorigo M, Gambardella LM, Birattari M (eds), Ant Colony Optimization and Swarm Intelligence. Springer (Berlin, Heidelberg), 247–258, 2006. |
[21] | Xue LC, , Rodrigues JP, , Kastritis PL , PRODIGY: A web server for predicting the binding affinity of protein–protein complexes, Bioinformatics, 32: : 3676–3678, (2016) . |
[22] | Vangone A, , Schaarschmidt J, , Koukos P , Large-scale prediction of binding affinity in protein-small ligand complexes: The PRODIGY-LIG web server, Bioinforma Oxf Engl, 35: : 1585–1587, (2019) . |
[23] | Daina A, , Michielin O, , Zoete V, SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci Rep, 7: : 42717, (2017) . |
[24] | Patlewicz G, , Jeliazkova N, , Safford RJ , An evaluation of the implementation of the Cramer classification scheme in the Toxtree software, SAR QSAR Environ Res, 19: : 495–524, (2008) . |
[25] | Lipinski CA, Drug-like properties and the causes of poor solubility and poor permeability, J Pharmacol Toxicol Methods, 44: : 235–249, (2000) . |
[26] | Damião MCFCB, , Pasqualoto KFM, , Polli MC , To be drug or prodrug: Structure-property exploratory approach regarding oral bioavailability, J Pharm Pharm Sci Publ Can Soc Pharm Sci Soc Can Sci Pharm, 17: : 532–540, (2014) . |
[27] | Filimonov DA, , Lagunin AA, , Gloriozova TA , Prediction of the biological activity spectra of organic compounds using the pass online web resource, Chem Heterocycl Compd, 50: : 444–457, (2014) . |
[28] | Istyastono EP, , Radifar M, , Yuniarti N , PyPLIF HIPPOS: A molecular interaction fingerprinting tool for docking results of AutoDock Vina and PLANTS, J Chem Inf Model, 60: : 3697–3702, (2020) . |
[29] | Liu K, , Kokubo H, Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: A cross-docking study, J Chem Inf Model, 57: : 2514–2522, (2017) . |
[30] | Demšar J, , Curk T, , Erjavec A , Orange: Data mining toolbox in python, J Mach Learn Res, 14: : 2349–2353, (2013) . |
[31] | Pfeffer CM, , Singh ATK, Apoptosis: A target for anticancer therapy, Int J Mol Sci, 19: : 448, (2018) . |
[32] | Kim KM, , Ahn A-R, , Park HS , Clinical significance of p53 protein expression and TP53 variation status in colorectal cancer, BMC Cancer, 22: : 940, (2022) . |
[33] | Powell E, , Piwnica-Worms D, , Piwnica-Worms H, Contribution of p53 to metastasis, Cancer Discov, 4: : 405–414, (2014) . |
[34] | Gloria NF, , Soares N, , Brand C , Lycopene and beta-carotene induce cell-cycle arrest and apoptosis in human breast cancer cell lines, Anticancer Res, 34: : 1377–1386, (2014) . |
[35] | Kim MJ, , Kim H, Anticancer effect of lycopene in gastric carcinogenesis, J Cancer Prev, 20: : 92–96, (2015) . |
[36] | Qi WJ, , Sheng WS, , Peng C , Investigating into anti-cancer potential of lycopene: Molecular targets, Biomed Pharmacother Biomedecine Pharmacother, 138: : 111546, (2021) . |


