
Nicotinamide Mononucleotide Prevents Cisplatin-Induced Mitochondrial Defects in Cortical Neurons Derived from Human Induced Pluripotent Stem Cells
Abstract
Background:
Chemotherapy-induced cognitive impairment (CICI) is a neurotoxic side effect of chemotherapy that has yet to have an effective treatment.
Objective:
Using cisplatin, a platinum-based chemotherapy together with excitatory cortical neurons derived from human induced pluripotent cells (iPSCs) to model of CICI, our recent study demonstrated that dysregulation of brain NAD+ metabolism contributes to cisplatin-induced impairments in neurogenesis and cognitive function, which was prevented by administration of the NAD+ precursor, nicotinamide mononucleotide (NMN). However, it remains unclear how cisplatin causes neurogenic dysfunction and the mechanism by which NMN prevents cisplatin-induced cognitive impairment. Given that mitochondrial dysfunction is thought to play a prominent role in age-related neurodegenerative disease and chemotherapy-induced neurotoxicity, we sought to explore if NMN prevents chemotherapy-related neurotoxicity by attenuating cisplatin-induced mitochondrial damage.
Results:
We demonstrate that cisplatin induces neuronal DNA damage, increases generation of mitochondrial reactive oxygen species (ROS) and decreases ATP production, all of which are indicative of oxidative DNA damage and mitochondrial functional defects. Ultrastructural analysis revealed that cisplatin caused loss of cristae membrane integrity and matrix swelling in human cortical neurons. Notably, pretreatment with NMN prevents cisplatin-induced defects in mitochondria of human cortical neurons.
Conclusion:
Our results suggest that increased mitochondrial oxidative stress and functional defects play key roles in cisplatin-induced neurotoxicity. Thus, NMN may be an effective therapeutic strategy to prevent cisplatin-induced deleterious effects on mitochondria, making this organelle a key factor in amelioration of cisplatin-induced cognitive impairments.
INTRODUCTION
Chemotherapy-induced cognitive impairment (CICI, also known as chemobrain) encompasses a broad range of neurological problems, including impairments in short- and long-term memory, attention, clarity of thought, executive functioning, and information processing speed [1, 2]. CICI is a major clinical concern as up to 60% of patients experience persistent cognitive problems years after completion of chemotherapy [3]. Unfortunately, there are no effective treatments currently available as the underlying pathogenic mechanisms of CICI are not well understood.
Mitochondria are the power generators of the cell, converting oxygen and nutrients into adenosine triphosphate (ATP). Damaged mitochondria are known to generate reactive oxygen species (ROS), release cytochrome c, apoptosis-inducing factor (AIF) and other proapoptotic proteins to promote cell death [4, 5]. Therefore, mitochondrial dysfunction is thought to play a prominent role in a number of neurological and neurodegenerative diseases including Alzheimer’s disease [6]. Similarly, previous studies have demonstrated that cisplatin induces neuronal mitochondrial dysfunction in vivo and in vitro [7]. For example, cisplatin, a platinum-based chemotherapy commonly used for lung, testicular, ovarian and breast cancers, crosses the blood-brain barrier (BBB) at sufficient levels to cause DNA damage, form adducts with mitochondrial DNA (mtDNA), while inhibiting mtDNA replication and mitochondrial gene transcription [8, 9]. These neurotoxic effects by cisplatin can lead to defects in neural stem cell proliferation as well as hippocampal neurons [10]. In addition, studies examining the mechanisms of cisplatin-induced peripheral neuropathy in dorsal root ganglion (DRG) neurons and ototoxicity in the cochlea show that cisplatin induces mtDNA damage and ROS generation, thus emphasizing the dose-limiting toxicities associated with cisplatin treatment [11–13].
Furthermore, cisplatin reduced mitochondrial membrane potential can lead to abnormal mitochondrial morphology, impair mitochondrial function, and induce autophagy in DRG neurons [14, 15]. Taken together, these findings suggest that cisplatin-induced mitochondrial damage is a key pathological mechanism driving neuronal dysfunction. Therefore, identification of interventions that prevent cisplatin-induced mitochondrial damage will be essential to develop therapeutic strategies.
Nicotinamide adenine dinucleotide (NAD+) plays an important role in energy metabolism, mitochondrial function, gene expression, calcium homeostasis, aging and cell death [16–18]. Importantly, age-related impairments are attenuated by increasing NAD+ through NAD+ precursors such as nicotinamide mononucleotide (NMN) or nicotinamide riboside. Both have been reported as promising therapeutic compounds known to delay the aging process and extend the lifespan [19, 20]. Using cisplatin to model CICI, our recent study demonstrated that depletion of brain metabolic NAD+ contributes to cisplatin-induced impairments in adult neurogenesis and cognitive function [21]. Notably, these cisplatin-induced impairments were fully prevented by increasing NAD+ levels through administration of NMN [21], suggesting a potential neuroprotective role for NMN in cisplatin-induced cognitive impairment. However, the cellular mechanisms by which NMN plays a preventative role in cisplatin-induced neurotoxicity remain unclear. Given that cisplatin impairs mitochondrial function in the adult brain [7], while conversely, NMN is known to restore mitochondrial function, ameliorate inflammation, and protectively prevent synaptic loss against neuronal cell death after ischemia [22–24] and intracerebral hemorrhage [24], we sought to explore whether NMN prevents cisplatin-induced neurotoxicity via attenuation of mitochondrial defects in human cortical neurons derived from hiPSCs.
METHODS
All experiments were performed in two or three independent experiments. All experiments and analyses were performed blind to treatment.
Human iPSC culture
Human iPSCs (004-BIOTR-0002) derived from a 16-year-old male without any detectible neurological diagnosis or without exposed to chemotherapy were obtained from the Biotrust at the Mayo Clinic Center for Regenerative Medicine. As described in our recent study [21], iPSCs were grown in Essential 8TM medium (ThermoFisher Scientific, A1517001) which was exchanged daily onto recombinant human vitronectin (rhVTN; Peprotech, AF-140-09)-coated T-25 flasks. Cultures were passaged routinely (at ∼60% confluency) using hypertonic citrate buffer (4.4 g/L sodium citrate, 24 g/L potassium chloride; osmolality: 630–640 mOsmol/kg) by first washing the iPSCs cells with hypertonic citrate buffer and then incubating with the same hypertonic citrate buffer (37°C, 10 min) until colony detachment occurred. Colonies were processed into smaller fragments by trituration and dissociation was quenched by addition of Essential 6TM medium (ThermoFisher Scientific, A1516401). Suspensions were collected into 15 mL conical tubes and centrifuged (3 min, 500 g). Supernatants were aspirated and pellets resuspended into an appropriate volume of Essential 8TM medium with a split ratio at 1:8 to 1:10. Cultures were carefully redistributed onto rhVTN-coated plates in Essential 8TM medium passaged every 3 days.
Differentiation of iPSC into cortical neurons
For neural induction, as shown in Fig. 1A, iPSCs cell cultures were passaged and plated onto rhVTN-coated 6 well plates in Essential 8TM medium. After 15–24 hours, the medium was exchanged for Essential 6™ medium supplemented with 2μM dorsomorphin (Sigma-Aldrich, P5499) and 6μM SB-431542 (Tocris, 1614) for 24 hours. 24 hours following neural induction, medium was exchanged daily for Essential 6™ medium supplemented with 2μM dorsomorphin (Sigma Aldrich, P5499), 6μM SB-431542 (Tocris, 1614), and 2μM XAV-939 (Tocris, 3748) for 4 days. On day 6, neural induction medium was then replaced for DMEM/F12 (ThermoFisher Scientific, 110–39021) supplemented with 20 ng/ml fibroblast growth factor (FGF; R&D Systems, 233-FB), N2 (CTS, 100X; ThermoFisher Scientific, A13707-01) and B27 (serum free, 50X, ThermoFisher Scientific, 17504044) for 24 hrs. To promote differentiation of the neural progenitors into cortical neurons, the neural medium was treated with 20 ng/ml brain-derived neurotrophic factor (BDNF; Peprotech, 450-02) and 20 ng/ml Neurotrophin-3 (NT3, Peprotech, 450-03) for 10 days (until day 18) with medium changes every 2 days. At Day 18, cultures were passaged with 0.5 mM EDTA (0.02% DPBS) and plated onto rhL521 (Life Science, 354222)-coated MatTek dishes/plates in DMEM/F12 supplemented with N2 and B27 containing 20 ng/ml BDNF (Peprotech, 450-02) and 20 ng/ml NT3 (Peprotech, 450-03) for 2 days (until day 20).
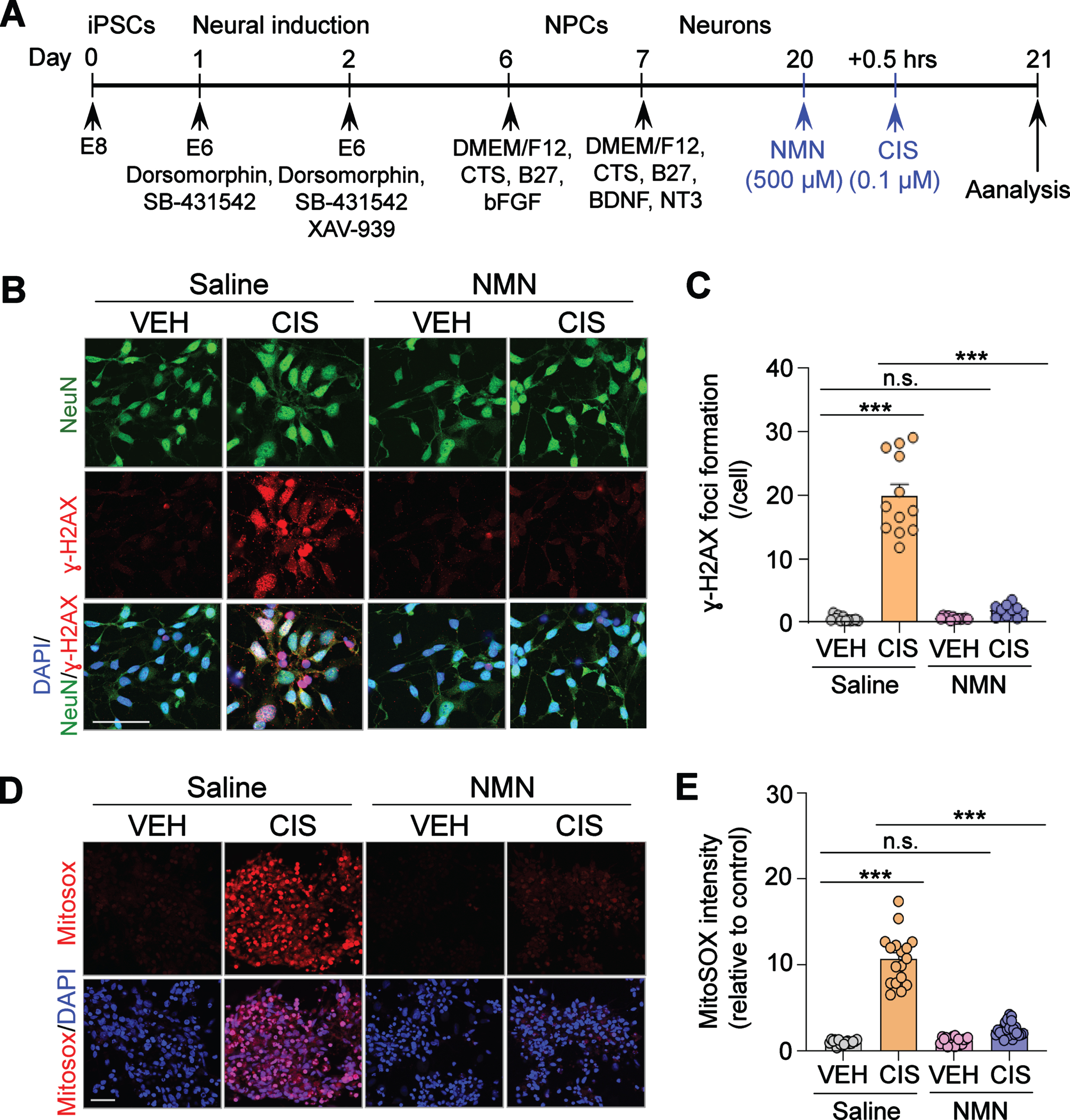
Fig. 1
Neuroprotective effects of NMN in cisplatin-induced DNA damage and mitochondrial reactive oxygen species (ROS) overproduction in human cortical neurons derived from iPSCs. (A) Schematic diagram of the iPSC differentiation procedure. (B) Representative confocal images for NeuN (Green; neuron marker), and γ-H2AX (Red, marker of DNA double-stranded breaks). Nuclei in cell are visualized using DAPI (Blue). The γ-H2AX foci formation was observed by confocal microscopy. Scale bar: 100μm. (C) Quantification of γ-H2AX+ foci in NeuN+ human neurons in each treatment group. (D) Representative confocal images for Mitosox (Red; mitochondrial ROS indicator), and DAPI (Blue, nuclei staining). Mitochondrial ROS generation was observed via confocal microscopy. Scale bar: 50μm. (E) Quantification of Mitosox intensity in each treatment group. All experiments were performed in two or three independent experiments. Data represent mean±SEM. One-way ANOVA followed by Tukey’s post-hoc corrections. ***: P < 0.001, n.s.: not significant. VEH: Vehicle, CIS: Cisplatin, NMN: nicotinamide mononucleotide.
Immunofluorescence staining
For immunofluorescence staining, human cortical neurons were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 30 min, and blocked with 0.1 M Tris-buffered saline (pH 7.5, TBS) containing 10% goat serum (Abcam, ab138478), and 0.3% Triton X-100 (Sigma Aldrich, T8787) for 1 hour at 25°C. The fixed cells were incubated at 4°C overnight with primary antibodies specific to neuronal nuclei (NeuN; mouse, 1:500, EMD Millipore, Cat# MAB377) and phospho H2AX (phospho-Ser139; 1:300, Cell Signaling, Cat# 9718S) used in this study. After washing with TBS, the cells were incubated with Alexa Fluor 488 and Alexa Fluor 555-conjugated secondary antibodies at 25°C for 60 min. To visualize nuclei, the cells were mounted with ProLong Gold antifade mounting medium with 4′,6-diamidino-2-phenylindole (DAPI, Thermo fisher Scientific, P36931). Fluorescence was visualized using an LSM780 confocal laser microscope (Carl Zeiss).
Immunofluorescence analysis and detection of mitochondrial reactive oxygen species (ROS)
Mitochondrial ROS level was estimated via mitochondrial ROS indicator MitoSOX (Thermo fisher Scientific, M36008) using confocal microscopy to visualize ROS production shown in Fig. 2. Briefly, 50μg of MitoSOX™ mitochondrial superoxide indicator was dissolved in 13μL of dimethylsulfoxide (DMSO) to make a 5 mM MitoSOX™ reagent stock solution. After that, 5 mM MitoSOX™ reagent stock solution (prepared above) wsa diluted in DMEM/F12 basal medium (no phenol red) to make a 5μM MitoSOX™ reagent working solution. Human cortical neurons were loaded with MitoSOX Red (5μmol/l) to detect mitochondrial ROS levels, incubated at 37°C for 30 min, and washed five times in ice-cold PBS. After being washed, the cells were fixed in 4% formaldehyde, washed in PBS and mounted with ProLong Gold antifade mounting medium with DAPI (Thermo fisher Scientific, P36931). Fluorescence was visualized using an LSM780 confocal laser microscope (Carl Zeiss). To quantify fluorescence intensity of ROS in mitochondria, Image J software was utilized and fold change relative to vehicle group was calculated.
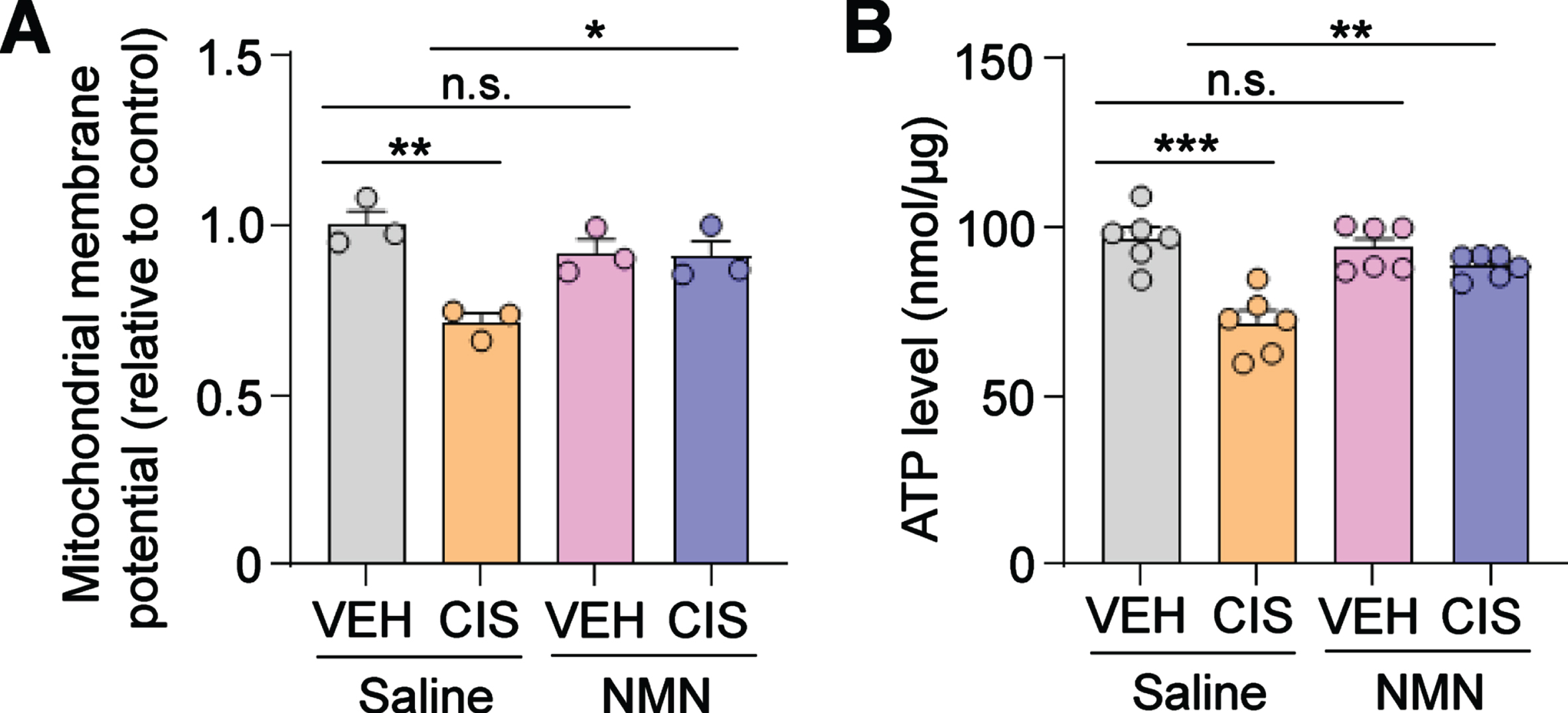
Fig. 2
NMN prevents cisplatin-induced reductions in mitochondrial membrane potential and ATP production in human cortical neurons. (A) The mitochondrial membrane potential (relative to vehicle- and saline-treated group) was evaluated as described in the Methods section. (B) ATP levels were estimated by using the ATP bioluminescent assay kit. Circles in bar graphs represent analysis of an individual well. All experiments were performed in two or three independent experiments. Data represent mean±SEM. One-way ANOVA followed by Tukey’s post-hoc corrections. *: P < 0.5, **: P < 0.01, ***: P < 0.001, n.s.: not significant. VEH: Vehicle, CIS: Cisplatin, NMN: nicotinamide mononucleotide.
Determination of mitochondrial membrane potential
The mitochondrial membrane potential of human cortical neurons was assessed with a 5,5,6,6’-tetrachloro-1,1’,3,3’ tetraethylbenzimi-dazoylcarbocyanine iodide (JC-1), a cationic dye that accumulates in energized mitochondira that is widely used as a sensitive marker for MMP [25] using mitochondrial membrane potential assay kit (Abcam, ab113850) according to the manufacturer’s instructions. The cells were incubated with 10μmol/L JC-1 dye for 20 minutes at 37°C and then washed twice with 1X dilution buffer. The samples were assessed on a fluorescent plate reader (Molecular Device) with a set excitation wavelength of 535 nm and an emission wavelength of 590 nm.
Measurement of intracellular ATP content
For assessment of intracellular ATP levels from cortical neurons, 7×104 human cortical neurons were cultured in 0.5 mL media unless specified otherwise. Cultured human cortical neurons (day 20) were treated with NMN for 30 min with or without the 24 hrs pretreatment with cisplatin and subsequently cells were lysed in lysis buffer containing protease inhibitor cocktail (Cell Signaling) on ice for 5 min followed by centrifugation at 10,000 rpm for 5 min at 4°C. The supernatant protein concentrations were measured by the BCA assay kit (Pierce, Cat# 23227). Equal amounts of protein concentration from cell lysates were used for ATP assay. ATP concentration was determined using the ATP bioluminescent assay kit supplied by Promega (Cat # G9241) according to the manufacturer’s instructions. Briefly, cells were lysed using a lysis buffer including protease inhibitor cocktail (Cell Signaling), and added a volume of CellTiter-Glo® 2.0 reagent equal to the volume of cell supernatant onto a 96-well plate. To mix the contents for 2 minutes on an orbital shaker to induce cell lysis, and allowed the plate to incubate at room temperature for 10 minutes to stabilize the luminescent signal. The intracellular ATP concentration was measured luminescence on a luminometer by using an integration time of 0.25–1 second per well as a manufacturer guideline. Subtract the value of the ATP blank sample form the raw data. The ATP content was calculated as nmoles of ATP per microgram protein.
Transmission electron microscopy
Human cortical neurons were plated onto a glass coverslip in a six-well plate. Cells were fixed in 4% paraformaldehyde +1% glutaraldehyde in 0.1 M phosphate buffer, pH 7.2 for 12 hours at 4°C. Further processing was performed with the aid of a PELCO BioWave® laboratory microwave oven (Ted Pella, Inc., Redding, CA) operating at 250 W. Fixed cells were microwaved under vacuum in 1% osmium tetroxide in 0.1 M phosphate buffer for the sequence: 2 min on; 2 min off; 2 min on; 15 min off. Cells were rinsed in nH2O and microwaved in 2% uranyl acetate using the previous sequence. Following another nH2O rinse, cells were dehydrated using the graded ethanol series (70%, 80%, 95%, 100%, 100% acetone). For each dehydration step, cells were microwaved for 40 sec and allowed to rest at RT for 2 min. Cells were then infiltrated with Embed 812/Araldite resin (EMS, Hatfield, PA) by microwaving under vacuum 2 min on, 2 min off, 2 min on, 30 min off, in 2:1, 1:1 and 3:1 ratio of acetone:resin sequentially. After a final incubation in 100% resin for 12 hours at room temperature, cells were embedded by inverting resin filled embedding molds atop the monolayer and allowing the resin to polymerize for 24 hours at 60°C. Ultrathin sections (0.1 micron) were stained with lead citrate 5 min at room temperature. Micrographs were acquired using a JEOL 1400+ Transmission Electron Microscope (JEOL, Inc., Peabody, MA) at 80 kV equipped with a Gatan Orius camera (Gatan, Inc., Warrendale, PA).
Statistical analysis
One-way ANOVA with Tukey’s post-hoc test for multiple comparisons as appropriate for each experiment. Statistical significance was defined as P < 0.05. All exact P values and statistical analysis information is provided in the Supplementary Table 1.
RESULTS
NMN attenuates cisplatin-induced DNA damage and oxidative stress in human excitatory cortical neurons
Cisplatin is known to accumulate in mitochondria to form adducts with mitochondrial DNA (mtDNA) leading to mtDNA damage and oxidative stress [9, 26]. We therefore tested whether NMN also prevents cisplatin-induced DNA damage in human neurons. As shown in Fig. 1A, human cortical neurons were generated from hiPSCs in vitro and exposed to NMN (500μM) for 30 min followed by treatment with cisplatin (0.1μM) for 24 hours. These cortical neurons were examined by immunocytochemistry with the expression of γ-H2AX, which is a marker for DNA double-stranded breaks [27, 28]. The doses of NMN and cisplatin were chosen based on our previous report [21], demonstrating that cisplatin concentrations as low as 0.1μM significantly decreases cell viability and neurite outgrowth. These neurotoxic effects were significantly ameliorated by NMN at doses ranging from 250 to 1,000μM, with the greatest efficacy at 500μM. First, we confirmed that cisplatin significantly increases DNA damage as we detected increased formation of γ-H2AX foci. In contrast, cisplatin-induced increases in γ-H2AX foci were significantly mitigated by pre-treatment of NMN (Fig. 1B and C), suggesting a neuroprotective effect of NMN in cisplatin-induced DNA damage in human cortical neurons.
Mitochondria are the main sources of cellular ROS. Because oxidative stress causes ROS overproduction which plays a pathogenic role in cisplatin-induced toxicities, including nephrotoxicity [29], ototoxicity [30], and peripheral neuropathy [31], we next asked whether NMN administration can prevent cisplatin-induced increases in mitochondria ROS production. Using fluorescent confocal microscopy to detect the mitochondrial superoxide MitoSOX, we reveal increased mROS in human neurons that were treated with cisplatin compared to vehicle, and as expected, NMN pre-treatment significantly reduced mROS production (Fig. 1D and E). Collectively, these results suggest a preventative effect of NMN in cisplatin-induced ROS overproduction in human cortical neurons.
NMN prevents cisplatin-induced mitochondrial defects in human excitatory cortical neurons
Elevated oxidative stress within mitochondria leads to the loss of mitochondrial membrane potential [32]. Therefore, we next investigated the effect of cisplatin and NMN on mitochondrial membrane potential in human neurons using the cationic by fluorescence dye tetraethylbenzimidazolylcarbocyanine iodide (JC-1), a sensitive marker for mitochondrial membrane potential [25, 33]. Our analysis showed that cisplatin significantly reduced mitochondrial membrane potential in human cortical neurons treated with cisplatin while conversely, NMN prevented the loss of membrane potential (Fig. 2A). This result suggests that NMN is efficacious in preventing cisplatin-induced loss of mitochondrial membrane potential in human neurons.
Adequate maintenance of mitochondrial membrane potential is a critical process required for neuroprotective ATP production. However, this process can be disrupted by oxidative stress and aberrant ROS generation, both which can suppress ATP production and are reported to contribute to neuropathology in a wide spectrum of neurodegenerative diseases including Alzheimer’s disease and Parkinson disease [34, 35]. In line with these studies, we found a significant decrease of ATP levels in human cortical neurons treated with cisplatin, whereas pre-treatment with NMN prevented these adverse effects (Fig. 2B). Taken together, these results suggest that NMN prevents cisplatin-induced loss of mitochondria membrane potential and ATP depletion in human cortical neurons.
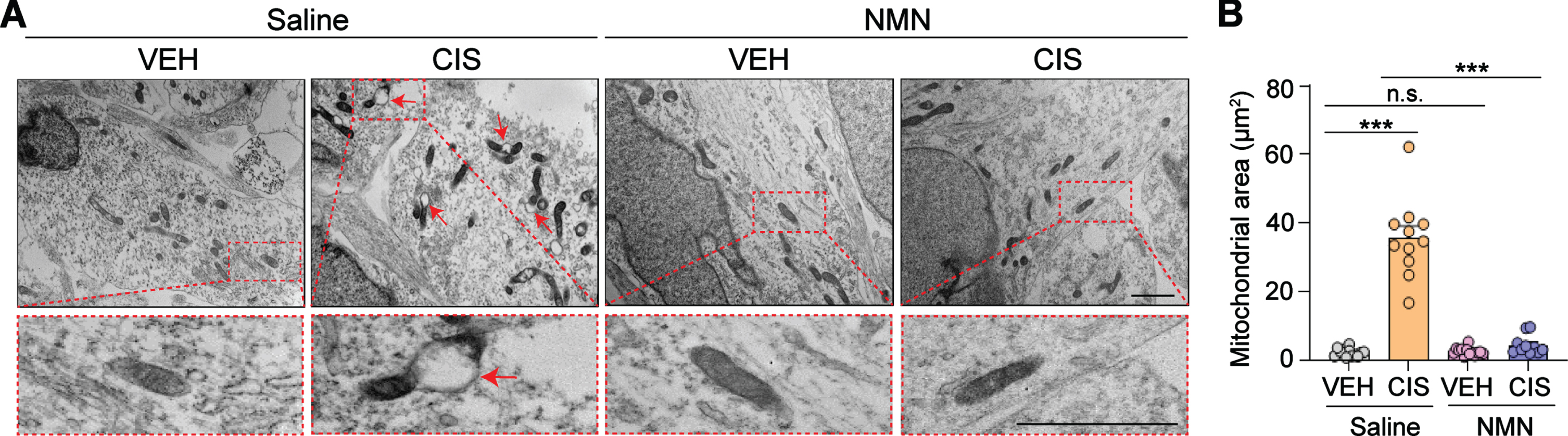
NMN protects against cisplatin-induced mitochondrial swelling in human excitatory cortical neurons
Mitochondrial swelling is one of the earlies and most striking ultrastructural changes reported in several neural injuries (e.g. ischemia) due to membrane permeability transition initiated by a variety of stimuli [14, 36]. Therefore, we used transmission electron microscopy (TEM) to examine the effect of cisplatin and NMN on mitochondrial ultrastructural changes in human cortical neurons derived from hiPSCs. TEM examination of human neurons treated with cisplatin indicated a significant expansion of mitochondrial space lacking a well-defined regular matrix structure (Fig. 3A and B), while remarkably, these aberrant structural defects were prevented by NMN treatment. These results suggest that NMN effectively preserves proper mitochondrial structure, function, and morphology during oxidative stress conditions associated with cisplatin exposure.
Fig. 3
NMN prevents cisplatin-induced increases in mitochondrial vacuolization and swelling in human cortical neurons. (A) Ultrastructural features in mitochondria were observed under transmission electron microscopy. Abnormal areas of mitochondria are indicated with red arrows and show swollen mitochondria by evidence of severely disrupted cristae. Insets outlined by red dashed lines show a more detailed examination of the detrimental effects of cisplatin and the neuroprotective effect of NMN on cisplatin-induced mitochondrial impairments. All scale bars: 2μm. (B) Quantification of mitochondria area in each treatment group was analyzed by Image J. Circles in bar graphs represent analysis of an individual mitochondria. n = 10–13 mitochondria/treatment derived from triplicate wells/treatment group. All experiments were performed in two or three independent experiments. Data represent mean±SEM. One-way ANOVA followed by Tukey’s post-hoc corrections. ***: P < 0.001, n.s.: not significant. VEH: Vehicle, CIS: Cisplatin, NMN: nicotinamide mononucleotide.
DISCUSSION
Cisplatin has been used to treat a number of cancers including ovarian, testicular, lung, breast, and bladder. It has also been reported to induce adverse neurotoxic side effects such as cognitive dysfunction [37] as well as peripheral neuropathy [38] in cancer survivors. Therefore, understanding the mechanisms of how cisplatin induces neurotoxicity will be critical for development of therapies to prevent or treat cisplatin-induced neurotoxicity. Mitochondrial dysfunction has been linked to the pathogenesis of neurodegenerative diseases including Parkinson’s and Alzheimer’s diseases, leading to neurodegeneration and related cognitive and behavioral abnormalities [34, 39, 40]. Previous studies have also reported that cisplatin causes mitochondria dysfunction resulting in cognitive impairment [7]. However, whether the neuroprotective effects of NMN on mitochondria observed in this study is mediated through prevention and/or protecting dysfunctional mitochondria resulting from cisplatin-induced neurotoxicity is not known. Using human excitatory cortical neurons derived from hiPSCs, our evidence demonstrates that NMN prevents cisplatin-induced mitochondrial dysfunction and structural changes, consequently advancing our understanding of the mechanisms by which NMN exerts its neuroprotective effects in CICI. Hence, our findings put forth important implications regarding the development of CICI.
Growing evidence suggests that NMN has beneficial neuroprotective effects in a variety of animal models of neurological and age-related neurodegenerative disorders. For example, NMN attenuates brain injury after intracerebral hemorrhage by suppressing neuroinflammation and oxidative stress [24]. NMN administration also ameliorates brain mitochondrial respiratory functional deficits in an animal model of Alzheimer’s disease [41]. Furthermore, NMN improves cognitive function in aged mice by restoring mitochondrial bioenergetics and reducing mitochondrial ROS production [42]. In line with these previous observations, our recent study also revealed a beneficial effect of NMN against cisplatin-induced impairments in neurogenesis and cognitive function. The current studies in this report also showed a neuroprotective role of NMN in cisplatin-induced oxidative stress and mitochondria dysfunction, leading to recovery of cisplatin-induced impairment of neurite outgrowth of human cortical neurons [21]. Therefore, NMN may serve as a general therapeutic option for preventing cisplatin-induced cognitive impairments as well as other cognitive dysfunctions associated with neurological and neurodegenerative illnesses.
In addition, our findings also identify underlying mechanism of how NMN may plays a neuroprotective role in cisplatin-induced neurotoxicity. Mitochondria are essential organelles within the cell which bioenergetically produce high levels of ATP through oxidative phosphorylation. Mitochondrial dysfunction results in reduced ATP production and conversely, increased ROS production, both of which are known to contribute to the loss of mitochondrial membrane potential. These impairments are hypothesized to be prominent features in brain aging and neurodegeneration [34, 43]. Similarly, we observed that cisplatin causes increased ROS production, loss of mitochondrial membrane potential, and reduced ATP production, along with loss of cristae membrane structure and matrix swelling in human cortical neurons. Intriguingly, NMN significantly attenuated these cisplatin-induced mitochondrial defects, supporting our hypothesis which states that NMN exerts antioxidant properties. Therefore, we suggest that NMN, through prevention of oxidative stress and mitochondrial dysfunction, provides therapeutic efficacy against cisplatin-induced neurotoxicity. However, while our results reinforce the hypothesis stating that NMN confers neuroprotection against cisplatin-induced defects on mitochondrial function, we emphasize that our use of cortical cultures derived from a single donor iPSC line presents a limitation of our experimental approach. Future studies will have to identify whether additional cell lines (e.g., hippocampal) derived from human iPSCs can similarly reflect our findings.
In summary, NAD+ precursors such as NMN and nicotinamide riboside are actively being tested for aging and age-related diseases in clinical trials. Therefore, increasing NAD+ levels using NMN supplementation during chemotherapy may represent a promising therapeutic strategy that is rapidly and safely applicable to prevent chemotherapy-induced neurotoxicity, thus improving quality of life for cancer patients.
AUTHOR CONTRIBUTION
M.A.R., A.O., Y.S.K. and M.H.J. designed the study. M.A.R. performed the experiments and data analysis and interpretation. M.A.R., A.O., Y.S.K. and M.H.J. wrote the manuscript. All authors reviewed and edited the manuscript.
CONFLICT OF INTEREST
The authors have declared that no conflict of interest exists.
ACKNOWLEDGMENTS
We would like to thank Dr. Zachary Resch and Lindsey A. Kirkeby at Biotrust at Mayo Clinic Center for Regenerative Medicine for providing hiPSC line.
FUNDING
This work was supported by NIH (R01CA242158, R01AG058560), Regenerative Medicine Minnesota (RMM091718DS005), Eagles 5th District Cancer Telethon Funds and Rutgers Cancer Institute of New Jersey (CINJ) survivorship award to M.H.J., NIH (R01DE026677, R01DE031477), UTHSA Startup from University of Texas system and Rising STAR Award from University of Texas system to Y.S.K., and the Bosarge Family Foundation-Waun Ki Hong Scholar Award from the American Association for Cancer Research (19-40-60-OLIV) and the Rutgers CINJ Pediatric Cancer and Blood Disorders Research Center to A.O.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/BPL-220143.
REFERENCES
[1] | Argyriou AA , Assimakopoulos K , Iconomou G , Giannakopoulou F , Kalofonos HP . Either called “chemobrain” or “chemofog,” the long-term chemotherapy-induced cognitive decline in cancer survivors is real. J Pain Symptom Manage. (2011) ;41: (1):126–39. |
[2] | Weiss B . Chemobrain: A translational challenge for neurotoxicology. Neurotoxicology. (2008) ;29: (5):891–8. |
[3] | Ahles TA , Saykin AJ . Candidate mechanisms for chemotherapy-induced cognitive changes. Nat Rev Cancer. (2007) ;7: (3):192–201. |
[4] | Circu ML , Aw TY . Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. (2010) ;48: (6):749–62. |
[5] | Garrido C , Galluzzi L , Brunet M , Puig PE , Didelot C , Kroemer G . Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. (2006) ;13: (9):1423–33. |
[6] | Wang W , Zhao F , Ma X , Perry G , Zhu X . Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol Neurodegener. (2020) ;15: (1):30. |
[7] | Lomeli N , Di K , Czerniawski J , Guzowski JF , Bota DA . Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radic Biol Med. (2017) ;102: :274–86. |
[8] | Todd RC , Lippard SJ . Inhibition of transcription by platinum antitumor compounds. Metallomics. (2009) ;1: (4):280–91. |
[9] | Yang Z , Schumaker LM , Egorin MJ , Zuhowski EG , Guo Z , Cullen KJ . Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: Possible role in apoptosis. Clin Cancer Res. (2006) ;12: (19):5817–25. |
[10] | Andres AL , Gong X , Di K , Bota DA . Low-doses of cisplatin injure hippocampal synapses: A mechanism for ‘chemo’ brain? Exp Neurol. (2014) ;255: :137–44. |
[11] | Areti A , Yerra VG , Naidu V , Kumar A . Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. (2014) ;2: :289–95. |
[12] | Kim HJ , Lee JH , Kim SJ , Oh GS , Moon HD , Kwon KB , et al. Roles of NADPH oxidases in cisplatin-induced reactive oxygen species generation and ototoxicity. J Neurosci. (2010) ;30: (11):3933–46. |
[13] | Podratz JL , Knight AM , Ta LE , Staff NP , Gass JM , Genelin K , et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. (2011) ;41: (3):661–8. |
[14] | Maj MA , Ma J , Krukowski KN , Kavelaars A , Heijnen CJ . Inhibition of mitochondrial p53 accumulation by PFT-mu prevents cisplatin-induced peripheral neuropathy. Front Mol Neurosci. (2017) ;10: :108. |
[15] | Melli G , Taiana M , Camozzi F , Triolo D , Podini P , Quattrini A , et al. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol. (2008) ;214: (2):276–84. |
[16] | Xia W , Wang Z , Wang Q , Han J , Zhao C , Hong Y , et al. Roles of NAD(+) / NADH and NADP(+) / NADPH in cell death. Curr Pharm Des. (2009) ;15: (1):12–9. |
[17] | Ying W . NAD+ and NADH in cellular functions and cell death. Front Biosci. (2006) ;11: :3129–48. |
[18] | Ying W . NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid Redox Signal. (2008) ;10: (2):179–206. |
[19] | Lautrup S , Sinclair DA , Mattson MP , Fang EF . NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. (2019) ;30: (4):630–55. |
[20] | Stein LR , Imai S . Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. (2014) ;33: (12):1321–40. |
[21] | Yoo KH , Tang JJ , Rashid MA , Cho CH , Corujo-Ramirez A , Choi J , et al. Nicotinamide mononucleotide prevents cisplatin-induced cognitive impairments. Cancer Res. (2021) ;81: (13):3727–37. |
[22] | Park JH , Long A , Owens K , Kristian T . Nicotinamide mononucleotide inhibits post-ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol Dis. (2016) ;95: :102–10. |
[23] | Wei CC , Kong YY , Hua X , Li GQ , Zheng SL , Cheng MH , et al. NAD replenishment with nicotinamide mononucleotide protects blood-brain barrier integrity and attenuates delayed tissue plasminogen activator-induced haemorrhagic transformation after cerebral ischaemia. Br J Pharmacol. (2017) ;174: (21):3823–36. |
[24] | Wei CC , Kong YY , Li GQ , Guan YF , Wang P , Miao CY . Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci Rep. (2017) ;7: (1):717. |
[25] | Lee YJ , Kim GH , Park SI , Lim JH . Down-regulation of the mitochondrial i-AAA protease Yme1L induces muscle atrophy via FoxO3a and myostatin activation. J Cell Mol Med. (2020) ;24: (1):899–909. |
[26] | Olivero OA , Chang PK , Lopez-Larraza DM , Semino-Mora MC , Poirier MC . Preferential formation and decreased removal of cisplatin-DNA adducts in Chinese hamster ovary cell mitochondrial DNA as compared to nuclear DNA. Mutat Res. (1997) ;391: (1-2):79–86. |
[27] | Mah LJ , El-Osta A , Karagiannis TC . gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia. (2010) ;24: (4):679–86. |
[28] | Sun X , Wang S , Gai J , Guan J , Li J , Li Y , et al. SIRT5 promotes cisplatin resistance in ovarian cancer by suppressing DNA damage in a ROS-dependent manner via regulation of the Nrf2/HO-1 pathway. Front Oncol. (2019) ;9: :754. |
[29] | Volarevic V , Djokovic B , Jankovic MG , Harrell CR , Fellabaum C , Djonov V , et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: A balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci. (2019) ;26: (1):25. |
[30] | Yu D , Gu J , Chen Y , Kang W , Wang X , Wu H . Current strategies to combat cisplatin-induced ototoxicity. Front Pharmacol. (2020) ;11: :999. |
[31] | Calls A , Torres-Espin A , Navarro X , Yuste VJ , Udina E , Bruna J . Cisplatin-induced peripheral neuropathy is associated with neuronal senescence-like response. Neuro Oncol. (2021) ;23: (1):88–99. |
[32] | Cabreiro F , Picot CR , Perichon M , Castel J , Friguet B , Petropoulos I . Overexpression of mitochondrial methionine sulfoxide reductase B2 protects leukemia cells from oxidative stress-induced cell death and protein damage. J Biol Chem. (2008) ;283: (24):16673–81. |
[33] | Park SJ , Shin JH , Jeong JI , Song JH , Jo YK , Kim ES , et al. Down-regulation of mortalin exacerbates Abeta-mediated mitochondrial fragmentation and dysfunction. J Biol Chem. (2014) ;289: (4):2195–204. |
[34] | Misrani A , Tabassum S , Yang L . Mitochondrial dysfunction and oxidative stress in Alzheimer’s disease. Front Aging Neurosci. (2021) ;13: :617588. |
[35] | Wang X , Wang W , Li L , Perry G , Lee HG , Zhu X . Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. (2014) ;1842: (8):1240–7. |
[36] | Zhao XY , Lu MH , Yuan DJ , Xu DE , Yao PP , Ji WL , et al. Mitochondrial dysfunction in neural injury. Front Neurosci. (2019) ;13: :30. |
[37] | Chovanec M , Vasilkova L , Setteyova L , Obertova J , Palacka P , Rejlekova K , et al. Long-term cognitive functioning in testicular germ-cell tumor survivors. Oncologist. (2018) ;23: (5):617–23. |
[38] | Staff NP , Grisold A , Grisold W , Windebank AJ . Chemotherapy-induced peripheral neuropathy: A current review. Ann Neurol. (2017) ;81: (6):772–81. |
[39] | Falabella M , Vernon HJ , Hanna MG , Claypool SM , Pitceathly RDS . Cardiolipin, mitochondria, and neurological disease. Trends Endocrinol Metab. (2021) ;32: (4):224–37. |
[40] | Norat P , Soldozy S , Sokolowski JD , Gorick CM , Kumar JS , Chae Y , et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. NPJ Regen Med. (2020) ;5: (1):22. |
[41] | Long AN , Owens K , Schlappal AE , Kristian T , Fishman PS , Schuh RA . Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer’s disease-relevant murine model. BMC Neurol. (2015) ;15: :19. |
[42] | Tarantini S , Valcarcel-Ares MN , Toth P , Yabluchanskiy A , Tucsek Z , Kiss T , et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. (2019) ;24: :101192. |
[43] | Grimm A , Eckert A . Brain aging and neurodegeneration: From a mitochondrial point of view. J Neurochem. (2017) ;143: (4):418–31. |

