
Autophagy and Adult Neurogenesis: Discoveries Made Half a Century Ago Yet in their Infancy of being Connected
Abstract
Within the brain, the physiological and pathological functions of autophagy in development and throughout the lifespan are being elucidated. This review summarizes recent in vitro and in vivo results that are defining the role of autophagy-related genes during the process of adult neurogenesis. We also discuss the need for future experiments to determine the molecular mechanism and functional significance of autophagy in the different neural stem cell populations and throughout the stages of adult neurogenesis.
INTRODUCTION
Macroautophagy (hereafter referred to as autophagy) is a cellular process in which organelles and portions of the cytoplasm are engulfed in double membrane vesicles called autophagosomes to allow for recycling of the components through lysosomal degradation pathways. Autophagy occurs constitutively in all cells and there is growing appreciation for its significance in physiological and pathological conditions, including in the development and progression of several neurodegenerative diseases [1].
In this review, we discuss the in vitro and in vivo studies that have highlighted the differential roles for genes in the autophagy pathway within neural stem cells (NSCs) and their progeny. These studies have primarily examined adult neurogenesis, following the modulation of autophagy related genes (Atg) during early development using conditional mouse models. In contrast, studies modulating autophagy related genes using an inducible model to target adult neurogenic cells are limited. Although there is no doubt that autophagy is a regulator of adult neurogenesis, much remains to be understood about the specific functions of autophagy within the adult and during the multiple stages of the process of adult neurogenesis. Additionally, understanding the mechanism by which adult neurogenesis and autophagy interact will open new avenues to modulate endogenous stem cells within the brain for regenerative medicine.
ADULT NEUROGENESIS
Over 50 years have passed since neurogenesis was discovered to occur in the adult mammalian brain [2]. Prior to this discovery, it was believed that, during development, the process of neurogenesis created all the neurons one would have within a lifetime, and that no new neurons could be generated in the adult brain. It is now well accepted that in most mammals, including humans, the adult brain contains both NSCs and neural progenitor cells (NPCs), and that adult-generated neurons contribute to behavior and have relevance to disease [3–7]. This section provides a brief overview of the process of adult neurogenesis and updates in the field that are relevant to the analysis of autophagy and adult neurogenesis.
Adult neurogenesis occurs in neurogenic niches that include the subventricular zone (SVZ) that lines the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus (DG) in the hippocampus [3–7]. Both of these complex microenvironments support many different cell populations that include NSCs, NPCs, ependymal cells, vascular cells, astrocytes, microglia and interneurons. Within these niches, NSCs progress through distinct stages of development to generate mature functional cells. For example, dividing radial glia-like (Type B) cells in the SVZ generate the transient amplifying (Type C) cells that give rise to neuroblasts (Type A). These neuroblasts migrate in chains along the rostral migratory stream (RMS) to reach the olfactory bulb (OB), where they differentiate into granule cells and periglomerular neurons. In the second neurogenic niche, the SGZ, the relatively slowly dividing radial-glia like (Type-1) NSCs generate rapidly proliferating intermediate NPCs (Type-2 cells), which in turn differentiate into neuroblasts (Type 3 cells) and finally into granule neurons. Although the SVZ and SGZ were the first identified and most characterized neurogenic niches, a series of studies initiated in 2012 have since demonstrated that the median eminence ependymal layer of the third ventricle in the hypothalamus is also a neurogenic niche that retains the capacity for neurogenesis in the adult brain [8–10]. No studies have yet examined whether autophagy regulates the radial glia-like cells termed tanycytes that can proliferate and generate new hypothalamic neurons. Given the known connections between autophagy and hypothalamic function [11], this is an area ripe for future investigation.
The regulation of neurogenesis has been studied in the SGZ and SVZ using both in vitro and in vivo experimental models. The most frequently used assay for NSC self-renewal and differentiation potential in vitro involves the expansion of NSCs derived from SGZ or SVZ as neurospheres, or as adherent monolayer cultures [12–16]. The long-term expansion of primary stem cells as neurospheres, isolated from the neurogenic niches, has been particularly useful in determining stem cell potential, but also has its limitations [17]. For example, multiple populations of stem cells and their progeny can make neurospheres, which requires the cells to be grown in saturating levels of exogenous growth factors that may bias their differentiation potential. The requirement for growth factors is also a limitation for the commercially available cell line derived from female rat hippocampal neural stem/progenitor cells (HCNs, also referred to as adult hippocampal neural stem/progenitor cells AHPs), which are not utilized as often as primary cells [13, 15, 16, 18]. Despite these limitations, the in vitro findings from primary cultures and AHPs have synergized with in vivo strategies to dissect the intrinsic properties and molecular regulators of adult NSCs and NPCs.
In vivo studies of adult neurogenesis have utilized a variety of well-established methods to label, quantify and manipulate NSCs, NPCs and their progeny. These methods include, but are not limited to, the use of thymidine analogues, endogenous cell-cycle proteins, a variety of lineage commitment markers and lineage tracing through the use of transgenic mice and retroviral vectors [19, 20]. Collectively, these methods have enabled visualization of NSCs and NPCs and identification of the different stages of cell maturation, as well as the cellular and molecular mechanisms that are important in the different stages of neurogenesis. For example, they can highlight factors that are involved in NSC self-renewal, proliferation, survival, and differentiation, and for the final integration into neural circuitry. The host of inducible transgenic mouse models has also been particularly useful for dissecting the spatial and temporal regulation of gene expression during the different stages of adult neurogenesis. Well-characterized inducible and conditional mouse models used to modify gene expression in adult NSCs including for example glial fibrillary acidic protein (GFAP), glutamate aspartate transporter (GLAST), and numerous nestin constructs [20–22] for promoters. These transgenic models have provided a better appreciation of the complex heterogeneity of NSCs in vivo [23, 24]. The diversity of the NSC population is also supported by findings from studies utilizing in vivo imaging approaches [25] and single-cell gene expression analysis [26]. Thus, overall these methods have allowed for the elucidation of mechanisms governing the neurogenic process in the adult brain and our understanding of how adult-generated neurons contribute to brain function in health and disease.
AUTOPHAGY
In 1963, two years prior to the discovery of adult neurogenesis, the term “autophagy” was coined by Christian de Duve following high-resolution transmission electron microscopic (TEM) observations of the mammalian cell [27]. Similar in many ways to the history of the discovery of adult neurogenesis, there was a long-time span after the initial observation of autophagy before critical advancements firmly established the significance of this pathway in cell biology. Indeed, autophagy research reached its summit in 2016 when Yoshinori Ohsumi received the Nobel Prize in Physiology or Medicine for his seminal work that defined the molecular principles governing autophagy and led to the understanding of its broad physiological roles and relevance to health and disease [28–30]. Different types of autophagy processes have been described including macroautophagy, microautophagy and chaperone-mediated autophagy [28–30]. In this review we will focus on macroautophagy, as this is the process that has been studied in the context of adult neurogenesis.
As shown in Fig. 1 the basic autophagy pathway consists of 1) an isolation membrane that forms upon autophagy induction, 2) the enclosing of the isolation membrane to form a double-membraned autophagosome and finally 3) fusion of the autophagosome with a lysosome (autolysosome) for degradation of enclosed contents [31]. During each of these phases of autophagosome formation key autophagy-associated proteins are required, and those that have been examined in relation to adult neurogenesis [1, 32, 33] are briefly described here. The initiation of autophagy is regulated through the phosphorylation of the unc-51-like kinase 1 (ULK1) complex, that includes family kinase-interacting protein of 200 kDa (FIP200). During nucleation of the isolation membrane the class III phosphatidylinositol 3-kinase complex (PtdIns3K) is activated to form phosphatidylinositol-3-phosphate. The PtdIns3K complex is comprised of the vacuolar protein sorting 34 (VPS34), whose activation depends on the formation of a complex that include VPS15, Ambra1, ATG14 and Bax-interacting factor 1 (BIF1), as well as Beclin1. The availability of Beclin1 is important in regulating the induction of the phagophore, with the loss of the interaction between Beclin1 and BCL-2 required for activation of VPS34. The elongation of the isolation membrane proceeds through two ubiquitin-like conjugation pathways that result in the formation of the autophagosome. In the first pathway, a covalent conjugation forms between ATG12 and ATG5 through the actions of ATG7 and ATG10. ATG12-ATG5 then interacts with ATG16L1 (ATG16-like 1) and the tri-protein complex in turn can exhibit an E3 ligase-like activity to promote the lipidation of the light chain 3 (LC3) protein. In the second ubiquitin-like pathway there is the conversion of LC3 I to LC3 II due to conjugation of phosphatidylethanolamine (PE) to LC3-I via sequential action of ATG4, ATG7 and ATG3. The conjugation of PE to LC3 facilitates the closure of the isolation membrane. ATG4 removes lipidated LC3 (LC3-II) from the outer surface of the autophagosome while LC3 on the inner surface is degraded when the autophagosome fuses with the lysosome to form an autolysosome.
Fig.1
The mammalian autophagy pathway. The key steps involved in autophagy and critical members of the autophagy pathway are illustrated. The members of the autophagy pathway that have thicker black circles are those that have been studied in relation to autophagy and adult neurogenesis, as listed in Table 2, and described within the text.
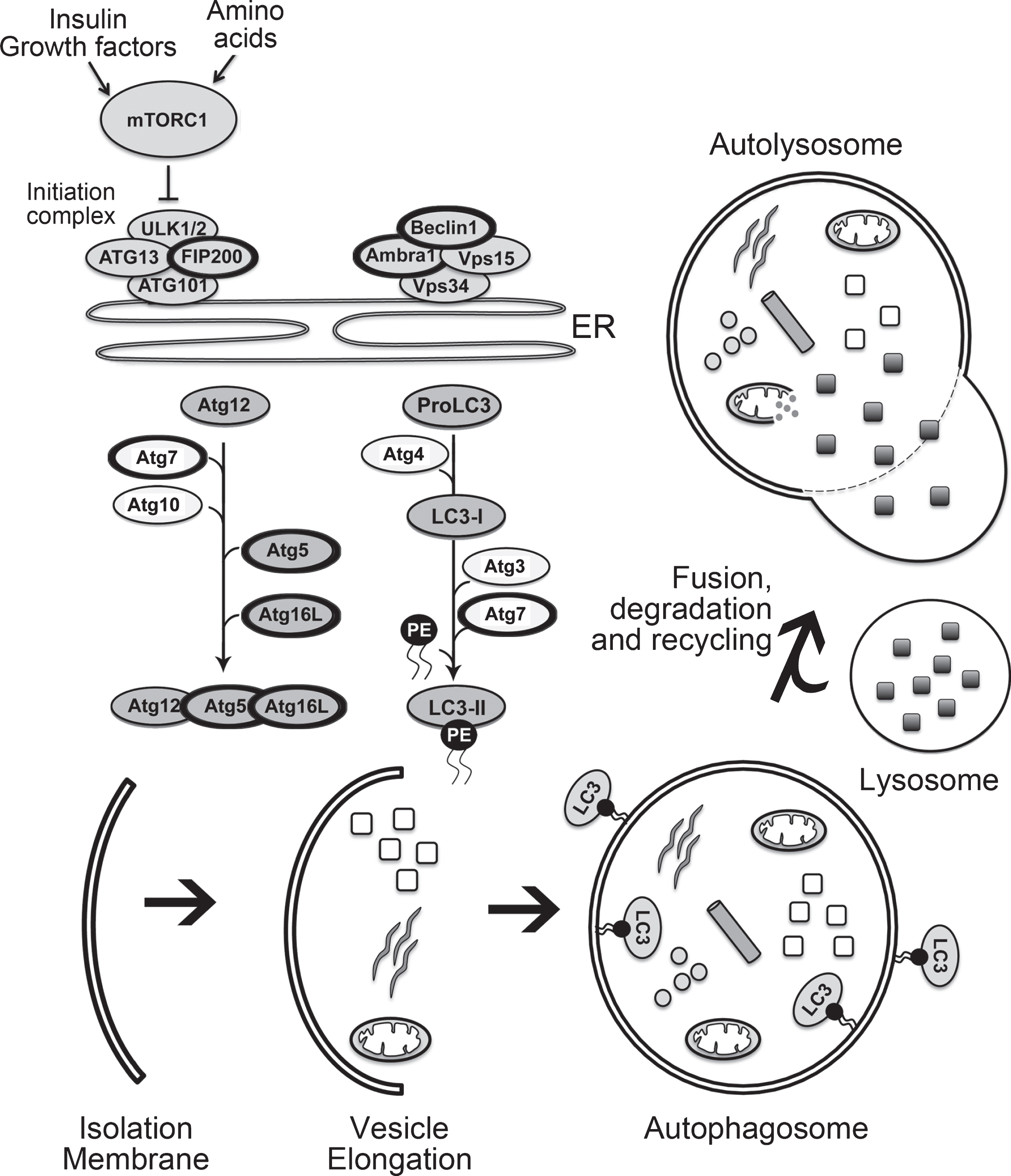
One of the challenges in the autophagy research is accurately measuring this process. Given that there is currently no “gold standard” for detecting and quantifying autophagy, multiple methods are used and guidelines have been drawn up to standardize these methods in different cellular contexts [34]. A common approach is to quantify the number or volume of different components of the autophagy process, such as the autophagosomes or autolysosomes. Although this method is very popular, caution is advised when interpreting these outcomes as “percentage change” in the amount of autophagy, since the appearance of more autophagosomes in a given cell does not always reflect more autophagic activity [35, 36]. This is because autophagosomes can accumulate as a result of blocking their degradation, without a change in autophagosome biogenesis. To be accurate, interpretation of an outcome as a change in autophagy requires a measure of the flux in the amount and rate of cargo sequestered and degraded.
Given this concern with “static” measures of autophagy, techniques have been refined or created that allow for monitoring of autophagy flux, which is an indication of the amount and rate of cargo sequestered and degraded [34, 36]. For example, an increase in the accumulation of LC3-II on a western blot is now commonly used as a measure of flux when measured in conjunction with lysosomal degradation inhibitors. Due to confounds with the use of lysosomal inhibitors, there is also increasing use of tandem florescent constructs such as mRFP/mCherry-GFP-tagged LC3 to measure flux [34, 37]. This fluorescence assay allows for simultaneous measurement of the induction of autophagy and flux through the autophagic compartments. Flux is monitored by different emission signals depending on the environment, which allows for a decrease in green/red (yellow) fluorescence (phagophores, autophagosomes) and an increase in red fluorescence (autolysosomes). Given that this method requires single cell analysis, a variety of flow and multispectral imaging cytometric methods are being developed that allow for medium- to high-throughput analysis [34, 38]. A caveat with this approach is that in the absence of permeabilization of the cells these methods cannot accurately evaluate localization of the autophagosomes or lipidation of LC3-II. Ongoing efforts are also focused on the optimized detection of autophagy flux in vivo or in organs, due to the relatively less developed methods compared to those that exist for use in cell culture [34]. The main methods used in vivo include GFP-LC3 mice or transgenic strains or viral methods that utilize a GFP-LC3 or mRFP/mCherry-GFP-tagged LC3 plasmid [39, 40]. Enhancements in the ability to measure autophagy in vivo is particularly important for the study of autophagy in adult neurogenesis given the caveats of in vitro methods to measure adult neurogenesis and effects of the neurogenic niche on adult neurogenesis. The methods that have been specifically used to monitor autophagy in adult neurogenesis studies, as well as their caveats, are additionally summarized in Table 1 and discussed below in greater detail for each study.
Table 1
Methods used to monitor autophagy in adult neurogenesis studies
| Assay | In vitro or | Readout | Caveats of technique | Reference |
| In vivo | (from Klionsky DJ et al., 2016) | |||
| Transmission Electron Microscopy (TEM) | In vitro | Vacuoles containing cytoplasmic contents (autophagosomes or autolysosomes) | Difficult to quantify using unbiased method. Variability in cell areas sampled. | [55] |
| Detection of LC3-I and LC3-II | In vitro | Increased band intensity of LC3-II and increased ratio of LC3-II over LC3-I | Difficult to distinguish between upregulation of autophagosome formation or blockage of autophagic degradation without using lysosomal inhibitors. | [55] |
| [56] | ||||
| [57] | ||||
| [58] | ||||
| Detection of LC3-II and p62 in presence of chloroquine | In vitro | Lower LC3-II and higher p62 accumulation | Changes in p62 levels can be specific to cell-type and context being studied. | [47] |
| [50] | ||||
| p62 sequestosome/degradation assay | In vitro | Accumulation of p62 in presence of proteasome inhibitor | Not always a correlation between increase in autophagy and decrease in P62 levels. | [57, 58] |
| LC3 immunofluorescence | In vitro | Reduced in number of LC3 puncta | Not all LC3 puncta represent LC3-II and correspond to autophagosomes. | [50] |
| GFP-LC3 puncta | In vitro | Increase in number of GFP-LC3 puncta | High levels of GFP-LC3 can associate with ubiquitinated aggregates to lead to misinterpretation about the number of autophagic structures. Reflects only a snapshot of the numbers of autophagy-related structures. | [56] |
| GFP-LC3 transgenic mice and detection of LC3-II in presence of chloroquine | In vitro and in vivo | Phenotype cells expressing GFP-LC3 puncta. Lower LC3-II accumulation | GFP-LC3 expression driven by CAG promoter, thus intensity of the GFP signal may represent CAG promoter activity, not autophagic activity. | [49] |
| mRFP-GFP-LC3 tandem construct | In vitro | Increases in percentage of mRFP-only positive LC3 puncta. | Colocalization of GFP and mRFP might be seen in the case of impaired proteolytic degradation within autolysosomes, or altered lysosomal pH | [58] |
| mCherry-EGFP-LC3 retrovirus | In vivo | mCherry+ puncta (autolysosomes) around cell body. | Difficult to distinguish between reduced autophagosome production or enhanced autolysosomal degradation without using lysosomal inhibitors. | [40] |
AUTOPHAGY AND NEUROGENESIS
Lethality associated with embryonic knockout of autophagy
The function of autophagy in adult NSCs and adult neurogenesis is in its infancy compared to the knowledge about the role of autophagy in mammalian development [29, 41]. Research into the function of autophagy in development was ignited by the first knockout mouse made for Atg5 in 2004 [42]. While the Atg5 knockout mice were able to produce offspring, they were neonatal lethal, highlighting the essential role of autophagy for neonatal survival in the developing animal. Similar phenotypes have been reported for other Atg genes such as Atg3, Atg7, Atg9 and Atg16L1, whereas Beclin1, Ambra1, FIP200 mice are all unable to produce homozygous offspring due to embryonic lethality [29, 41]. Although these developmental abnormalities support the requirement of autophagy for nutrient recycling, the different phenotypes observed for the various autophagy gene knockout mouse models remain only partly understood. There are many possible explanations for these effects including varying degrees of redundancy between different Atg proteins, different levels of compensatory mechanisms, and potential confounds due to autophagy-independent functions for these genes [41].
Effects of conditional embryonic disruption of autophagy on adult neurogenesis
Lethality in the knockout mouse models led to the creation of tissue-specific conditional autophagy knockout models that enhanced the understanding of the function of autophagy. In 2006 it was reported that removal of Atg5 from NSCs using a nestin promoter driving Cre recombinase led to accumulation of cytoplasmic inclusion bodies, neurodegeneration, and progressive deficits in motor function starting at 3 weeks of age [43]. A similar progressive neurodegeneration phenotype was also reported in 2006 for the neuronal-specific Atg7 knockout mice [44]. These mice had a more severe phenotype, with massive loss of pyramidal neurons in the cortex and Purkinje cells in the cerebellum. Notably, there was no reported phenotype within the neurogenic niche of the hippocampus or olfactory bulb. In these studies using the Nestin-Cre conditional mice, however, the ablation of Atg5 or Atg7 was first initiated within the nestin-expressing NSCs at embryonic day 10.5 [43, 44]. The observed phenotypes thus resulted from Atg-null stem and progenitors that generated Atg-null mature neurons for a lifespan. Therefore, these findings were seminal in highlighting that autophagy has an important intracellular quality control function in non-dividing neurons over their lifespan. Interestingly, this role also extends to axons, as demonstrated by Atg7-deficient Purkinje axons exhibiting progressive dystrophy and degeneration of terminals that proceeded neurodegeneration [45]. As extensively reviewed by others in detail [46], the role of autophagy in quality control within the brain is even more essential when autophagy is required for the elimination of misfolded or damaged proteins, such as pathogenic aggregate-prone proteins, which has numerous implications for neurodegenerative diseases.
The implication of autophagy regulating adult neurogenesis following conditional embryonic disruption of autophagy, was first reported in 2013 following ablation of FIP200. As summarized in Table 2, this study was subsequently followed by others that made mouse models that ablated or modified either Ambra 1, Atg5, Atg7, Atg16L1, Beclin 1 or FIP200, which are all highlighted in Fig. 1 as members of the autophagy pathway. In general, these studies used in vitro and in vivo approaches, utilized different measures to assess autophagy (Table 1), examined the SVZ and/or SGZ neurogenic cells, and have showed differential effects on the processes of NSC self-renewal, proliferation, survival and differentiation. Each of these studies will be briefly described below.
Table 2
Studies examining the role of autophagy in adult neurogenesis
| Gene | Model | Age at time | Region | Effect (increase ↑, decrease ↓, | Reference | ||||
| of disruption | SGZ/SVZ | no change ↔, not assessed (na)) | |||||||
| Embryo (E) | Stem Cell | Proliferation | Survival | Differentiation | |||||
| Adult (A) | Renewal | ||||||||
| Ambra1 | In vitro | Neurosphere Assay Ambra1 –/+ KO | E | SVZ | ↓ | ↓ | ↓ | ↓ | [49] |
| Atg5 | In vivo | Retroviral Cre Floxed Atg5 | A | SGZ | na | ↔ | ↓ | ↔ | [40] |
| In vivo | Retroviral Cre Floxed Atg5/BAX KO | na | ↔ | rescue | ↔ | ||||
| In vitro | Neurosphere Assay | E | SGZ, SVZ | ↔ | ↔ | ↔ | ↔ | [50] | |
| In vivo | Atg5GFAP cKO | ||||||||
| Atg7 | In vitro | Neurosphere Assay | E | SGZ, SVZ | ↔ | ↔ | ↔ | ↔ | [50] |
| In vivo | Atg5GFAP cKO | ||||||||
| Atg16L1 | In vitro | Neurosphere assay | E | SGZ, SVZ | ↔ | ↔ | ↔ | ↔ | [50] |
| In vivo | Atg16L1GFAP cKO | ||||||||
| In vivo | ATG16L1 hypomorph | E | SVZ | na | ↓ | ↔ | ↔ | [51] | |
| Beclin1 | In vitro | Neurosphere assay | E | SVZ | ↓ | ↓ | ↓ | ↓ | [49] |
| In vivo | Beclin1 –/+ | ||||||||
| FIP200 | In vitro | Neurosphere assay | E | SGZ, SVZ | ↓ | ↓ | ↓ | ↓ | [47] |
| In vivo | FIP200GFAP cKO | ||||||||
| In vivo | FIP200;Trp53 cKO | rescue | rescue | ↔ | ↔ | ||||
| In vivo | FIP200;p62 cKO | rescue | rescue | rescue | rescue | [50] | |||
Wang et al. in 2013 [47] discovered that embryonic disruption of autophagy had a robust effect of ablating adult neurogenesis in a GFAP-Cre:FIP200f/f mouse (hereafter referred to as FIP200GFAP cKO mouse). In this mouse, the human GFAP promoter drives the expression of Cre recombinase to knock out FIP200 in radial glia NSCs beginning at approximately embryonic day 10.5. FIP200 functions as a component of the multiprotein complex ULK1/Atg13/FIP200/Atg101, which is required for induction of autophagy in mammalian cells (Fig. 1) [48]. The FIP200GFAP cKO mice had a dramatic aging phenotype characterized by a reduction in NSCs, NPCs, neuroblasts and adult-generated neurons in the olfactory bulb and the dentate gyrus. The loss of FIP200 was confirmed to reduce autophagy through examination of the lysates from the SVZ of the cKO mice treated with the lysosomal inhibitor chloroquine, with lower LC3-II levels and accumulation of p62 observed (Table 1). The progressive depletion of NSCs was accompanied by increased apoptosis of the adult-generated cells and significant reduction in proliferation and self-renewal of the NSCs as demonstrated by in vitro and in vivo methods. Ablation of FIP200 not only inhibited autophagy but also led to an accumulation of mitochondria and an increase in the levels of reactive oxygen species (ROS) in NSCs. Suppression of the aberrant ROS and consequent elevation in p53 could rescue the NSC phenotype. Together these results support the hypothesis that autophagy helps mitigate metabolic stress in neuronal cells by recycling damaged organelles and proteins, as observed in several other cell types [31].
This seminal study also raised many questions. Firstly, despite the depletion of FIP200 occurring in the embryonic NSCs, there was no effect on the embryonic NSCs. In this model, the phenotype occurred between P0 to 4 weeks of age, which was in part attributed to a relatively low amount of autophagy occurring in the embryonic NSCs. This conclusion was based on quantification of the amount of LC3-II and p62 in the cortex (Table 1), leaving future work to determine if these findings translate into a reduction in autophagic flux specifically in the embryonic versus postnatal NSCs and NPCs. Secondly, there was increased astrogenesis (nestin-/GFAP+ cells) in the FIP200GFAP cKO mice that could be rescued by scavenging ROS. This occurred due to FIP200 being removed using the GFAP-Cre mice, raising the need for future work to determine whether removal of FIP200, or a reduction in autophagy would alter adult neurogenesis, independent of alterations in astrogenesis. Lastly, the potential for differential effects of the loss of FIP200 in the different NSC populations, such as nestin-expressing NSCs, within the adult brain remains to be addressed.
The question of whether adult neurogenesis was differentially altered by embryonic conditional removal of other members of the autophagy pathway was addressed subsequently through examination of mice that were a heterozygous embryonic knockout for Beclin1 and Ambra1 (activating molecule in Beclin 1-regulated autophagy) [49]. These two proteins are involved in the complex promoting nucleation (Fig. 1) and these mice had reductions in adult neurogenesis. This work showed in vitro basal levels of autophagy increasing in concomitance with the early steps of neuronal differentiation, using the GFP-LC3 mice combined with the chloroquine treatment to inhibit lysosome-mediated proteolysis. Both Beclin1 and Ambra1 were also shown to be required to sustain self-renewal, proliferation, survival and the differentiation of the SVZ-derived neurospheres (Table 1). Additionally, in vivo high expression of Beclin1 and Ambra1 was observed in cells in the SVZ. Beclin1+/– mice also had a significant reduction in Ki67-expressing proliferating NPCs and Tuj1-expressing immature neurons, accompanied by increased apoptosis in the SVZ. Although these findings do not include in vivo analysis of Ambra1 mice, nor is there examination of the hippocampus, overall, they support the idea that early autophagy regulators, like Ambra1 and Beclin 1, are similar to FIP200, in that their absence has detrimental effects on all stages of adult neurogenesis.
Given the consistent reduction in adult neurogenesis in autophagy-deficient mouse models, it was counterintuitive when in 2016 Wang et al. [50] published that ablation of three genes required for autophagosome elongation, Atg5, Atg16L and Atg7, had no impact on adult neurogenesis. These findings came from mice created using the GFAP promoter to drive Cre expression that had previously been used to produce the FIP200GFAP cKO mice that had an increase in mitochondrial mass and ROS. Similar to FIP200GFAP cKO mice, the Atg5, Atg16L1, and Atg7 cKO mice also had postnatal deficiencies in autophagic flux and increased functional and dysfunctional mitochondria mass. Given that none of the three models had abnormal neurogenesis in vitro or in vivo, these results suggested that an increase in mitochondria mass and elevation of ROS alone is not sufficient to induce NSC and NPC deficits in adult neurogenesis. The disparity between the phenotype of the FIP200 cKO and the Atg5, Atg16L1, and Atg7 cKO models was in part resolved through the additional discovery that the FIP200-null cells, compared to either Atg5 or Atg16L1-null cells, had significantly higher accumulation of the adaptor protein p62. This ultimately resulted in superoxide O2•– accumulation caused by decreased cytoplasmic SOD1 level that inhibited neurogenesis. In agreement with this hypothesis that the elevation in p62 was causally related to the reduction in neurogenesis, the concurrent ablation of p62 was able to completely rescue the neurogenesis phenotype in the FIP200 cKO mice, in the absence of altered increases in mitochondrial mass. The question of what triggers the increase in p62 levels that leads to the deficits in NSC maintenance and differentiation remains to be determined. One potential reason could be the implication of both the canonical and noncanonical functions occurring independently of the effect of Fip200 on autophagy induction. Overall, however, this seminal study revealed that, in contrast to downstream autophagy genes, the autophagy induction gene FIP200 was required within postnatal NSCs following its removal during early development.
Whereas the Atg16L1 cKO mice showed a lack of phenotype, mice with a hypomorphic mutation in Atg16L1 were shown to have modest impairments in autophagy along with deficits in Notch degradation [51]. In this model, the consequence of hyperactive Notch signaling was associated with a significant developmental retention in neurogenesis and haematopoiesis in the gut villi. Specifically, within the neurogenic regions there was a significant reduction in BrdU-labeled cells that could be attenuated by pharmacological inhibition of Notch. Although it remains to be determined which neurogenic cell types (NSCs, NPCs) were specifically affected in Atg16L1 hypomorphic mutation, it is probable that active Notch signaling led to repression of pro-neural gene expression and maintenance of NSCs, as previously described for Notch signaling [52]. Thus, this study also makes an important contribution in highlighting that modest impairments in autophagy can have different effects, mediated through Notch signaling, compared to a complete inhibition of autophagy.
Together, these studies highlight that there is a delicate balance in how members of the autophagy machinery can alter adult NSCs following embryonic disruption of autophagy. They also raise a variety of questions about mechanisms that regulate these phenotypes and point to the need to understand whether some of these in vivo findings are only found in GFAP-expressing NSCs or are more global effects that occur in diverse NSC and NPC populations [23, 24]. Furthermore, they extend the challenge to determine whether the effects on adult neurogenesis are due to disruption of autophagy or the loss of other non-autophagy functions of these genes, which is a common challenge in all autophagy research.
Requirement of autophagy in adult-born NSCs and their progeny
It is increasingly recognized that embryonic and adult neurogenesis have distinct mechanisms that can control NSC proliferation, survival and differentiation [53, 54], making it necessary for autophagy to be specifically measured in the context of adult neurogenesis. As described below, this work includes a variety of studies using HCN cells, as well as in our published work examining the effect of removing Atg5 within the adult dividing NPCs.
A number of studies by Dr. Yu’s laboratory have utilized the in vitro HCNs to establish the function of autophagy in the context of cell death. When cells were cultured under conditions of insulin withdrawal, HCNs underwent autophagic cell death (ACD) instead of apoptosis [55]. Importantly, knockdown of Atg7 suppressed ACD under these conditions. Three separate studies using similar growth conditions showed that indirect modulation of autophagy could affect cell death [56, 57]. Glycogen synthase kinase 3β (GSK-3β) was identified as a positive regulator of autophagic flux and ACD. Knockdown of Atg7 could reverse GSK-3β induced ACD, implicating ACD as the primary cell death mechanism in insulin-deficient cells [56]. Cell death could also be controlled by the expression of Calpains 1 and 2, with ACD enhanced following Calpain 2 inhibition and reduced following ectopic expression of Calpain1. This work identified Calpains as potential regulators of a switch between ACD and apoptosis [57]. Finally, Chung et al. [58] described the Type 3 Ryanodine Receptor (RyR3) as a mediator of ACD by showing that treatment with RyR agonist enhanced ACD, and that this effect was blocked by Atg7 knockdown. Together these in vitro studies suggest ACD as a means of eliminating dysfunctional HCNs when insulin signaling is impaired, supporting the hypothesis that autophagy is a quality control mechanism in the adult stem and progenitor cells [59].
To address whether autophagy is required for adult dividing NPCs in vivo, we measured autophagic flux during the development of adult-generated cells and showed flux with autolysosomes in both the soma and the dendrites of developing NPCs [40]. Given that autophagy can regulate postnatal cortical spine pruning [60], and the in vivo imaging work demonstrating that pruning occurs in the developing dendrites of adult-born cells [61], our findings raise the interesting hypothesis that autophagy may be a player that can regulate the pruning of adult-born cells. We also observed a reduction in the number of autolysosomes with cell maturation [40], which contrasts the increase in flux found concurrent with differentiation in an in vitro study using the LC3-GFP reporter mice [49].
To examine the function of autophagy in adult-generated neurons, we ablated Atg5 from dividing NPCs using a retroviral delivery of GFP-Cre in Atg5fl/fl mice and showed a reduction in autophagic flux in the NPCs [40]. Atg5-null cells also had a significant reduction in survival during a critical time period of 3 to 7 days old, which is the time that they are most likely to undergo death [5]. Unlike all of the developmental disruptions of autophagy reviewed above, this delay in maturation was not accompanied by any significant differences in proliferation. Also, although the cells were delayed in differentiation, those that did mature formed granule hippocampal neurons. These results are in contrast to the early embryonic requirement for Atg5 in differentiation of cortical NPCs and astrocytes, as well as the lack of adult neurogenic phenotype following removal of Atg5 from embryonic GFAP-expressing NSCs [62, 63]. Given the heterogeneity of NSCs in the adult neurogenic niche, this further strengthens the need for future studies to use different adult inducible transgenic mice to alter and examine autophagy regulation in the different neurogenic populations [23, 64].
Adult neurogenesis can be regulated by several physiological stimuli at almost every stage of the neurogenic process (for review see [7]). Physical exercise such as voluntary running is just one example that stimulates neurogenesis by enhancing proliferation and survival [65] and can also induce autophagy in the brain [66]. Supporting the suggestion that autophagy may have a mechanistic role in mediating running-induced neurogenesis, we found Atg5 is required for the running-induced increase in survival of NPCs [40]. Whether other essential genes of the autophagy pathway are required for running-induced neurogenesis remains unknown, and future work will also need to determine if autophagy is involved in modulation of neurogenesis by other physiologic stimuli, such as environmental enrichment.
CONCLUSIONS AND FUTURE DIRECTIONS
We are just beginning to unravel the connections between autophagy and adult neurogenesis. The evidence to date suggests that certain key members of the autophagy pathway are necessary for NSCs to produce adult-generated neurons, however many questions remain unanswered. These include the physiological significance of autophagy in the different stages of adult neurogenesis, identification of the content of the autophagosomes and autolysosomes, and dissection of the molecular mechanisms that regulate autophagic flux during adult neurogenesis.
Elucidation of the means by which changes in adult neurogenesis and autophagy are connected to cellular changes in metabolism also requires future work. In hematopoietic stem cells, loss of autophagy induces accumulation of mitochondria and an activated metabolic state that accelerates myeloid differentiation and impairs stem-cell self-renewal activity and regenerative potential with aging [67]. In NSCs there is also a growing appreciation for how metabolic programs and mitochondria dynamics are functionally linked to distinct developmental steps within adult neurogenesis [68–70]. For example, manipulation of mitochondrial structure impairs NSC self-renewal, with consequent age-dependent depletion and neurogenesis defects that occur through a ROS-mediated process [71]. Perturbation of mitochondrial complex function impairs lineage progression of adult NSCs, and enhancement of mitochondrial function can promote neurogenesis in the aging hippocampus [68]. These results, when combined with the connections between autophagy and adult neurogenesis reviewed here and the connections between autophagy and metabolism [72], make it highly likely that the autophagic and metabolic pathways coordinate the life-long addition of adult-generated neurons to the brain.
Recent technical advances in single-cell sequencing have enabled identification of genome-wide molecular transitions that occur at different stages of stem cell development [73]. For example, in the field of adult neurogenesis, the transcriptional changes that occur when the quiescent NSCs are activated and neurogenesis occurs are being identified [26]. Similarly, in the field of autophagy, the changes in transcriptional activity of autophagy-related genes in hematopoietic stem cell formation have been identified during mouse embryogenesis [74]. These methodological advances can now be applied to drive forward the next phase of discoveries on how autophagy regulates adult neurogenesis. Discovering the regulation of the autophagy gene network under both physiological and pathological conditions during the different stages of adult neurogenesis will foreseeably lead to novel insights for therapeutic targets to stimulate brain repair.
ACKNOWLEDGMENTS
We would like to thank Alexander Sanghwan for research assistance. Our research is supported by a National Sciences and Engineering Research Council of Canada (NSERC), Canadian Institute of Health Research (CIHR) and the Canadian Partnership for Stroke Recovery (CPSR).
REFERENCES
[1] | Yamamoto A , Yue Z . Autophagy and its normal and pathogenic states in the brain. Annu Rev Neurosci. (2014) ;37: :55–78. |
[2] | Altman J , Das GD . Autoradiographic and histological evidence ofpostnatal hippocampal neurogenesis in rats. J Comp Neurol. (1965) ;124: :319–35. |
[3] | Lledo PM , Valley M . Adult olfactory bulb neurogenesis. Cold Spring Harb Perspect Biol. (2016) ;8. |
[4] | Song J , Olsen RH , Sun J , Ming GL , Song H . Neuronal circuitry mechanisms regulating adult mammalian neurogenesis. Cold Spring Harb Perspect Biol. (2016) ;8. |
[5] | Gonçalves JT , Schafer ST , Gage FH . Adult neurogenesis in the hippocampus: From stem cells to behavior. Cell. (2016) ;167: :897–914. |
[6] | Lim DA , Alvarez-Buylla A . The Adult Ventricular-Subventricular Zone (V-SVZ) and Olfactory Bulb (OB) Neurogenesis. Cold Spring Harb Perspect Biol. (2016) ;8. |
[7] | Ming GL , Song H . Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron. (2011) ;70: :687–702. |
[8] | Lee DA , Bedont JL , Pak T , Wang H , Song J , Miranda-Angulo A , Takiar V , Charubhumi V , Balordi F , Takebayashi H , et al. Tanycytes of the hypothalamic median eminence form a diet-responsive neurogenic niche. Nature Neuroscience. (2012) ;15: :700–2. |
[9] | Kokoeva MV , Yin HL , Flier JS . Neurogenesis in the hypothalamus of adult mice: Potential role in energy balance. Science. (2005) ;310: :679–83. |
[10] | Robins SC , Stewart I , McNay DE , Taylor V , Giachino C , Goetz M , Ninkovic J , Briancon N , Maratos-Flier E , Flier JS , et al. alpha-Tanycytes of the adult hypothalamic third ventricle include distinct populations of FGF-responsive neural progenitors. Nat Commun. (2013) ;4: :2049. |
[11] | Kim MS , Quan W , Lee MS . Role of hypothalamic autophagy in the control of whole body energy balance. Rev Endocr Metab Disord. (2013) ;14: :377–86. |
[12] | Palmer TD , Markakis EA , Willhoite AR , Safar F , Gage FH . Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. (1999) ;19: :8487–97. |
[13] | Palmer TD , Takahashi J , Gage FH . The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. (1997) ;8: :389–404. |
[14] | Reynolds BA , Weiss S . Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. (1992) ;255: :1707–10. |
[15] | Song HJ , Stevens CF , Gage FH . Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat Neurosci. (2002) ;5: :438–45. |
[16] | Takahashi J , Palmer TD , Gage FH . Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol. (1999) ;38: :65–81. |
[17] | Pastrana E , Silva-Vargas V , Doetsch F . Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell. (2011) ;8: :486–98. |
[18] | Suhonen JO , Peterson DA , Ray J , Gage FH . Differentiation of adult hippocampus-derived progenitors into olfactory neurons in vivo. Nature. (1996) ;383: :624–7. |
[19] | Kuhn HG , Eisch AJ , Spalding K , Peterson DA . Detection and phenotypic characterization of adult neurogenesis. Cold Spring Harb Perspect Biola. (2016) ;8: :025981. |
[20] | Enikolopov G , Overstreet-Wadiche L , Ge S . Viral and transgenic reporters and genetic analysis of adult neurogenesis. Cold Spring Harb Perspect Biol. (2015) ;7: :a018804. |
[21] | Dhaliwal J , Lagace DC . Visualization and genetic manipulation of adult neurogenesis using transgenic mice. Eur J Neurosci. (2011) ;33: :1025–36. |
[22] | Sun MY , Yetman MJ , Lee TC , Chen Y , Jankowsky JL . Specificity and efficiency of reporter expression in adult neural progenitors vary substantially among nestin-CreER(T2) lines. J Comp Neurol. (2014) ;522: :1191–208. |
[23] | DeCarolis NA , Mechanic M , Petrik D , Carlton A , Ables JL , Malhotra S , Bachoo R , Götz M , Lagace DC , Eisch AJ . In vivo contribution of nestin- and GLAST-lineage cells to adult hippocampal neurogenesis. Hippocampus. (2013) ;23: :708–19. |
[24] | Bonaguidi MA , Stadel RP , Berg DA , Sun J , Ming GL , Song H . Diversity of neural precursors in the adult mammalian brain. Cold Spring Harb Perspect Biol. (2016) ;8: :a018838. |
[25] | Calzolari F , Michel J , Baumgart EV , Theis F , Götz M , Ninkovic J . Fast clonal expansion and limited neural stem cell self-renewal in the adult subependymal zone. Nat Neurosci. (2015) ;18: :490–492. |
[26] | Shin J , Berg DA , Zhu Y , Shin JY , Song J , Bonaguidi MA , Enikolopov G , Nauen DW , Christian KM , Ming GL , Song H . Single-cell RNA-seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell. (2015) ;17: :360–72. |
[27] | Eskelinen EL , Reggiori F , Baba M , Kovács AL , Seglen PO . Seeing is believing: The impact of electron microscopy on autophagy research. Autophagy. (2011) ;7: :935–56. |
[28] | Tooze SA , Dikic I . Autophagy captures the nobel prize. Cell. (2016) ;167: :1433–5. |
[29] | Ohsumi Y . Historical landmarks of autophagy research. Cell Res. (2014) ;24: :9–23. |
[30] | Mizushima N . The exponential growth of autophagy-related research: From the humble yeast to the Nobel Prize. FEBS Lett. (2017) ;591: :681–9. |
[31] | Mizushima N , Komatsu M . Autophagy: Renovation of cells and tissues. Cell. (2011) ;147: :728–741. |
[32] | Fullgrabe J , Klionsky DJ , Joseph B . The return of the nucleus: Transcriptional and epigenetic control of autophagy. Nature Reviews Molecular Cell Biology. (2014) ;15: :65–74. |
[33] | Nixon RA . The role of autophagy in neurodegenerative disease. Nat Med. (2013) ;19: :983–97. |
[34] | Klionsky DJ , Abdelmohsen K , Abe A , Abedin MJ , Abeliovich H , Acevedo Arozena A , Adachi H , Adams CM , Adams PD , Adeli K , et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) ;12: :1–222. |
[35] | Klionsky DJ , Abdalla FC , Abeliovich H , Abraham RT , Acevedo-Arozena A , Adeli K , Agholme L , Agnello M , Agostinis P , Aguirre-Ghiso JA , et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. (2012) ;8: :445–544. |
[36] | Tanida I , Minematsu-Ikeguchi N , Ueno T , Kominami E . Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. (2005) ;1: :84–91. |
[37] | Kimura S , Noda T , Yoshimori T . Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. (2007) ;3: :452–60. |
[38] | Eng KE , Panas MD , Karlsson Hedestam GB , McInerney GM . A novel quantitative flow cytometry-based assay for autophagy. Autophagy. (2010) ;6: :634–41. |
[39] | Mizushima N . Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. (2009) ;452: :13–23. |
[40] | Xi Y , Dhaliwal JS , Ceizar M , Vaculik M , Kumar KL , Lagace DC . Knockout of Atg5 delays the maturation and reduces the survival of adult-generated neurons in the hippocampus. Cell Death Dis. (2016) ;7: :e2127. |
[41] | Mizushima N , Levine B . Autophagy in mammalian development and differentiation. Nat Cell Biol. (2010) ;12: :823–30. |
[42] | Kuma A , Hatano M , Matsui M , Yamamoto A , Nakaya H , Yoshimori T , Ohsumi Y , Tokuhisa T , Mizushima N . The role of autophagy during the early neonatal starvation period. Nature. (2004) ;432: :1032–6. |
[43] | Hara T , Nakamura K , Matsui M , Yamamoto A , Nakahara Y , Suzuki-Migishima R . Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. (2006) ;441: :885–9. |
[44] | Komatsu M , Waguri S , Chiba T , Murata S , Iwata J , Tanida I , Ueno T , Koike M , Uchiyama Y , Kominami E , Tanaka K . Loss of autophagy inthe central nervous system causes neurodegeneration inmice. Nature. (2006) ;441: :880–4. |
[45] | Komatsu M , Wang QJ , Holstein GR , Friedrich VL Jr , Iwata J , Kominami E , Chait BT , Tanaka K , Yue Z . Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. (2007) ;104: :14489–94. |
[46] | Menzies FM , Fleming A , Caricasole A , Bento CF , Andrews SP , Ashkenazi A , Füllgrabe J , Jackson A , Jimenez Sanchez M , Karabiyik C , et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. (2017) ;93: :1015–34. |
[47] | Wang C , Liang CC , Bian ZC , Zhu Y , Guan JL . FIP200 is required for maintenance and differentiation of postnatal neural stem cells. Nat Neurosci. (2013) ;16: :532–42. |
[48] | Hara T , Takamura A , Kishi C , Iemura SI , Natsume T , Guan JL , Mizushima N . FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. Journal of Cell Biology. (2008) ;181: :497–510. |
[49] | Yazdankhah M , Farioli-Vecchioli S , Tonchev AB , Stoykova A , Cecconi F . The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell Death Dis. (2014) ;5: :e1403. |
[50] | Wang CR , Chen S , Yeo S , Karsli-Uzunbas G , White E , Mizushima N , Virgin HW , Guan JL . Elevated p62/SQSTM1 determines the fate of autophagy-deficient neural stem cells by increasing superoxide. Journal of Cell Biology. (2016) ;212: :545–60. |
[51] | Wu X , Fleming A , Ricketts T , Pavel M , Virgin H , Menzies FM , Rubinsztein DC . Autophagy regulates Notch degradation and modulates stem cell development and neurogenesis. Nat Commun. (2015) ;7: :10533. |
[52] | Imayoshi I , Sakamoto M , Yamaguchi M , Mori K , Kageyama R . Essential roles of notch signaling in maintenance of neural stem cells in developing and adult brains. Journal of Neuroscience. (2010) ;30: :3489–98. |
[53] | Urban N , Guillemot F . Neurogenesis in the embryonic and adult brain: Same regulators, different roles. Front Cell Neurosci. (2014) ;8: :396. |
[54] | Gotz M , Nakafuku M , Petrik D . Neurogenesis in the developing and adult brain-similarities and key differences. Cold Spring Harb Perspect Biol. (2016) ;8. |
[55] | Yu SW , Baek SH , Brennan RT , Bradley CJ , Park SK , Lee YS , Jun EJ , Lookingland KJ , Kim EK , Lee H , et al. Autophagic death of adult hippocampal neural stem cells following insulin withdrawal. Stem Cells. (2008) ;26: :2602–10. |
[56] | Ha S , Ryu HY , Chung KM , Baek SH , Kim EK , Yu SW . Regulation of autophagic cell death by glycogen synthase kinase-3beta in adult hippocampal neural stem cells following insulin withdrawal. Mol Brain. (2015) ;8: :30. |
[57] | Chung KM , Park H , Jung S , Ha S , Yoo SJ , Woo H , Lee HJ , Kim SW , Kim EK , Moon C , Yu SW . Calpain determines the propensity of adult hippocampal neural stem cells to autophagic cell death following insulin withdrawal. Stem Cells. (2015) ;33: :3052–64. |
[58] | Chung KM , Jeong EJ , Park H , An HK , Yu SW . Mediation of autophagic cell death by type 3 ryanodine receptor (RyR3) in adult hippocampal neural stem cells. Front Cell Neurosci. (2016) ;10. |
[59] | Hong CJ , Park H , Yu SW . Autophagy for the quality control of adult hippocampal neural stem cells. Brain Res. (1649) 166–72. |
[60] | Tang G , Gudsnuk K , Kuo SH , Cotrina ML , Rosoklija G , Sosunov A , Sonders MS , Kanter E , Castagna C , Yamamoto A , et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron. (2014) ;83: :1131–43. |
[61] | Gonçalves JT , Bloyd CW , Shtrahman M , Johnston ST , Schafer ST , Parylak SL , Tran T , Chang T , Gage FH . In vivo imaging of dendritic pruning in dentate granule cells. Nat Neurosci. (2016) ;19: :788–91. |
[62] | Wang S , Li B , Qiao H , Lv X , Liang Q , Shi Z , Xia W , Ji F , Jiao J . Autophagy-related gene Atg5 is essential for astrocyte differentiation in the developing mouse cortex. EMBO Re. (2014) ;15: :1053–61. |
[63] | Lv X , Jiang H , Li B , Liang Q , Wang S , Zhao Q , Jiao J . The crucial role of Atg5 in cortical neurogenesis during early brain development. Sci Re. (2014) ;4: :6010. |
[64] | Gebara E , Bonaguidi MA , Beckervordersandforth R , Sultan S , Udry F , Gijs PJ , Lie DC , Ming GL , Song H , Toni N . Heterogeneity of radial glia-like cells in the adult hippocampus. Stem Cells. (2016) ;34: :997–1010. |
[65] | van Praag H , Kempermann G , Gage FH . Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci. (1999) ;2: :266–70. |
[66] | He C , Sumpter R Jr , Levine B . Exercise induces autophagy in peripheral tissues and in the brain. Autophagy. (2012) ;8: :1548–51. |
[67] | Ho TT , Warr MR , Adelman ER , Lansinger OM , Flach J , Verovskaya EV , Figueroa ME , Passegué E . Autophagy maintains the metabolism and function of young and old stem cells. Nature. (2017) ;543: :205–10. |
[68] | Beckervordersandforth R , Ebert B , Schäffner I , Moss J , Fiebig C , Shin J , Moore DL , Ghosh L , Trinchero MF , Stockburger C , et al. Role of mitochondrial metabolism in the control of early lineage progression and aging phenotypes in adult hippocampal neurogenesis. Neuron. (2017) ;93: :560–73.e566. |
[69] | Knobloch M , Jessberger S . Metabolism and neurogenesis. Curr Opin Neurobiol. (2017) ;42: :45–52. |
[70] | Zheng X , Boyer L , Jin M , Mertens J , Kim Y , Ma L , Hamm M , Gage FH , Hunter T . Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife. (2016) ;5. |
[71] | Khacho M , Clark A , Svoboda DS , Azzi J , MacLaurin JG , Meghaizel C , Sesaki H , Lagace DC , Germain M , Harper ME , et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. (2016) ;19: :232–47. |
[72] | Galluzzi L , Pietrocola F , Levine B , Kroemer G . Metabolic control of autophagy. Cell. (2014) ;159: :1263–76. |
[73] | Wen L , Tang F . Single-cell sequencing in stem cell biology. Genome Biol. (2016) ;17: :71. |
[74] | Hu Y , Huang Y , Yi Y , Wang H , Liu B , Yu J , Wang D . Single-cell RNA sequencing highlights transcription activity of autophagy-related genes during hematopoietic stem cell formation in mouse embryos. Autophagy. (2017) ;13: (4):770–771. |

