
Spectrum of FGFR2/3 Alterations in Cell-Free DNA of Patients with Advanced Urothelial Carcinoma
Abstract
Detecting genomic alterations (GAs) in advanced urothelial carcinoma (aUC) can expand treatment options by identifying candidates for targeted therapies. Erdafitinib is FDA-approved for patients with platinum-refractory aUC with activating mutation or fusion in FGFR2/3. We explored the prevalence and spectrum of FGFR2/3 GAs identified with plasma cfDNA NGS testing (Guardant360) in 997 patients with aUC. FGFR2/3 GAs were detected in 201 patients (20%) with characterized activating GAs in 141 (14%). Our results indicate the Guardant360-based FGFR2/3 GA detection rate is similar to those described from previous studies employing tumor tissue testing, suggesting that plasma-based cfDNA NGS may non-invasively identify candidates for anti-FGFR targeted therapies.
A recent wave of new agents has been introduced to the arsenal of treatments for patients with aUC, including immune checkpoint inhibitors, targeted therapies and antibody-drug conjugates. In April 2019, erdafitinib, a potent tyrosine kinase inhibitor (TKI) of FGFR1–4, received FDA accelerated approval for adult patients with locally advanced or metastatic urothelial carcinoma exhibiting susceptible activating mutations or fusions in the FGFR2 or FGFR3 (FGFR2/3) genes who had progression on platinum-containing chemotherapy. Other FGFR inhibitors, including infigratinib, pemigatinib, rogaratinib and Debio-1347, are under investigation in urothelial cancer.
Platinum-based chemotherapy remains the first-line standard of care for patients with aUC, with median overall survival noted between 9 and 15 months in older trials [1, 2]. However, for patients who have progression following this initial treatment, the median overall survival may only be estimated at 5–7 months [3]. Erdafitinib demonstrated effectiveness as a subsequent therapy following platinum-based chemotherapy with a reported 40%ORR in patients with FGFR2/3 GAs in the BLC2001 single arm phase II trial [4]. Specifically, the ORR in patients with FGFR3 mutation and FGFR2/3 fusion were 49%and 16%, respectively. Median PFS and OS were 5.5 and 13.8 months, respectively. Unfortunately, adverse events, including, but not limited to, hyperphosphatemia, anorexia, fatigue, skin, gastrointestinal and ocular events remain a clinical challenge, requiring proper education, early recognition and multi-specialty approach.
NCCN Guidelines recommend conducting molecular genomic testing for FGFR2/3 GAs in patients with stage IIIB-IVB urothelial cancer to identify patients who may benefit from targeted therapies [5]. Additionally, studies have reported that patients with FGFR3 mutation or fusion seem less likely to respond to immune checkpoint inhibitors compared to those without such alterations, while a correlation has been proposed between FGFR3 activation and the immune-depleted phenotype in UC. However, this interesting concept is still under evaluation and debate [4, 6–8]. Overall, genomic profiling of aUC tumors has a significant role in selecting patients for erdafitinib and also for clinical trials (e.g. NCT04197986); however, there are several logistical challenges with tumor tissue testing, including difficult or not feasible biopsy procedures, patient unwillingness to undergo repeat or additional procedures, insufficient quantity or quality of tissue specimen for genomic analysis, and limited ability to assess tumor heterogeneity.
Genomic profiling with plasma cell-free DNA (cfDNA) next-generation sequencing (NGS) is now FDA approved and increasingly used to non-in-vasively identify potentially targetable GAs in patients with advanced cancer, often faster than or in conjunction with tumor tissue testing [9–11]. Plasma cfDNA NGS may identify biomarkers of response to targeted therapies, and also suggest mechanisms of resistance. Our retrospective study explored the prevalence and spectrum of FGFR2/3 GAs identified by plasma cfDNA NGS analysis in a large cohort with standard of care cfDNA testing. We hypothesized that the detection rate of FGFR2/3 GAs would be similar to historical data from tumor tissue genomic testing.
Clinical genomic testing results from Guardant360 (G360; Guardant Health, Inc, Redwood City, CA, USA) plasma cfDNA NGS analysis between 10/19/15–8/28/19 were queried for all consecutive patients with aUC diagnosis in accordance with ethics standards and institutional review board approval which waived the need for informed consent to analyze deidentified data (Advarra IRB Pro00034566/CR00218935). G360 detects single nucleotide variants (SNVs), indels, copy number amplifications, and fusions in up to 74 genes with a turnaround time of approximately seven days. All panel versions included in the study cohort included assessment for FGFR2/3 fusions and sequencing of all critical exons of FGFR2/3 harboring sensitizing SNVs. Guardant360 testing is validated and intended for the detection of somatic alterations.
During the specified study time frame, a total of 1,349 samples from 1,096 unique patients with aUC (a number of patients had testing performed at multiple time points) had associated G360 results for analysis. Median patient age was 69 (range 28–93); 28%were female.
Presumed somatic GAs were identified in 1,192 (88%) cfDNA tests from 997 unique patients. Of these, 201 (20%) patients with aUC had ≥1 fusion and/or nonsynonymous SNV in FGFR2/3; 141 patients (14%) had at least one characterized activating FGFR2/3 GA while the remainder of GAs were functionally uncharacterized variants of uncertain significance (VUS); VUS are generally not used to recommend treatment with targeted agents based on lack of clear functional significance, but could possibly be reclassified in the future with additional investigation and generated data. Of patients with FGFR2/3 GAs, the median age was 70 (range 39–92) and 27%were female, similar to the entire cohort.
GAs were observed more often in FGFR3 (n = 125) than FGFR2 (n = 30). The majority (82%) of the observed GAs in FGFR3 were predicted to be activating, while most FGFR2 GAs (86%) were VUS (Figs. 1a, b). However, there was a greater diversity of unique variants in FGFR2 (n = 59) vs. FGFR3 (n = 30) (Figs 1c, d); for the latter, this was primarily driven by the S249C variant, which comprised 46%of nonsynonymous FGFR3 SNV observations (Fig. 2a). The second most common FGFR3 non-synonymous SNV, Y373C, accounted for 16%of cases. There were no unique significantly recurrent FGFR2 variants. In both genes, VUS were individually uncommon.
Fig. 1
Fig 1 Occurrences and unique variants in FGFR2/3. The number and proportion of both characterized and VUS GAs observed in cfDNA in FGFR3 (a) and FGFR2 (b) across the aUC cohort. Excluding occurrences of the same variant seen in multiple patients, the number and proportion of both characterized and VUS unique variants present in FGFR3 (c) and FGFR2 (d) across the cfDNA aUC cohort. VUS, variant of uncertain significance.
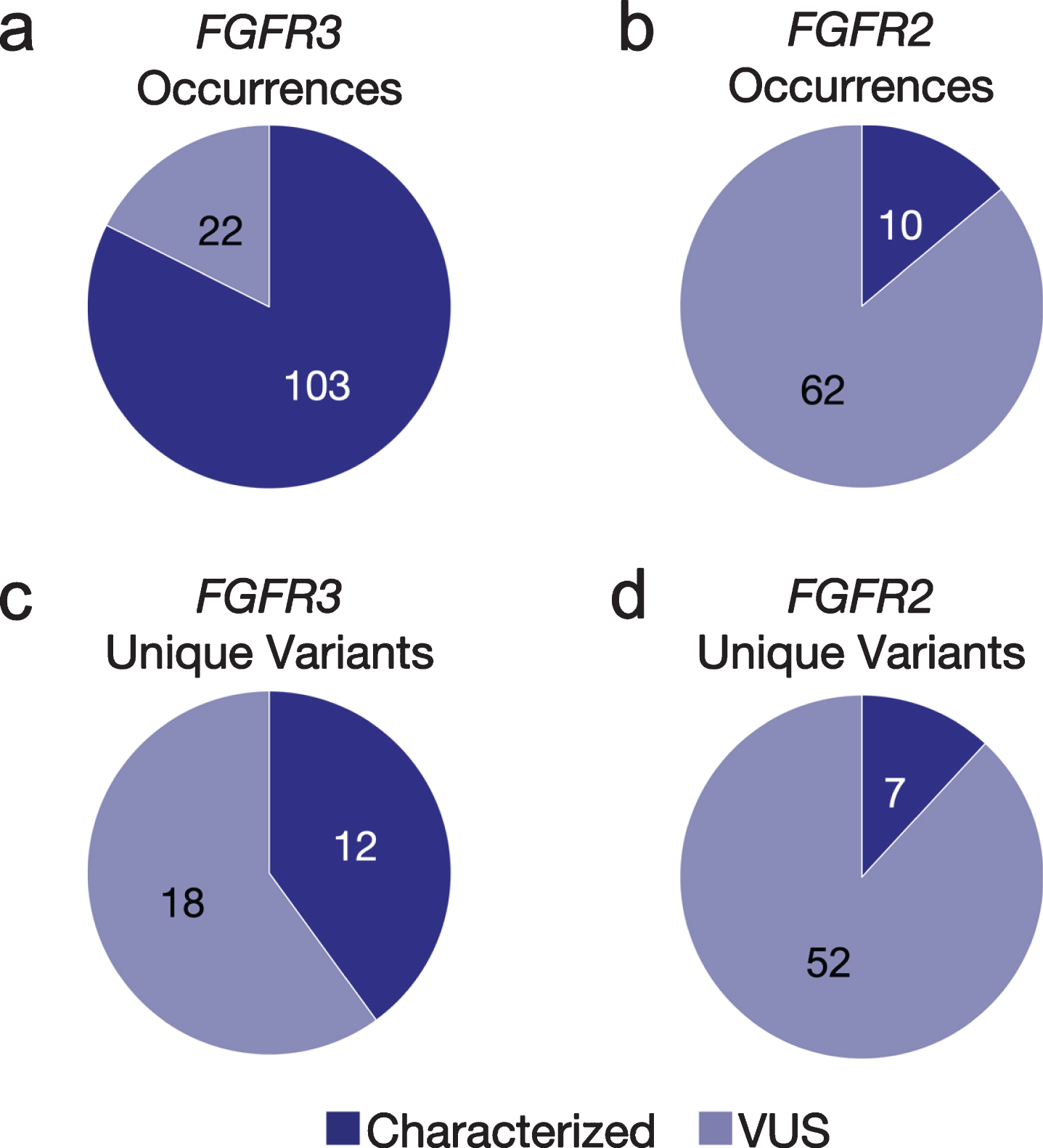
Fig. 2
Mutational spectrum and clonality assessment of nonsynonymous FGFR2/3 SNVs. Lollipop plots show the location and number of observed GAs in FGFR3 (a) and FGFR2 (c), by characterized vs. VUS status. Clonality plots (right) show the copy number-adjusted clonality and relative frequency of the spectrum of clonality of both characterized and VUS GAs observed in FGFR3 (b) and FGFR2 (d). VUS, variant of uncertain significance.
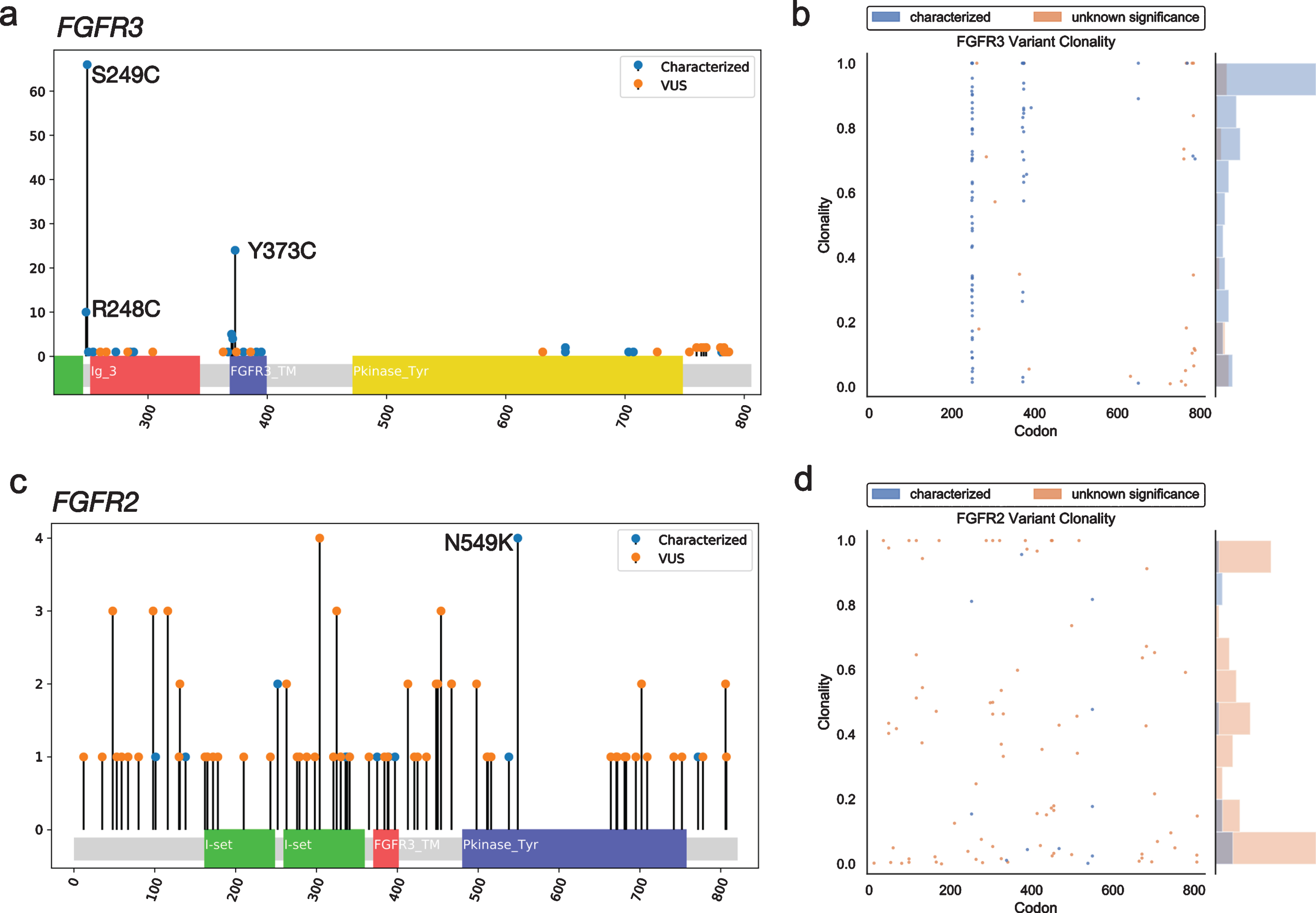
FGFR3 fusions were identified in 34 (3.1%) patients. The vast majority (n = 32, 94%) of these involved TACC3 as the fusion partner, but JAKMIP1 and TNIP2 were detected as fusion partners in one patient each.
Copy number-adjusted clonality was determined by calculating the relative variant allele fraction (VAF) and correcting for copy number, as previously described [12]. The median copy number-adjusted clonality of nonsynonymous SNVs was higher in FGFR3 (0.80) than FGFR2 (0.20); this remained true when limiting to characterized activating mutations (0.84 vs. 0.17) (Fig. 2b).
When examining the co-occurrence of characterized activating GAs, there was very little overlap between activation of FGFR2 and FGFR3. However, two patients were identified in the cohort whose samples contained activating point mutations in both genes. One patient’s sample had an overall maximum VAF of 8.2%and a total of 17 GAs identified including FGFR2 N549K and FGFR3 S249C at VAFs of 1.5%and 1.4%, respectively. The other patient’s sample had an overall maximum VAF of 2.5%and a total of 9 GAs identified including FGFR2 S252W and FGFR3 G370C at VAF of 2.0%and 0.7%, respectively. A recent study utilizing tissue NGS across multiple cancer types identified a degree of co-occurrence in FGF/FGFR GAs, but observed mutual exclusivity between FGFR2 and FGFR3 in particular [13]. While no clinical information is available regarding these patients to better understand this intriguing phenomenon, the ability of cfDNA sampling to capture potential genomic heterogeneity may provide unique further insights into co-occurrence vs mutual exclusivity of biologically relevant pathways in future work.
This study demonstrates that cfDNA NGS analysis identifies fusions and a broad spectrum of SNVs in FGFR2/3 at a similar rate to historically reported tumor tissue testing, with previous studies demonstrating that tissue analysis detects FGFR3 mutations in up to 15%–20%of advanced/metastatic urothelial cancers [4, 14–19]. FGFR2/3 fusions are less common at a frequency of 2.2%–2.4%in patients with aUC who had tissue testing, [18, 20] with TACC3 being the most common fusion partner, consistent with the current study [4, 21]. FGFR3-TACC3 fusions are reported to be enriched in younger (≤50) patients (12%), Asians (13%), and never smokers (5.6%) [20].
Concordance of plasma cfDNA results with tumor tissue across solid tumors has been previously researched; overall concordance can vary depending on a variety of factors, including timing of sample collection and overlap of assay panels utilized, but in studies controlling for these variables, analytical concordance is quite high [9, 10, 22, 23]. Such comparisons are limited specifically for FGFR2/3 GAs in aUC; a cohort comparison study found a similar overall frequency of FGFR3 GAs between plasma and tissue-based datasets in lower tract aUC [24] and a pilot concordance study for patients with metastatic urinary tract cancer found that plasma genomic profiling detected FGFR2/3 GAs in 4 of 5 patients in whom they were detected by tissue testing [25].
This study explored both fusions and point mutations across both FGFR2/3 with an analysis of functionally activating and uncharacterized (VUS) GAs in both. It should be noted that the BLC2001 study of erdafitinib included only activating mutations and fusions in both FGFR2/3; this study cohort did not include patients with activating FGFR2 point mutations [4]. While the present analysis suggests that FGFR2 point mutations known to have functionally activating properties appear rare in advanced urothelial cancer, they may be potential drug targets. However, it is not clear whether these specific GAs would be ideal targets for anti-FGFR therapeutics and further exploration of this question is warranted.
The difference in copy number-adjusted clonality between FGFR2 (0.20) and FGFR3 (0.80) suggests that FGFR3 GAs are frequently early truncal mutations, whereas FGFR2 GAs may arise later in a heterogeneous fashion [12]. While further study is needed to understand the therapeutic impact of subclonal alterations, especially those with low relative VAF, there may be potential clinical benefit for matching targeted therapies and there is much more to learn regarding response to immunotherapy. Therefore, capturing the components of dynamic tumor heterogeneity over time with serial samples may prove very important.
Especially given that tumors can increase in genomic complexity over time and with previous exposure to systemic treatment, cfDNA NGS offers the opportunity to assess the current genomic profile of the evolving disease versus “snapshot” testing of archival tissue. Peripheral blood samples for cfDNA analysis can be obtained in a non-invasive manner at the time of consideration of post-progression therapy compared to tissue acquisition, which can be invasive, costly, not feasible, and logistically challenging. Moreover, cfDNA NGS testing using plasma also provides the opportunity to track the evolution of GAs across multiple time points in the treatment course, exploring response to treatment as well as the emergence and types of resistance mechanisms.
The present study has several inherent limitations, including the potential for selection and confounding biases, retrospective nature, and absence of matched tumor tissue testing and clinical outcomes data, highlighting the need for prospective validation of our generated hypotheses. In the absence of available matched tissue specimens or tissue testing information from this particular patient cohort, we compared observed alteration frequencies to previously published analyses of these GAs in cohorts with tissue testing performed, but we cannot rule out potential cohort differences, which could affect this comparison. These are common limitations when studying such cohorts derived through standard of care clinical testing genomic databases; however, outcomes-focused studies are ongoing [26].
To conclude, genomic profiling via cfDNA is a fast, minimally invasive tool that may be used to help inform decision-making for patients with advanced solid tumors. The results from our large retrospective study indicate a detection rate of FGFR2/3 GAs identified in plasma cfDNA using the G360 assay that is similar to historical rates based on conventional tumor tissue testing. Our hypothesis-generating results can contribute to ongoing discussions about identification of patients who can benefit from erdafitinib and emerging therapies in clinical trials.
ACKNOWLEDGMENTS
The authors have no acknowledgments.
FUNDING
Data and personnel support were provided by Guardant Health.
AUTHOR CONTRIBUTIONS
All authors contributed to careful interpretation of data and writing the article. Additionally, PG/LAK/RAT contributed to conception, LAK/AIH contributed to performance of work, and PG/LAK/AIH had access to data.
CONFLICTS OF INTEREST
Petros Grivas MD PhD reports grants and consulting/advisory board involvement from AstraZeneca, Bayer, Bristol-Myers Squibb, Clovis Oncology, GlaxoSmithKline, Genentech, Immunomedics, Merck, Mirati Therapeutics, Pfizer, and QED Therapeutics; grants from Bavarian Nordic, Debiopharm, Kure It Cancer Research, and Oncogenex; and consulting/advisory board involvement from Driver, Dyania Health, EMD Serono, Exelixis, Foundation Medicine, Genzyme, Heron Therapeutics, Janssen, Roche, and Seattle Genetics, outside the submitted work.
Lesli A. Kiedrowski MS MPH reports employment and stock ownership from Guardant Health during the conduct of the study.
Guru P. Sonpavde MD reports grants, personal fees, non-financial support, and advisory board/com-mittee involvement from AstraZeneca; grants and advisory board/committee involvement from Sanofi; non-financial support and advisory board/committee involvement from Bristol-Meyers Squibb; personal fees and advisory board/committee involvement from EMD Serono; advisory board/committee involvement from Genentech, Merck, Seattle Genetics, Astellas, Exelixis, Janssen, Bicycle Therapeutics, Pfizer, Bavarian Nordic, and QED; and personal fees from Physicians Education Resource, Onclive, Research to Practice, Medscape, Up To Date, Elsevier, and Debiopharm, outside the submitted work.
Sumati V. Gupta MD reports grants and non-financial support from QED; grants from BMS, Rexahn, Incyte, Novartis, LSK, Five Prime, Mirati, Debiopharm, Merck, Pfizer, AstraZeneca, MedImmune, Clovis, and Daiichi Sankyo; and spouse stock ownership in Salarius Pharmaceuticals, outside the submitted work.
Roby A. Thomas MD reports personal fees and advisory board involvement from Integra Connect, outside the submitted work.
Theodore S. Gourdin MD reports grants from Ferring Pharmaceuticals and advisory board involvement from EMD Serono and Exelixis, outside the submitted work.
Aaron I. Hardin PhD reports employment and stock ownership from Guardant Health during the conduct of the study.
Kimberly M. Hamann MS reports employment and stock ownership from Guardant Health during the conduct of the study.
Bishoy M. Faltas MD reports grants from Eli Lilly and personal fees from Immunomedics, Merck, and Urotoday, outside the submitted work.
Nicholas J. Vogelzang MD reports personal fees, non-financial support, and advisory board/consulting involvement from Bayer, Genentech, and Pfizer; non-financial support and advisory board/consulting involvement from AstraZeneca; personal fees and advisory board/consulting involvement from Astellas and Clovis; personal fees and non-financial support from Sanofi; grants and non-financial support from US Oncology; advisory board/consulting involvement and stock ownership from Caris Life Sciences; grants and advisory board/consulting involvement from Merck; grants from Endocyte and Suzhou Kintor; personal fees from Bristol-Meyers Squibb, Novartis, Seattle Genetics, and Up To Date; and non-financial support from Exelixis, MedImmune, and Onyx, outside the submitted work.
REFERENCES
[1] | von der Maase H , Sengelov L , Roberts JT , Ricci S , Dogliotti L , Oliver T , et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol. (2005) ;23: (21):4602–608. |
[2] | De Santis M , Bellmunt J , Mead G , Kerst JM , Leahy M , Maroto P , et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 6. J Clin Oncol. (2012) ;30: (2):191–9. |
[3] | Bellmunt J , Théodore C , Demkov T , Komyakov B , Sengelov L , Daugaard G , et al. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract [published correction appears in J Clin Oncol. 2010;28(1):182. Winquist, Eric. J Clin Oncol. (2009) ;27: (27):4454–61. |
[4] | Loriot Y , Necchi A , Park SH , Garcia-Donas J , Huddart R , Burgess E , et al. BLC2001 Study Group (2019). Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. (2019) ;381: (4):338–48. |
[5] | National Comprehensive Cancer Network.Bladder Cancer (Version 6.2020). https://www.nccn.org/professionals/physician_gls/pdf/bladder.pdf. Accessed September 25, 2020. |
[6] | Joerger M , Cassier PA , Penel N , Cathomas R , Richly H , Schostak M , et al. Rogaratinib in patients with advanced urothelial carcinomas prescreened for tumor FGFR mRNA expression and effects of mutations in the FGFR signaling pathway. J Clin Oncol. (2018) ;36: (25). |
[7] | Robinson BD , Vlachostergios PJ , Bhinder B , Liu W , Li K , Moss T , et al. Upper tract urothelial carcinoma has a luminal-papillary T-cell depleted contexture and activated FGFR3 signaling. Nat Commun. (2019) ;10: (1):2977. |
[8] | Wang L , Gong Y , Saci A , Szabo PM , Martini A , Necchi A , et al. Fibroblast Growth Factor Receptor 3 Alterations and Response to PD-1/PD-L1 Blockade in Patients with Metastatic Urothelial Cancer. Eur Urol. (2019) ;76: (5):599–603. |
[9] | Makarem M , Leighl NB . Molecular testing for lung adenocarcinoma: Is it time to adopt a “plasma-first” approach? Cancer. (2020) ;126: (14):3176–80. |
[10] | Turner NC , Kingston B , Kilburn LS , Kernaghan S , Wardley AM , Macpherson IR , etc. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): a multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. (2020) :S1470–2045(20)30444-7. |
[11] | Leighl NB , Page RD , Raymond VM , Daniel DB , Divers SG , Reckamp KL , , et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin Cancer Res. (2019) ;25: (15):4691–700. |
[12] | Zill OA , Banks KC , Fairclough SR , Mortimer SA , Vowles JV , Mokhtari R , et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin Cancer Res. (2018) ;24: (15):3528–38. |
[13] | Zuo W , He Y , Li W , Wu H , Zhao Z , Zhang Y , et al. Landscape of FGF/FGFR Alterations in 12,372 Chinese Cancer Patients. J Cancer. (2020) ;11: (22):6695–9. |
[14] | Knowles MA , Hurst CD . Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. (2015) ;15: (1):25–41. |
[15] | Billerey C , Chopin D , Aubriot-Lorton MH , Ricol D , Medina SGDM , Rhijn BV , et al . Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. (2001) ;158: (6):1955–9. |
[16] | Kimura T , Suzuki H , Ohashi T , Asano K , Kiyota H , Eto Y . The incidence of thanatophoric dysplasia mutations in FGFR3 gene is higher in low-grade or superficial bladder carcinomas [published correction appears in Cancer. 2002;94(7):2117].. Cancer. (2001) ;92: (10):2555–61. |
[17] | Tomlinson DC , Baldo O , Harnden P , Knowles MA . FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J Pathol. (2007) ;213: (1):91–8. |
[18] | Ross JS , Wang K , Khaira D , Ali SM , Fisher HAG , Mian B , et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer. (2016) ;122: (5):702–11. |
[19] | Kim YS , Kim K , Kwon GY , Lee SJ , Park SH . Fibroblast growth factor receptor 3 (FGFR3) aberrations in muscle-invasive urothelial carcinoma. BMC Urol. (2018) ;18: (1):68. |
[20] | Nassar AH , Lundgren K , Pomerantz M , Van Allen E , Harshman L , Choudhury AD , et al. Enrichment of FGFR3-TACC3 fusions in patients with bladder cancer who are young, Asian, or have never smoked. JCO Precis. Published online May 16, 2018. |
[21] | Robertson AG , Kim J , Al-Ahmadie H , Bellmunt J , Guo G , Cherniack AD , et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer [published correction appears in Cell. 2018;174(4):1033]. Cell. (2017) ;171: (3):540–556.e25. |
[22] | Lanman RB , Mortimer SA , Zill OA , Sebisanovic D , Lopez R , Blau S , et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLoS One. (2015) ;10: (10):e0140712. |
[23] | Gupta R , Othman T , Chen C , Sandhu J , Ouyang C , Fakih M . Guardant360 Circulating Tumor DNA Assay Is Concordant with FoundationOne Next-Generation Sequencing in Detecting Actionable Driver Mutations in Anti-EGFR Naive Metastatic Colorectal Cancer. Oncologist. (2020) ;25: (3):235–43. |
[24] | Agarwal N , Pal SK , Hahn AW , Nussenzveig R , Pond GR , Gupta SV , et al. Characterization of metastatic urothelial carcinoma via comprehensive genomic profiling of circulating tumor DNA. Cancer. (2018) ;124: (10):2115–24. |
[25] | Barata PC , Koshkin VS , Funchain P , Sohal D , Pritchard A , Klek S , et al. Next-generation sequencing (NGS) of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: a pilot assessment of concordance. Ann Oncol. (2017) ;28: (10):2458–63. |
[26] | Grivas P , Lalani AA , Pond GR , Nagy R , Faltas B , Agarwal N , et al. Circulating Tumor DNA Alterations in Advanced Urothelial Carcinoma and Association with Clinical Outcomes: A Pilot Study. Eur Urol Oncol. (2020) ;3: (5):695–9. |


