
Antibody-Drug Conjugates in Bladder Cancer
Abstract
Urothelial carcinoma (UC) is characterized by expression of a plethora of cell surface antigens, thus offering opportunities for specific therapeutic targeting with use of antibody-drug conjugates (ADCs). ADCs are structured from two major constituents, a monoclonal antibody (mAb) against a specific target and a cytotoxic drug connected via a linker molecule. Several ADCs are developed against different UC surface markers, but the ones at most advanced stages of development include sacituzumab govitecan (IMMU-132), enfortumab vedotin (ASG-22CE/ASG-22ME), ASG-15ME for advanced UC, and oportuzumab monatox (VB4-845) for early UC. Several new targets are identified and utilized for novel or existing ADC testing. The most promising ones include human epidermal growth factor receptor 2 (HER2) and members of the fibroblast growth factor receptor axis (FGF/FGFR). Positive preclinical and early clinical results are reported in many cases, thus the next step involves further improving efficacy and reducing toxicity as well as testing combination strategies with approved agents.
INTRODUCTION
Urothelial carcinoma (UC) can arise along any part of the urinary tract lined with transitional epithelium, with the majority of cases (90% – 95%) in the lower tract (bladder, urethra) and the remainder (5% – 10%) in the upper tract (renal pelvis, ureter) [1, 2]. UC of the bladder is the fifth most common malignancy in the US, with a projected incidence of 79,030 new cases in 2017 and responsible for 16,890 deaths [3]. For patients with metastatic UC, cisplatin-based combination chemotherapy is the standard of care, conferring a median overall survival of 13– 16 months [4–6]. Most patients eventually relapse and recent phase II/III data favor immune checkpoint inhibitors (ICIs) over chemotherapy following platinum-based treatment [7–11]; ICIs are also available in the first-line setting, in cisplatin-unfit patients [12]. Nevertheless, response rates are approximately 20% in unselected patients [7–12]. This underlines the need for expanding the armamentarium of effective therapies for advanced and metastatic UC.
One class of agents which has gained interest in several tumor types, including UC, is antibody-drug conjugates (ADCs). ADCs are monoclonal antibodies (mAbs) specific for tumor antigens, which are conjugated to cytotoxic agents. To provide a comprehensive overview of the current place of ADCs in the treatment of UC, we performed a Pubmed-search using the following terms: “antibody-drugconjugate”, “urothelial cancer”, “IMMU-132”, “ASG-15ME”, “ASG-22ME”, “DS-8201a“, “VB4-845”, “TDM-1”, “JAA-F11”, “fibroblast growth factor”, “prostate-specific membrane antigen”.
ADCs represent an attractive treatment modality due to selective targeting of cancer cells, thus limiting cytotoxic side effects to normal tissues and increasing drug concentration to target tissues [13, 14]. The first ADC that received FDA approval was gemtuzumab ozogamicin for acute myeloid leukemia in 2000 [15, 16]. The drug was created by conjugation of the antitumor antibiotic calicheamicin to an anti-CD33 antibody. However, it was withdrawn from US market in 2010 due to a combination of toxicity and efficacy issues, including persistent thrombocytopenia due to off-target effects, vaso-occlussive disease and lack of confirmatory phase II results in the phase III setting, respectively. Use of a low fractionated dose and combination with chemotherapy has recently resulted in a new FDA approval [17]. Hence, the development of effective and well tolerated ADCs has proven to be challenging. At present, three additional ADCs are approved for clinical use after showing clinical benefit compared to standard of care, including: trastuzumab emtansine for HER2-positive metastatic breast cancer [18, 19], brentuximab vedotin for refractory Hodgkin’s lymphoma [20] and systemic anaplastic large-cell lymphoma [21], and inotuzumab ozogamicin for relapsed or refractory B-cell precursor acute lymphoblastic leukemia [22].
A better understanding of the properties of ADCs has led to the design of a plethora of new agents, which are currently in different phases of clinical development for various cancer types. In this review we discuss the preclinical and clinical evidence on the targets, efficacy and toxicity of ADCs in UC.
ADC CHARACTERISTICS
The structure of an ADC involves three major components, the mAb, the cytotoxic drug (payload) and a linker molecule [13, 14]. The antibody binds to a specific antigen on the surface of cancer cells, followed by internalization of the conjugate by endocytosis and release of its cytotoxic payload after lysosomal degradation [13, 14]. To ensure appropriate delivery of the drug to the target, it is important to ensure that the target is specific, thus expressed at high levels in tumor tissues and sparing as much as possible normal cells. High binding affinity and ability of the antigen-antibody complex to internalize after binding are also crucial steps, otherwise risk of toxicity from release to neighboring non-target cells increases [13, 14]. Homogenous expression of the antigen within tumors as well as accessibility of the antigen from the bloodstream are important to maximize the availability of ADC to the target [13, 14]. Immunogenicity of the antibody has been a limitation before due to mouse-derived antibodies; however development of humanized and fully human antibodies has minimized this concern [13].
Certain characteristics of the linker are also critical for the success of ADC-based therapy. Stability of the linker in the bloodstream enables the drug to be released only after binding to the target and avoid systemic toxicities. Depending on their intracellular stability and way of degradation, linkers are divided into cleavable and non-cleavable [13]. Cleavable linkers become unstable under the low pH, protease-enriched environment of lysosomes or high intracellular level of glutathione. In contrast, non-cleavable linkers are stable and solely depend on lysosomal degradation to release their payload[13, 23].
The payload is the cytotoxic drug which is responsible for the antitumor effect and reaches high concentrations in the tumor. There are several different payloads depending on the mechanism of cytotoxicity but in general they can be divided in two groups, those affecting DNA (calicheamicins, duocarmycins, pyrrolobenzodiazepines, SN-38), and those interfering with tubulin formation (auristatins, maytansines) [13, 14, 23]. The auristatins, including monomethyl auristatin E (MMAE) and F (MMAF) and maytansines, including emtasine (DM1) and ravtansine (DM4) represent the majority of cytotoxic payloads in ADCs [24]. In addition to the direct cytotoxic effect of the payload at the cellular level, the antitumor effect can be mediated through additional mechanisms involving signal transduction inhibition from direct binding, antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) [13, 14, 23]. Common class toxicities for ADCs are hematologic (anemia, neutropenia, thrombocytopenia), hepatic, ocular, and peripheral neuropathy [24]. However, depending on the payload the incidence of severe (grade 3/4) toxicities varies among different agents. Based on a recent metaanalysis of ADC studies with reported adverse events, ocular toxicity is most pronounced in MMAF (16%) whereas MMAE and DM4 mainly cause neutropenia (16.4% , 11.7%) followed by thrombocytopenia (9.7% , 7.3%) and anemia (6.8% , 6.4%) [24]. Peripheral neuropathy is also higher in MMAE (6.5%). DM1 may lead to thrombocytopenia (9.3%) and hepatotoxicity (7.2%) [24].
ADC AGENTS TESTED IN ADVANCED UC
While there are no currently approved ADCs for treatment of advanced UC, several agents are in advanced phases of clinical development and have shown evidence of tolerable toxicity and clinical activity. It is interesting that while presence of liver metastases in advanced UC patients treated with ICIs resulted in a lower objective response rate (ORR) compared to patients without liver metastases (5 vs. 19%) [8], extensive clinical experience with approved ADCs, such as TDM-1 in metastatic breast cancer, supports high ORRs (44%) for liver lesions [25]. This may suggest that ADCs as a class of agents might be better suited for patients with hepatic metastases compared to ICIs, and is also reflected in early data from ADC use in UC, discussed below.
Sacituzumab govitecan (IMMU-132)
Trop-2 (human trophoblast cell-surface antigen) is a cell-surface glycoprotein with broad expression in many epithelial cancers [26]. The invariable upregulation of Trop-2 in many cancers as compared to normal tissues, combined with Trop-2-dependent stimulation of growth in a variety of cancer cells provided the rational for Trop-2 targeting [27]. In bladder UC, Trop-2 tissue expression is higher in invasive UC compared to normal urothelium and non-invasive UC and correlates with stage [28]. These findings prompted the design of an ADC called sacituzumab govitecan (IMMU-132), in which an anti-Trop-2 mAb is conjugated with SN-38, the active metabolite of irinotecan, for selective delivery in cancer tissues [29]. In a comparative study of irinotecan versus IMMU-132 administration in nude mice tumor xenografts, the area under the curve (AUC) analysis demonstrated a 20-fold higher delivery of SN-38 to tumors while simultaneously 9-fold lower intestinal concentrations with IMMU-132 compared to irinotecan [30].
The first-in-human Phase I trial of IMMU-132 (sacituzumab govitecan) enrolled 25 patients with metastatic tumors from several different primaries, including one with UC [29]. Among 8, 10, 12, and 18 mg/kg doses tested, 12 mg/kg was declared the maximum tolerated dose (MTD), with no dose-limiting toxicities (DLT) out of 9 patients at 12 mg/kg and two grade 4 dose limiting neutropenias out of 3 patients at 18 mg/kg [29]. A starting dose of 8 or 10 mg/kg, administered on days 1 and 8 of a 21-day cycle was considered acceptable for further evaluation and 10 mg/kg was selected for the subsequent Phase II trial. The most common severe, grade 3 or higher toxicities were neutropenia (n = 9, but with 7/9 receiving high doses of 12 mg/kg and 18 mg/kg), febrile neutropenia (n = 2), diarrhea (n = 3) and fatigue (n = 3). Overall, the drug exhibited high anti-tumor activity at non-toxic doses, without any preselection of patients based on tumor Trop-2 expression [29]. A more expanded study in a larger group of patients with diverse metastatic cancers who received IMMU-132 at 8 mg/kg (n = 81) and 10 mg/kg (n = 97) confirmed the safety of the drug and revealed a spectrum of activity with ORR as high as 30% in different tumor types, including colorectal, small cell lung, non-small cell lung, and breast cancers [31].
In the early phase clinical development of IMMU-132, investigators observed that the ADC appeared effective in a small group of 6 patients with advanced, heavily pretreated UC, enrolled in the “pan-cancer” Phase I/II study (NCT01631552). Two patients achieved a partial response (PR), demonstrating a 38% reduction in target lesions as best response, while one patient had stable disease (SD) and 3 patients had progressive disease (PD). Responses were translated into a progression-free survival (PFS) of 6.7– 8.2 months and an overall survival (OS) of 7.5+ to 11.4+ months [32]. A follow-up report of an expanded number of 41 UC patients of the same study reported an ORR of 39% (including 5/13, 39% with liver metastases), PFS of 7.1 months, OS of 16.1 months in the intention-to-treat (ITT) population [33]. These patients had durable responses (median duration 12.6 months) and tolerated treatment well, with grade ≥3 adverse event rates similar to what was previously described, consisting of 39% neutropenia, 10% anemia, 7% fatigue, 7% diarrhea, and 5% febrile neutropenia [33]. Hence, IMMU-132 has preliminary efficacy and tolerability at phase I/II level in patients with treatment-refractory UC. In view of the drug’s significant activity in metastatic triple-negative breast cancer with ORR as high as 30% in phase II setting [34], IMMU-132 was recently granted FDA breakthrough therapy designation for this indication and will be submitted for Biologics License Application (BLA) [35]. It is anticipated that a similar planning approach will be followed for metastatic UC, which continues to represent a disease with unmet needs beyond cisplatin-based chemotherapy andICIs.
Enfortumab vedotin (ASG-22CE/ASG-22ME)
Nectin-4, also known as poliovirus receptor-related protein 4 (PVRL4), is a type I transmembrane protein [36]. Copy number gain of the PVRL4 gene is a frequent event in carcinogenesis and promotes epithelial-to-mesenchymal transition, invasion and metastasis through integrin-, PI3K/Akt- and Wnt/β-catenin-signaling pathways [37, 38]. While nectin-4 protein is expressed in several tumors, it is particularly prevalent in breast cancer and UC [36]. Out of 2,394 bladder, breast, non-small cell lung, pancreatic, ovarian, head/neck and esophageal tumor specimens, immunohistochemical (IHC) analysis revealed that over half (69%) express nectin-4, with moderate to strong staining (H-score ≥100) seen in bladder (60%) and breast (53%) tumor tissues [36]. In the tissue microarray of UC samples, nectin-4 was expressed in 83% of total samples [36]. Nectin-4 expression in normal tissue is limited, which renders it a valuable target in epithelial cancers. A fully human mAb, enfortumab vedotin, was developed through two main lines, hybridoma (ASG-22ME) and Chinese hamster ovary (ASG-22CE), and created using Amgen’s transgenic system (XenoMouse). It consists of a human anti-nectin-4 antibody conjugated to the anti-mitotic agent MMAE, which disrupts microtubules and induces apoptosis [36]. Mouse and patient xenograft models were used to test enfortumab vedotin’s antitumor activity in human breast, bladder, pancreatic and lung cancers [36]. Inhibition of tumor growth in all cancerous tissues and tumor regression of bladder and breast tumors [36] supported further early clinical testing of enfortumab vedotin.
Enfortumab vedotin was tested at phase I level in patients with metastatic UC previously treated with at least one prior chemotherapy regimen. Patients’ tumors were tested for nectin-4 IHC expression and enrolled if H-score ≥150 [39]; however this prescreening was subsequently aborted as 97% of the tissue samples tested showed nectin-4 overexpression [40]. Enfortumab vedotin has a half-life of 1.5 days and was studied in 4 dose schedules 0.5, 0.75, 1, or 1.25 mg/kg administered intravenously (IV) every 3 out of every 4 wks [39]. An interim analysis of the study was presented in 2016, reporting a tolerable toxicity profile with fatigue being the most common adverse event (38%) and ORR of 30% was reported in 33/42 response-evaluable UC patients [39]. Updated results from 68 patients (as of 3 Jan 2017) demonstrated limited grade ≥3 toxicity (urinary tract infection 10% , hypophosphatemia 9%) at the recommended phase II dose (RP2D) of 1.25 mg/kg [41]. A drug-related rash may be seen; notably nectin-4 is expressed in skin and sweat glands. Hyperglycemia has also been reported. In terms of efficacy, the ORR was 40% among 60 evaluable patients, with 3 complete responses (CR). Notably, a significant proportion of patients (40%) had received ICI as prior therapy, suggesting the activity of the drug in this setting which represents an unmet need. Updated results were recently presented on 81 patients with metastatic UC that were treated with enfortumab vedotin [42]. Seventy-one patients had evaluable responses, of whom 29 patients (41%) had an objective response (OR), consisting of three (4%) CR and 26 (37%) PR. Of the 30 patients treated at the RP2D level, 16 patients (53%) had an OR with one (3%) CR and 15 (50%) PR. Twenty-two patients (73%) achieved SD. Out of 19 patients with liver metastases, 9 (47%) had an OR, with one patient (5%) achieving a CR and eight (42%) PR. Thirteen patients (68%) had SD [42]. A dose expansion of the study is also ongoing in 3 cohorts: metastatic UC with severe renal insufficiency (at a dose of 1 mg/kg escalating to 1.25 mg/kg), non-small cell lung cancer and ovarian cancer (both at 1.25 mg/kg) [42]. In 55 evaluable metastatic UC patients previously treated with ICI, ORR was as high as 53% (confirmed PR 31%) and disease control rate (DCR) was 72% [42]. Hyponatremia (6%) and vomiting (3%) were the most common grade 3 adverse events, while a new safety finding emerged with hyperglycemia, that led to amendment of the study protocol [42].
The phase II trial of single-agent enfortumab vedotin (EV-201) began in September 2017 and is currently enrolling (NCT0329333). Patients will receive enfortumab three weeks on, one week off at a dose of 1.25 mg/kg. Primary outcome is ORR by an independent review facility with secondary outcome of duration of response. Eligible patients must have history of ICI in locally advanced or metastatic UC setting and prior treatment with platinum-based chemotherapy or to be ineligible to receive cisplatin at the time of enfortumab vedotin treatment initiation [43]. A phase III randomized study versus second-line chemotherapy has also been activated. Additionally, a phase Ib study exploring the efficacy of enfortumab vedotin in cisplatin-ineligiblechemo-naïve UC patients combined with pembrolizumab or atezolizumab (EV-103) is currently enrolling (NCT03288545). The aim of this study is to investigate the utility of enfortumab vedotin in the first-line setting. Primary outcome measures include safety and tolerability of the combination while secondary endpoints are the RP2D of enfortumab in combination with ICI, ORR, PFS, and OS [44].
Overall, enfortumab vedotin is a promising agent in advanced UC and has recently received breakthrough designation which further supports its potential for future FDA-approval as a post-immunotherapy option in metastatic UC patients. More mature results from further exploration of its efficacy as monotherapy, as well as of in combination with other drugs are eagerly awaited.
ASG-15ME
The Slit- and Trk-like (SLITRK) family represents a group of type I transmembrane proteins that share leucine-rich repeat domains and are primarily found in the brain [45]. SLITRK6 is a member of this family and although it plays a crucial role in the development of normal hearing as well as vision in humans and in mice [46], its function in cancer is poorly understood. SLITRK6 was discovered to be expressed at high levels by IHC on UC tissue microarrays. SLITRK6 is expressed in 88% of bladder tumors, with 90% seen in transitional cell carcinoma cases, 100% in metastatic UC samples, and 54% in other histological subtypes [47]. SLITRK6 is also overexpressed in upper tract UC, where its presence is of an even greater magnitude [47]. This consists a significant advantage of this target protein, given its universal overexpression in tumors of both lower and upper urinary tract.
ASG-15ME is an ADC that was developed to target SLITRK6. ASG-15ME is composed of a SLITRK6-specific human gamma 2 antibody (ASG-15 C) conjugated to MMAE, a microtubule-disrupting agent. It is linked via a protease-cleavable linker. ASG-15C was chosen among seven anti-SLITRK6 mAbs and MMAE was chosen due to its efficacy in tumor inhibition and regression [48]. Upon binding to its target (SLITRK6), ASG-15ME is rapidly internalized and trafficked to lysosomes and early endosomes. Administered in a biweekly schedule at a dose of 5 mg/kg in UC cell line-derived and patient-derived xenograft models, ASG-15ME caused a significant tumor regression of 49 and 45% , respectively [47].
In view of these encouraging results, the first phase I trial was initiated in metastatic UC patients, unselected for SLITRK6 IHC expression and previously treated with chemotherapy [48]. ASG-15ME was administered IV on a weekly basis every 3 out of 4 weeks and six dose-escalating levels were studied, including 0.1, 0.25, 0.5, 0.75, 1.0, and 1.25 mg/kg. An interim analysis of the study reported results from 49 evaluable patients, of whom one had CR and 17 had a PR with an ORR of 38% , including 43% with prior ICI treatment. Notably, 6/13 (46%) patients with liver metastases achieved an OR. Fatigue was the most common adverse event (44%), but ≥grade 3 drug-related events were limited to 20% of patients and involved anemia (6%), increased lipase (6%), urinary tract infection (6%). Median duration on ASG-15ME was 13 weeks. The RP2D was 1.0 mg/kg given 2 patients experienced DLT (one with myocardial infarction, and one with grade 4 neutropenia with multi-organ failure) at 1.25 mg/kg. All events resolved with appropriate management. At the MTD the ORR was 50% [48]. The study is currently in active, non-recruiting status. While still in the early phases of development, ASG-15ME has shown promise as a treatment option for metastatic UC that received prior chemotherapy or/and immunotherapy.
ADC AGENTS TESTED IN EARLY STAGE UC
Oportuzumab monatox (VB4-845)
The transmembrane glycoprotein epithelial cell-adhesion molecule (EpCAM) is overexpressed in many epithelial tumors, showing markedly high expression in UC [49]. Notably, urinary EpCAM has independent prognostic utility in UC [50]. Additionally, circulating tumor cells (CTCs) using CellSearch, which is based on detection of EpCAM expression, may serve as an independent marker of UC progression after radical cystectomy [51]. Thus, EpCAM represents a viable target in UC and this prompted the design of oportuzumab monatox (VB4-845), a recombinant fusion protein that targets EpCAM-positive cancer cells. It consists of an anti-EpCAM humanized single-chain variable fragment (scFv) linked to a truncated form of Pseudomonas exotoxin A (ETA252– 608) that lacks the cell-binding domain [52]. Administration of this agent in different Ep-CAM-positive cancer cells and xenograft tumors resulted in growth inhibition and regression [52], supporting the advancement of the drug to phase I testing.
The first phase I study of VB4-845 was addressed to patients with non-muscle-invasive bladder UC, refractory to or intolerant of bacillus Calmette-Guerin (BCG). Ascending doses ranging between 0.1 and 30.16 mg were administered for 6 consecutive weeks via bladder instillation to 64 patients with Grade 2 or 3, stage Ta or T1 UC or carcinoma in situ (CIS), refractory to intolerant of intravesicular BCG [53]. At the 12-week timepoint, a MTD was not determined as the drug was well tolerated, and 39% of patients achieved a CR [53]. A subsequent phase II study enrolled 46 patients in total, who underwent one induction cycle of 6 (cohort 1) or 12 (cohort 2) weekly intravesical oportuzumab monatox (VB4-845) instillations of 30 mg, followed by up to three maintenance cycles of 3 weekly administrations every 3 months. A significant proportion of the study population (44%) achieved CR, with 16% remaining disease-free after a follow up of 18-25 months. The median time to recurrence was shorter in cohort 1 compared to cohort 2 (274 vs. 408 days) but all patients tolerated treatment well, with only mild to moderate, reversible bladder adverse events (e.g. discomfort, pain, spasm, infections, hematuria, incontinence, retention) [54]. Collectively, oportuzumab monatox appears promising as a future option in a previously not well-targeted patient subgroup of BCG-refractory or – intolerant patients. Further to this, an interesting phase I combination of the drug with the ICI durvalumab is being tested, in an effort to maximize local responses and further prolong recurrence-free intervals in these patients (NCT03258593).
Efficacy and toxicity data from ADCs which are at the most advanced stages of clinical development for treatment of UC (early and advanced) are listed in Table 1.
Table 1
Antibody drug-conjugates in advanced phase of development in UC
| Sacituzumab Govitecan (IMMU-132) | Enfortumab vedotin (ASG-22CE/ASG-22ME) | ASG-15ME | Oportuzumab monatox (VB4-845) | |
| Study | Tagawa et al. [33] | Petrylak et al. [42] | Petrylak et al. [48] | Kowalski et al [54] |
| Phase | II | I | I | I/II |
| N | 41 | 67 | 42 | 46 |
| Setting | mUC after ≥1 prior chemo/ICI | mUC after ≥2 prior chemo/ICI | mUC after ≥1 prior chemo/ICI | NMIBC, BCG- refractory/intolerant |
| Target | Trop2 | Nectin-4 | SLITRK6 | EpCAM |
| Dosing/Frequency | 10 mg/kg | Cohort 1 : 0.5 mg/kg | Cohort 1 : 0.1 mg/kg | 1 induction cycle of 6 (cohort 1) or 12 (cohort 2) weekly intravesical instillations of 30 mg, followed by up to three maintenance cycles of 3 weekly administrations every 3 months |
| Days 1 and 8 every 3 weeks | Cohort 2 : 0.75 mg/kg | Cohort 2 : 0.25 mg/kg | ||
| Cohort 3 : 1.0 mg/kg | Cohort 3 : 0.5 mg/kg | |||
| Cohort 4 : 1.25 mg/kg | Cohort 4 : 0.75 mg/kg | |||
| Days 1, 8, and 15 every 4 weeks | Cohort 5 : 1.0 mg/kg | |||
| Cohort 6 : 1.25 mg/kg | ||||
| weekly for 3 out of every 4 weeks | ||||
| ORR | 34% | 31% | 33% | 44% (CR) |
| Median PFS/DFS Median OS (months) | 7.2 | 4.2 | 4 | 9 (cohort 1), 13 (cohort 2) |
| 15.5 | ||||
| Grade 3/4 treatment-related AEs | Neutropenia (39%) | Hyponatremia (6%) | Fatigue (44%) | None |
| Anemia (10%) | Anemia (3%) | 23 pts (50%) had Grade 3/4 AEs, 9 (20%) of which were considered related. | ||
| Diarrhea (7%) | Vomiting (3% | Ocular (1%) | ||
| Fatigue (7%) | Diarrhea (2%) | |||
| Febrile neutropenia (5%) | Decreased appetite (2%) | |||
| Rash (2%) | ||||
| Fatigue (2%) | ||||
| Most common AEs | Diarrhea (63%) | Fatigue (55%) | Ten pts had reversible ocular AEs. 4 pts had protocol defined dose limiting toxicities. | Urinary (63%): |
| Nausea (56%) | Nausea (48%) | dysuria, hematuria, incontinence | ||
| Neutropenia (49%) | Decreased appetite (45%) | General (26%): | ||
| Fatigue (49%) | Alopecia (43%) | asthenia, catheter site related reaction, fatigue | ||
| Constipation (37%) | Diarrhea (43%) | |||
| Rash (32%) | Dysgeusia (33%) | |||
| Anemia (29%) | Rash (25%) |
EMERGING TARGETS AND ADC THERAPEUTICS IN UC
Accumulating experience from testing of ADCs in several different tumor types has fueled further research for identification of new targets in UC. Another strategy lies in exploitation of targets expressed in UC, which are already paired to a particular ADC in use for a different malignancy. One such example is the receptor tyrosine-protein kinase erbB-2, also known as HER2. Although seen highest in breast and lung cancer tissue, the frequency of HER2 overexpression was also found to be significant in UC tissue, commonly as a result of gene amplifications or/and activating mutations, particularly in luminal subtypes [55, 56]. HER2 represents a strategic target because its expression is low or undetectable in normal urothelium but increases in early UC and becomes more pronounced as the disease progresses [57, 58].
Trastuzumab emtansine (T-DM1) is an ADC consisting of the mAb trastuzumab linked to the microtubule poison emtansine (DM1). It is currently FDA-approved for treatment of advanced HER2-positive, taxane/transtuzumab-pretreated breast cancer patients, based on PFS and OS benefit over lapatinib/capecitabine shown in the EMILIA phase III randomized trial [18]. Hence, T-DM1 may constitute a potential candidate for treating HER2-positive UC patients as well. Preclinical testing of T-DM1 in orthotopic xenograft UC models demonstrated tumor growth inhibition one week after the first treatment, which was significantly more pronounced compared to treatment with control IgG and trastuzumab alone in cisplatin-resistant UC cells [59]. These encouraging data mere further confirmation in UC patients. Although a clinical trial dedicated to UC is currently lacking, there are two ongoing phase II studies testing the drug in patients with HER2-overexpressing tumors, including UC (NCT02999672, NCT02675829).
A second HER2-targeting ADC, named DS-8201a, was recently developed as a more potent ADC with a higher drug-to-antibody-ratio compared to T-DM1, by virtue of its enzymatically cleavable peptide linker and exatecan-derivative topoisomerase I inhibitor (DXd) [60]. These characteristics enable DS-8201a to be active in low HER2-expressing and HER2-resistant gastric and breast cancer cells and xenograft tumors as well as in T-DM1 resistant patient-derived xenograft models of breast cancer [61–63]. Early phase I results in heavily pretreated patients with metastatic tumors from different primaries, including breast, gastric, colorectal, salivary, and non-small cell lung cancer, showed an impressive ORR of 40% in 73 evaluable patients including 14 with low HER2 expression [64]. The drug was well tolerated, with no DLT at initial dose of 8 mg/kg, which was then followed by dose escalation of 6.4 and 5.4 mg/kg IV every 3 weeks. One third of patients (29%) experienced ≥grade 3 AEs (grade 3 : 25% grade 4 : 4%). Overall, the most common adverse events were nausea (62%), anorexia (56%) and thrombocytopenia (28%). These compelling data led to a Fast Track designation to DS-8201 for the treatment of HER2-positive unresectable and/or metastatic breast cancer in patients who have progressed after prior treatment with HER2-targeted therapies including T-DM1 [64]. Additionally, several phase II studies of the drug are ongoing in advanced breast cancer (NCT03248492) and gastric cancer patients (NCT03329690). DS-8201a hold promise for testing in HER2-expressing UC, potentially even in tumors with low level of HER2 expression. This rational led to the design of a Phase Ib multicenter, open-label study combining DS-8201 with nivolumab in chemotherapy pretreated UC and breast cancer patients, with enrollment being planned during the first quarter of2018 [64].”
Another potential target protein with significant expression in UC is the Thomsen-Fridenreich antigen (TF-Ag). TF-Ag is a disaccharide hidden on normal cells but is selectively expressed on the surface of approximately 80% of UC cells along with breast, colon, and prostate cancer cells [65]. TF-Ag plays an important role in cancer cell adhesion and metastasis by interacting with lectins at metastatic sites [65]. JAA-F11 was developed as a highly specific mAb against TF-Ag. The bulk of in vitro evidence for antitumor activity of JAA-F11 emanates from breast cancer cell lines and mouse models [66]. The ability to conjugate JAA-F11 to 124I has enabled visualization of drug internalization and of resulting growth inhibition [66]. Development of a humanized JAA-F11 (hJAA-F11), which binds only to TF-Ag alpha-linked structures and not to TF-Ag beta-linked structures, further increased the drug’s efficacy in lung and breast cancer cell lines [66]. Binding of JAA-F11 to the tumor-associated Gal-β-(1– 3)-GalNAc α-linked structure and not to the corresponding β-linked structure, which is expressed in normal cells, renders the drug an excellent choice for efficient and effective drug delivery. More importantly, conjugation of the drug with DM1 (hJAA-F11-DM1) led to suppression of tumor growth in a human breast cancer xenograft model [65]. In view of the success of hJAA-F11-DM1 in breast cancer in vivo, this ADC warrants testing in UC.
The fibroblast-growth factor receptor (FGFR) pathway is essential for growth, invasion and migration progression of several tumor types, including UC. There are 22 known FGF ligands identified, with 4 highly conserved transmembrane tyrosine kinase receptors (FGFR1-4) that mediate their effects [67]. Particularly FGFR3 mutations, fusions and amplification are a hallmark of the luminal-papillary subtype of UC, which occurs in 35% of muscle-invasive disease [56]. FGFR targeting with use of a pan-FGFR small molecule inhibitor (JNJ-42756493 or erdafitinib) has shown promising efficacy and tolerability results in phase I/II setting and received breakthrough therapy designation in patients with metastatic or unresectable UC that harbor FGFR gene alterations [68, 69]. Although there is no currently available ADC using a pan-FGFR inhibitor as payload, specific FGFR1 targeting was successful via design of an engineered variant of fibroblast growth factor 1 (FGF1 V), which was conjugated to the potent cytotoxic drug, MMAE [70]. Focusing on FGFR1 signaling, it is effected through FGF2, involves MAPK activation and can be reversed by selective FGFR1 knockdown in vitro. This FGF1V– valine– citrulline– MMAE conjugate resulted in reduced cell viability of FGFR-expressing cell lines [70]. An improved FGF1-vcMMAE conjugate featuring selective endocytosis as compared with FGFR1-negative cells was developed [72] and further testing of the drug in vivo is awaited. An alternative approach within the same pathway targets the ligand itself, FGF2, instead of the receptor. FGF2 conjugates containing three molecules of MMAE showed effective targeting of FGFR1-expressing cells, with adequate internalization upon FGFR1-mediated endocytosis, and, high cytotoxicity, which was also associated with the FGFR1 expression level [73]. Collectively, further development of FGF/FGFR axis ADCs is eagerly awaited given promising early in vitroresults.
The fibroblast growth factor (FGF)-inducible 14 (Fn14) receptor is another relevant target, based on its low expression level in normal tissues but significant upregulation in UC and other tumor types [74]. An ADC consisting of a high-affinity anti-Fn14 mAb (ITEM-4) conjugated to a highly cytotoxic ribosome-inactivating N-glycosidase, named recombinant gelonin (rGel), was developed. The ITEM4-rGel ADC induced apoptosis and growth suppression in UC cells and mouse xenografts, respectively [74]. Design of second generation payloads and of a humanized version of this ADC were planned [74] but have yet to be reported.
Fig.1
111In-J591 single-photon-emission tomography (SPECT) in a patient with metastatic bladder urothelial carcinoma (UC). From left to right: coronal, sagittal and transaxial views from SPECT/CT obtained 72 hours after the administration of 5.55 mCi In-111 DOTA J-591. (A) CT images of the head showing a focal hyperintense lesion with surrounding edema in the right hemisphere. (B) SPECT images of the head in grayscale and (C) in color scale, showing multiple foci of increased uptake in the brain, skull, mandibles and cervical nodes.
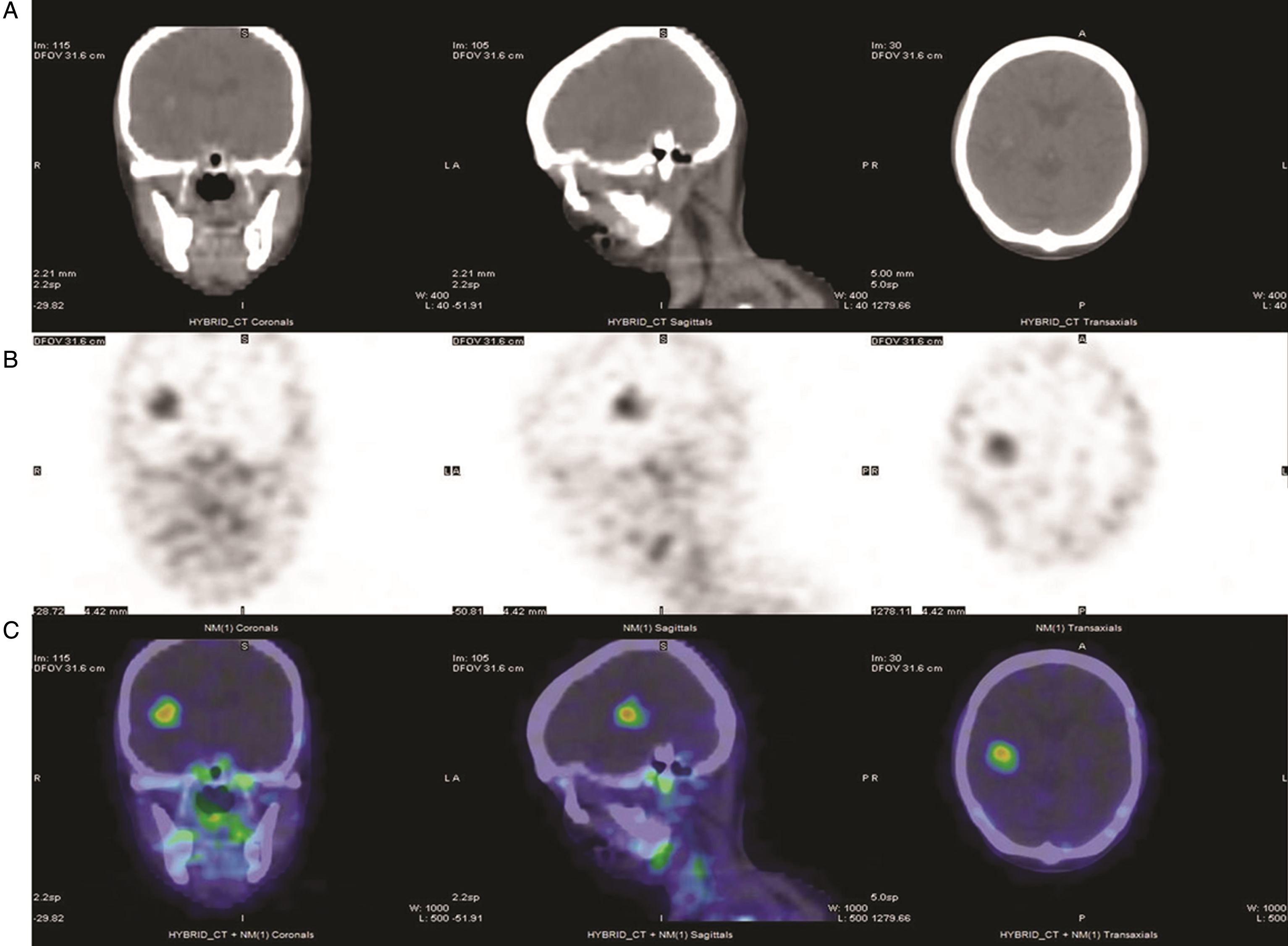
Of particular interest to the development of ADC agents is simultaneously achieving target-specific cytotoxicity and ability to detect this via imaging modalities (usually by specific PET studies). A novel ADC that fulfils these criteria was designed to target interleukin 5 receptor alpha subunit (IL5Rα), which plays an important role in orchestrating the migration of UC cells through increased MMP-9 expression [75, 76]. The ADC mAb A14, and a corresponding radiolabeled immunoconjugate (RIC) were developed by conjugation of IL5Rα to vinblastine and to the positron emitter 64Cu, respectively [76]. In muscle-invasive UC tumor-bearing mice, 64Cu-A14 PET was able to detect IL5Rα tumors while vinblastine-A14 was cytotoxic against them [76], thus providing the proof for further advancing this ADC into the clinical phase ofdevelopment.
Another ADC already in advanced phase of development in another malignancy which is of potential value in UC uses prostate-specific membrane antigen (PSMA) as target. PSMA is a transmembrane protein expressed in high amounts at the cell surface of prostate cancer cells [77], and its targeting via the mAb J591 has shown promising imaging and therapeutic results in phase I and II clinical trials utilizing PSMA mAb J591, radiolabeled with 90Y, 177Lu, 111In, or 89Zr [78–80]. While PSMA was originally thought to be prostate-specific, subsequent studies have indicated the protein is also expressed in multiple tumor types, both malignant and benign, including UC [81]. Although PSMA IHC expression was infrequent in UC (3/96 cases; 3%), all UC tumors demonstrated PSMA expression in the tumor vasculature, which may indicate a potential role in facilitating new vessel growth [81]. Angiogenesis is a key hallmark of UC and several targeted approaches including mAbs (anti-VEGFA: bevacizumab) and tyrosine kinase inhibitors (sunitinib, sorafenib, vandetaib, cabozantinib) were tested, commonly in combination with chemotherapy, with modest results and frequent toxicity limitations [82]. Addition of ramucirumab, a mAb against vascular endothelial growth factor receptor (VEGFR2) to docetaxel resulted in PFS prolongation compared to docetaxel alone in platinum-refractory advanced UC patients [82, 83]. Therefore PSMA, which is expressed in UC tumor vessels, may be a potential ADC target for inhibiting UC angiogenesis. While tissue studies consistently result in high frequencies of PSMA expression in tumor neovasculature, it is unclear if the levels of expression are high enough to be detected reliably with molecular imaging [84, 85]. However, 111In-J591 imaging with whole body gamma camera has a high detectability of tumor lesions and vasculature in various cancer types, including UC (Fig. 1) [86].
CONCLUSIONS
Several ADCs against epithelial antigens are in different stages of clinical development. They are generally well-tolerated and their ORR in heavily-pretreated UC patients compares favorably to historical controls. BCG-refractory patients with early UC is another setting where ADCs can be useful. These results provide justification to prospectively assess their efficacy in various combinations with cisplatin-based chemotherapy and ICIs. The choice and sequencing of systemic therapies in the post-chemotherapy setting is becoming increasingly challenging with several phase II single agent ICI positive trials [8–11], as well as initial positive phase III results from the ramucirumab/docetaxel combination [83]. The preliminary efficacy of several ADCs in advanced UC may present an additional treatment option in the future. In absence of prospective randomized comparisons, decision making should be determined after evaluation of both drug and disease characteristics as well as the level of evidence. For example, the observation that, unlike ICIs, ADCs may maintain a high efficacy level in poor prognostic groups such as patients with hepatic metastases could favor using of ADCs in this setting. In the era of precision oncology, a deeper understanding of the molecular subtypes of UC can offer additional predictive information if available. Associations of gene expression analyses and outcomes from large datasets of advanced UC patient cohorts treated with chemotherapy or ICIs have yielded interesting observations on the impact of biology on ORR and PFS. Patients with basal tumors appear to have the highest benefit (compared to luminal and claudin-low tumors) in cohorts treated with neoadjuvant platinum-chemotherapy [87]. In the prospective phase II study of atezolizumab in advanced, post-platinum UC patients, the ICI demonstrated the lowest response rates in the luminal-papillary (cluster I) subtype compared to other subtypes [8]. In another phase II ICI trial, response to nivolumab was lower in the luminal 1 (cluster I) UC tumors showing low expression of interferon-gamma signature genes [11]. Taken together, these findings suggest this subtype may be relatively insensitive to ICIs and provide a strong biological rationale and window of opportunity for evaluating ADCs in patients with tumors of luminalgenotype.
Although there is still a significant amount of knowledge to be learned, ADCs have the potential to become an additional treatment option in the armamentarium of available therapies for UC at various stages. While efforts in improving their efficacy-toxicity ratio should continue, development of surrogate markers of response will also be critical for better assessing their activity and best window of opportunity for use in the context of treatment sequencing.
CONFLICT OF INTEREST
STT has served as a paid consultant to Immunomedics. Weill Cornell Medicine has received funding for clinical trials from Immunomedics, Seattle Genetics, Genentech, Janssen, Astellas. The patent for J591 is assigned to the Cornell Center for Technology Licensing.
REFERENCES
[1] | Alfred Witjes J , Lebret T , Compérat EM , Cowan NC , De Santis M , Bruins HM, et al. Updated 2016 EAU guidelines on muscle-invasive and metastatic bladder cancer. Eur Urol. (2017) ;71: (3):462–75. |
[2] | Rouprêt M , Babjuk M , Compérat E , Zigeuner R , Sylvester RJ , Burger M, et al. European association of urology guidelines on upper urinary tract urothelial cell carcinoma: 2015 update. Eur Urol. (2015) ;68: (5):868–79. |
[3] | Siegel RL , Miller KD , Jemal A . Cancer statistics, 2016. CA Cancer J Clin. (2016) ;66: (1):7–30. |
[4] | Loehrer PJ , Einhorn LH , Elson PJ , Crawford ED , Kuebler P , Tannock I, et al. A randomized comparison of cisplatin alone or in combination with methotrexate, vinblastine, and doxorubicin in patients with metastatic urothelial carcinoma: A cooperative group study. J Clin Oncol. (1992) ;10: (7):1066–73. |
[5] | von der Maase H , Sengelov L , Roberts JT , Ricci S , Dogliotti L , Oliver T, et al. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J Clin Oncol. (2005) ;23: (21):4602–8. |
[6] | Bellmunt J , von der Maase H , Mead GM , Skoneczna I , De Santis M , Daugaard G, et al. Randomized phase III study comparing paclitaxel/cisplatin/gemcitabine and gemcitabine/cisplatin in patients with locally advanced or metastatic urothelial cancer without prior systemic therapy: EORTC Intergroup Study 30987. J Clin Oncol. (2012) ;30: (10):1107–13. |
[7] | Bellmunt J , de Wit R , Vaughn DJ , Fradet Y , Lee JL , Fong L, et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N Engl J Med. (2017) ;376: (11):1015–26. |
[8] | Rosenberg JE , Hoffman-Censits J , Powles T , van der Heijden MS , Balar AV , Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet. (2016) ;387: (10031):1909–20. |
[9] | Powles T , O’Donnell PH , Massard C , Arkenau HT , Friedlander TW , Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: Updated results from a phase 1/2 open-label study. JAMA Oncol. (2017) ;3: (9):e172411. |
[10] | Apolo AB , Infante JR , Balmanoukian A , Patel MR , Wang D , Kelly K, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: Results from a multicenter, phase Ib study. J Clin Oncol. (2017) ;35: (19):2117–24. |
[11] | Sharma P , Retz M , Siefker-Radtke A , Baron A , Necchi A , Bedke J , et al., Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. (2017) ;18: (3):312–22. |
[12] | Balar AV , Galsky MD , Rosenberg JE , Powles T , Petrylak DP , Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet. (2017) ;389: (10064):67–76. |
[13] | Nagayama A , Ellisen LW , Chabner B , Bardia A . Antibody-Drug Conjugates for the Treatment of Solid Tumors: Clinical Experience and Latest Developments. Target Oncol. (2017) ;12: (6):719–39. |
[14] | Klute K , Nackos E , Tasaki S , Nguyen DP , Bander NH , Tagawa ST . Microtubule inhibitor-based antibody-drug conjugates for cancer therapy. Onco Targets Ther. (2014) ;7: :2227–36. |
[15] | Sievers EL , Larson RA , Stadtmauer EA , Estey E , Löwenberg B , Dombret H, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. (2001) ;19: (13):3244–54. |
[16] | Bross PF , Beitz J , Chen G , Chen XH , Duffy E , Kieffer L, et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. (2001) ;7: (6):1490–6. |
[17] | U.S. Food and Drug Administration. FDA Approves Gemtuzumab Ozogamicin for CD33-positive AML. Available from: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm574518.htm. |
[18] | Verma S , Miles D , Gianni L , Krop IE , Welslau M , Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. (2012) ;367: (19):1783–91. |
[19] | Krop IE , Kim SB , Martin AG , LoRusso PM , Ferrero JM , Badovinac-Crnjevic T, et al. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. (2017) ;18: (6):743–54. |
[20] | Moskowitz CH , Nademanee A , Masszi T , Agura E , Holowiecki J , Abidi MH, et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2015) ;385: (9980):1853–62. |
[21] | Pro B , Advani R , Brice P , Bartlett NL , Rosenblatt JD , Illidge T, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J Clin Oncol. (2012) ;30: (18):2190–6. |
[22] | Kantarjian HM , DeAngelo DJ , Stelljes M , Martinelli G , Liedtke M , Stock W, et al. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. (2016) ;375: (8):740–53. |
[23] | Lambert JM , Morris CQ . Antibody-drug conjugates (ADCs) for personalized treatment of solid tumors: A review. Adv Ther. (2017) ;34: (5):1015–35. |
[24] | Masters JC , Nickens DJ , Xuan D , Shazer RL , Amantea M . Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Invest New Drugs. (2018) ;36: (1):121–35. |
[25] | Fabi A , De Laurentiis M , Caruso M , Valle E , Moscetti L , Santini D, et al. Efficacy and safety of T-DM1 in the ‘common-practice’ of HER2+ advanced breast cancer setting: A multicenter study. Oncotarget. (2017) ;8: (38):64481–9. |
[26] | Fornaro M , Dell’Arciprete R , Stella M , Bucci C , Nutini M , Capri MG, et al. Cloning of the gene encoding Trop-2, a cell-surface glycoprotein expressed by human carcinomas. Int J Cancer. (1995) ;62: (5):610–8. |
[27] | Trerotola M , Cantanelli P , Guerra E , Tripaldi R , Aloisi AL , Bonasera V, et al. Upregulation of Trop-2 quantitatively stimulates human cancer growth. Oncogene. (2013) ;32: (2):222–33. |
[28] | Avellini C , Licini C , Lazzarini R , Gesuita R , Guerra E , Tossetta G, et al. The trophoblast cell surface antigen 2 and miR-125b axis in urothelial bladder cancer. Oncotarget. (2017) ;8: (35):58642–53. |
[29] | Starodub AN , Ocean AJ , Shah MA , Guarino MJ , Picozzi VJ Jr , Vahdat LT, et al. First-in-human trial of a novel anti-trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res. (2015) ;21: (17):3870–8. |
[30] | Sharkey RM , McBride WJ , Cardillo TM , Govindan SV , Wang Y , Rossi EA, et al. Enhanced delivery of SN-38 to human tumor xenografts with an anti-trop-2-SN-38 antibody conjugate (sacituzumab govitecan). Clin Cancer Res. (2015) ;21: (22):5131–8. |
[31] | Ocean AJ , Starodub AN , Bardia A , Vahdat LT , Isakoff SJ , Guarino M, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer. (2017) ;123: (19):3843–54. |
[32] | Faltas B , Goldenberg DM , Ocean AJ , Govindan SV , Wilhelm F , Sharkey RM, et al. Sacituzumab govitecan, a novel antibody–drug conjugate, in patients with metastatic platinum-resistant urothelial carcinoma. Clin Genitourin Cancer.e. (2016) ;14: (1):75–9. |
[33] | Tagawa ST , Faltas B , Lam E , Saylor P , Bardia A , Hajdenberg J, et al. Sacituzumab govitecan (IMMU-132) for patients with pretreated metastatic urothelial cancer (UC): Interim results. Ann Oncol. (2017) ;28: (suppl_5): v295–v329. |
[34] | Bardia A , Mayer IA , Diamond JR , Moroose RL , Isakoff SJ , Starodub AN, et al. Efficacy and safety of anti-trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J Clin Oncol. (2017) ;35: (19):2141–8. |
[35] | ADC Review. Sacituzumab Govitecan Receives Breakthrough Therapy Designation for Triple-negative Breast Cancer., [Published on 08th February 2018]. Available from: https://adcreview.com/news/sacituzumab-govitecan-receives-breakthrough-therapy-designation-for-triple-negative-breast-cancer. |
[36] | Challita-Eid PM , Satpayev D , Yang P , An Z , Morrison K , Shostak Y, et al. Enfortumab vedotin antibody-drug conjugate targeting nectin-4 is a highly potent therapeutic agent in multiple preclinical cancer models. Cancer Res. (2016) ;76: (10), 3003–13. |
[37] | Pavlova NN , Pallasch C , Elia AE , Braun CJ , Westbrook TF , Hemann M , Elledge SJ . A role for PVRL4-driven cell-cell interactions in tumorigenesis. Elife. (2013) ;2: :e00358. |
[38] | Siddharth S , Goutam K , Das S , Nayak A , Nayak D , Sethy C, et al. Nectin-4 is a breast cancer stem cell marker that induces WNT/β-catenin signaling via Pi3k/Akt axis. Int J Biochem Cell Biol. (2017) ;89: :85–94. |
[39] | Rosenberg JE , Heath E , Perez R , Merchan J , Lang J , Ruether D, et al. Interim analysis of a phase I dose escalation trial of ASG-22CE (ASG-22ME; enfortumab vedotin), an antibody drug conjugate (ADC), in patients (Pts) with metastatic urothelial cancer (mUC). Ann Oncol. (2016) ;27: (6):266–95. |
[40] | [No authors listed] Targeting Nectin-4 in Bladder Cancer. Cancer Discov. (2017) ;7: (8):OF3. |
[41] | Petrylak DP , Perez RP , Zhang J , Smith DC , Ruether JD , Sridhar SS, et al. A phase I study of enfortumab vedotin (ASG-22CE; ASG-22ME): Updated analysis of patients with metastatic urothelial cancer. J Clin Oncol. 35: (suppl 15; abstr 106). |
[42] | Petrylak DP , Smith DC , Flaig TW , Zhang J , Sridhar SS , Ruether JD, et al. Enfortumab vedotin (EV) in patients (Pts) with metastatic urothelial carcinoma (mUC) with prior checkpoint inhibitor (CPI) failure: A prospective cohort of an ongoing phase 1 study. J Clin Oncol. (2018) ;36: (suppl 6S;abstr 431). |
[43] | ADC Review. Phase I Data of Enfortumab Vedotin in Metastatic Urothelial Cancer Supports Start of Pivotal Monotherapy Phase II Trial., [Published on 07th June]. Available from: https://adcreview.com/news/phase-data-enfortumab-vedotin-metastatic-urothelial-cancer-supports-start-pivotal-monotherapy-phase-ii-trial/. |
[44] | ADC Review. Promising Phase I Data for Endortumab Vendotin (ASG-22ME) and ASG-15ME in Metastatic Urothelial Cancer Presented at the ESMO Congress.. [Published on 17th October 2016]. Available from: https://adcreview.com/news/promising-phase-data-enfortumab-vedotin-asg-22me-asg-15me-metastatic-urothelial-cancer-presented-2016-esmo-congress/. |
[45] | Aruga J , Yokota N , Mikoshiba K . Human SLITRK family genes: Genomic organization and expression profiling in normal brain and brain tumor tissue. Gene. (2003) ;317: :87–94. |
[46] | Tekin M , Chioza BA , Matsumoto Y , Diaz-Horta O , Cross HE , Duman D, et al. SLITRK6 mutations cause myopia and deafness in humans and mice. J Clin Invest. (2013) ;123: (5):2094–102. |
[47] | Morrison K , Challita-eid PM , Raitano A , An Z , Yang P , Abad JD, et al. Development of ASG-15ME, a Novel Antibody-Drug Conjugate Targeting SLITRK6, a New Urothelial Cancer Biomarker. Mol Cancer Ther. (2016) ;15: (6):1301–10. |
[48] | Petrylak D , Heath E , Sonpavde G , George S , Morgans AK , Eigl BJ, et al. Interim analysis of phase 1 dose escalation trial of the antibody-drug conjugate (ADC) ASG15E (ASG-15ME) in patients (Pts) with metastatic urothelial cancer (mUC). Ann Oncol. (2016) ;27: (6):266–95. |
[49] | Fong D , Seeber A , Terracciano L , Kasal A , Mazzoleni G , Lehne F, et al. Expression of EpCAM(MF) and EpCAM(MT) variants in human carcinomas. J Clin Pathol. (2014) ;67: (5):408–14. |
[50] | Bryan RT , Regan HL , Pirrie SJ , Devall AJ , Cheng KK , Zeegers MP, et al. Protein shedding in urothelial bladder cancer: Prognostic implications of soluble urinary EGFR and EpCAM. Br J Cancer. (2015) ;112: (6):1052–8. |
[51] | Abrahamsson J , Aaltonen K , Engilbertsson H , Liedberg F , Patschan O , Rydén L, et al. Circulating tumor cells in patients with advanced urothelial carcinoma of the bladder: Association with tumor stage, lymph node metastases, FDG-PET findings, and survival. Urol Oncol. (2017) ;35: (10):606.e9–606.e16. |
[52] | Di Paolo C , Willuda J , Kubetzko S , Lauffer I , Tschudi D , Waibel R, et al. A recombinant immunotoxin derived from a humanized epithelial cell adhesion molecule-specific single-chain antibody fragment has potent and selective antitumor activity. Clin Cancer Res. (2003) ;9: (7):2837–48. |
[53] | Kowalski M , Entwistle J , Cizeau J , Niforos D , Loewen S , Chapman W, et al. A Phase I study of an intravesically administered immunotoxin targeting EpCAM for the treatment of nonmuscle-invasive bladder cancer in BCG refractory and BCG-intolerant patients. Drug Des Devel Ther. (2010) ;4: :313–20. |
[54] | Kowalski M , Guindon J , Brazas L , Moore C , Entwistle J , Cizeau J, et al. A phase II study of oportuzumab monatox: An immunotoxin therapy for patients with noninvasive urothelial carcinoma in situ previously treated with bacillus Calmette-Guérin. J Urol. (2012) ;188: (5):1712–8. |
[55] | Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. (2014) 507: (7492):315–22. |
[56] | Robertson AG , Kim J , Al-Ahmadie H , Bellmunt J , Guo G , Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. (2017) ;171: (3):540–556.e25. |
[57] | Fleischmann A , Rotzer D , Seiler R , Studer UE , Thalmann GN . Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur Urol. 201. 60: (2):350–7. |
[58] | Tschui J , Vassella E , Bandi N , Baumgartner U , Genitsch V , Rotzer D, et al. Morphological and molecular characteristics of HER2 amplified urothelial bladder cancer. Virchows Arch. (2015) ;466: (6):703–10. |
[59] | Hayashi T , Seiler R , Oo H . Z, Jäger W, Moskalev I, Awrey S, et al. Targeting HER2 with T-DM1, an antibody cytotoxic drug conjugate, is effective in HER2 over expressing bladder cancer. J Urol. (2015) ;194: (4):1120–31. |
[60] | Takegawa N , Nonagase Y , Yonesaka K , Sakai K , Maenishi O , Ogitani Y, et al. DS-a, a new HER2-targeting antibody-drug conjugate incorporating a novel DNA topoisomerase I inhibitor, overcomes HER2-positive gastric cancer T-DM1 resistance. Int J Cancer. (2017) ;141: (8):1682–9. |
[61] | Ogitani Y , Hagihara K , Oitate M , Naito H , Agatsuma T . Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. (2016) ;107: (7):1039–46. |
[62] | Ogitani Y , Aida T , Hagihara K , Yamaguchi J , Ishii C , Harada N, et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin Cancer Res. (2016) ;22: (20):5097–108. |
[63] | Doi T , Iwata H , Tsurutani J , Takahashi S , Park H , Redfern CH , et al. Single agent activity of DS-8201a, a HER2-targeting antibody-drug conjugate, in heavily pretreated HER2 expressing solid tumors. J Clin Oncol. (2017) ;35: (15 suppl; abstr 108). |
[64] | ADC Review. Updated analysis from ongoing phase I trial of DS-8201 confirms significant benefit for HER2+ breast cancer patients pretreated with Ado-transtuzumab emtasine. [Published on 7th December (2017) ]. Available from: https://adcreview.com/news/updated-analysis-ongoing-phase-trial-ds-8201-confirms-significant-benefit-her2-breast-cancer-patients-pretreated-ado-trastuzumab-emtansine/ |
[65] | Tati S , Fisk J.C. , Abdullah J , Karacosta L , Chrisikos T , Philbin P, et al. Humanization of JAA-F11, a highly specific anti-thomsen-friedenreich pancarcinoma antibody and in vitro efficacy analysis. Neoplasia. (2017) ;19: :716–33. |
[66] | Ferguson K , Yadav A , Morey S , Abdullah J , Hrysenko G , Eng JY, et al. Preclinical studies with JAA-F11 anti-Thomsen-Friedenreich monoclonal antibody for human breast cancer. Future Oncol. (2014) ;10: (3):385–99. |
[67] | Chae YK , Ranganath K , Hammerman PS , Vaklavas C , Mohindra N , Kalyan A, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget. (2017) ;8: (9):16052–74. |
[68] | Tabernero J , Bahelda R , Dienstmann R , Infante JR , Mita A , Italiano A, et al. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. (2015) ;33: (30):3401–8. |
[69] | Siefker-Radtke AO , Mellado B , Decaestecker K , Burke JM , O’Hagan A , Avadhani A, et al. Ongoing phase 2 study of erdafitinib (JNJ-42756493), a pan-fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in patients (pts) with metastatic or unresectable urothelial carcinoma (M/UR UC) and FGFR gene alterations. Ann Oncol. (2016) ;27: (suppl_6):845TiP. |
[70] | Szlachcic A , Zakrzewska M , Lobocki M , Jakimowicz P , Otlewski J . Design and characteristics of cytotoxic fibroblast growth factor 1 conjugate for fibroblast growth factor receptor-targeted cancer therapy. Drug Des Devel Ther. (2016) ;10: :2547–60. |
[71] | Tomlinson DC , Lamont FR , Shnyder SD , Knowles MA . Fibroblast growth factor receptor 1 promotes proliferation and survival via activation of the mitogen-activated protein kinase pathway in bladder cancer. Cancer Res. (2009) ;69: (11):4613–20. |
[72] | Lobocki M , Zakrzewska M , Szlachcic A , Krzyscik MA , Sokolowska-Wedzina A , Otlewski J . High-Yield Site-Specific Conjugation of Fibroblast Growth Factor 1 with Monomethylauristatin E via Cysteine Flanked by Basic Residues. Bioconjug Chem. (2017) ;28: (7):1850–8. |
[73] | Krzyscik MA , Zakrzewska M , Sørensen ørensen V , Sokolowska-Wedzina A , Lobocki M , Swiderska KW, et al. Cytotoxic conjugates of fibroblast growth factor 2 (FGF2) with monomethyl auristatin e for effective killing of cells expressing FGF receptors. ACS Omega. (2017) ;2: (7):3792–805. |
[74] | Zhou H , Mohamedali KA , Gonzalez-Angulo AM , Cao Y , Migliorini M , Cheung LH, et al. Development of human serine protease-based therapeutics targeting Fn14 and identification of Fn14 as a new target overexpressed in TNBC. Mol Cancer Ther. (2014) ;13: (11):2688–705. |
[75] | Lee EJ , Park SS , Kim WJ , Moon SK . IL-5-induced migration via ERK1/2-mediated MMP-9 expression by inducing activation of NF-κB in HTcells. Oncol Re. (2012) ;28: (3):1084–90. |
[76] | Paquette M , Vilera-Perez LG , Beaudoin S , Ekindi-Ndongo N , Boudreaut PL , Bonin MA, et al. Targeting IL-5Rα with antibody-conjugates reveals a strategy for imaging and therapy for invasive bladder cancer. Oncoimmunology. (2017) ;6: (10):e1331195. |
[77] | Vlachostergios PJ , Galletti G , Palmer J , Lam L , Karir BS , Tagawa ST . Antibody therapeutics for treating prostate cancer: Where are we now and what comes next? Expert Opin Biol Ther. (2017) ;17: (2):135–49. |
[78] | Bander NH , Milowsky MI , Nanus DM , Kostakoglu L , Vallabhajosula S , Goldsmith SJ . Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J Clin Oncol. (2005) ;23: (21):4591–601. |
[79] | Tagawa ST , Milowsky MI , Morris M , Vallabhajosula S , Christos P , Akhtar NH, et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin Cancer Res. (2013) ;19: (18):5182–91. |
[80] | Pandit-Taskar N , O’Donoghue JA , Durack JC , Lyashchenko SK , Cheal SM , Beylergil V, et al. A phase I/II study for analytic validation of 89Zr-J591 immunoPET as a molecular imaging agent for metastatic prostate cancer. Clin Cancer Res. (2015) ;21: (23):5277–85. |
[81] | Samplaski MK , Heston W , Elson P , Magi-Galluzzi C , Hansel DE . Folate hydrolase (prostate-specific antigen) 1 expression in bladder cancer subtypes and associated tumor neovasculature. Mod Pathol. (2011) ;24: (11):1521–9. |
[82] | Vlachostergios PJ , Lee A , Thomas C , Walsh R , Tagawa ST . A critical review on ramucirumab in the treatment of advanced urothelial cancer. Future Oncol. (2017) . doi: 10.2217/fon-2017-0473 |
[83] | Petrylak DP , de Wit R , Chi KN , Drakaki A , Sternberg CN , Nishiyama H , et al. Ramucirumab plus docetaxel versus placebo plus docetaxel in patients with locally advanced or metastatic urothelial carcinoma after platinum-based therapy (RANGE): A randomised, double-blind, phase 3 trial. Lancet. (2017) ;390: (10109):2266–77. |
[84] | Campbell SP , Baras AS , Ball MW , Kates M , Hahn NM , Bivalacqua TJ , et al. Low levels of PSMA expression limit the utility of 18F-DCFPyL PET/CT for imaging urothelial carcinoma. Ann Nucl Med. (2018) ;32: (1):69–74. |
[85] | Roy SG , Parida GK , Tripathy S , Singhal A , Tripathi M , Bal C . In vivo demonstration of PSMA expression in adenocarcinoma urinary bladder using 68Ga-PSMA 11 PET/CT. Clin Nucl Med. (2017) ;42: (7):542–3. |
[86] | Pandit-Taskar N , O’Donoghue JA , Divgi CR , Wills EA , Schwartz L , Gönen M , et al. Indium 111-labeled J591 anti-PSMA antibody for vascular targeted imaging in progressive solid tumors. EJNMMI Res. (2015) ;5: :28. |
[87] | Seiler R , Ashab HAD , Erho N , van Rhijn BWG , Winters B , Douglas J, et al. Impact of Molecular subtypes in muscle-invasive bladder cancer on predicting response and survival after neoadjuvant chemotherapy. Eur Urol. (2017) ;72: (4):544–54. |


