
Bladder Cancer Genetic Susceptibility. A Systematic Review
Abstract
Background:
The variant/gene candidate approach to explore bladder cancer (BC) genetic susceptibility has been applied in many studies with significant findings reported. However, results are not always conclusive due to the lack of replication by subsequent studies.
Objectives:
To identify all epidemiological investigations on the genetic associations with BC risk, to quantify the likely magnitude of the associations by applying metaanalysis methodology and to assess whether there is a potential for publication/reporting bias.
Methods:
To address our aims, we have catalogued all genetic association studies published in the field of BC risk since 2000. Furthermore, we metaanalysed all polymorphisms with data available from at least three independent case-control studies with subjects of Caucasian origin analyzed under the same mode of inheritance.
Results:
The characterization of the genetic susceptibility of BC is composed of 28 variants, GWAS contributing most of them. Most of the significant variants associated with BC risk are located in genes belonging to chemical carcinogenesis, DNA repair, and cell cycle pathways. Causal relationship was also provided by functional analysis for GSTM1-null, NAT2-slow, APOBEC-rs1014971, CCNE1-rs8102137, SLC14A1-rs10775480, PSCA-rs2294008, UGT1A-rs1189203, and TP63-rs35592567.
Conclusions:
Genetic susceptibility of BC is still poorly defined, with GWAS contributing most of the strongest evidence. The systematic review did not provide evidence of further genetic associations. The potential public health translation of the existing knowledge on genetic susceptibility on BC is still limited.
INTRODUCTION
The role of genetic susceptibility in bladder cancer (BC) carcinogenesis has been well recognized for a long time ago, the first studies dating from the early eighties and mainly focusing on xenobiotic metabolic genetic heterogeneity [1]. Because of the difficulties in applying molecular genotyping analysis, these first reports used phenotypic approaches to assess bladder cancer risk such as the caffeine tests to assess NAT2 metabolic variability.
Technological advances allowed the determination of genetic variants early in the 1990 s and bladder cancer was one of the first cancers benefiting from these advances; and, again, it was associated with xenobiotic metabolic genes variation [1]. The reason for focusing the attention on the genes participating in both phase 1 and phase 2 carcinogen metabolism was based upon the well-known risk factors for bladder cancer: smoking and occupational exposure to aromatic and heterocyclic amines. Therefore, the main hypothesis driving these preliminary studies focused on the candidate genetic variants that could modify the risk of bladder cancer conferred by the abovementioned carcinogens. Indeed, N-acetyl transferase 2 (NAT2) and Glutathione-s-transferase M1 (GSTM1) variants were the first showing a gene*smoking interaction with bladder cancer risk [2].
The variant/gene candidate approach to explore bladder cancer genetic susceptibility has been applied in many studies and significant findings have been reported. However, results are not always conclusive due to the lack of replication by subsequent studies or to the lack of reports on a specific variant/gene, a fact that may point to a potential publication bias. The candidate pathway approach in bladder cancer genetic susceptibility has mainly focused on DNA repair and cell cycle pathways [3–9].
The first genome-wide association study (GWAS) in bladder cancer was published in 2008 [10]. Together with seven additional GWAS published in the following years, a total of 23 loci distributed in 18 regions of 13 chromosomes were identified, in addition to confirm the GSTM1-null genotype in 1p13.3 [reviewed in 11].
While the GWAS methodology is robust enough to provide conclusive results and quantify the risk of the identified variants, there is a need for a quantitative summary for genetic variants identified through candidate approaches. Therefore, the goals of this systematic review were to identify all epidemiological investigations on the genetic associations with bladder cancer risk, to quantify the likely magnitude of the associations by applying metaanalysis methodology and to assess whether there is a potential for publication/reporting bias.
MATERIAL AND METHODS
To address the aims of this systematic review, we have catalogued all genetic association studies published in the field of bladder cancer risk since 2000. Furthermore, for each polymorphism, we metaanalyzed all summary data available from at least three independent case-control studies of Caucasian origin using the same mode of inheritance (MOI).
Literature search strategy
We combined searches from PubMed and ISI Web of Knowledge (WoK) electronic databases. Search strategies used keywords relevant to genetic association studies and bladder cancer risk for the period Jan 1, 2000 – May 1, 2017. A detailed search strategy was developed for use in Pubmed: a combination of genetic and phenotype keywords and Medical Subject Headings (MeSH) terms: (polymorphism OR SNP OR genetic) AND (“urinary bladder neoplasm” [MeSH] AND (risk OR susceptibility). Since Web Knowledge of Science does not accept MeSH terms, the search strategy designed for Pubmed was then adapted for Web Knowledge of Science database: Topic: ((“urinary bladder neoplasm” OR “bladder neoplasm” OR “bladder tumor” OR “bladder tumour” OR “urinary bladder cancer” OR “malignant tumor of urinary bladder” OR “malignant tumour of urinary bladder” OR “cancer of the bladder” OR “bladder cancer” OR “cancer of bladder”) AND (“snp” OR “polymorphism” OR “genetics”) AND (“risk” OR “susceptibility”)) AND Year of publication: (2000–2017) AND Language: (English). Then, since the search system in Web of Knowledge of Science database does not allow the selection of papers by month of publication, the papers published after May 1, 2017 were then discarded.
Selection of eligible articles
Studies were included in this review if they: 1) were published in a peer-reviewed journal in English; 2) included adult, Caucasian human subjects; and 3) reported associations between SNPs and urothelial bladder cancer risk, our outcome of interest, using both case-control and cross-sectional designs. We included association studies testing for any genetic polymorphism at the nucleotide level, including SNPs, deletions, duplications, and copy-number variants, but excluded larger microscopic variants at the karyotype level. Reviews, metaanalyses, editorials, letters to the editor, book reviews, commentaries and proceedings to congresses were excluded. When it was not clear whether the study met these criteria, the reviewer gathered the full text. Two reviewers (ELM and MR) independently assessed the eligibility of all potentially eligible full-text studies, and any discrepancies were resolved through discussion with a third reviewer NM. We used the metagear R package [12] to assess the eligibility of the studies.
Information extraction
Co-authors ELM, MR, CA and OS independently abstracted data from all studies selected according to the inclusion criteria. The reviewers were not blinded to the names of authors, journal or institutions. Data extracted included information on the setting of each study, details of the sampling strategy and sampled population (age, gender, ethnicity), overall and by cases and controls sample size, the definition of cases and controls, genotyping method, gene, polymorphism, MOI considered, and risk allele.
Quality assessment
Independent duplication of steps in the review process was performed, particularly in the selection of studies and extraction of data, reducing biases and minimizing errors. After the first screening of the papers, both eligible and not eligible papers were reviewed by independent reviewers. Each polymorphism was assigned to the corresponding dbSNP rs IDs using the BiomaRt R package, which is an interface to BioMart databases according to the GRCh37.p13 version [13, 14]. If a SNP ID (rs number) was merged with another, the most updated ID was used. Genes and pathways were annotated using VEP and the KEGG API, respectively.
Metaanalysis
Multiple reports of the same study were merged to obtain a single best estimator for the study prior to inclusion in the metaanalysis. For polymorphisms assessed in at least three studies using the same MOI, we conducted both random and fixed effects metaanalyses using the metafor R package [15]. The I2 statistic was used as a measure of between-studies heterogeneity. In addition, we assessed the funnel plot asymmetry to test possible reporting biases or heterogeneity [16, 17]. Moreover, we used contour-enhanced funnel plots, which are funnel plots centered at 0 and with various levels of statistical significance of the points/studies indicated by the shaded regions. In particular, the unshaded (i.e., white) region in the middle corresponds to p-values greater than 0.10, the grey-shaded region corresponds to p-values between 0.10 and 0.05, the dark grey-shaded region corresponds to p-values between 0.05 and 0.01, and the region outside of the funnel corresponds to p-values below 0.01. Funnel plots drawn in this way are more useful for detecting publication bias due to the suppression of non-significant findings.
Reporting of this review complies with recommendations of the PRISMA statements [18].
RESULTS
Article selection
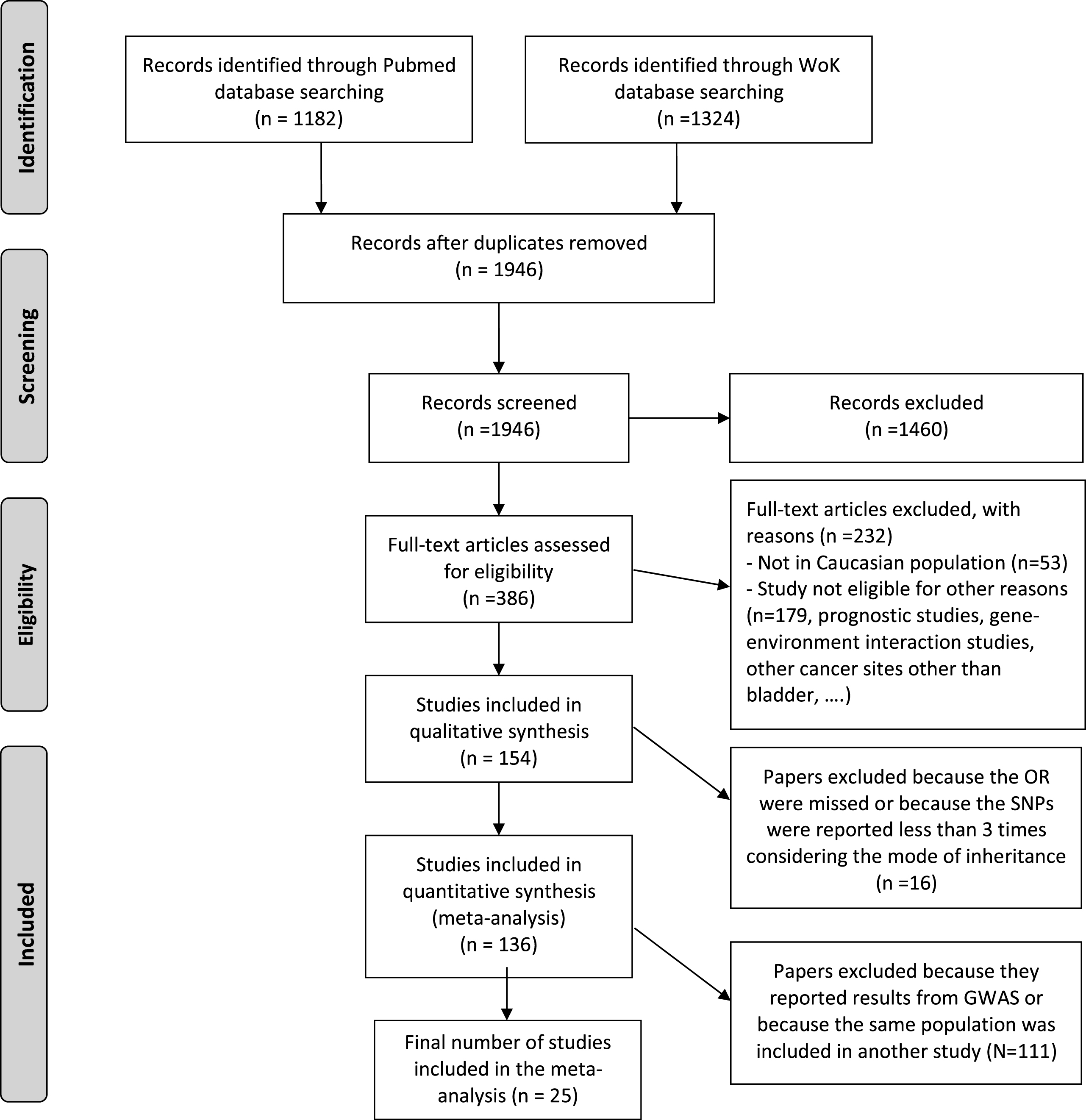
Figure 1 shows the flow diagram detailing the article selection steps according to PRISMA guidelines. The search strategy generated 1946 records after removing the duplicates produced when the searches from the two electronic databases (Pubmed and WoK) were pooled. From these abstracts, 386 articles were fully reviewed for eligibility. Overall, 8,865 variants were considered for association in the 154 papers considered to be eligible for metaanalysis and for which we retrieved the full-text. A list with the references of these articles is provided as Supplementary Material and the list of all those variants, together with their annotated genes, is provided in Supplementary Table 1.
Fig.1
PRISMA 2009 Flow Diagram detailing the article selection steps to guide the systematic review and metaanalysis on genetic variants associated with bladder cancer based on a candidate approach.
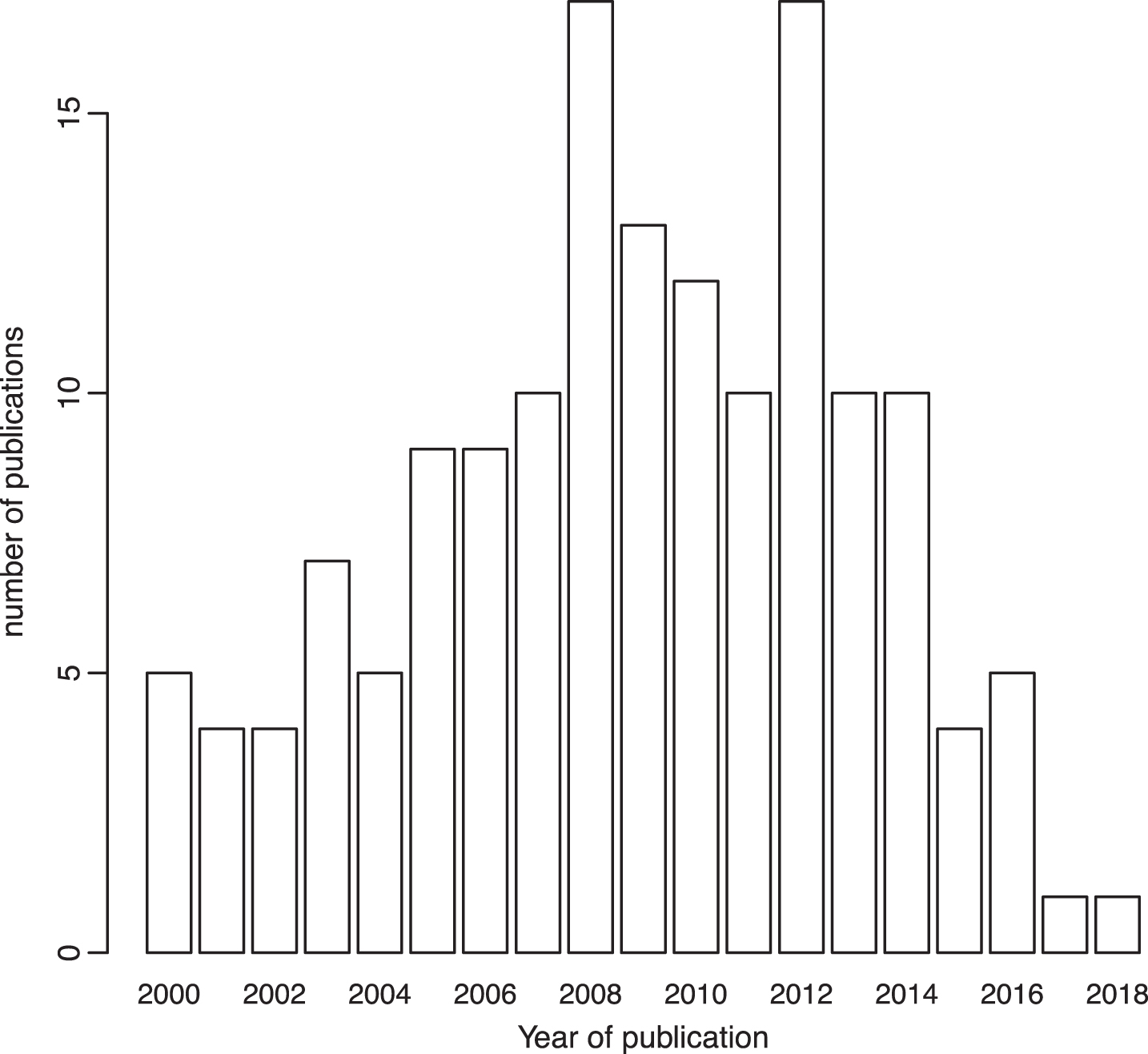
After excluding the variants for which the OR for the association with bladder cancer was not reported, those without a risk allele specified, and those that appeared less than three times considering their MOI, quantitative syntheses were possible for 58 polymorphisms in or near 62 genes from 136 papers. Only the last estimate of repetitive studies conducted in the same population was included in the metaanalysis. Variants detected in GWAS studies were not considered in the metaanalysis because of their high level of evidence and we took their results for granted. Finally, only 17 out of 58 polymorphisms reported in 25 papers were considered for metaanalysis. Most of the studies were published before 2014 (Fig. 2) and they considered both men and women. Each metaanalysis included at most 16 studies or subgroups. Codominant was the most used MOI, followed by the dominant MOI. GSTT1 was the only polymorphism included in the metaanalysis that was analyzed using a recessive MOI.
Fig.2
Number of published papers considered in the systematic review according to their year of publication.
Publication bias and selective analysis
A visual inspection of the funnel plots suggested that, in most cases when asymmetry is present, it is due to the small sample size of the studies considered rather than to a substantial publication bias. Besides, tests for funnel plots asymmetry are known to have low power and, even when a test did not provide evidence of asymmetry, bias could not be excluded [17]. For example, the funnel plots for the variant rs2228001 in XPC showed evidence of statistically significant asymmetry under a codominant MOI, where the estimates that drove the asymmetry corresponded to the study of Fontana et al. [19], which showed the largest confidence interval due to the small sample size of the study (51 cases and 45 controls). Similar considerations can be extended to the funnel plot of GSTP1, under a codominant MOI. Overall, in most cases, only few studies were included in the metaanalysis and it was hard to detect asymmetry in the plots.
Metaanalyses
Here, we report the results from the metaanalysis for the selected variants according to their annotation in genes and KEGG pathways. Most association studies in bladder cancer genetic susceptibility focused on variation in genes involved in chemical carcinogenesis, DNA repair, and cell cycle pathways. In Table 1 we list the significant variants identified by GWAS together with those explored in the pooled analysis [20] and those included in the metaanalysis. In bold, we highlight those significantly associated with bladder cancer. Only GSTT1 metaanalyses was based in >10 publications.
Table 1
Annotated pathways and genes for the variants selected in this metaanalysis (MA), and those identified by the bladder cancer GWAS and pooled analysis (PA). In bold, the variants that provided statistical significant meta-risks
| Gene | VARIANT | Other pathways |
| MAIN PATHWAY : Chemical carcinogenesis | ||
| GSTM1 | Null/Present (GWAS) | Glutathione metabolism; Metabolism of xenobiotics by cytochrome P450; Drug metabolism – cytochrome P450; Platinum drug resistance; Pathways in cancer; Hepatocellular carcinoma; Fluid shear stress and atherosclerosis |
| GSTT1 | Null/Present (MA) | Glutathione metabolism; Metabolism of xenobiotics by cytochrome P450; Drug metabolism – cytochrome P450; Platinum drug resistance; Pathways in cancer; Hepatocellular carcinoma; Fluid shear stress and atherosclerosis |
| GSTP1 | I105V (MA) | Glutathione metabolism; Metabolism of xenobiotics by cytochrome P450; Drug metabolism – cytochrome P450; Platinum drug resistance; Pathways in cancer; Prostate cancer; Hepatocellular carcinoma; Fluid shear stress and atherosclerosis |
| NAT2 | rs1495741 (GWAS) | Caffeine metabolism; Drug metabolism – other enzymes |
| NAT1 | *4/*10 (MA) | Caffeine metabolism; Drug metabolism – other enzymes |
| CYP1B1 | rs1056836 (MA) | |
| CLK3/CYP1A2 | rs11543198 (GWAS) | Steroid hormone biosynthesis; Caffeine metabolism; Tryptophan metabolism; Linoleic acid metabolism; Retinol metabolism; Metabolism of xenobiotics by cytochrome P450; Drug metabolism – cytochrome P450; Metabolic pathways |
| UGT1A8/UGT1A10 | rs11892031 (GWAS) | Pentose and glucuronate interconversions; Ascorbate and aldarate metabolism; Steroid hormone biosynthesis; Retinol metabolism; Porphyrin and chlorophyll metabolism; Metabolism of xenobiotics by cytochrome P450; Drug metabolism – cytochrome P450; Drug metabolism – other enzymes; Metabolic pathways |
| MAIN PATHWAY : One carbon pool by folate | ||
| MTHFR | rs1801131 (MA) rs1801133 (MA) | Carbon metabolism; Antifolate resistance; Metabolic pathways |
| PATHWAY : Base excision repair | ||
| APEX1 | rs1130409 (MA + PA) | |
| XRCC1 | rs25487 (MA + PA) rs1799782 (PA) | |
| MAIN PATHWAY : Nucleotide excision repair | ||
| ERCC2 | rs13181 (MA + PA) rs1799793 (MA + PA) | Basal transcription factors |
| ERCC4 | rs1799801 (PA) | Fanconi anemia pathway |
| ERCC5 | rs17655 (MA) | |
| XPC | rs2228000(PA) rs2228001 (MA + PA) poly AT (MA) | |
| MAIN PATHWAY : Homologous recombination | ||
| XRCC3 | rs861539 (MA + PA) | |
| NBN | rs1805794 (PA) | Cellular senescence |
| MAIN PATHWAY : Cell cycle | ||
| CCND1 | rs9344 (MA) | Endocrine resistance; p53 signalling pathway; PI3KAkt signalling pathway; AMPK signalling pathway; Cellular senescence; Wnt signalling pathway; Hedgehog signalling pathway; Apelin signalling pathway; Hippo signalling pathway; FoxO signalling |
| CCNE1 | rs8102137 (GWAS) | Cell cycle; Oocyte meiosis; p53 signaling pathway; PI3K-Akt signaling pathway; Cellular senescence; Hepatitis B; Measles; Human papillomavirus infection; Pathways in cancer; Viral carcinogenesis; MicroRNAs in cancer; Prostate cancer; Small cell lung cancer; Gastric cancer; MODULE: Cell cycle – G1/S transition |
| MYC/POU5F1B/PVT1 | rs10094872 (GWAS) | MYC: MAPK signalling pathway; ErbB signalling pathway; Cell cycle; PI3K-Akt signalling pathway; Cellular senescence; Wnt signalling pathway; TGF-beta signalling pathway; Hippo signalling pathway; Signalling pathways regulating pluripotency of stem cells; Jak-STAT signalling pathway; Thyroid hormone signalling pathway; Hepatitis B; HTLV-I infection; Kaposi’s sarcoma-associated herpesvirus infection; Epstein-Barr virus infection; Pathways in cancer; Transcriptional misregulation in cancer; Proteoglycans in cancer; MicroRNAs in cancer; Colorectal cancer; Endometrial cancer; Thyroid cancer; Bladder cancer; Chronic myeloid leukaemia; Acute myeloid leukaemia; Small cell lung cancer; Breast cancer; Hepatocellular carcinoma; Gastric cancer; Central carbon metabolism in cancer POU5F1B: Signalling pathways regulating pluripotency of stem cells |
| MYC/BC042052/CASC11 | rs9642880 (MA+GWAS) | MAPK signalling pathway; ErbB signalling pathway; Cell cycle; PI3K-Akt signalling pathway; Cellular senescence; Wnt signalling pathway; TGF-beta signalling pathway; Hippo signalling pathway; Signalling pathways regulating pluripotency of stem cells; Jak-STAT signalling pathway; Thyroid hormone signalling pathway; Hepatitis B; HTLV-I infection; Kaposi’s sarcoma-associated herpesvirus infection; Epstein-Barr virus infection; Pathways in cancer; Transcriptional misregulation in cancer; Proteoglycans in cancer; MicroRNAs in cancer; Colorectal cancer; Endometrial cancer; Thyroid cancer; Bladder cancer; Chronic myeloid leukaemia; Acute myeloid leukaemia; Small cell lung cancer; Breast cancer; Hepatocellular carcinoma; Gastric cancer; Central carbon metabolism in cancer |
| MAIN PATHWAY : Peroxisome | ||
| SOD2 | rs4880 (MA) | FoxO signalling pathway; Longevity regulating pathway; Huntington’s disease |
| MAIN PATHWAY : Ubiquinone and other terpenoid-quinone biosynthesis | ||
| NQO1 | rs1800566 (MA) | Ubiquinone and other terpenoidquinone biosynthesis; Hepatocellular carcinoma; Fluid shear stress and atherosclerosis |
| OTHER PATHWAYS | ||
| ACTRT3/MYNN/TERC/LRRC34 | rs10936599 (GWAS) | Pathways in cancer; Hepatocellular carcinoma; Gastric cancer |
| TERT/CLPTM1L | rs401681 (GWAS) | Human papillomavirus infection; HTLV-I infection; Pathways in cancer; Hepatocellular carcinoma; Gastric cancer |
| TP63 | rs710521 (GWAS) | MicroRNAs in cancer |
| JAG1 | rs62185668 (GWAS) | Endocrine resistance; Notch signalling pathway; Apelin signalling pathway; Th1 and Th2 cell differentiation; TNF signalling pathway; Human papillomavirus infection; Pathways in cancer; Breast cancer |
| TMEM129/TACC3/FGFR3 | rs798766 (GWAS) | EGFR tyrosine kinase inhibitor resistance; MAPK signalling pathway; Ras signalling pathway; Rap1 signalling pathway; Endocytosis; PI3K-Akt signalling pathway; Signalling pathways regulating pluripotency of stem cells; Regulation of actin cytoskeleton; Pathways in cancer; MicroRNAs in cancer; Bladder cancer; Central carbon metabolism in cancer RNA transport (TACC3) |
| NO PATHWAY ANNOTATED | ||
| CWC27 | rs2042329 (GWAS) | |
| NR | rs7747724 (GWAS) rs5003154 (GWAS) rs4907479 (GWAS) | |
| CDKAL1 | rs4510656 (GWAS) | |
| JRK/PSCA | rs2294008 (GWAS) | |
| LSP | rs907611 (GWAS) | |
| SLC14A1 | rs17674580 (GWAS) rs7238033 (GWAS) | |
| SLC14A2 | rs10775480 (GWAS) | |
| C20orf187/ LOC339593 | rs6104690 (GWAS) | |
| CBX6/APOBEC3A | rs1014971 (GWAS) | |
Chemical carcinogenesis
The variants included in the metaanalysis and annotated in this pathway were: GSTT1-null (16 studies: metaOR = 1.06, 95% CI 0.88–1.27, I2 = 46.5%), GSTP1-rs1695 (4 studies for the dominant and 5 the codominant MOI), NAT1-slow (5 studies), and CYP1B1-rs1056836 (6 studies analyzed the codominant MOI). None of them showed a significant association with bladder cancer. Asymmetries in the forest plots may be due to the small number of papers published on this topic or to a potential publication/reporting bias (Supplementary Figure 1). On the contrary, GSTM1-null and NAT2-slow variants have been reported to be associated with bladder cancer by all approaches, including GWAS [20]. The other two variants identified through GWAS were CLK3/CYP1A2-rs11543198, UGT1A8/UGT1A10-rs1189203.
Base-excision DNA repair
APEX1-rs1130409 (3 studies) was metaanalized using a dominant MOI, although the association was not significant. XRCC1-rs25487 and XRCC1- rs1799782 were metaanalized (4 studies for both dominant and codominant MOI) and were also analyzed in the pooled study by Stern et al., although the associations were not significant in either approach [21].
Nucleotide-excision DNA repair
ERCC2-rs13181 (3 studies for the codominant and 6 for the dominant MOI), ERCC2-rs1799793 (3 studies for the codominant MOI), ERCC5-rs17655 (3 studies for the codominant MOI), XPC-rs2228001 (4 studies for the codominant, 3 for the dominant, and 3 for the recessive MOI), and XPC-polyAT (4 studies for the codominant MOI) were included in the metaanalysis. None of the metaOR were significant. Noteworthy, Stern et al. found significant associations between ERCC2-rs13181, ERCC2-rs1799793 and BC risk [21]. The same authors also analyzed ERCC4- rs1799801 and XPC-rs2228001, although none of these variants were significantly associated with bladder cancer risk [21].
Homologous recombination DNA repair
The variant XRCC3-rs861539 was analyzed using a codominant (4 studies) and a dominant (3 studies) MOI, although none of the estimates reached the significance threshold. This variant was also reported as not associated with BC risk in Stern et al. [21]. Interestingly, NBN-rs1805794 was found significantly associated with BC risk under the dominant and the additive MOI in the pooled analysis [21].
Cell cycle
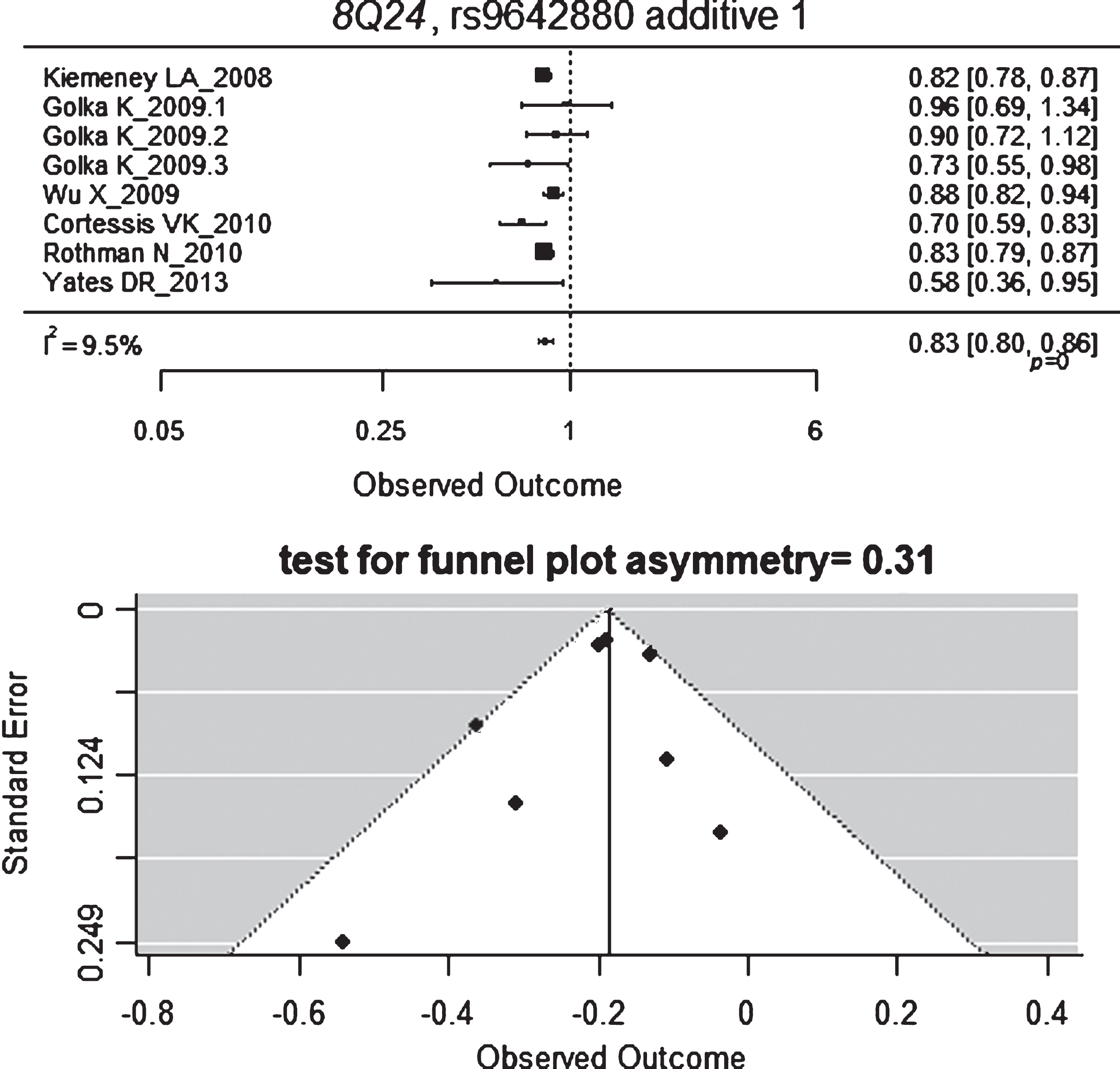
The only variant annotated in this pathway that was considered for the metaanalysis was CCND1-rs9344 (3 studies applied a codominant MOI) and its association with BC risk was not found to be significant. The following hits in GWAS studies also annotated in the cell cycle pathway are: CCNE1-rs8102137, MYC/POU5F1B/PVT1-rs10094872, MYC/BC042052/CASC11-rs9642880. Interestingly, the latter variant was the only one that provided significant results when we metaanalyzed the non-GWAS available data with a random-effect model (Fig. 3) [22–24]. Three estimates were reported by Golka et al. [22] from three independent series and two reported negative but not significant OR, with a quite low estimate for heterogeneity (I = 30%). Negative and significant estimate and similar in magnitude was obtained with a fixed effect model (metaOR = 0.77, 95% CI 0.69, 0.86). The funnel plot showed no evidence for asymmetry. The study at the bottom left of the plot corresponds to Yates et al. [24], which is the one with the lowest sample size.
Fig.3
Forest and funnel plots for MYC/BC042052/CASC11-rs9642880.
One carbon pool by folate
MTHFR-rs1801131 (5 studies applied the codominant and 3 the dominant MOI) and MTHFR-rs1801133 (5 studies applied the codominant MOI) on this pathway were included in the metaanalysis, although none of them showed a significant association with BC risk.
Peroxisome
Only SOD2-rs4880 with 3 studies applying a codominant MOI could be metaanalysed and the result showed no significant association with BC risk.
Ubiquinone and other terpenoid-quinone biosynthesis
Four studies applying the codominant MOI for NQO1-rs1800566 were considered in the metaanalysis showing a non-significant association with BC risk.
The GWAS reported variants ACTRT3/MYNN/TERC/LRRC34-rs10936599, TERT/CLPTM1L- rs401681, TP63-rs710521, JAG1-rs62185668, TMEM129/TACC3/FGFR3-rs798766 associated with BC risk were annotated in other pathways as “Pathways in cancer”, whereas CWC27-rs2042329, NR-rs7747724, NR-rs5003154, NR-rs4907479, CDKAL1-rs4510656, JRK/PSCA- rs2294008, LSP- rs907611, SLC14A1-rs17674580, SLC14A1-rs7238033, SLC14A2-rs10775480, C20orf187/ LOC339593-rs6104690, and CBX6/APOBEC3A-rs1014971 were not annotated in any KEGG pathway.
DISCUSSION
A total number of 386 studies were considered in this systematic review. After applying inclusion criteria, 154 studies assessing 8,865 unique SNPs remained (Supplementary Table 1). Candidate variant-based studies were published between 2000 to 2017. We included in this systematic review the pooled study by Stern et al. [21]. However, we excluded previous metaanalyses performed on variants in GSTM1, NAT2 [2, 25–27], GSTP1 [25, 28], hOGG1 [29], NQO1 [25, 30–32], TP53 [33], XRCC1 [34–37], XRCC3 [25, 38], XPC [25], XPD [25], PSCA [32], SDF-1 [39], TRAIL-R1 [40], E-cadherin [41], and TGFBR1 [42].
The systematic review did not provide enough evidences for additional candidate variants associated with bladder cancer risk further from those already reported in the GWAS (N = 24) and the pooled analysis (N = 4) [21] (Table 1). This limited characterization of the genetic susceptibility of BC, in comparison to that of other cancer such as breast and prostate [https://www.ebi.ac.uk/gwas] is intriguing, being BC a paradigm of complex diseases. The fact that this cancer is largely environmentally-driven, may partly explain the low number of variants identified at present. Either a gene*environment interaction or an extreme-phenotype approaches are probably needed to identify those missing or hidden heritability of BC, as suggested by Lopez-Maturana et al. [43]. In this already published study, we provided evidence on the importance of applying multi-marker/SNP analyses towards a better characterization of the BC genetic risk.
It is interesting to observe that an important proportion of significant variants with strong evidence of involvement in BC carcinogenesis belong to genes involved in chemical carcinogenesis (GSTM1, NAT2, CLK3/CYP1A2, and UGT1A8/UGT1A10), DNA repair pathways (ERCC2, XPC, and NBN), and cell cycle pathway (CCNE1, MYC/POU5F1B/PVT1, and MYC/BC042052/CASC1). GWAS also identified variants in genes involved in many other pathways as well as in yet not-annotated pathways. A recent pathway analysis based on GWAS data further identified variants in 9 novel genes (RAPGEF1, SKP1, HERPUD1, CACNB2, CACNA1C, CACNA1S, COL4A2, SRC, and CACNA1C) [44]. Functional analyses have also provided evidences of causality for GSTM1-null, NAT2-slow, APOBEC-rs1014971, CCNE1-rs8102137, SLC14A1-rs10775480, PSCA-rs2294008, UGT1A-rs1189203, and TP63-rs35592567 [45–52]. These signals should act as clues to further explore the gene and pathway variation in association with BC risk. Actually, a rapid consultation of the variants included in Supplementary Table 1 using GWAS data would provide conclusive results for most of them.
Pleiotropy at all levels is noteworthy with most variants, genes, and pathways being shared with genetic susceptibility of other cancers and non-malignant diseases. This fact would support a holistic genetic susceptibility approach across diseases (malignant and non-malignant) using a competing-risk analysis to identify the common and the specific actors promoting and preventing their development.
Except for GWAS results, the knowledge of genetic susceptibility scenario of BC has not changed in the last 10 years. Extending the publication period to 1990–1999 did not provide further genetic variants suitable to contribute to this metaanalysis in addition to those in GSTM1, GSTT1, NAT2, and NAT1 that were already included. Despite the thousands of candidate genetic variants being analyzed, only 6 have provided consistent results of an association with BC risk in Caucasian population-based studies, including GSTM1-null and NAT2-slow acetylation. The lack of replication of most of the results from candidate genetic variant based-studies should preclude the conduction of such type of studies and, instead, move towards the GWAS approach.
With the exception of GWAS, the quality of the evidence provided by this systematic review on the null-associated variants is limited. The validity of our metaanalysis relies upon the validity of the studies included within it and most studies are small and heterogeneous. In addition, since effect magnitudes in genetic association studies are generally small, small biases may be relevant in concluding about their role on genetic susceptibility of BC. We are also aware of potential biases related to the studies considered, which could be related to the case definition (histologically confirmed urothelial bladder carcinoma), population stratification, and methods in the collection and processing of DNA as well as the determination of genotypes. In addition, potential biases related to the collection of studies as a whole may be possible though we explored two sources of publications, as well as reporting biases (including publication biases and selective reporting), and bias in the selection of genetic variants to study in each paper. Particularly in GWAS and also in some candidate gene approaches, only the significant associations are reported in the papers. Asymmetries were difficult to be identified in the funnel plots paired to each forest plots in Supplementary Figure 1 because of the small number of papers included in each metaanalysis rather than indicating lack of bias. Also, the investigation of a polymorphism-disease association maybe based on already published results, and interesting findings are the most likely targets for replication [17, 53].
The above-mentioned evidences regard to low penetrance variants frequent in the population. Nevertheless, it is worth noting that a higher risk of BC has also been reported in germline MSH2 mutation carriers and first degree relatives of Lynch syndrome families [54, 55], in Costello syndrome (HRAS germline mutations) affected individuals [56, 57], and in retinoblastoma (RB1 germline mutations) affected survivors [58, 59], the latter ones showing the higher risk for BC. While germline mutations of these genes are rare in the population, somatic mutations of them are common in BC tumors.
In conclusion, the characterization of the genetic susceptibility of BC is still limited, with 28 variants showing strong evidence of association, mainly through GWAS. The systematic review did not provide evidences of further genetic associations. Most of the significant variants associated with BC risk are located in genes belonging to chemical carcinogenesis, DNA repair, and cell cycle pathways. Causal relationship has been also provided by functional analysis for GSTM1-null, NAT2-slow, APOBEC-rs1014971, CCNE1-rs8102137, SLC14A1-rs10775480, PSCA-rs2294008, UGT1A-rs1189203, and TP63-rs35592567.
There is a vast list of potentially associated variants, genes and pathways suggested by candidate-based approaches that can act as clues to further explore the genetic variation in association with BC risk using already available GWAS data. While the public health application of the existing knowledge on genetic susceptibility on BC based on individual SNPs is limited, the short-term perspectives to analyze them through system approaches will probably allow to identify genetic susceptibility signatures with a higher translational potential.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS INCLUDING SOURCES OF SUPPORT
The work was partially supported by the Asociación Contra el Cáncer (AECC, GCX140027); Fondo de Investigación Sanitaria, Instituto de Salud Carlos III, Spain (PI09-02102, G03/174); Red Temá-tica de Investigación Cooperativa en Cáncer (RTICC, #RD12/0036/0050), Spanish Ministry of Economy and Competitiveness & European Regional Development Fund (FEDER) “Una manera de hacer Europa”); and Mujeres por África Foundation that funded CA.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/BLC-170159.
REFERENCES
[1] | d’Errico A , Malats N , Vineis P , Boffetta P . Review of studies of selected metabolic polymorphisms and cancer. IARC Sci Publ No 148. (1999) ;323–93. |
[2] | García-Closas M , Malats N , Silverman D , Dosemeci M , Kogevinas M , et al. NAT2 slow acetylation, GSTM1 null genotype, and risk of bladder cancer: Results from the Spanish Bladder Cancer Study and meta-analyses. Lancet. (2005) ;366: :649–59. |
[3] | Wu X , Gu J , Grossman HB , Amos CI , Etzel C , et al. Bladder cancer predisposition: A multigenic approach to DNA-repair and cell-cycle-control genes. Am J Hum Genet. (2006) ;78: :464–79. |
[4] | García-Closas M , Malats N , Real FX , Welch R , Kogevinas M , et al. Genetic variation in the nucleotide excision repair pathway and bladder cancer risk. Cancer Epidemiol Biomarkers Prev. (2006) ;15: :536–42. |
[5] | Figueroa JD , Malats N , Real FX , Silverman D , Kogevinas M , et al. Genetic variation in the base excision repair pathway and bladder cancer risk. Hum Genet. (2007) ;121: :233–42. |
[6] | Figueroa JD , Malats N , Rothman N , Real FX , Silverman D , et al. Evaluation of genetic variation in the double-strand break repair pathway and bladder cancer risk. Carcinogenesis. (2007) ;28: :1788–93. |
[7] | Xing J , Dinney CP , Shete S , Huang M , Hildebrandt MA , et al. Comprehensive pathway-based interrogation of genetic variations in the nucleotide excision DNA repair pathway and risk of bladder cancer. Cancer. (2012) ;118: :205–15. |
[8] | Xie H , Gong Y , Dai J , Wu X , Gu J . Genetic variations in base excision repair pathway and risk of bladder cancer: A case-control study in the United States. Mol Carcinog. (2015) ;54: :50–7. |
[9] | Corral R , Lewinger JP , Van Den Berg D , Joshi AD , Yuan JM , et al. Comprehensive analyses of DNA repair pathways, smoking and bladder cancer risk in Los Angeles and Shanghai. Int J Cancer. (2014) ;135: :335–47. |
[10] | Kiemeney LA , Thorlacius S , Sulem P , Geller F , Aben KKH , et al. Sequence variant on 8q24 confers susceptibility to urinary bladder cancer. Nature Genetics. (2008) ;40: :1307–12. |
[11] | López-Maturana E , Malats N . Future perspective in bladder cancer: Genetic testing, genetic variation and genetic susceptibility. In: Hyeon Ku J. Bladder Cancer. Elsevier Science. (2017) . https://books.google.es/books?id=KTNHDgAAQBAJ. |
[12] | Lajeunesse MJ . Facilitating systematic reviews, data extraction and meta-analysis with the metagear package for R. Methods in Ecology and Evolution. (2016) ;7: :323–30. |
[13] | Durinck S , Moreau Y , Kasprzyk A , Davis S , De Moor B , et al. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics. (2005) ;21: :3439–40. |
[14] | Durinck S , Spellman P , Birney E and Huber W, et al. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature Protocols. (2009) ;4: :1184–91. |
[15] | Viechtbauer W . Conducting meta-analyses in R with the meta for package. J Stat Software. (2010) ;36: :1–48. |
[16] | Egger M , Davey Smith G , Schneider M , Minder C , et al. Bias in meta-analysis detected by a simple, graphical test. BMJ. (1997) ;315: :629–34. |
[17] | Sterne JA , Sutton AJ , Ioannidis JP , Terrin N , Jones DR , et al. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomized controlled trials. BMJ. (2011) ;343: :d4002. |
[18] | Moher D , Liberati A , Tetzlaff J , Altman DG , The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. (2009) ;6: :e1000097. |
[19] | Fontana L , Bosviel R , Delort L , Guy L , Chalabi N , Kwiatkowski F , Satih S , Rabiau N , Boiteux JP , Chamoux A , Bignon YJ , Bernard-Gallon DJ . DNA repair gene ERCC2, XPC, XRCC1, XRCC3 polymorphisms and associations with bladder cancer risk in a French cohort. Anticancer Res. (2008) ;28: (3B):1853–6. PubMed PMID: 18630471. |
[20] | Rothman N , Garcia-Closas M , Chatterjee N , Malats N , Wu X , et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nature Genetics. (2010) ;42: :978–84. |
[21] | Stern MC , Lin J , Figueroa JD , Kelsey KT , Kiltie AE , et al. Polymorphisms in DNA repair genes, smoking, and bladder cancer risk: Findings from the international consortium of bladder cancer. Cancer Res. (2009) ;69: :6857–64. |
[22] | Golka K , Hermes M , Selinski S , Blaszkewicz M , Bolt HM , et al. Susceptibility to urinary bladder cancer: Relevance of rs9642880[T], GSTM1 0/0 and occupational exposure. Pharmacogenet Genomics. (2009) ;19: :903–6. |
[23] | Cortessis VK , Yuan JM , Van Den Berg D , Jiang X , Gago-Dominguez M , et al. Risk of urinary bladder cancer is associated with 8q24 variant rs9642880[T] in multiple racial/ethnic groups: Results from the Los Angeles-Shanghai case-control study. Cancer Epidemiol Biomarkers Prev. (2010) ;19: :3150–6. |
[24] | Yates DR , Rouprêt M , Drouin SJ , Audouin M , Cancel-Tassin G , et al. Genetic polymorphisms on 8q24.1 and 4p16.3 are not linked with urothelial carcinoma of the bladder in contrast to their association with aggressive upper urinary tract tumours. World J Urol. (2013) ;31: :53–9. |
[25] | Dong LM , Potter JD , White E , Ulrich CM , Cardon LR , et al. Genetic susceptibility to cancer: The role of polymorphisms in candidate genes. JAMA. (2008) ;299: :2423–36. |
[26] | Moore LE , Baris DR , Figueroa JD , Garcia-Closas M , Karagas MR , et al. GSTM1 null and NAT2 slow acetylation genotypes, smoking intensity and bladder cancer risk: Results from the New England bladder cancer study and NAT2 meta-analysis. Carcinogenesis. (2011) ;32: :182–9. |
[27] | Green J , Banks E , Berrington A , Darby S , Deo H , et al. N-acetyltransferase 2 and bladder cancer: An overview and consideration of the evidence for gene-environment interaction. Br J Cancer. (2000) ;83: :412–7. |
[28] | Kellen E , Hemelt M , Broberg K , Golka K , Kristensen VN , et al. Pooled analysis and meta-analysis of the glutathione S-transferase P1 Ile105Val polymorphism and bladder cancer: A HuGE-GSEC review. Am J Epidemiol. (2007) ;165: :1221–30. |
[29] | Zhong DY , Chu HY , Wang ML , Ma L , Shi DN , et al. Meta-analysis demonstrates lack of association of the hOGG1 Ser326Cys polymorphism with bladder cancer risk. Genet Mol Res. (2012) ;11: :3490–6. |
[30] | Guo ZJ , Feng CL . The NQO1 rs1800566 polymorphism and risk of bladder cancer: Evidence from 6,169 subjects. Asian Pac J Cancer Prev. (2012) ;13: :6343–8. |
[31] | Zhang Y , Yang D , Zhu JH , Chen MB , Shen WX , et al. The association between NQO1 Pro187Ser polymorphism and urinary system cancer susceptibility: A meta-analysis of 22 studies. Cancer Invest. (2015) ;33: :39–40. |
[32] | Yang S , Jin T , Su HX , Zhu JH , Wang DW , et al. The association between NQO1 Pro187Ser polymorphism and bladder cancer susceptibility: A meta-analysis of 15 studies. PLoS One. (2015) ;10: :e0116500. |
[33] | Liu ZH , Bao ED . Quantitative assessment of the association between TP53 Arg72Pro polymorphism and bladder cancer risk. Mol Biol Rep. (2013) ;40: :2389–95. |
[34] | Hu Z , Ma H , Chen F , Wei Q , Shen H . XRCC1 polymorphisms and cancer risk: A meta-analysis of 38 case-control studies. Cancer Epidemiol Biomarkers Prev. (2005) ;14: :1810–8. |
[35] | Li S , Peng Q , Chen Y , You J , Chen Z , et al. DNA repair gene XRCC1 polymorphisms, smoking, and bladder cancer risk: A meta-analysis. PLoS One. (2013) ;8: :e73448. |
[36] | Mao Y , Xu X , Lin Y , Chen H , Wu J , et al. Quantitative assessment of the associations between XRCC1 polymorphisms and bladder cancer risk. World J Surg Oncol. (2013) ;11: :58. |
[37] | Yang D , Liu C , Shi J , Wang N , Du X , et al. Association of XRCC1 Arg399Gln polymorphism with bladder cancer susceptibility: A meta-analysis. Gene. (2014) ;534: :17–23. |
[38] | Han S , Zhang HT , Wang Z , Xie Y , Tang R , et al. DNA repair gene XRCC3 polymorphisms and cancer risk: A meta-analysis of 48 case-control studies. Eur J Hum Genet. (2006) ;14: :1136–44. |
[39] | Tong X , Ma Y , Deng H , Wang X , Liu S , et al. The SDF-1 rs1801157 Polymorphism is Associated with Cancer Risk: An Update Pooled Analysis and FPRP Test of 17,876 Participants. Sci Rep. (2016) ;6: :27466. |
[40] | Chen B , Liu S , Wang XL , Xu W , Li Y , et al. TRAIL-R1 polymorphisms and cancer susceptibility: An evidence-based meta-analysis. Eur J Cancer. (2009) ;45: :2598–605. |
[41] | Wang M , Wang XJ , Ma YF , Ma XB , Dai ZM , et al. PSCA rs2294008C>T polymorphism contributes to gastric and bladder cancer risk. Ther Clin Risk Manag. (2015) ;11: :237–45. |
[42] | Kaklamani VG , Hou N , Bian Y , Reich J , Offit K , et al. TGFBR1*6A and cancer risk: A meta-analysis of seven case-control studies. J Clin Oncol. (2003) ;21: :3236–43. |
[43] | de Maturana EL , Chanok SJ , Picornell AC , Rothman N , Herranz J , et al. Whole genome prediction of bladder cancer risk with the Bayesian LASSO. Genet Epidemiol. (2014) ;38: :467–76. |
[44] | Chen M , Rothman N , Ye Y , Gu J , Scheet PA , et al. Pathway analysis of bladder cancer genome-wide association study identifies novel pathways involved in bladder cancer development. Genes Cancer. (2016) ;7: :229–39. |
[45] | Vineis P , Malats N , Lang M , d’Errico A , Caporaso N , Cuzick J , Boffetta P . Metabolic Polymorphisms and Susceptibility to Cancer. IARC Scientific Publication No. 148, (1999) . |
[46] | Tang W , Fu YP , Figueroa JD , Malats N , Garcia-Closas M , et al. Mapping of the UGT1A locus identifies an uncommon coding variant that affects mRNA expression and protects from bladder cancer. Hum Mol Genet. (2012) ;21: :1918–30. |
[47] | Fu YP , Kohaar I , Rothman N , Earl J , Figueroa JD , et al. Common genetic variants in the PSCA gene influence gene expression and bladder cancer risk. Proc Natl Acad Sci U S A. (2012) ;109: :4974–9. |
[48] | Koutros S , Baris D , Fischer A , Tang W , Garcia-Closas M , et al. Differential urinary specific gravity as a molecular phenotype of the bladder cancer genetic association in the urea transporter gene, SLC14A1. Int J Cancer. (2013) ;133: :3008–13. |
[49] | Fu YP , Kohaar I , Moore LE , Lenz P , Figueroa JD , et al. The 19q12 bladder cancer GWAS signal: Association with cyclin E function and aggressive disease. Cancer Res. (2014) ;74: :5808–18. |
[50] | Middlebrooks CD , Banday AR , Matsuda K , Udquim KI , Onabajo OO , et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat Genet. (2016) ;48: :1330–8. |
[51] | Pattison JM , Posternak V , Cole MD . Transcription Factor KLF5 Binds a Cyclin E1 Polymorphic Intronic Enhancer to Confer Increased Bladder Cancer Risk. Mol Cancer Res. (2016) ;14: :1078–86. |
[52] | Wang M , Du M , Ma L , Chu H , Lv Q , et al. A functional variant in TP63 at 3q28 associated with bladder cancer risk by creating an miR-140-5p binding site. Int J Cancer. (2016) ;139: :65–74. |
[53] | Sagoo GS , Little J , Higgins JP . Systematic reviews of genetic association studies. Human Genome Epidemiology Network. PLoS Med. (2009) ;6: :e28. |
[54] | van der Post RS , Kiemeney LA , Ligtenberg MJ , Witjes JA , Hulsbergen-van de Kaa CA , et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. (2010) ;47: :464–70. |
[55] | Skeldon SC , Semotiuk K , Aronson M , Holter S , Gallinger S , et al. Patients with Lynch syndrome mismatch repair gene mutations are at higher risk for not only upper tract urothelial cancer but also bladder cancer. Eur Urol. (2013) ;63: :379–85. |
[56] | Beukers W , Hercegovac A , Zwarthoff EC . HRAS mutations in bladder cancer at an early age and the possible association with the Costello Syndrome. Eur J Hum Genet. (2014) ;22: (6), 837–9. |
[57] | Gripp KW , Lin AE . Costello Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, Eds. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2018. |
[58] | Marees T , Moll AC , Imhof SM , de Boer MR , Ringens PJ , van Leeuwen FE . Risk of second malignancies in survivors of retinoblastoma: More than 40 years of follow-up. J Natl Cancer Inst. 100: :1771–9. |
[59] | Fletcher O , Easton D , Anderson K , Gilham C , Jay M , Peto J . Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst. (2004) ;96: :357–63. |


