
The New Immortalized Uroepithelial Cell Line HBLAK Contains Defined Genetic Aberrations Typical of Early Stage Urothelial Tumors
Abstract
Background: Cell culture models of normal urothelial cells are important for studying differentiation, disease mechanisms and anticancer drug development. Beyond primary cultures with their limitations in lifespan, interindividual heterogeneity and supply, few conditionally immortalized cell lines with limited applicability due to partial transformation or impaired differentiation capacity are available. We describe characteristics of the new spontaneously immortalized cell line HBLAK derived from a primary culture of uroepithelial cells.
Objective: To characterize utility and limitations of HBLAK cells as an urothelial cell culture model.
Methods: Differentiation markers were investigated by immunofluorescence and RT-PCR, genetic changes by standard karyotyping, array-CGH, PCR, RT-PCR and exome sequencing; expression of p53 and p21 by Western blotting.
Results: HBLAK cells proliferated for >50 passages without senescing. They expressed cytokeratins of basal urothelial cells. Terminal differentiation markers appeared only after induction of differentiation by specific protocols. The karyotype was stable, with few chromosomal changes, especially gains of chromosomes 5 and 20 and a chromosome 9p21 deletion resulting in p16INK4A loss. A C228T TERT promoter mutation was present, but no other mutation typical of urothelial carcinoma. TP53 was wild-type and the cell cycle was arrested in response to genomic stress.
Conclusions: HBLAK cells retain some differentiation potential and respond to cytotoxic agents similar to normal urothelial cells, but contain genetic changes contributing to immortalization in urothelial tumors. HBLAK may be valuable for evaluating the tumor specificity of novel cancer drugs, but may also be applied as an urothelial in vitro carcinogenesis model.
ABBREVIATIONS
CK | Cytokeratin |
aCGH | Array comparative genomic hybridization |
UC | Urothelial carcinoma |
FBS | Fetal bovine serum |
NHUC | Normal human urothelial cells |
MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
SA-β-Gal | Senescence-associated, acidic β-galactosidase |
qPCR | Quantitative polymerase chain reaction |
HPV | Human papilloma virus |
INTRODUCTION
Cell culture models of normal urothelial cells are valuable for studying urothelial physiology and differentiation, for investigating the function of genetic and epigenetic changes found in urothelial carcinoma and as controls in the evaluation of the tumor specificity of novel cancer drugs. Primary cultures of urothelial cells, usually established from healthy ureters removed during tumor nephrectomy, provide one valuable model [1]. Importantly, these cultures can be induced to differentiate into urothelium-like epithelia by several protocols [2, 3]. However, they have a finite lifespan, display large interindividual variation and are not broadly available. Thus, several immortalized cell lines have been generated from such cultures by introducing viral oncogenes (SV-HUC1, UROtsa) or telomerase (TERT-NHUC) [4, 5]. Whereas viral oncogenes like the SV40 largeT-antigen elicit partial transformation by interfering with cell cycle control and checkpoints by inactivating RB1 and p53, urothelial cell lines immortalized by telomerase retain proper cell cycle control and are generally genetically stable, but, over time, they lose the ability to differentiate, which is associated with changes in the expression of important chromatin regulator proteins [6, 7].
We obtained the cell line HBLAK from a company supplying primary cultures of normal urothelial cells, who noted that one batch of these cells did not senesce as usual after several passages. Having confirmed this observation, we characterized this cell line described as “spontaneously immortalized” by the distributor in detail, investigating its expression of urothelial markers, its ability to differentiate, its karyotype and typical alterations associated with urothelial transformation, as well as its suitability as a normal control when investigating new therapeutic compounds. Although this cell line originates from ostensibly normal bladder tissue and shares many characteristics with normal urothelial cells, we discovered some genetic changes thought to contribute to immortalization of urothelial cells. Taken together, our results suggest that HBLAK cells retain many phenotypic characteristics of normal urothelial cells, but also harbor a limited number of genetic changes known to contribute to immortalization of normal and cancerous urothelial cells.
MATERIALS AND METHODS
Cell culture and induction of differentiation
The human urothelial cell line HBLAK, spontaneously outgrown from a standard lot of normal human uroepithelial cells, was kindly provided by CELLnTEC (Switzerland), from where the cell line is commercially available. This culture was established from morphologically inconspicuous primary urothelium taken from the urinary bladder of an 80 years old male patient who underwent surgery for a disease other than genitourinary cancer. The patient was not diagnosed for bladder cancer at the time of the surgery. Due to ethical guidelines no further follow-up data is available for the donor. HBLAK cells were cultured according to the manufacturer’s recommendation in serum-free CnT-Prime Epithelial Culture Medium (CELLnTEC) at 37°C and with 5% CO2.
To study their differentiation capacity the cells were seeded subconfluently into 6-well plates and subsequently treated according to various published protocols [2, 3]. In one protocol, EGFR inhibition and PPARγ activation were achieved by treatment with 1 μM PD153035 (Merck, Germany) and 1 μM Troglitazone (Cayman, USA) in CnT-Prime Epithelial Culture Medium (ready to use and supplemented with EGF, CELLnTEC) or in KFSM medium without supplemented EGF (Life Technologies, Germany) for seven days, during which the medium was changed twice. For some experiments 5% FBS (Biochrom, Germany) was added to cells in CnT-Prime Medium. For a second, calcium-based protocol near-confluent cultures were maintained in CnT-Prime Medium supplemented with CaCl2 to a final concentration of 2 mM, with additional 5% FBS in some experiments, for a period of 10–14 days. The medium was changed every 3 days.
For comparison, primary cultures of normal urothelial cells (NHUC) were established from healthy ureters removed during tumor nephrectomy. These samples were collected with informed consent of the patients and their use was approved by the Ethics Committee of the Medical Faculty of the Heinrich-Heine-University, study number 1788. The cells were cultured as published previously [8] in KFSM medium supplemented with 5 ng/ml EGF and 50 μg/ml bovine pituitary extract (Life Technologies).
TERT-NHUC cells, kindly provided by Prof. M.A. Knowles, University of Leeds, UK, were cultured as previously described [9].
Cell proliferation analysis
For calculating cell doubling time, cell viability was determined over a period of four days by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide dye reduction assay (MTT, Sigma Aldrich, Germany). IC50 concentrations for Cisplatin (Accord Healthcare, Germany) were determined by treatment of different passages of HBLAK, different passages of primary NHUC cells and triplicate analysis of TERT-NHUC cells in their respective standard media at various concentrations for 72 h.
Senescent cells were identified by acidic β-galactosidase staining. Following fixation for 5 minutes in 2% formaldehyde and 0.2% glutaraldehyde and washing, cells were stained with fresh SA-β-Gal staining solution (1 mg/ml X-Gal, 150 mM NaCl, 2 mM MgCl2, 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6) for 4 h at 37°C. Images were taken using the NIS-Elements software with a Nikon Eclipse TE2000-S microscope (Nikon, Germany).
Measurements of urothelial markers
Expression of urothelial markers was determined by qRT-PCR or immunofluorescence. RNA was extracted by the RNeasy Mini Kit as recommended by the manufacturer (Qiagen, Germany). One μg RNA was reverse transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen), but with an extended incubation time of 30 min at 42 °C. qRT-PCR was performed with QuantiTect SYBR Green RT-PCR Kit (Qiagen) according to the manufacturer’s instructions. Expression of Cytokeratins (CK) 14 and 20, Uroplakin 2 (UPK2), p53, TERT and p16INK4A mRNAs was determined using primers as listed in Table 1. The housekeeping gene TATA-box binding protein (TBP) was used as a reference gene. Real time qPCR was performed using initial denaturation at 95°C for 15 min and 45 cycles of amplification including denaturation at 94°C for 15 s, annealing for 30 s (temperatures see Table 1) and elongation at 72°C for 30 s. Reactions were performed with the LightCycler 96 platform (Roche, Germany).
For immunofluorescence staining, cells were seeded onto glass slides and allowed to grow for 24 h. After washing with PBS, cells were fixed with 4% formaldehyde in PBS for 30 min and permeabilized by incubation with 0.5% Triton X-100 in PBS for 3 min. Slides were washed, blocked with 1% BSA in PBS for 30 min. Primary antibodies (CK5 1:100, ab-53121; CK14 1:50, ab-7800; CK7 1:200, ab68459; all Abcam, UK; CK7 1:50, MSK032 Zytomed Systems, Germany) and Alexa 488-conjugated or Alexa 594-conjugated secondary antibodies (1:500; Life Technologies) were applied for 1 h each. Nuclei were stained by DAPI (1 μg/ml; Roche) for 5 min. Images were taken using a Nikon Eclipse 400 microscope with the NIS Elements D2-30 software.
Karyotyping, DNA extraction, STR profiling, HPV testing and arrayCGH analysis
STR profiling was performed by standard DNA fingerprint analysis. Cytogenetic characterization was performed as previously described [10]. The karyotype was designated according to the International System for Human Cytogenetic Nomenclature [11]. Schematic presentations of the karyotypic findings were generated by the cytogenetic data analysis system (CyDAS, Germany) [12].
DNA was extracted by a standard proteinase K/phenol/chloroform extraction technique. Comparative genomic hybridization (CGH) was performed using an array of 60mer DNA-oligonucleotide probes with a median overall probe spacing of 13 kb (SurePrint G3 Human CGH Microarray 180 K, Agilent Technologies, USA). Array CGH (aCGH) was done and evaluated essentially according to the protocol of the manufacturer as described elsewhere [10].
HPV testing was performed by means of the digene® HC2 HPV DNA Test (Qiagen) according to the manufacturer’s protocol.
Mutation analysis by sequencing
For p53 mutation analysis, cDNA was amplified by PCR to yield a 1100 bp amplicon containing exons 1-10 of TA-p53 as described [10] (primers # 15 and 16). Mutations in the telomerase reverse transcriptase (TERT) promoter region were detected following PCR amplification. PCR products were purified using the DNA Clean and Concentrator Kit (Zymo Research, USA) and analyzed by Sanger sequencing. Deletion of p16INK4A was detected by amplification of exon 3 from genomic DNA. Primer sequences are detailed in Table 1. Mutation analysis for FGFR3 was performed as described [13].
Library preparation, next generation exome sequencing and data analysis
The exome NGS library was generated from HBLAK high quality genomic DNA with the Ion AmpliSeq™ Exome RDY Kit (Life Technologies, Germany). Amplification and adapter ligation were performed according to the manufacturer’s protocol (MAN0010084 Rev.C, Life Technologies). The library was barcoded during the ligation reaction using the Ion Xpress™ Barcode Adapter Kit (Life Technologies). Library concentration was determined by Qubit measurement (Life Technologies) and library fragment length was analyzed using the 2100 Bioanalyzer (Agilent Technologies, Germany). The library was diluted to a final concentration of 8 pM. Emulsion-PCR and enrichment were performed on the Ion OneTouch™ 2 System (Life Technologies) using the Ion PI™ Hi-Q™ OT2 200 Kit following the manufacturer’s protocol (MAN0010857 Rev.B, Life Technologies). Sequencing was performed on the Ion ProtonTM System (Life Technologies) using Ion PI™ v2 chip with the Ion PI™ Hi-Q™ Sequencing 200 Kit according to the manufacturer’s protocol MAN0010947 Rev.B, Life Technologies).
Amplicon sequences were aligned to the human reference genome GRCh37 (hg19) in the target region of the AmpliSeq™ exome using the Torrent SuiteTM software 4.4.3 with the implemented TMAP algorithm (Life Technologies). Subsequently, variants were detected using the Torrent Variant Caller Plugin v4.4.3.3 and the predefined parameter set ‘somatic low stringency’. Detailed annotations to each detected variant were collected in the databases of the NCBI Reference Sequence Database (RefSeq, version from 2015/03/22), Exome Sequencing Project (ESP6500), 1000 Genomes Project (1000 g), ClinVar (2015/06/29) and dbSNP142 using the open source software tool ANNOVAR [14]. Further information was added by the implemented prediction tools MutationTaster [15–19]. The Integrative Genomics Viewer (IGV 2.3) was used to visualize the read alignments and to check possible errors due to e.g. homopolymeric stretches [20]. Further information on filtering parameters and according numbers of variant is given in supplementary file 1, sheet 1.
Western blotting
Western blot analysis was used to determine p53 and p21 expression subsequent to 24 h of genotoxic stress by treatment with 5 μM/ 10 μM Cisplatin, 100 μM Etoposide, 1 μM Doxorubicin or irradiation with UV light (40 J/m2). Total protein was extracted by lysing the cells in a buffer containing 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 1% Nonidet P-40, 0.1% SDS, 1 mM EDTA, 50 mM Tris (pH 7.6), protease inhibitor cocktail (P-8340, Sigma-Aldrich) and phosphatase inhibitor cocktail (P-0044, Sigma Aldrich) for 30 min on ice. Protein concentration was determined by BCA protein assay (Pierce, Germany). Equal amounts of protein were separated in SDS-PAGE gels and transferred to PVDF membranes (Millipore, Germany). The membrane was blocked with 5% non-fat milk in TBS-T (150 mM NaCl, 10 mM Tris, pH 7.4, and 0.1% Tween-20). Primary antibodies (p53 Ab-6, 1:1000, OP43-100UG, Oncogene Science, USA; p21 Sx118, 1:1000, 556430, BD Biosciences, Germany) were applied overnight at 4°C. α-Tubulin was used as a loading control (1:10000, ab4074, Abcam). After washing, the membrane was incubated with the suitable horseradish peroxidase-conjugated secondary antibody (Santa Cruz, USA) for 1 h and exposed using Super SignalTM West Femto (Thermo Fisher Scientific, Germany) or WesternBright Quantum Kit (Biozym, Germany) according to the manufacturer’s recommendations. Signal intensities were recorded using a LI-COR C-Digit scanner (LI-COR, Germany).
RESULTS
Phenotype of HBLAK cells
HBLAK cells were received from the company and then regularly expanded at a 1:4 ratio for at least 50 further passages. No evidence of diminishing proliferation or morphological transformation was noted. In particular, we did not observe indications of a senescent phenotype during continuous passaging (Fig. 1a). Population doubling time was estimated at about 30 h (Fig. 1b).
Similar to primary cultures of normal human urothelial cells in low-calcium, serum-free KFSM medium (NHUC; Fig. 1c left panel), HBLAK cells cultured as monolayers in low-calcium, serum-free CnT-Prime medium displayed a proliferating and non-differentiated epithelial phenotype with prominent cell-cell contacts (Fig. 1c right panel). To study their differentiation state, RNA expression of urothelial differentiation markers in HBLAK cells was compared to that of three independent cultures of primary uroepithelial cells. CK14, a marker of precursor basal urothelial cells [21] was significantly more strongly expressed in HBLAK cells than in the NHUC cultures (Fig. 1d). Markers of differentiated urothelial cells such as UPK2 and CK20 were variably expressed among the three NHUC cultures, but undetectable in HBLAK. Analysis of protein expression by immunofluorescence revealed that all HBLAK cells expressed CK5 (Fig. 1e, right panel), a marker of basal and intermediate urothelial cells. CK14 was also broadly expressed, but not in all cells (Fig. 1e, left panel). Co-staining displayed that CK14 negative cells were still positive for CK5 (Fig. 1f). HBLAK cells stained also positive for CK7, a common marker of urothelial cells (Fig. 1g). Like CK5, CK7 was expressed in all HBLAK cells, but the staining appeared less homogeneous in intensity (Fig. 1h). Taken together, HBLAK cells phenotypically resemble primary urothelial cell cultures, but consist more homogeneously of basal-like cells compared to NHUC cultures.
Differentiation capacity of HBLAK cells
NHUC cells can be induced to differentiate by two established protocols [2, 3]. In one protocol, simultaneous inhibition of epidermal growth factor receptor (EGFR) signaling with PD153035 and activation of PPARγ by its cognate agonist Troglitazone led to significant induction of the differentiation markers UPK2 and CK20 after one week (Fig. 2a), whereas expression of the precursor marker CK14 was concomitantly lost. In the second protocol, differentiation was induced by increased calcium concentration over a period of two weeks. In addition to induction of terminal differentiation markers, visible stratification of the cultures was observed with this protocol (Fig. 2b bottom). However, induction of differentiation markers appeared to be not as pronounced as in the first protocol, but could be augmented by addition of 5% FBS, which also diminished expression of squamous differentiation markers [3] (data not shown).
Although differentiation markers CK20 and UPK2 were not induced in HBLAK cells by treatment with the EGFR inhibitor and the PPARγ agonist, if the differentiation experiment was performed in the standard CnT-Prime culture medium, which is supplemented with EGF by the manufacturer (Fig. 2c), changes in the morphology of HBLAK cells were similar compared to those of NHUC (Fig. 2d center left). Induction of UPK2 and phenotypical changes could be achieved by further addition of 5% FBS (Fig. 2d center right). In a different low-calcium medium (KFSM), where EGF supplementation is optional, the addition of PD153035 and Troglitazone resulted in a significant induction of CK20 and UPK2 in the absence of EGF (Fig. 2c).
Treatment of HBLAK cells by the high-calcium protocol altered the morphological appearance of the monolayer (Fig. 2d bottom left), but no significant stratification was observed, unlike in NHUC cells (Fig. 2b bottom). In HBLAK cells treated with high calcium, CK14 expression increased, but neither UPK2 nor CK20 became induced (Fig. 2c). Addition of 5% FBS led to a more urothelial-like expression pattern (Fig. 2d bottom right) with diminished CK14 and induction of UPK2, albeit not CK20.
Genetic changes in HBLAK cells
For further characterization of the HBLAK cell line a STR profile was generated as a reference for future analyses and in order to prove that the cell line is unique (Table 2).
Standard karyotyping revealed a hyperdiploid karyotype with trisomies of chromosomes 5 and 20 in all metaphases; two subclones were characterized by additional gains of chromosomes 7 or 9, respectively. No structural chromosomal changes were discernible. The formal description of the karyotype is 48-50, XY, +5, +20[9]/49, XY, +5, +9, +20[5]/50, XY,+5, +7, +20, +20[4]. These findings were confirmed by array-CGH, which also revealed further smaller gains and deletions (Table 3). Notable among these were several distinct deletions on chromosome 9, which was overall gained (i.e. trisomic in many metaphases). The arrayCGH data are best explained by the assumption that all copies of chromosome 9 lack a small 9p21.3 fragment, whereas only one copy contains additional deletions at 9p22.2, 9q21.3 and 9q33-4. The homozygous deletion at 9p21.3 affected a 60 kb region including part of the CDKN2A locus, the MTAP gene and part of the non-coding RNA ANRIL gene. Deletion of CDKN2A/p16INK4 was further confirmed by PCR for CDKN2A exon 3 (Fig. 3a) and for p16 mRNA on samples obtained from different passage numbers. By real time qRT-PCR p16 mRNA expression was undetectable compared to controls (data not shown).
The commonly gained chromosome 5 contains the telomerase reverse transcriptase (TERT) locus at 5p15.33. Mutations in the TERT promoter region leading to elevated telomerase expression are highly prevalent in urothelial carcinoma [22]. Indeed, sequencing of the TERT promoter region in HBLAK revealed the most common C228T mutation (Fig. 3b). TERT mRNA expression was determined by real time qRT-PCR in different passages of HBLAK cells and other cell lines with wildtype (HEK293, 253J) or mutant TERT promoter (VM-CUB1) as well as in TERT-NHUC cells [22] (Fig. 3c). In HBLAK cells, across all passages tested, TERT mRNA was well detectable, other than in normal urothelial cells (NHUC), but was lower than in UC cell lines and in TERT-NHUC cells with engineered telomerase overexpression.
Gains of chromosome 20 are observed in urothelial tumors, but also quite consistently in NHUC cell lines immortalized by HPV oncogenes [23]. We therefore ascertained the lack of HPV DNA in HBLAK by a clinical routine assay.
As p53 inactivation is observed in normal cells immortalized by viral oncogenes and also in many cancerous urothelial cell lines [24, 25], we investigated its status in HBLAK. Expression of the mRNA encoding full-length p53 was well detectable, but not as strongly as in the UC cell lines J82 and VM-CUB1 with mutant p53 (Fig. 3d). Sequencing revealed a wild-type sequence heterozygous for the well-characterized codon 72 G⟶C (Arg⟶Pro) polymorphism. Typical of wild-type p53, the protein was weakly expressed under standard growth conditions (Fig. 4a).
For further characterization of the genetic background of HBLAK cells next generation exome sequencing was performed. Exome sequencing confirmed the deletion of CDKN2A/p16INK4A exon 3 and the lack of p53 mutations. In brief, after filtering, as described in the supplementary file sheet 1, 1104 variants affecting 967 genes were detected compared to the hg19 reference genome (supplementary file, sheet 2). Type and total quantity of variants across the genome are summarized in Table 4. Quantity and type of variants per chromosome are displayed in Fig. 3e. On every chromosome, nonsynonymous single-nucleotide variants (SNV) accounted for the majority of differences, while frameshift and nonframeshift variants were infrequent. Chromosome 1 harbored the most nonsynonymous SNVs, chromosomes 13 and 21 were the least affected. No mutations were detected in genes commonly mutated in urothelial carcinoma (supplementary file, sheets 2 and 3). In particular, FGFR3, the most commonly mutated gene in non-invasive UC and especially in low-grade papillary tumors neither harbored point mutations nor displayed loss of exons indicative of gene fusions, as confirmed by an independent SNaPshot assay.
Sensitivity of HBLAK cells to cytotoxic treatment
Finally, we investigated whether HBLAK cells retained normal responses to cytotoxic treatment. Western blot analysis revealed induction of p53 expression following a 24 h treatment with cytotoxic compounds or UV-irradiation. Induction of p21 expression was also detectable (Fig. 4a).
HBLAK cells were also included in dose response analyses for various pharmaceutical compounds. Exemplarily, we illustrate the sensitivity of HBLAK cells towards treatment with cisplatin for 72 h in Fig. 4b. IC50 concentrations were about 5 μM, higher than those of a primary non-immortalized NHUC, but lower than those of TERT-NHUC cells.
DISCUSSION
Previous reports on artificially immortalized uroepithelial cells have demonstrated altered expression of genes particularly involved in the regulation of differentiation, self-renewal and tumorigenesis, limiting their suitability as models to study these processes [6]. Thus, we characterized the utility and limitations of the new HBLAK cell line, reported as “spontaneously immortalized”, as a cell culture model for normal uroepithelial cells.
Data presented here and in previous studies [26, 27] demonstrate that HBLAK cells behave more like normal primary (NHUC) or telomerase-immortalized urothelial cells (TERT-NHUC) than like urothelial carcinoma (UC) cell lines in drug sensitivity assays and can serve as a convenient control in a similar fashion as TERT-NHUC. One reason for this behavior should be that, in contrast to most urothelial carcinoma cells, HBLAK cells express wildtype p53, which can be induced by treatment with cytotoxic compounds and retain more normal checkpoint signaling. Accordingly, p21 induction can be observed.
The expression pattern of cytokeratins in HBLAK cells corresponds to that of basal or early intermediate urothelial cells, which express CK5 and less regularly CK14, but never CK20 [28]. Accordingly, UPK2 expression, which is weak in intermediate cells and maximal in terminally differentiated umbrella cells, was absent. Importantly, differentiation markers are more homogeneous in HBLAK than in primary NHUC cultures. The homogeneous and reproducible differentiation state of the cell line, in addition to its unlimited propagation, represents another advantage in several applications.
Like primary NHUC cells, HBLAK cells can be induced to differentiate. However, their ability to differentiate is more strongly dependent on the particular protocol than that of NHUC cultures and they do not stratify well. Accordingly, treatment with PD153035 and Troglitazone [2, 3] induced the urothelial differentiation marker UPK2 only in medium without EGF. Likewise, induction of differentiation by high calcium strictly required addition of FBS, which in NHUC enhances calcium-induced urothelial differentiation, but is not as obligatory. Moreover, even after inclusion of FBS in the protocol HBLAK cells retained considerable CK14 expression, indicating incomplete differentiation or a shift towards squamous differentiation, which occurs also when NHUC differentiate in culture under suboptimal conditions [2, 29, 30]. Similar results have been reported for a spontaneously immortalized keratinocyte cell line named HaCaT, which can be induced to differentiate accompanied by the according expression changes of early differentiation markers, but which lost its ability for stratification [31].
A crucial question is how HBLAK became immortalized. Experimentally, NHUC, like many other cell types, can be immortalized by transformation with SV40 largeT-antigen [4] or human papillomavirus (HPV16) E7 [23, 32, 33], which bind and inactivate p53 and RB1 (T-antigen) or RB1 (E7) [24]. Inactivation of p53 and RB1 is also common in invasive UC and accordingly, in many UC cell lines, but essentially absent in papillary UC. Immortalization of NHUCs by DNA virus oncogenes is accompanied by accumulation of additional genetic mutations and restored telomerase activity. Notably, immortalization of NHUC by HPV E7 resulted reproducibly in gain of chromosome 20q, which was also found in HBLAK cells [23]. In a few cell lines derived by HPV E7 transfer, an additional amplification event occurred involving the 20q13.2 region. Other than chromosome 20q gain per se, this amplification was associated with genomic instability, despite retention of wild-type p53 [23]. Interestingly, both 20q gain and 20q13.2 amplification have been reported for various cancer types [33] including UC [34–36], where amplifications of chromosome 20q generally appear to be associated with immortalization, oncogenic transformation and malignant progression. Whereas high level amplifications correlated with tumor progression in breast cancer [37], low-level amplifications appeared to be rather associated with early stage ovarian carcinoma [38]. Similarly, 20q gain also occurred as an early event in normal prostatic cells immortalized by TERT overexpression, where a number of oncogenes in the 20q13.2 region cooperate to support immortalization [39]. Gains at 20q thus appear to represent an early event in the evolution of a malignant phenotype [39]. Taken together, these findings suggest that the gain chromosome 20 in HBLAK cells is not incidental, but contributes to the immortalization of the cell line. However, in HBLAK cells this gain is unrelated to HPV oncogenes as the cell line tested negative for HPV.
Likely, one mechanism underlying spontaneous immortalization of HBLAK cells is telomerase reactivation. In UC, activation of telomerase is a common early event and is typically caused by mutations in the TERT gene promoter [22, 36, 40]. Further, gain of chromosome 5p15.33, harboring the TERT gene, was found to be significantly associated with disease progression in UC [41]. Intriguingly, E7 transformed NHUC cells also gained chromosome 5, which may explain their telomerase overexpression. Accordingly, NHUC can be immortalized by exogenous TERT overexpression [7, 9], like several other cell types [39, 42, 43]. Gain of chromosome 5, including 5p15.33, represents the second consistent chromosomal change in the HBLAK cell line. Moreover, sequencing of the TERT promoter in HBLAK revealed the most common TERT promoter mutation in urothelial carcinoma and TERT mRNA was moderately overexpressed compared to normal urothelial cells, albeit not as strongly as in UC cell lines.
In addition to these obvious chromosomal alterations, further chromosomal alterations in HBLAK cells were not seen across all metaphases or were revealed only by array-CGH. In particular, metaphases contained two or three copies of chromosome 9 with various alterations. All deletions detected in HBLAK can also occur in UC, especially in papillary tumors [44]. Loss of chromosome 9 is the most common chromosomal aberration in UC and the most consistently deleted region is 9p21.3 encompassing CDKN2A encoding the cell cycle inhibitor p16INK4. As loss of p16INK4 contributes to senescence bypass, the homozygous deletion at 9p21.3 removing the 3’-end of CDKN2A in HBLAK cells is likely a third factor permitting their immortalization. Thus, the TERT mutation in conjunction with gain of chromosomes 5 and chromosome 20q and with loss of p16INK4 may concomitantly contribute to immortalization of the HBLAK cell line. Of note, since the original tissue is not available, we do not know whether these changes were already present in the original tissue or arose under culture conditions. However, a number of previous reports on a spontaneously immortalized keratinocyte cell line (HaCaT), immortalization experiments with human mammary epithelial cells (HMEC) and normal prostatic cells suggest that normal epithelial cells can spontaneously acquire the above mentioned alterations in the course of immortalization and that these alterations are sufficient to immortalize normal, non-cancerous cells [31, 48, 49]. Notably, these studies also demonstrate clearly that cells with this limited extent of genetic changes can retain an otherwise normal epithelial phenotype and do not necessarily undergo transformation towards a malignant phenotype. Cells with such genetic alterations therefore do not necessarily originate from cancer cells, but may be susceptible to transformation by further genetic changes.
This interpretation is further supported by the lack of further mutations in genes associated with UC documented by whole exome sequencing and exemplified by the results on two other genes frequently mutated in UC, but unaltered in HBLAK. FGFR3 is the most commonly mutated gene in non-invasive UC and especially in low-grade papillary tumors [50], whereas p53 mutations are more common in invasive UC. In HBLAK, p53 protein size and expression, basal and following DNA damage, fit to a wild-type protein. While MDM2 amplifications do occur in papillary urothelial carcinoma [51], none was found in HBLAK. Similarly, neither arrayCGH nor exome sequencing revealed any relevant alterations in the FGFR3 gene.
Taken together, HBLAK cells harbor some genetic alterations contributing to immortalization of urothelial carcinoma cells (TERT mutation, p16INK4A loss), but lack additional genetic changes characteristic for this tumor type. Notably, we did not obtain evidence for genetic changes characteristic of invasive UC such as p53 mutation and general chromosomal instability. Also a more detailed analysis of the genetic background by exome sequencing confirmed that HBLAK cells harbor only a limited number of genetic changes and, especially, that genes found to be frequently mutated in comprehensive genomic studies of urothelial carcinoma are unaltered in HBLAK. Although neither further primary tissue nor clinical follow-up of the donor is available, our results better support the interpretation that HBLAK cells represent a preneoplastic state of urothelial cells, rather than being derived from an undetected urothelial carcinoma.
In conclusion, HBLAK cells retained, on one hand, many characteristics of normal urothelial cells, including response to cytotoxic agents, allowing its application in studies on tumor-specific effects of novel antineoplastic agents without the limitations by interindividual heterogeneity, limited lifespan and irregular supply of primary cultures. On the other hand, no immortalized epithelial cell line can be truly normal. Interestingly, the immortalization of HBLAK cells appears to be brought about by specific genetic alterations that are thought to lead to immortalization during urothelial carcinogenesis. However, the cells do not contain genetic alterations typical of invasive UC and not even an FGFR3 mutation, suggesting that they represent a preneoplastic stage of urothelial carcinoma. In this regard, they may provide a unique in vitro carcinogenesis model to study factors involved in the stepwise progression of preneoplastic, immortalized urothelial cells towards cancer, alike to the HaCaT cell line used in research on squamous cell carcinoma progression [48]. Although we do not have access to donor tissue in order to ascertain whether the observed genetic changes were already present in the original tissue, none of our results from various analyses indicates that any of the observed genetic alterations was acquired in later passages in culture. We also consider it unlikely that three independent genetic changes favoring immortalization were acquired and fixed in a generally genetically stable cell culture. Accordingly, we propose that immortalized clones of urothelial cells may exist in vivo that contain genetic changes analogous to those experimentally introduced in order to immortalize NHUC.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest. In particular, they did not receive financial support by the CELLnTEC Company and the company did not exert any influence on the results or conclusions of this study.
ACKNOWLEDGMENTS
We are very grateful to Dr. Peter Girling, CELLnTEC, Bern, Switzerland for providing HBLAK cells and donor information. We gratefully acknowledge help with cell culturing by Marija Lenzen. We thank Dr. Tanja Arent for performing the STR profiling, Dr. Robert Stöhr for the additional FGRF3 mutation analysis and Ursula Grolik for HPV testing. M.P. and G.N. were financially supported by a grant from the Deutsche Forschungsgemeinschaft, E.K. is a postdoctoral fellow of the Bodossaki foundation, and M.A.S. is supported by a fellowship of the Duesseldorf School of Oncology (funded by the Comprehensive Cancer Centre Duesseldorf/ Deutsche Krebshilfe and the Medical Faculty of the Heinrich-Heine-University Duesseldorf).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/BLC-160065.
REFERENCES
[1] | Southgate J , Hutton KA , Thomas DF , Trejdosiewicz LK . Normal human urothelial cells in vitro: Proliferation and induction of stratification. Lab Invest (1994) ;71: (4):583–94. |
[2] | Cross WR , Eardley I , Leese HJ , Southgate J . A biomimetic tissue from cultured normal human urothelial cells: Analysis of physiological function. Am J Physiol Renal Physiol (2005) ;289: (2):F459–468. |
[3] | Varley CL , Stahlschmidt J , Lee WC , Holder J , Diggle C , Selby PJ , Trejdosiewicz LK , Southgate J . Role of PPARgamma and EGFR signalling in the urothelial terminal differentiation programme. J Cell Sci (2004) ;117: (Pt 10):2029–36. |
[4] | Kao C , Huang J , Wu SQ , Hauser P , Reznikoff CA . Role of SV40 T antigen binding to pRB and p53 in multistep transformation in vitro of human uroepithelial cells. Carcinogenesis (1993) ;14: (11):2297–302. |
[5] | Rossi MR , Masters JR , Park S , Todd JH , Garrett SH , Sens MA , Somji S , Nath J , Sens DA . The immortalized UROtsa cell line as a potential cell culture model of human urothelium. Environ Health Perspect (2001) ;109: (8):801–8. |
[6] | Chapman EJ , Kelly G , Knowles MA . Genes involved in differentiation, stem cell renewal, and tumorigenesis are modulated in telomerase-immortalized human urothelial cells. Mol Cancer Res (2008) ;6: (7):1154–68. |
[7] | Georgopoulos NT , Kirkwood LA , Varley CL , MacLaine NJ , Aziz N , Southgate J . Immortalisation of normal human urothelial cells compromises differentiation capacity. Eur Urol (2011) ;60: (1):141–9. |
[8] | Swiatkowski S , Seifert HH , Steinhoff C , Prior A , Thievessen I , Schliess F , Schulz WA . Activities of MAP-kinase pathways in normal uroepithelial cells and urothelial carcinoma cell lines. Exp Cell Res (2003) ;282: (1):48–57. |
[9] | Chapman EJ , Hurst CD , Pitt E , Chambers P , Aveyard JS , Knowles MA . Expression of hTERT immortalises normal human urothelial cells without inactivation of the p16/Rb pathway. Oncogene (2006) ;25: (36):5037–45. |
[10] | Koch A , Hatina J , Rieder H , Seifert HH , Huckenbeck W , Jankowiak F , Florl AR , Stoehr R , Schulz WA . Discovery of TP53 splice variants in two novel papillary urothelial cancer cell lines. Cell Oncol (Dordr) (2012) ;35: (4):243–57. |
[11] | Brothman AR , Persons DL , Shaffer LG . Nomenclature evolution: Changes in the ISCN from the 2005 to the 2009 edition. Cytogenet Genome Res (2009) ;127: (1):1–4. |
[12] | Hiller B , Bradtke J , Balz H , Rieder H . CyDAS: A cytogenetic data analysis system. Bioinformatics (2005) ;21: (7):1282–3. |
[13] | van Oers JM , Lurkin I , van Exsel AJ , Nijsen Y , van Rhijn BW , van der Aa MN , Zwarthoff EC . A simple and fast method for the simultaneous detection of nine fibroblast growth factor receptor 3 mutations in bladder cancer and voided urine. Clin Cancer Res (2005) ;11: (21):7743–8. |
[14] | Wang K , Li M , Hakonarson H . ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res (2010) ;38: (16):e164. |
[15] | Schwarz JM , Rodelsperger C , Schuelke M , Seelow D . MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods (2010) ;7: (8):575–6. |
[16] | Kumar P , Henikoff S , Ng PC . Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc (2009) ;4: (7):1073–81. |
[17] | Adzhubei IA , Schmidt S , Peshkin L , Ramensky VE , Gerasimova A , Bork P , Kondrashov AS , Sunyaev SR . A method and server for predicting damaging missense mutations. Nat Methods (2010) ;7: (4):248–9. |
[18] | Chun S , Fay JC . Identification of deleterious mutations within three human genomes. Genome Res (2009) ;19: (9):1553–61. |
[19] | Shihab HA , Gough J , Cooper DN , Stenson PD , Barker GL , Edwards KJ , Day IN , Gaunt TR . Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat (2013) ;34: (1):57–65. |
[20] | Robinson JT , Thorvaldsdottir H , Winckler W , Guttman M , Lander ES , Getz G , Mesirov JP . Integrative genomics viewer. Nat Biotechnol (2011) ;29: (1):24–6. |
[21] | Ho PL , Kurtova A , Chan KS . Normal and neoplastic urothelial stem cells: Getting to the root of the problem. Nat Rev Urol (2012) ;9: (10):583–94. |
[22] | Borah S , Xi L , Zaug AJ , Powell NM , Dancik GM , Cohen SB , Costello JC , Theodorescu D , Cech TR . Cancer. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science (2015) ;347: (6225):1006–10. |
[23] | Savelieva E , Belair CD , Newton MA , DeVries S , Gray JW , Waldman F , Reznikoff CA . 20q gain associates with immortalization: 20q13.2 amplification correlates with genome instability in human papillomavirus 16 E7 transformed human uroepithelial cells. Oncogene (1997) ;14: (5):551–60. |
[24] | Bryan TM , Reddel RR . SV40-induced immortalization of human cells. Crit Rev Oncog (1994) ;5: (4):331–57. |
[25] | Crallan RA , Georgopoulos NT , Southgate J . Experimental models of human bladder carcinogenesis. Carcinogenesis (2006) ;27: (3):374–81. |
[26] | Pinkerneil M , Hoffmann MJ , Deenen R , Kohrer K , Arent T , Schulz WA , Niegisch G . Inhibition of class I histone deacetylases 1 and 2 promotes urothelial carcinoma cell death by various mechanisms. Mol Cancer Ther (2016) ;15: (2):299–312. |
[27] | Skowron MA , Niegisch G , Fritz G , Arent T , van Roermund JG , Romano A , Albers P , Schulz WA , Hoffmann MJ . Phenotype plasticity rather than repopulation from CD90/CK14+cancer stem cells leads to cisplatin resistance of urothelial carcinoma cell lines. J Exp Clin Cancer Res (2015) ;34: :144–. |
[28] | Southgate J , Harnden P , Trejdosiewicz LK . Cytokeratin expression patterns in normal and malignant urothelium: A review of the biological and diagnostic implications. Histol Histopathol (1999) ;14: (2):657–64. |
[29] | Harnden P , Southgate J . Cytokeratin 14 as a marker of squamous differentiation in transitional cell carcinomas. J Clin Pathol (1997) ;50: (12):1032–33. |
[30] | Choi W , Porten S , Kim S , Willis D , Plimack ER , Hoffman-Censits J , Roth B , Cheng T , Tran M , Lee IL , Melquist J , Bondaruk J , Majewski T , Zhang S , Pretzsch S , Baggerly K , Siefker-Radtke A , Czerniak B , Dinney CP , McConkey DJ . Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell (2014) ;25: (2):152–65. |
[31] | Cerezo A , Stark HJ , Moshir S , Boukamp P . Constitutive overexpression of human telomerase reverse transcriptase but not c-myc blocks terminal differentiation in human HaCaT skin keratinocytes. J Invest Dermatol (2003) ;121: (1):110–19. |
[32] | Carmean N , Kosman JW , Leaf EM , Hudson AE , Opheim KE , Bassuk JA . Immortalization of human urothelial cells by human papillomavirus type 16 E6 and E7 genes in a defined serum-free system. Cell Prolif (2007) ;40: (2):166–84. |
[33] | Cuthill S , Agarwal P , Sarkar S , Savelieva E , Reznikoff CA . Dominant genetic alterations in immortalization: Role for 20q gain. Genes Chromosomes Cancer (1999) ;26: (4):304–11. |
[34] | Grammatico P , Lombardo A , Governatori M , Mordenti C , Poscente M , Del Porto G . Trisomy 20 in a papillary urothelial carcinoma of the ureter. Cancer Genet Cytogenet (1996) ;90: (2):132–34. |
[35] | Kallioniemi A , Kallioniemi OP , Citro G , Sauter G , DeVries S , Kerschmann R , Caroll P , Waldman F . Identification of gains and losses of DNA sequences in primary bladder cancer by comparative genomic hybridization. Genes Chromosomes Cancer (1995) ;12: (3):213–19. |
[36] | Pinto-Leite R , Carreira I , Melo J , Ferreira SI , Ribeiro I , Ferreira J , Filipe M , Bernardo C , Arantes-Rodrigues R , Oliveira P , Santos L . Genomic characterization of three urinary bladder cancer cell lines: Understanding genomic types of urinary bladder cancer. Tumour Biol (2014) ;35: (5):4599–617. |
[37] | Tanner MM , Tirkkonen M , Kallioniemi A , Collins C , Stokke T , Karhu R , Kowbel D , Shadravan F , Hintz M , Kuo WL , et al. Increased copy number at 20q13 in breast cancer: Defining the critical region and exclusion of candidate genes. Cancer Res (1994) ;54: (16):4257–60. |
[38] | Iwabuchi H , Sakamoto M , Sakunaga H , Ma YY , Carcangiu ML , Pinkel D , Yang-Feng TL , Gray JW . Genetic analysis of benign, low-grade, and high-grade ovarian tumors. Cancer Res (1995) ;55: (24):6172–80. |
[39] | Tabach Y , Kogan-Sakin I , Buganim Y , Solomon H , Goldfinger N , Hovland R , Ke XS , Oyan AM , Kalland KH , Rotter V , Domany E . Amplification of the 20q chromosomal arm occurs early in tumorigenic transformation and may initiate cancer. PLoS One (2011) ;6: (1):e14632. |
[40] | Kinde I , Munari E , Faraj SF , Hruban RH , Schoenberg M , Bivalacqua T , Allaf M , Springer S , Wang Y , Diaz LA Jr , Kinzler KW, Vogelstein B, Papadopoulos N, Netto GJ. TERT promoter mutations occur early in urothelial neoplasia and are biomarkers of early disease and disease recurrence in urine. Cancer Res (2013) ;73: (24):7162–67. |
[41] | Yamamoto Y , Chochi Y , Matsuyama H , Eguchi S , Kawauchi S , Furuya T , Oga A , Kang JJ , Naito K , Sasaki K . Gain of 5p15.33 is associated with progression of bladder cancer. Oncology (2007) ;72: (1-2):132–8. |
[42] | Lewis CM , Herbert BS , Bu D , Halloway S , Beck A , Shadeo A , Zhang C , Ashfaq R , Shay JW , Euhus DM . Telomerase immortalization of human mammary epithelial cells derived from a BRCA2 mutation carrier. Breast Cancer Res Treat (2006) ;99: (1):103–15. |
[43] | Puttmann S , Senner V , Braune S , Hillmann B , Exeler R , Rickert CH , Paulus W . Establishment of a benign meningioma cell line by hTERT-mediated immortalization. Lab Invest (2005) ;85: (9):1163–71. |
[44] | Prat E , Bernues M , Caballin MR , Egozcue J , Gelabert A , Miro R . Detection of chromosomal imbalances in papillary bladder tumors by comparative genomic hybridization. Urology (2001) ;57: (5):986–92. |
[45] | Kim WY , Sharpless NE . The regulation of INK4/ARF in cancer and aging. Cell (2006) ;127: (2):265–75. |
[46] | Florl AR , Franke KH , Niederacher D , Gerharz CD , Seifert HH , Schulz WA . DNA methylation and the mechanisms of CDKN2A inactivation in transitional cell carcinoma of the urinary bladder. Lab Invest (2000) ;80: (10):1513–22. |
[47] | Chapman EJ , Harnden P , Chambers P , Johnston C , Knowles MA . Comprehensive analysis of CDKN2A status in microdissected urothelial cell carcinoma reveals potential haploinsufficiency, a high frequency of homozygous co-deletion and associations with clinical phenotype. Clin Cancer Res (2005) ;11: (16):5740–7. |
[48] | Fusenig NE , Boukamp P . Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol Carcinog (1998) ;23: (3):144–58. |
[49] | Garbe JC , Vrba L , Sputova K , Fuchs L , Novak P , Brothman AR , Jackson M , Chin K , LaBarge MA , Watts G , Futscher BW , Stampfer MR . Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle (2014) ;13: (21):3423–35. |
[50] | Jebar AH , Hurst CD , Tomlinson DC , Johnston C , Taylor CF , Knowles MA . FGFR3 and Ras gene mutations are mutually exclusive genetic events in urothelial cell carcinoma. Oncogene (2005) ;24: (33):5218–25. |
[51] | Nord H , Segersten U , Sandgren J , Wester K , Busch C , Menzel U , Komorowski J , Dumanski JP , Malmstrom PU , Diaz de Stahl T . Focal amplifications are associated with high grade and recurrences in stage Ta bladder carcinoma. Int J Cancer (2010) ;126: (6):1390–402. |
Figures and Tables
Fig.1
Phenotypic characterization of HBLAK cells. (a) HBLAK cells stained for senescence-associated (acidic) β-galactosidase; staining is negative indicating lack of senescence (left panel 4x magnification, right panel 10x magnification). (b) Results from MTT -assays performed over a period of 96 h were used to calculate population doubling time (29.9±3.4 h). (c) Comparison of morphology of HBLAK cells (right panel, cultured in CnT-Prime medium) to proliferating, non-differentiated primary normal human urothelial cells (NHUC, left panel, cultured in KFSM). (d) mRNA expression of differentiation markers CK14, CK20 and UPK2 as determined by qRT-PCR for HBLAK cells and three independent primary NHUC cultures derived from different patients. TBP was used as a reference gene. (e) Expression of CK5 and CK14 (green, Alexa-488) as visualized by single immunofluorescence stainings (all 10x magnification). Nuclei were counterstained by DAPI (blue). (f) Simultaneous expression of CK14 (red, Alexa-594) and of CK5 (green, Alexa-488) in HBLAK cells was illustrated by co-staining. Negative controls were only stained with the respective secondary antibody and counterstained with DAPI. (g) Expression of the common urothelial marker CK7 displayed by green fluorescence. (h) Simultaneous expression of CK7 (red, Alexa-594) and of CK5 (green, Alexa-488) in HBLAK cells evidenced by co-staining.
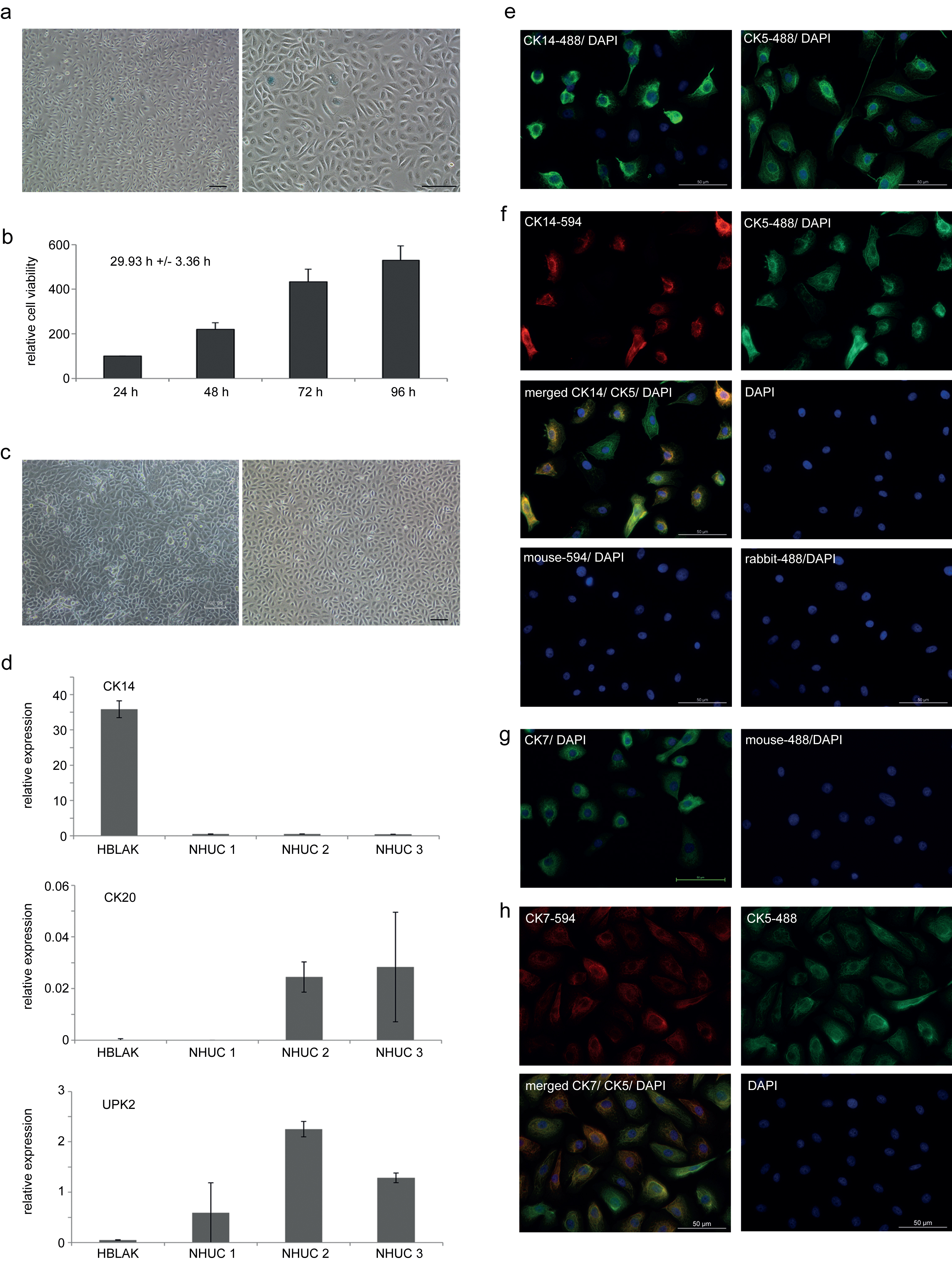
Fig.2
Differentiation capacity of HBLAK cells. (a) qRT-PCR results for mRNA expression changes of urothelial differentiation markers and (b) morphological changes following induction of differentiation in a primary NHUC culture by the indicated protocols (10x magnification). Note in particular stratification in the bottom panel of (b). (c) Representative qRT-PCR results for mRNA expression changes of urothelial differentiation markers and (d) morphological changes following induction of differentiation in HBLAK cells by the indicated protocols.
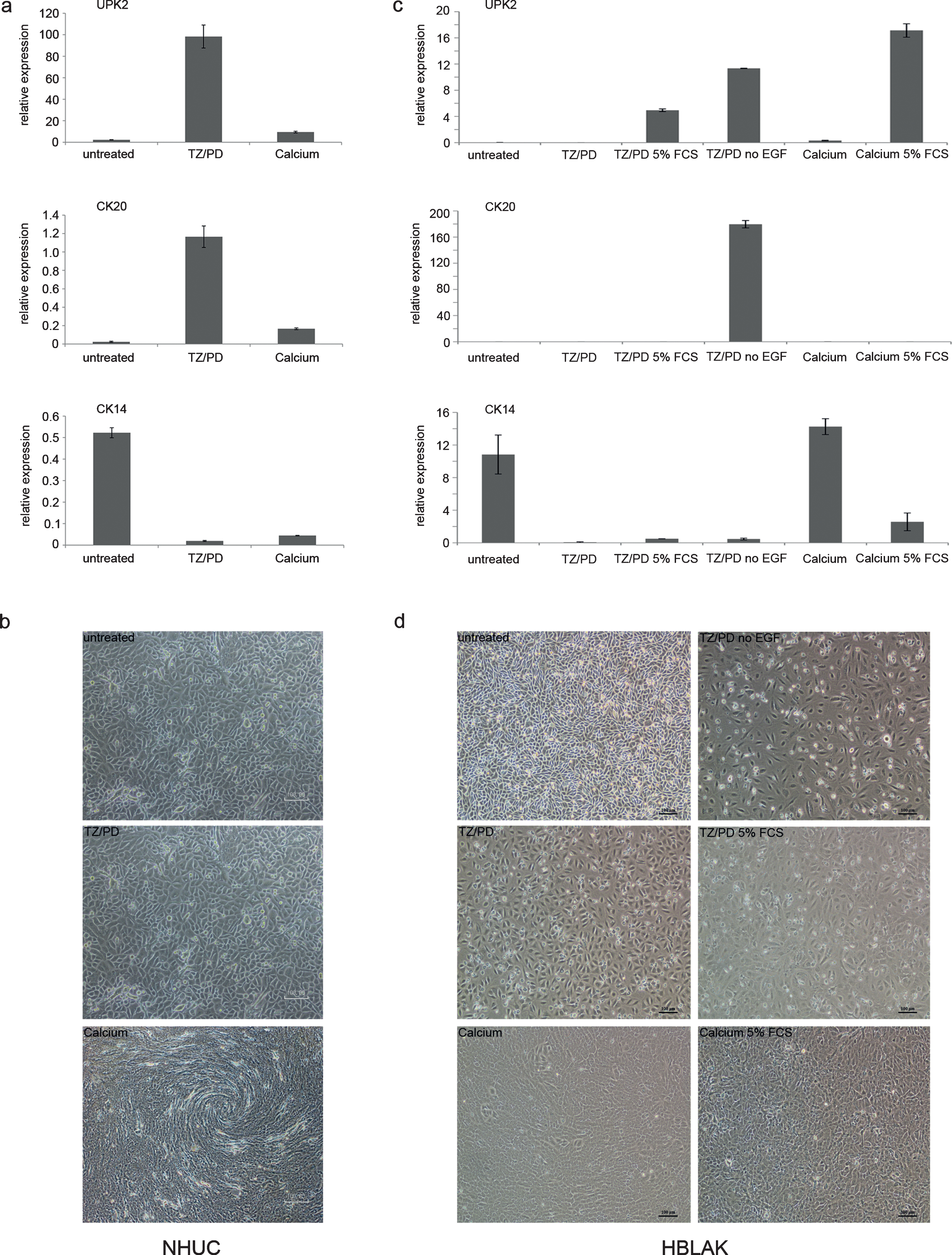
Fig.3
Characterization of genetic changes in HBLAK cells. (a) Deletion of CDKN2A/p16INK4 exon 3 confirmed by PCR amplification of genomic DNA of various HBLAK passages. Urothelial carcinoma cell lines retaining expression of p16INK4 (VM-CUB1, 639-V) and experimentally immortalized normal NHUC cells (TERT-NHUC) served as positive controls. The UC cell line RT-112 with a homozygous deletion of CDKN2A served as a negative control. (b) Sanger sequencing revealed the most common TERT promoter mutation (C228T) in HBLAK, whereas C250 was retained. (c) TERT mRNA expression was determined by real time qRT-PCR in different passages of HBLAK cells compared to non-immortalized NHUC cells of different patients, TERT-NHUC and further cell lines with wildtype (HEK293, 253J) and mutant (VM-CUB1) TERT promoter. TBP was used as a reference gene. Note that TERT expression in TERT-NHUC is much higher than that of all other cell lines (76.0). (d) The full-length transcript of p53 was detectable in HBLAK by RT-PCR, but weakly expressed compared to urothelial carcinoma cell lines J82 and VM-CUB1. The amplification product was used for subsequent Sanger sequencing of the amplicon revealing that HBLAK cells contain wildtype p53. Left lane: size markers. (e) Overview of variant frequency by chromosome in HBLAK cells compared to the hg19 reference genome per chromosome as detected by exome sequencing. For each chromosome, frameshift (blue), nonframeshift (red), and nonsynonymous (green) variants are indicated. Other types are detailed in Table 4 and all variants are listed individually in supplementary file, sheet 2.
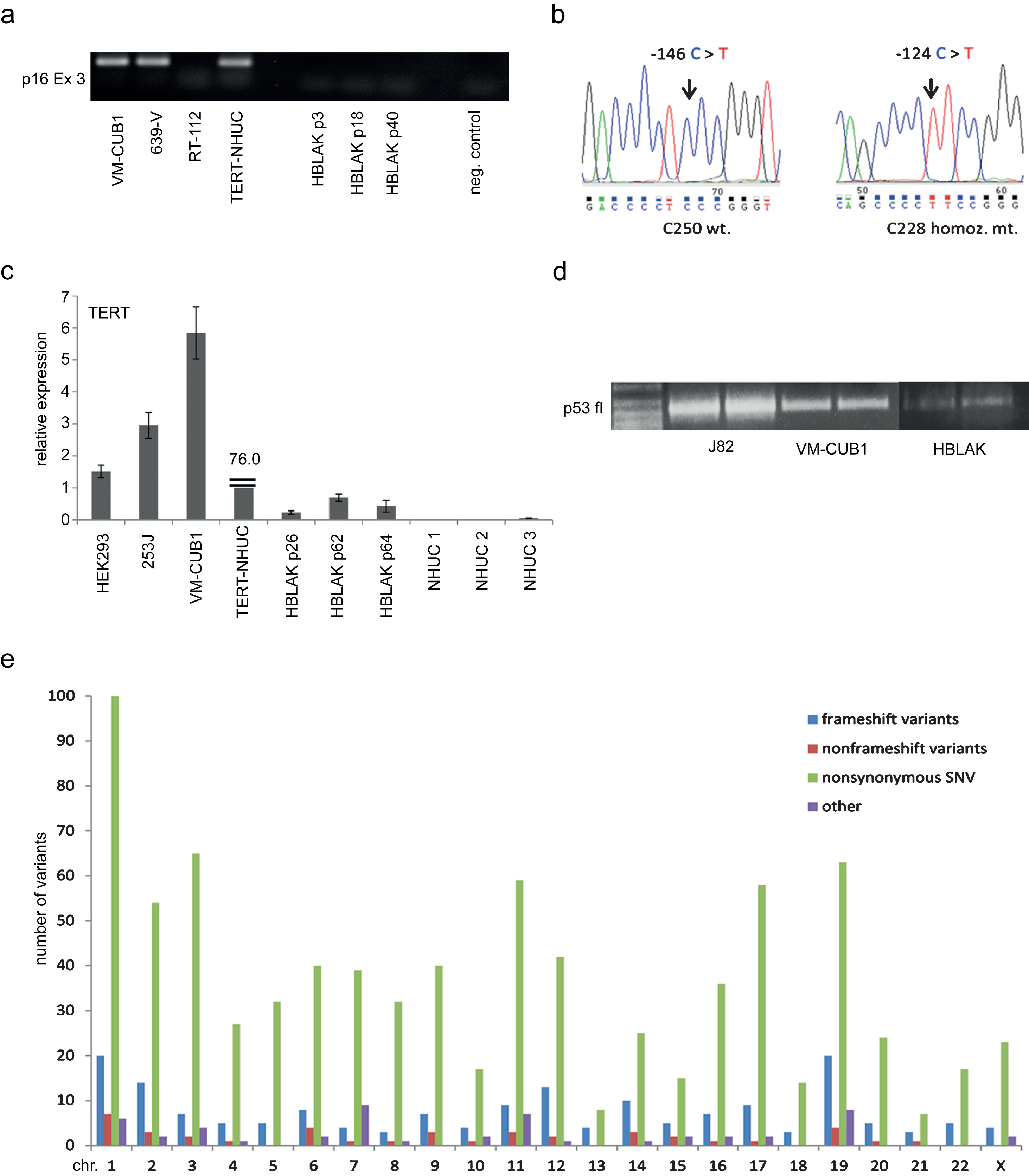
Fig.4
Sensitivity of HBLAK cells towards cytotoxic treatment. (a) Molecular response to cellular stress induced by UV radiation (40 J/m2) or the indicated concentrations of Cisplatin, Etoposide and Doxorubicin (all 24 h) by western blot analysis of p53 and p21. α-Tubulin was detected as a loading control. The UC cell line 5637 and cells transfected with a p21 plasmid served as positive controls for p53 and p21, respectively. (b) Sensitivity of HBLAK towards treatment with Cisplatin for 72 h determined by MTT assay. Mean results from triplicate assays are shown. IC50 concentrations ranged around 5 μM. For comparison TERT-NHUC cells and three different passages of a primary NHUC cultured were treated accordingly.
Table 1
Primer sequences and annealing temperature used for real time qPCR or amplicon sequencing
| Gene name | Sequence 5′-3′ | Annealing T [°C] | Use |
| TBP | Fwd: CAACAGCCTGCCACCTTA | 55 | qRT-PCR |
| Rev: GAATAGGCTGTGGGGTCAGT | |||
| CK14 | Fwd: GCGCACCATGCAGAACCTG | 59 | qRT-PCR |
| Rev: CCTCCACGCTGCCAATCATC | |||
| CK20 | Fwd: GAACTGAGGTTCAACTAACGGAGC | 57 | qRT-PCR |
| Rev: TGGCTAACTGGCTGCTGTAACG | |||
| UPK2 | Fwd: GACAGCCACTGAGTCCAGCAG | 59 | qRT-PCR |
| Rev: AGCACCGTGATGACCACCATG | |||
| p16INK4 | Fwd: CAACGCACCGAATAGTTACG | 55 | qRT-PCR |
| Rev: AGCACCACCAGCGTGTC | |||
| TERT | Fwd: CGGAAGAGTGTCTGGAGCAA | 56 | qRT-PCR |
| Rev: GGATGAAGCGGAGTCTGGA | |||
| p53 | Fwd: GTGACACGCTTCCCTGGAT | 58 | Sequencing |
| Rev: GAGTTCCAAGGCCTCATTCA | |||
| TERT | Fwd: CACCCGTCCTGCCCCTTCACCTT | 56 | Sequencing |
| Rev: GGCTTCCCACGTGCGCAGCAGGA | |||
| p16INK4 Exon 3 | Fwd: CCGGTAGGGACGGCAAGAGA | 60 | Sequencing |
| Rev: CTGTAGGACCCTCGGTGACTG |
Table 2
Results of STR profiling for HBLAK cells
| AM | XY |
| D3S1358 | 17–18 |
| D19S433 | 14–15 |
| D2S1338 | 19–25 |
| D22S1045 | 13–16 |
| D16S539 | 11–12 |
| D18S51 | 12–16 |
| D1S1656 | 15–18 |
| D10S1248 | 13–14 |
| D2S441 | 10–11 |
| THO1 | 6–7 |
| VWA | 13–18 |
| D21S11 | 30–33.2 |
| D12S391 | 17–21 |
| D8S1179 | 11–13 |
| FGA | 20–22 |
| SE33 | 24.2–27.2 |
| D7S820 | 10–12 |
| CSF1PO | 12 |
| D13S317 | 10–12 |
| TPOX | 8–11 |
| D5S818 | 11 |
Table 3
Chromosomal alterations of HBLAK cells detected by array CGH
| Gains | Cytoband | Start | Stop | Gain |
| chr1 | p31.1 | 73038943 | 75148461 | 0,517485 |
| chr4 | q32.1-q34.3 | 156657280 | 182136039 | 0,526251 |
| chr5 | p15.33-p11 | 132146 | 45908585 | 0,532254 |
| chr5 | q11.1-q35.3 | 49738768 | 180629412 | 0,522551 |
| chr6 | p21.32 | 32558677 | 32659387 | 0,853835 |
| chr9 | p24.3-p11.2 | 194193 | 43776424 | 0,265615 |
| chr9 | q12-q21.31 | 66245109 | 80376504 | 0,345433 |
| chr9 | q21.33-q33.3 | 87170456 | 126944819 | 0,327093 |
| chr9 | q34.2-q34.3 | 135139065 | 140138805 | 0,317787 |
| chr17 | q21.31 | 41527705 | 41700815 | 0,923838 |
| chr20 | p13-p11.1 | 15778 | 26142459 | 0,623331 |
| chr20 | q11.21-q13.33 | 29297270 | 62374945 | 0,599131 |
| Losses | Cytoband | Start | Stop | Loss |
| chr1 | q44 | 246794552 | 246875075 | –2,574785 |
| chr9 | p22.2 | 16684298 | 18368550 | –0,776048 |
| chr9 | p21.3 | 21892814 | 21957607 | –5,194027 |
| chr9 | p21.3 | 21958041 | 22076857 | –0,563264 |
| chr9 | q21.31-q21.33 | 80427340 | 87136770 | –0,559369 |
| chr9 | q33.3-q34.2 | 126955828 | 135063066 | –0,608385 |
| chr16 | p13.3 | 3427264 | 3687746 | –0,630671 |
| chr22 | q13.1-q13.31 | 38996009 | 42793438 | –0,683451 |
Table 4
Summary of genetic variants detected by exome sequencing of HBLAK cells
| Variant type | Quantity |
| Frameshift deletion | 18 |
| Frameshift insertion | 156 |
| Nonframeshift deletion | 13 |
| Nonframeshift insertion | 1 |
| Nonframeshift substitution | 27 |
| Nonsynonymous SNV | 836 |
| Stopgain | 11 |


