
The Qualification of an Enrichment Biomarker for Clinical Trials Targeting Early Stages of Parkinson’s Disease
Abstract
As therapeutic trials target early stages of Parkinson’s disease (PD), appropriate patient selection based purely on clinical criteria poses significant challenges. Members of the Critical Path for Parkinson’s Consortium formally submitted documentation to the European Medicines Agency (EMA) supporting the use of Dopamine Transporter (DAT) neuroimaging in early PD. Regulatory documents included a comprehensive literature review, a proposed analysis plan of both observational and clinical trial data, and an assessment of biomarker reproducibility and reliability. The research plan included longitudinal analysis of the Parkinson Research Examination of CEP-1347 Trial (PRECEPT) and the Parkinson’s Progression Markers Initiative (PPMI) study to estimate the degree of enrichment achieved and impact on future trials in subjects with early motor PD. The presence of reduced striatal DAT binding based on visual reads of single photon emission tomography (SPECT) scans in early motor PD subjects was an independent predictor of faster decline in UPDRS Parts II and III as compared to subjects with scans without evidence of dopaminergic deficit (SWEDD) over 24 months. The EMA issued in 2018 a full Qualification Opinion for the use of DAT as an enrichment biomarker in PD trials targeting subjects with early motor symptoms. Exclusion of SWEDD subjects in future clinical trials targeting early motor PD subjects aims to enrich clinical trial populations with idiopathic PD patients, improve statistical power, and exclude subjects who are unlikely to progress clinically from being exposed to novel test therapeutics.
BACKGROUND
Parkinson’s disease (PD) is the second most common neurodegenerative disease affecting 2% to 3% of people over 65 years of age [1] and has been recently reported to be the most rapidly growing neurological disease [2, 3]. Although symptomatic treatments are available for the disabling motor and some non-motor symptoms of the disease, these treatments have not been shown to slow the progression of the underlying disease pathology. Thus, the development of drugs that modify the course of the disease is an important aim being pursued by industry, academia and non-profit organizations. Recent advances in PD research include the discovery and development of promising targets [4, 5]. Yet drug developers face many challenges in advancing new treatments aimed at disease modification [6– 8]. PD is linked to toxic accumulation of aggregated forms of the normal synaptic protein, α-synuclein, in the brain and peripheral nervous system. α-synuclein is the major component of Lewy bodies and Lewy neurites, the hallmark pathology of PD. Rare point mutations and the presence of extra copies of the synuclein gene result in autosomal dominant PD [9, 10]. Novel therapeutic development efforts are underway targeting α-synuclein [11] and the two most common genetic causes of PD, glucocerebrosidase (GBA) deficiency [12], and mutated forms of leucine rich repeat kinase 2 (LRRK2) [4, 13]. Cellular processes considered to be valid targets for drug development include the molecular processes leading to α-synuclein accumulation (synthesis, aggregation and clearance), as well as mitochondrial dysfunction and neuroinflammation [14]. Recent PD clinical trials have focused on the following therapeutic targets: calcium flux (Isradipine), brain levels of the antioxidant, urate (Inosine), the c-Abl kinase (Nilotinib), and insulin signalling via the GLP-1 receptor (Exenatide) (reviewed by [5, 6]).
Regardless of molecular target, clinical studies of agents intended to slow disease progression in PD could benefit from application of biomarkers to help enrich clinical trial populations with the most appropriate patients for the study. This review highlights the formal qualification opinion issued by the European Medicines Agency (EMA) for the use of molecular neuroimaging of the dopamine transporter (DAT) as an enrichment biomarker for use in PD clinical trials. A manuscript focused on highlighting the data analyses and regulatory aspects of this project was recently published [62].
The need to identify clinical trial populations using biomarkers and pathology
There is an emerging consensus among PD researchers that, as with other neurodegenerative diseases, disease modifying treatments are most likely to be effective at an early stage of the disease, when delaying progression and disability will be most impactful. In fact, we have learned that motor symptoms are only apparent after profound loss of dopaminergic neurons in PD [15]. It can, however, be challenging to diagnose PD, especially in early stage disease when other neurological disorders can present with similar signs and symptoms [16, 17]. Identifying the right participants to enroll in clinical trials is critically important for early intervention and is particularly challenging based on clinical criteria alone [18]. One approach to identify the appropriate participants is to use biomarkers, particularly in light of increasing emphasis on precision medicine. Imaging biomarkers have been employed in PD clinical trials quite extensively primarily using dopaminergic ligands. At present there is strong interest in developing novel molecular imaging tracers, particularly those such as synuclein radiotracers [19, 20] that will enable confirmation of target engagement.
Defining enrichment
The use of biomarkers to enrich the clinical trial population, leading to a more homogenous treatment cohort, is likely to address this challenge. There are many examples to learn from; for example, in therapeutic development in Alzheimer’s disease [23, 24]. A biomarker may be applied as an inclusion or exclusion criterion to distinguish the targeted indication from another condition with a similar clinical presentation but a different cause. For example, in contemporary Alzheimer’s disease trials, an amyloid PET scan has been used to enrich the clinical trial population by excluding subjects without fibrillar amyloid deposits in their brain [55]. This results in a high screen failure rate but provided a more homogeneous population by excluding subjects whose memory impairment is unlikely to be caused by amyloid pathology.
Regulatory authorities define “enrichment” as “the prospective use of any patient characteristic to select a study population in which detection of a drug effect (if one is in fact present) is more likely than it would be in an unselected population” [21]. Health authorities in the US and Europe have encouraged the use of enrichment biomarkers in clinical trials to support drug development [22, 23– 25]. Biomarkers for enrichment are aimed at identifying a population of trial participants who are more likely to progress or respond to treatment. An example is the use of low baseline hippocampal volume as an enrichment biomarker to identify predementia subjects more likely to progress in a clinical trial for early Alzheimer’s disease [26].
Ensuring consensus and broad adoption: biomarker qualification
The selection of biomarkers for use in clinical trials conventionally merits a confidential discussion between the regulator and the study’s sponsor [5]. Regulators also offer biomarker qualification, a public and formal review process to accelerate the adoption of drug development tools across targets and sponsors [23, 27, 28]. Regulatory qualification provides a mechanism by which the regulators define and endorse a biomarker for use in a clearly defined way to support drug development. The development of a strategic pathway for biomarker qualification provides a uniform, consistent method for advancing specific biomarkers for specific contexts. The explicit methods of validation vary depending on the process mapped out, yet regulatory guidance on qualification affords clarity and transparency (21st Century Cures). The “context of use” refers to the specific application of the biomarker in drug development. Once the regulators have qualified a particular biomarker for a specified context of use, this biomarker generally does not require further regulatory review and approval for use by sponsors in drug development [28, 29].
Dopamine transporter imaging as an enrichment biomarker
For therapeutic trials in patients with PD, drug developers have an urgent need for effective biomarkers to enrich study patients and assess target engagement, disease prognosis, and clinical response [7– 30]. There is not yet a reliable biofluid or imaging biomarker of α-synuclein to enrich PD clinical trial populations [19, 20, 30, 57]. However, neuroimaging biomarkers, particularly ones probing the dopaminergic system in the brain, have been employed in PD clinical trials for many years [31, 32]. The dopamine transporter (DAT), a transporter protein that is located on the presynaptic terminal of dopaminergic neurons, can be imaged with specific molecular radiotracers [33]. Reductions of DAT radiotracer binding (DAT deficit) correlate with the disease duration from early stages of the disease [34], matching pathological findings from DAT immunohistochemistry [35]. Recent studies demonstrate DAT deficit may be augmented in synuclein mutation PD subjects [36].
The application of DAT in clinical trials can be used to detect the absence of DAT-deficiency, termed ‘scans without evidence of dopaminergic deficit’ (SWEDD) [37]. Subjects defined as ‘SWEDD’ had been identified at the end of specific clinical trials when unblinding the imaging results even though such subjects met clinically defined inclusion criteria for PD at the study recruitment phase. The Parkinson Progression Marker Initiative (PPMI) observational study elected to identify a specific cohort of SWEDD subjects at study onset and follow these subjects longitudinally [38]. Although the pathological basis for Parkinson-like symptoms in subjects identified as SWEDD remains uncertain and no correlative post-mortem data is yet available, data from multiple sources has indicated that motor deterioration in SWEDD subjects is much slower than in subjects whose scans show evidence of dopamine nerve terminal loss [39– 44]. Data to date when looked at collectively demonstrate that for cohorts enrolled within one year of diagnosis approximately 10 to 15% of subjects enrolled in PD clinical studies may lack imaging evidence of dopamine deficiency at baseline and that their disease progression will be significantly slower than those who do.
Figure 2 illustrates that the proportion of SWEDD subjects is greater in study populations closer to the date of diagnosis. As clinical trial sponsors aim to treat PD populations at earlier stages of the disease, the value of enrichment with DAT imaging therefore increases.
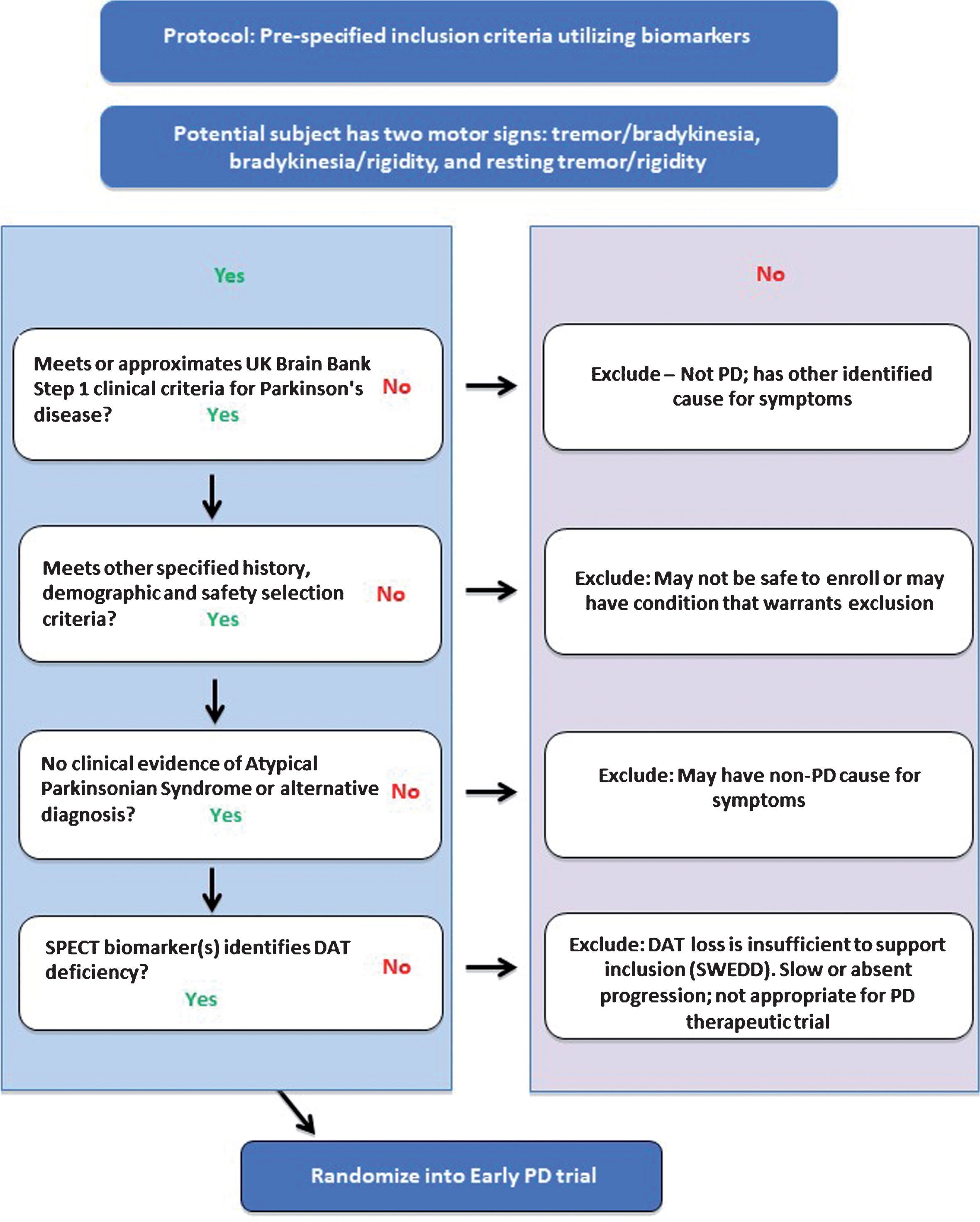
Fig.1
Proposed flow chart to apply DAT imaging as an enrichment biomarker in clinical trials targeting subjects with motor signs of early PD. Each of the four inclusion criteria / steps must be met for subjects to be successfully enrolled into the PD clinical trial. The clinical criteria must be met before subjects undergo DAT SPECT imaging (final step).
The radioligand (123I) β-CIT, developed as a selective tracer of the dopamine transporter, was shown to be useful in the evaluation of PD and has been used in several PD clinical research studies and trials. Fluoroalkyl analogues were subsequently introduced, which required shorter tracer uptake times and which were somewhat more selective [45]. One of these DAT imaging agents (the single photon emission tomography, SPECT, radiotracer Ioflupane or 123I FP-CIT) was developed further and subsequently, approved as a medical product by both FDA (DaTscan™) and EMA (DaTSCAN). It is available widely throughout the world. At present, this DAT imaging agent is approved for clinical use to assist in the evaluation of patients with suspected parkinsonian syndromes to distingusih PD from essential tremor [46].
Fig.2
The proportion of subjects with absence of DAT deficit in clinical trials increases with earlier stages of PD. A number of PD clinical studies have identified subjects defined as SWEDD by employing DAT imaging at baseline and follow up. The proportion of subjects defined as SWEDD decreases with the time since diagnosis for those in specific PD clinical studies targeting early stages of the disease. The proportion of SWEDD subjects is lower in studies enrolling subjects with longer duration of disease. As sponsors enroll PD subjects earlier in the disease (i.e., sooner after diagnosis), the value (benefit) of DAT imaging for enrichment increases. References for PD clinical studies: [38, 39, 41, 59– 61]. ELLDOPA: Levodopa; PRECEPT: Mixed Lineage Kinase Inhibitor CEP 1347; REAL-PD: Ropinirole; CALM-PD: Pramipexole; GPI1485: Immunophilin; PPMI: Parkinson’s Progression Markers Initiative.
![The proportion of subjects with absence of DAT deficit in clinical trials increases with earlier stages of PD. A number of PD clinical studies have identified subjects defined as SWEDD by employing DAT imaging at baseline and follow up. The proportion of subjects defined as SWEDD decreases with the time since diagnosis for those in specific PD clinical studies targeting early stages of the disease. The proportion of SWEDD subjects is lower in studies enrolling subjects with longer duration of disease. As sponsors enroll PD subjects earlier in the disease (i.e., sooner after diagnosis), the value (benefit) of DAT imaging for enrichment increases. References for PD clinical studies: [38, 39, 41, 59– 61]. ELLDOPA: Levodopa; PRECEPT: Mixed Lineage Kinase Inhibitor CEP 1347; REAL-PD: Ropinirole; CALM-PD: Pramipexole; GPI1485: Immunophilin; PPMI: Parkinson’s Progression Markers Initiative.](https://content.iospress.com:443/media/jpd/2019/9-3/jpd-9-3-jpd191648/jpd-9-jpd191648-g002.jpg)
The Critical Path for Parkinson’s (CPP) is a global public-private partnership to accelerate drug development in PD [47]. The Critical Path Institute in the United States and Parkinson’s UK jointly sponsor this collaboration. CPP is comprised of pharmaceutical companies, academic researchers, government agencies and advocacy research organizations collaborating to share data and development drug development tools for regulatory acceptance.
CPP has proposed the use of DAT imaging as a biomarker during the screening phase of clinical trials of novel PD therapies, to enrich the selected population, by excluding SWEDD subjects and thus provide a more homogeneous study population. Conducting a PD clinical study in such an enriched patient population, can optimize trial sample size, thereby reducing trial costs. Furthermore, such enrichment will direct the treatment to the intended patients for whom the evaluation of benefit and safety of the intervention is intended, without unnecessarily exposing others to therapeutic candidates.
In 2015 and 2016 respectively, the US FDA and the EMA issued letters of support for use of DAT imaging as a drug development tool as an enrichment biomarker in early PD clinical trials [48, 49]. The letters of support originated from the Critical Path Institute consortia, coalition Against Major Diseases (CAMD) and CPP. The purpose of these formal public letters issued is to communicate to the external community the growing evidence for the use of the biomarker and encourage sponsors to collect and share relevant data supporting the biomarker use in ongoing and prospective clinical trials.
Qualification of DAT imaging as an enrichment biomarker in early PD trials
The full qualification document including details of the data analyses and imaging methodology recommendations can be found on the EMA website [50]. The analyses that supported the qualification opinion has been published [50, 62] and was accepted for publication prior to the final regulatory qualification. The qualification document contains the operational details required to employ DAT SPECT imaging in a clinical trial of patients with early motor PD. The methodology outlining the foreseen use is based on visual reads and fully aligns with recommendations for use of DAT imaging in US [33] and EU [51]. Figure 1 illustrates how this enrichment biomarker is recommended for clinical trials based on the proposed context of use.
Biomarker qualification requires a clear definition of the application, stage of drug development and the population studied. The context-of-use of the PD imaging biomarker qualified by the EMA is outlined below:
Context-of-use (COU) statement
General Area
Enrichment biomarker for clinical trials in early motor Parkinson’s disease.
Target population for use
Patients with early motor PD, defined by the UK Brain Bank Criteria as outlined below:
Having at least two of the following: resting tremor, bradykinesia, rigidity (must have either resting tremor or bradykinesia); OR either asymmetric resting tremor or asymmetric bradykinesia. Based on above criteria, combinations could include: resting tremor/bradykinesia, bradykinesia/rigidity, and resting tremor/rigidity.
• Symptom(s) or signs may include bradykinesia, a 4– 6 Hz resting tremor, muscle rigidity, or postural instability not caused by primary visual, vestibular, cerebellar or proprioceptive dysfunction.
• Hoehn and Yahr Stage I or II at baseline. Although postural instability is a common feature in PD, based on the inclusion criterion of Hoehn and Yahr Stage I or II, postural instability would not be expected in the target population.
Stage of drug development for use
All clinical stages of early PD drug development, including proof of concept, dose-ranging through to confirmatory clinical trials. This is not intended for candidate therapies for more advanced stages of PD such as drugs to treat levodopa induced dyskinesias.
The predictive value of DAT imaging in a realistic clinical trial setting
To support the EMA biomarker qualification, CPP collected, standardized, and integrated data from two clinical studies that employed DAT imaging namely, PPMI [38] and PRECEPT [40, 52]. These studies had similar clinical inclusion criteria and target populations at baseline, with equivalent visual reads based on different DAT tracers. Two different versions of the clinical score were used to track disease progression (MDS-UPDRS part III in PPMI versus UPDRS part III in PRECEPT); these versions were homogenized to the MDS-UPDRS part III for analyses of the integrated data.
PRECEPT and PPMI represent uniquely rich cohorts of well characterized subjects with early stage PD where subject-level data was available to CPP for analyses to support regulatory science goals. Both studies include similar patient populations from multicenter global sites with application of DAT imaging at baseline and long term clinical follow up. The use of both observational and randomized clinical trial (RCT) populations aided in the confidence of predictability of the results to prospective trial populations that align with the proposed context-of-use.
Additional considerations worth noting are that SWEDD subjects in PPMI learned their imaging results after the baseline scans were interpreted. Subjects were offered the opportunity to remain enrolled in PPMI to advance the understanding of PD and many opted to continue. In PRECEPT, all subjects and clinicians remained blinded throughout the duration of the clinical trial. SWEDD status was not defined in PRECEPT until study completion.
The analysis of the integrated patient-level data from both studies took careful account of the heterogeneity in specific variables. This integrated analysis confirmed that, as a subpopulation, subjects who had no evidence of DAT deficit (SWEDD subjects) progressed more slowly than the subpopulation of those classified with DAT deficiency. The change from baseline of MDS-UPDRS part III at 24 months was 4.28 points (90% CI: 3.45, 5.08) for those with DAT deficit, and 1.12 points (90% CI: – 0.98, 3.1) for SWEDD subjects [53].
Patient-level longitudinal data of 672 early stage PD subjects in the PPMI observational study and the PRECEPT clinical trial were utilized in a linear mixed effects model analysis. The rate of worsening in the motor scores between subjects with and without DAT deficit was different both statistically and clinically. Since the application of the biomarker is intended for clinical trial enrichment based on predictions of continuous disease progression, examination of diagnostic sensitivity, specificity and predictive values was not required for the analyses as this would lack applicability in this context. Clinical trial simulations were carried out in order to understand the impact of enrichment on sample sizes. For illustration, a trial simulation analysis was based on the PPMI and PRECEPT data and simulated a 24-month clinical trial with or without enrichment with the DAT imaging biomarker for subjects recruited soon after diagnosis. This analysis showed that using the DAT imaging to include only subjects with confirmed dopamine deficit resulted in a potential gain in statistical power equivalent to a 24% reduction in sample size and reduced the heterogeneity of the study population. DAT imaging would thereby decrease the exposure of non-PD patients to the therapeutic intervention and increase the chance of successfully establishing whether an experimental medicine is effective, regardless of the drug’s specific mechanism of action.
Because the enrichment biomarker excludes subjects that show minimal to no decline in disease progression, this gain in statistical power is not specific to a therapeutic target but is generally applicable to clinical trials in early motor PD soon after diagnosis. It is important to note that the qualification is for the biomarker itself rather than a specific radiotracer or commercial product.
Limitations of the biomarker
The limitations to the use of this imaging biomarker include: 1) Any condition in which there is loss of dopaminergic nerve terminals in the striatum, such as progressive supranuclear palsy and multiple system atrophy, will show reduced signal using DAT imaging [32]. CPP is not proposing DAT imaging for use as a diagnostic biomarker to differentiate the various forms of degenerative Parkinsonism. 2) DAT imaging in the present context is not to be applied as a biomarker of target engagement in PD trials. DAT is not specific to α-synuclein and decreased DAT levels cannot be equated with the presence of synuclein pathology in the brain. 3) The operational use of the biomarker should be managed centrally in accordance with the FDA guidance on imaging endpoints for clinical trials [54], including the use of trained central readers and 4) the diagnostic criteria that are included in the qualification will need to be refined to align with new criteria recognizing multiple stages of early disease [55].
CONCLUSIONS
The EMA qualification opinion of DAT imaging for PD clinical trials represents the first biomarker qualified for use for PD clinical trials as a drug development tool and is aimed to encourage the advancement of therapies targeting early stages of the disease. The current target population is early motor PD, from 1 to 2 years of diagnosis. Given emerging data demonstrating that accurate identification of PD pathology poses significant challenges, particularly at early stages [16] new biomarkers are urgently needed for identifying the right subjects to recruit into clinical trials. Imaging of synucleinopathies will be a step forward for the field [19, 33] as well as the discovery and validation of novel biomarkers for use in clinical trials [56]. Continued work is needed to refine this tool as an enrichment marker in future trials of patients in the prodromal and even premotor stages of PD [57]. In addition, the use of longitudinal data from PPMI, combined with interventional data from PRECEPT, a legacy clinical trial conducted nearly 15 years ago, demonstrates that data sharing from past and ongoing PD clinical studies can optimize the opportunity of success of future PD clinical trials. The overall strategy aligns with recommendations for the future vision of PD aimed at biomarker driven phenotypes as the key strategy to enabling precision medicine [58].
Funding sources
Parkinson’s UK, Abbvie, Biogen, Eli Lilly, GE Healthcare, GSK, Lundbeck, Merck Sharp and Dohme, Pfizer, Denali, Sanofi, Takeda and UCB.
CONFLICTS OF INTEREST
Authors who are employed at pharmaceutical or diagnostic companies have respective conflicts of interest based on their affiliations.
MT, SV, DS, KR, DC, AR, JG, DD, SI have no conflicts of interest.
JS: Consultant (Biogen, Roche, LikeMinds, Life Molecular Imaging); Equity (Invicro).
DG: received grant funding from the Neurosciences Foundation, Michael’s Movers, and Parkinson’s UK, and honoraria from Bial Pharmaceuticals and GE Healthcare.
GS: received grant funding from NIH, Michael J Fox Foundation and honoraria from Biogen.
KM: received funding from the The Michael J. Fox Foundation, the US Department of Defense and is employed by Invicro and has received consultant fees from Pfizer, GE Healthcare, Lilly, BMS, Piramal, Biogen, Prothena, Roche, Neuropore,US Worldmeds, Neurophage, UCB, Oxford Biomedica, Lysosomal Therapetic, Inc, Neuroderm, Denali and the Michael J. Fox Foundation.
ACKNOWLEDGMENTS
The Critical Path Institute’s Critical Path for Parkinson’s (CPP) Consortium is funded by Parkinson’s UK and the following industry members: Abbvie, Biogen, Eli Lilly, GE Healthcare, Lundbeck, Merck Sharp and Dohme, Pfizer, Roche, Denali, Sanofi, GSK, Takeda and UCB. We also acknowledge additional CPP member organizations, including Parkinson’s Disease Foundation, Michael J. Fox Foundation, Davis Phinney Foundation, The Cure Parkinson’s Trust, University of Oxford, University of Cambridge, Newcastle University, University of Glasgow, Radboud University, NINDS, FDA, and EMA.
The Critical Path Institute is supported in part by grant number 5U18FD005320 from the Food and Drug Administration. CPP acknowledges the strong support of FDA liaison’s Dr. Gerald Podskalny and Dr. Billy Dunn, Division of Neurology Products.
CPP recognizes Teva for contributing the PRECEPT patient-level data, the PRECEPT study investigators for their role in leading the study; Institute of Neurodegenerative Disorders for their efforts in analysing the imaging results from both PRECEPT and PPMI; and the Michael J. Fox Foundation for funding of PPMI. Data were obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (http://www.ppmi-info.org/data). PPMI – a public-private partnership – is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners, including AbbVie, Avid, Biogen, Bristol-Myers Squibb, Convance, GE Healthcare, Genentech, GSK, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Sanofi Genzyme, Servier, TEVA, UCB, and Golub Capital. For up-to-date information on the study, visit http://www.ppmi-info.org.
The views expressed in this publication by the authors from the European Medicines Agency are the personal views of the authors and may not be understood nor quoted as being made on behalf of or reflecting the position of EMA or one of its committees or working parties or any of the national agencies.
CPP recognizes contributions of the individual members of the CPP imaging biomarker team and the CPP modelling and simulation team. CPP recognizes the consortium’s scientific advisors: Karl Kieburtz, Charles Venuto, E. Ray Dorsey, Bastiaan R. Bloem. The CPP consortium recognizes Dr. Ira Shoulson for his leadership as principal investigator of the PRECEPT study in addition to the Parkinson’s Study Group. The following individuals are acknowledged for their contributions to the project: Patricia Cole, Igor Grachev, Grace Crotty, Volker Kern, Steve Arneric, Mark Frasier, Alyssa Reimer and Jamie Eberling.
REFERENCES
[1] | Reeve A , Simcox E , Turnbull D ((2014) ) Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res Rev 14: , 19–30. |
[2] | Dorsey ER , Sherer T , Okun MS , Bloem BR ((2018) ) The emerging evidence of the Parkinson pandemic. J Parkinsons Dis 8: , S3–S8. |
[3] | Rocca WA ((2018) ) The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol 17: , 928–929. |
[4] | Sardi SP , Cedarbaum JM , Brundin P ((2018) ) Targeted therapies for Parkinson’s disease: From genetics to the clinic: Targeted Therapies for PD. Mov Disord 33: , 684–696. |
[5] | Kieburtz K , Katz R , Olanow C. Warren ((2017) ) New drugs for Parkinson’s disease: The regulatory and clinical development pathways in the United States. Mov Disord 33: , 920–927. |
[6] | Lang AE , Espay AJ ((2018) ) Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations: Disease modification in PD. Mov Disord 33: , 660–677. |
[7] | Cedarbaum JM ((2018) ) Parkinson’s disease and “proof of concept” clinical trials. Mov Disord 33: , 697–700. |
[8] | Stoessl AJ ((2017) ) Challenges and unfulfilled promises in Parkinson’s disease. Lancet Neurol 16: , 866–867. |
[9] | Konno T , Ross OA , Puschmann A , Dickson DW , Wszolek ZK ((2016) ) Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord 22: , S1–S6. |
[10] | Olgiati S , Thomas A , Quadri M , Breedveld GJ , Graafland J , Eussen H , Douben H , de Klein A , Onofrj M , Bonifati V ((2015) ) Early-onset parkinsonism caused by alpha-synuclein gene triplication: Clinical and genetic findings in a novel family. Parkinsonism Relat Disord 21: , 981–986. |
[11] | Kingwell K ((2017) ) Zeroing in on neurodegenerative α-synuclein. Nat Rev Drug Discov 16: , 371–373. |
[12] | Siebert M , Sidransky E , Westbroek W ((2014) ) Glucocerebrosidase is shaking up the synucleinopathies. Brain 137: , 1304–1322. |
[13] | Zimprich A , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Müller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , Gasser T ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |
[14] | Valera E , Masliah E ((2016) ) Therapeutic approaches in Parkinson’s disease and related disorders. J Neurochem 139: , 346–352. |
[15] | Hornykiewicz O ((1998) ) Biochemical aspects of Parkinson’s disease. Neurology 51: , S2–S9. |
[16] | Adler CH , Beach TG , Hentz JG , Shill HA , Caviness JN , Driver-Dunckley E , Sabbagh MN , Sue LI , Jacobson SA , Belden CM , Dugger BN ((2014) ) Low clinical diagnostic accuracy of early vs advanced Parkinson disease clinicopathologic study. Neurology 83: , 406–412. |
[17] | Rizzo G , Copetti M , Arcuti S , Martino D , Fontana A , Logroscino G ((2016) ) Accuracy of clinical diagnosis of Parkinson disease A systematic review and meta-analysis. Neurology 86: , 566–576. |
[18] | Beach TG , Adler CH ((2018) ) Importance of low diagnostic accuracy for early Parkinson’s disease: Low diagnostic accuracy for early PD. Mov Disord 33: , 1551–1554. |
[19] | Eberling JL , Dave KD , Frasier MA ((2013) ) α-synuclein imaging: A critical need for Parkinson’s disease research. J Parkinsons Dis 3: , 565–567. |
[20] | Shah M , Seibyl J , Cartier A , Bhatt R , Catafau AM ((2014) ) Molecular imaging insights into neurodegeneration: Focus on alpha-synuclein radiotracers. J Nucl Med 55: , 1397–1400. |
[21] | FDA ((2012) ) Guidance for Industry - Enrichment Strategies for Clinical Trials to Support Approval of Human Drugs and Biological Products. December 2012. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM332181.pdf. Accessed January 17, 2019. FDA (2019) Enrichment Strategies for Clinical Trials to Support Determination of Effectiveness of Human Drugs and Biological Products Guidance for Industry. March, 2019. |
[22] | FDA ((2018) ) Early Alzheimer’s Disease: Developing Drugs for Treatment; Guidance for Industry, Food and Drug Administration, Silver Spring, MD. Food and Drug Administration; 2018. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM596728.pdf. |
[23] | Isaac M , Gispen-de Wied C ((2015) ) CNS biomarkers: Potential from a regulatory perspective. Eur Neuropsychopharmacol 25: , 1003–1009. |
[24] | Jack CR , Bennett DA , Blennow K , Carrillo MC , Dunn B , Haeberlein SB , Holtzman DM , Jagust W , Jessen F , Karlawish J , Liu E , Molinuevo JL , Montine T , Phelps C , Rankin KP , Rowe CC , Scheltens P , Siemers E , Snyder HM , Sperling R , Elliott C , Masliah E , Ryan L , Silverberg N ((2018) ) NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement 14: , 535–562. |
[25] | Haas M , Mantua V , Haberkamp M , Pani L , Isaac M , Butlen-Ducuing F , Vamvakas S , Broich K ((2015) ) The European Medicines Agency’s strategies to meet the challenges of Alzheimer disease. Nat Rev Drug Discov 14: , 221–222. |
[26] | Hill DLG , Schwarz AJ , Isaac M , Pani L , Vamvakas S , Hemmings R , Carrillo MC , Yu P , Sun J , Beckett L , Boccardi M , Brewer J , Brumfield M , Cantillon M , Cole PE , Fox N , Frisoni GB , Jack C , Kelleher T , Luo F , Novak G , Maguire P , Meibach R , Patterson P , Bain L , Sampaio C , Raunig D , Soares H , Suhy J , Wang H , Wolz R , Stephenson D ((2014) ) Coalition Against Major Diseases/European Medicines Agency biomarker qualification of hippocampal volume for enrichment of clinical trials in predementia stages of Alzheimer’s disease. Alzheimers Dement 10: , 421–429.e3. |
[27] | Leptak C , Menetski JP , Wagner JA , Aubrecht J , Brady L , Brumfield M , Chin WW , Hoffmann S , Kelloff G , Lavezzari G ((2017) ) What evidence do we need for biomarker qualification? Sci Transl Med 9: , eaal4599. |
[28] | Manolis E , Koch A , Deforce D , Vamvakas S ((2015) ) The European Medicines Agency experience with biomarker qualification. In Clinical Proteomics: Methods and Protocols, Vlahou A , Makridakis M , eds. Springer New York, New York, NY. pp. 255–272 . |
[29] | Amur S , LaVange L , Zineh I , Buckman-Garner S , Woodcock J ((2015) ) Biomarker qualification: Toward a multiple stakeholder framework for biomarker development, regulatory acceptance, and utilization. Clin Pharmacol Ther 98: , 34–46. |
[30] | Merchant KM , Cedarbaum JM , Brundin P , Dave KD , Eberling J , Espay AJ , Hutten SJ , Javidnia M , Luthman J , Maetzler W , Menalled L , Reimer AN , Stoessl AJ , Weiner DM ((2019) ) A proposed roadmap for Parkinson’s disease proof of concept clinical trials investigating compounds targeting alpha-synuclein. J Parkinsons Dis 9: , 31–61. |
[31] | Brooks DJ , Pavese N ((2011) ) Imaging biomarkers in Parkinson’s disease. Prog Neurobiol 95: , 614–628. |
[32] | Brooks DJ ((2016) ) Molecular imaging of dopamine transporters. Ageing Res Rev 30: , 114–121. |
[33] | Djang DSW , Janssen MJR , Bohnen N , Booij J , Henderson TA , Herholz K , Minoshima S , Rowe CC , Sabri O , Seibyl J , Van Berckel BNM , Wanner M ((2012) ) SNM practice guideline for dopamine transporter imaging with 123I-Ioflupane SPECT 1.0. J Nucl Med 53: , 154–163. |
[34] | Benamer TS , Patterson J , Grosset DG , Booij J , de Bruin K , van Royen E , Speelman JD , Horstink MH , Sips HJ , Dierckx RA , Versijpt J , Decoo D , Van Der Linden C , Hadley DM , Doder M , Lees AJ , Costa DC , Gacinovic S , Oertel WH , Pogarell O , Hoeffken H , Joseph K , Tatsch K , Schwarz J , Ries V ((2000) ) Accurate differentiation of parkinsonism and essential tremor using visual assessment of [123I]-FP-CIT SPECT imaging: The [123I]-FP-CIT study group. Mov Disord 15: , 503–510. |
[35] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: , 2419–2431. |
[36] | Koros C , Simitsi A , Prentakis A , Beratis I , Papadimitriou D , Kontaxopoulou D , Fragkiadaki S , Papagiannakis N , Seibyl J , Marek K , Papageorgiou SG , Trapali XG , Stamelou M , Stefanis L ((2018) ) 123I-FP-CIT SPECT [(123) I-2β-carbo-methoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl) nortropane single photon emission computed tomography] Imaging in a p.A53T α-synuclein Parkinson’s disease cohort versus Parkinson’s disease: 123I-FP-CIT imaging in a P.A53T PD cohort. Mov Disord 33: , 1734–1739. |
[37] | Marek K , Jennings D , Seibyl J ((2003) ) Single photon emission tomography and dopmaim transporter imaging in Parkinson’s disease. Adv Neurol 91: , 183–191. |
[38] | Marek K , Jennings D , Lasch S , Siderowf A , Tanner C , Simuni T , Coffey C , Kieburtz K , Flagg E , Chowdhury S , Poewe W , Mollenhauer B , Klinik P-E , Sherer T , Frasier M , Meunier C , Rudolph A , Casaceli C , Seibyl J , Mendick S , Schuff N , Zhang Y , Toga A , Crawford K , Ansbach A , De Blasio P , Piovella M , Trojanowski J , Shaw L , Singleton A , Hawkins K , Eberling J , Brooks D , Russell D , Leary L , Factor S , Sommerfeld B , Hogarth P , Pighetti E , Williams K , Standaert D , Guthrie S , Hauser R , Delgado H , Jankovic J , Hunter C , Stern M , Tran B , Leverenz J , Baca M , Frank S , Thomas C-A , Richard I , Deeley C , Rees L , Sprenger F , Lang E , Shill H , Obradov S , Fernandez H , Winters A , Berg D , Gauss K , Galasko D , Fontaine D , Mari Z , Gerstenhaber M , Brooks D , Malloy S , Barone P , Longo K , Comery T , Ravina B , Grachev I , Gallagher K , Collins M , Widnell KL , Ostrowizki S , Fontoura P , Ho T , Luthman J , Brug M van der , Reith AD , Taylor P ((2011) ) The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 95: , 629–635. |
[39] | Parkinson Study Group ((2007) ) Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 69: , 1480. |
[40] | Marek K , Seibyl J , Eberly S , Oakes D , Shoulson I , Lang AE , Hyson C , Jennings D ((2014) ) Longitudinal follow-up of SWEDD subjects in the PRECEPT Study. Neurology 82: , 1791–1797. |
[41] | Whone AL , Watts RL , Stoessl AJ , Davis M , Reske S , Nahmias C , Lang AE , Rascol O , Ribeiro MJ , Remy P , Poewe WH , Hauser RA , Brooks DJ ; REAL-PET Study Group ((2003) ) Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol 54: , 93–101. |
[42] | Erro R , Schneider SA , Stamelou M , Quinn NP , Bhatia KP ((2016) ) What do patients with scans without evidence of dopaminergic deficit (SWEDD) have? New evidence and continuing controversies. J Neurol Neurosurg Psychiatry 87: , 319–323. |
[43] | Seibyl J , Marek K , Zubal IG ((2010) ) The role of the core imaging laboratory in multicenter trials. Semin Nucl Med 40: , 338–346. |
[44] | Ravina B , Marek K , Eberly S , Oakes D , Kurlan R , Ascherio A , Beal F , Beck J , Flagg E , Galpern WR , Harman J , Lang AE , Schwarzschild M , Tanner C , Shoulson I ((2012) ) Dopamine transporter imaging is associated with long-term outcomes in Parkinson’s disease. Mov Disord 27: , 1392–1397. |
[45] | Abi-Dargham A , Gandelman MS , DeErausquin GA , Zea-Ponce Y , Zoghbi SS , Baldwin RM , Laruelle M , Chamey DS , Hoffer PB , Neumeyer JL , Innis RB ((1996) ) SPECT imaging of dopamine transporters in human brain with iodine-123-fluoroalkyl analogs of n-CIT. J Nucl Med 37: , 1129–1134. |
[46] | Bajaj N , Hauser RA , Grachev ID ((2013) ) Clinical utility of dopamine transporter single photon emission CT (DaT-SPECT) with (123I) ioflupane in diagnosis of parkinsonian syndromes. J Neurol Neurosurg Psychiatry 84: , 1288–1295. |
[47] | Stephenson D , Hu MT , Romero K , Breen K , Burn D , Ben-Shlomo Y , Bhattaram A , Isaac M , Venuto C , Kubota K , Little MA , Friend S , Lovestone S , Morris HR , Grosset D , Sutherland M , Gallacher J , Williams-Gray C , Bain LJ , Avilés E , Marek K , Toga AW , Stark Y , Forrest Gordon M , Ford S ((2015) ) Precompetitive data sharing as a catalyst to address unmet needs in Parkinson’s disease. J Parkinsons Dis 5: , 581–594. |
[48] | EMA ((2016) ) Letter of support for molecular imaging of the dopamine transporter biomarker as an enrichment biomarker for clinical trials for early Parkinson’s disease. September 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2016/10/WC500213914.pdf. |
[49] | FDA ((2015) ) Biomarker Letter of Support. Parkinson’s Imaging Biomarker. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/UCM605357.pdf. |
[50] | EMA, Committee for Medicinal Products for Human Use (CHMP) ((2018) ) Qualification opinion on dopamine transporter imaging as an enrichment biomarker for Parkinson’s disease clinical trials in patients with early Parkinsonian symptoms. May 2018. https://www.ema.europa.eu/documents/regulatory-procedural-guideline/qualification-opinion-dopamine-transporter-imaging-enrichment-biomarker-parkinsons-disease-clinical_en.pdf. |
[51] | Darcourt J , Booij J , Tatsch K , Varrone A , Vander Borght T , Kapucu ÖL , Någren K , Nobili F , Walker Z , Van Laere K ((2010) ) EANM procedure guidelines for brain neurotransmission SPECT using 123I-labelled dopamine transporter ligands, version 2. Eur J Nucl Med Mol Imaging 37: , 443–450. |
[52] | Parkinson Study Group ((2002) ) Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on parkinson disease progression. JAMA 287: , 1653–1661. |
[53] | Conrado DJ , Nicholas T , Tsai K , Macha S , Sinha V , Stone J , Corrigan B , Bani M , Muglia P , Watson IA , Kern VD , Sheveleva E , Marek K , Stephenson DT , Romero K , on behalf of the Critical Path for Parkinson’s (CPP) Parkinson’s Disease Modeling and Simulation Working Group ((2017) ) Dopamine transporter neuroimaging as an enrichment biomarker in early Parkinson’s disease clinical trials: A disease progression modeling analysis: Dopamine transporter imaging enrichment biomarker. Clin Transl Sci 11: , 63–70. |
[54] | FDA, U.S. Department of Health and Human Services. Clinical Trial Imaging Endpoint Process Standards Guidance for Industry https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM268555.pdf. |
[55] | Postuma RB , Gagnon J-F , Pelletier A , Montplaisir JY ((2017) ) Insomnia and somnolence in idiopathic RBD: A prospective cohort study. NPJ Parkinsons Dis 3: , 9. |
[56] | Chen-Plotkin AS , Albin R , Alcalay R , Babcock D , Bajaj V , Bowman D , Buko A , Cedarbaum J , Chelsky D , Cookson MR , Dawson TM , Dewey R , Foroud T , Frasier M , German D , Gwinn K , Huang X , Kopil C , Kremer T , Lasch S , Marek K , Marto JA , Merchant K , Mollenhauer B , Naito A , Potashkin J , Reimer A , Rosenthal LS , Saunders-Pullman R , Scherzer CR , Sherer T , Singleton A , Sutherland M , Thiele I , Walt D , West A , Zhang J ((2018) ) Finding useful biomarkers for Parkinson’s disease. Sci Transl Med 10: , eaam6003. |
[57] | Jennings D , Siderowf A , Stern M , Seibyl J , Eberly S , Oakes D , Marek K , for the PARS Investigators ((2017) ) Conversion to Parkinson disease in the PARS Hyposmic and Dopamine Transporter-Deficit Prodromal Cohort. JAMA Neurol 74: , 933. |
[58] | Espay AJ , Schwarzschild MA , Tanner CM , Fernandez HH , Simon DK , Leverenz JB , Merola A , Chen-Plotkin A , Brundin P , Kauffman MA , Erro R , Kieburtz K , Woo D , Macklin EA , Standaert DG , Lang AE ((2017) ) Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials: Molecular phenotyping For PD. Mov Disord 32: , 319–324. |
[59] | NINDS NET-PD Investigators ((2007) ) NINDS NET-PD Investigators (2007) A randomized clinical trial of coenzyme Q10 and GPI-1485 in early PD. Neurology 68: , 20–8. |
[60] | Fahn S ((1999) ) Parkinson disease, the effect of levodopa, and the ELLDOPA trial. Earlier vs Later L-DOPA. Arch Neurol 56: , 529–535. |
[61] | Parkinson Study Group ((2000) ) A randomized controlled trial comparing pramipexole with levodopa in early Parkinson’s disease: Design and methods of the CALM-PD Study. Clin Neuropharmacol 23: , 34–44. |
[62] | Romero K , Conrado D , Burton J , Nicholas T , Sinha V , Macha S , Ahamadi M , Cedarbaum J , Seibyl J , Marek K , Basseches P , Hill D , Somer E , Gallagher J , Dexter DT , Roach A , Stephenson D ; Critical Path for Parkinson’s (CPP) Consortium; Parkinson’s Progression Markers Initiative (PPMI) ((2019) ) molecular neuroimaging of the dopamine transporter (DAT) as a patient enrichment biomarker for clinical trials for early Parkinson disease. Clin Transl Sci 12: , 240–246. |

