
Targeted Therapies for Skeletal Muscle Ion Channelopathies: Systematic Review and Steps Towards Precision Medicine
Abstract
Background:
Skeletal muscle ion channelopathies include non-dystrophic myotonias (NDM), periodic paralyses (PP), congenital myasthenic syndrome, and recently identified congenital myopathies. The treatment of these diseases is mainly symptomatic, aimed at reducing muscle excitability in NDM or modifying triggers of attacks in PP.
Objective:
This systematic review collected the evidences regarding effects of pharmacological treatment on muscle ion channelopathies, focusing on the possible link between treatments and genetic background.
Methods:
We searched databases for randomized clinical trials (RCT) and other human studies reporting pharmacological treatments. Preclinical studies were considered to gain further information regarding mutation-dependent drug effects. All steps were performed by two independent investigators, while two others critically reviewed the entire process.
Results:
For NMD, RCT showed therapeutic benefits of mexiletine and lamotrigine, while other human studies suggest some efficacy of various sodium channel blockers and of the carbonic anhydrase inhibitor (CAI) acetazolamide. Preclinical studies suggest that mutations may alter sensitivity of the channel to sodium channel blockers in vitro, which has been translated to humans in some cases. For hyperkalemic and hypokalemic PP, RCT showed efficacy of the CAI dichlorphenamide in preventing paralysis. However, hypokalemic PP patients carrying sodium channel mutations may have fewer benefits from CAI compared to those carrying calcium channel mutations. Few data are available for treatment of congenital myopathies.
Conclusions:
These studies provided limited information about the response to treatments of individual mutations or groups of mutations. A major effort is needed to perform human studies for designing a mutation-driven precision medicine in muscle ion channelopathies.
INTRODUCTION
Skeletal muscle ion channelopathies are a genetically heterogeneous family of skeletal muscle diseases due to mutations in ion channel genes [1, 2]. On one hand, mutations in different genes induce diseases with overlapping symptoms, which may render difficult the exact diagnosis. On the other hand, mutations in the same gene may result in diseases with opposite symptoms. These simple and general observations are sufficient to formulate the hypothesis that mutation driven pharmacological therapies may have great benefits to the affected patients. Up to now, most of the drugs used in ion channelopathies were tested in an empirical manner [3]. Only recently, a few randomized controlled trials have been performed eventually leading to the designation of orphan drugs and marketing authorization. These drugs however only address the symptoms, without considering the genetic background of the disease. We performed a systematic review to report the state of the art and verify whether literature data may support the future development of precision medicine in skeletal muscle ion channelopathies.
In skeletal muscle fibers, ion channels are critical for the modulation of muscle excitability, excitation-contraction coupling, and contraction. Gene mutations of these channels may have various impacts on muscle function, which span from sarcolemma inexcitability (flaccid paralysis, muscle weakness, fatigue) to over-excitability (myotonia, muscle stiffness, cramps), and on muscle integrity with various myopathic traits (Fig. 1). The current review is focused on the non-dystrophic myotonias (NDM) and periodic paralyses (PP), for which muscle wasting, if present, is not predominant (Table 1). In addition, the congenital myasthenic syndromes or congenital myopathies due to mutations in voltage-gated sodium and calcium channels have been investigated, because these diseases may present with symptoms overlapping myotonia or periodic paralysis in the adult life.
Fig. 1
Schematic description of current knowledge regarding skeletal muscle ion channelopathies considered in this systematic review. First line: mutated gene. Second line: mode of inheritance (AR, autosomal recessive; AD: autosomal dominant). Third line: disease names. Fourth line: main effect of mutations on channel function. Fifth line: main symptom. Sixth line: currently preferred symptomatic drugs. Seventh line: second choice drugs.
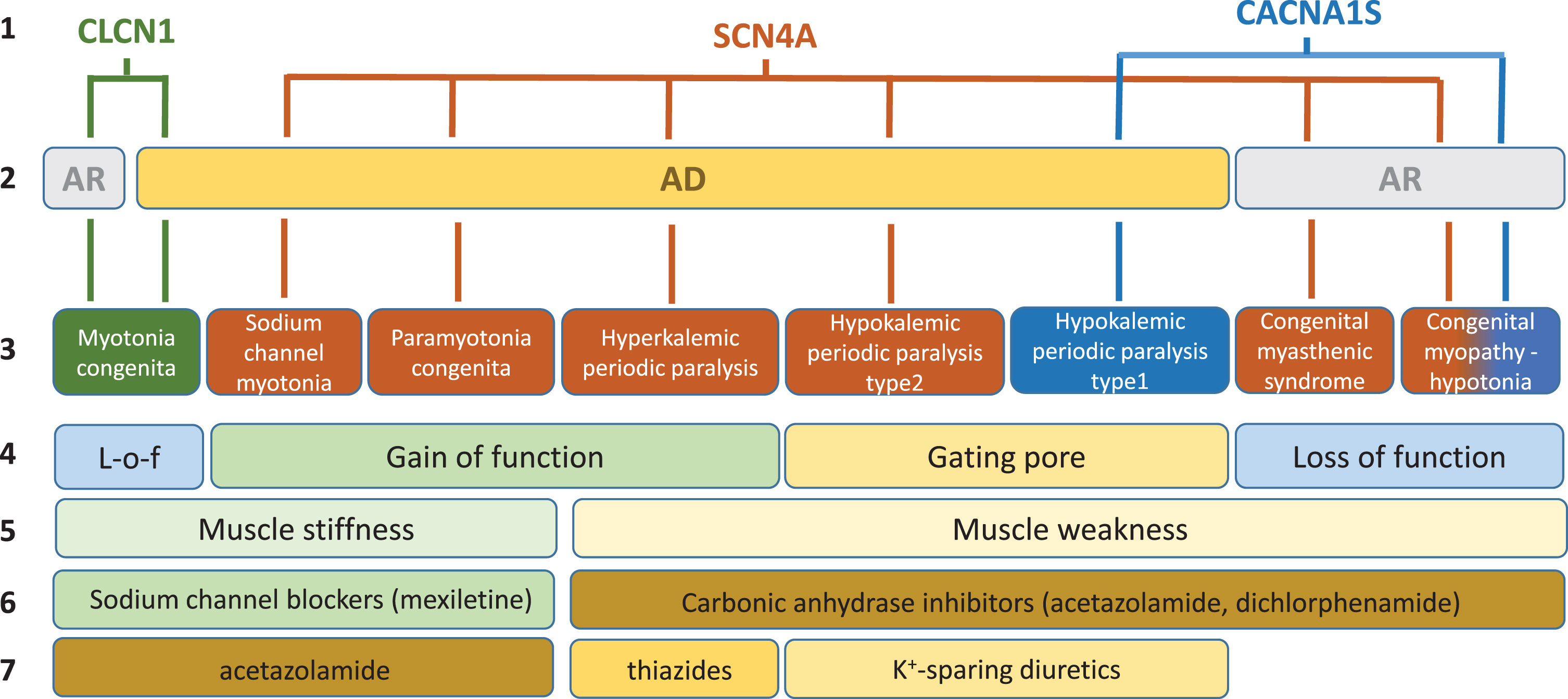
Table 1
Names of the diseases considered in the systematic review
| Group of diseases | Disease names | Phenotype MIM number | Mutated gene (protein) |
| Nondystrophic myotonias | myotonia congenita, autosomal recessive; Becker’s disease | 255700 | CLCN1 [118425] (ClC-1) |
| myotonia congenita, autosomal dominant; Thomsen’s disease | 160800 | CLCN1 [11842] (ClC-1) | |
| K+-aggravated myotonia; acetazolamide-responsive myotonia; sodium channel myotonia | 608390 | SCN4A [603967] (Nav1.4) | |
| paramyotonia congenita von Eulenburg | 168300 | SCN4A [603967] (Nav1.4) | |
| Periodic paralyses | hyperkalemic periodic paralysis; adynamia episodica hereditaria; Gamstorp disease | 170500 | SCN4A [603967] (Nav1.4) |
| hypokalemic periodic paralysis, type 2 (normokalemic periodic paralysis included) | 613345 | SCN4A [603967] (Nav1.4) | |
| hypokalemic periodic paralysis, type 1 | 170400 | CACNA1S [114208] (Cav1.1) | |
| Congenital myopathies | congenital myasthenic syndrome, type 16 | 614198 | SCN4A [603967] (Nav1.4) |
| congenital myopathy | n.a. | SCN4A [603967] (Nav1.4) | |
| congenital myopathy | n.a. | CACNA1S [114208] (Cav1.1) |
n.a.: not available; MIM: Mendelian Inheritance in Man (https://omim.org/).
The treatment of these diseases is mainly symptomatic, aimed at reducing muscle excitability in NDM or modifying triggers of paralytic attacks. In most countries, the sodium channel blocker, mexiletine, is considered the first drug choice for treatment of myotonia, whatever the culprit gene [4]. Other sodium channel blockers were reported efficient. Inhibition of sodium channels reduces the abnormal firing of action potentials responsible for sarcolemma over-excitability and muscle stiffness [5, 6]. A Cochrane review published in 2006 assessing drug treatment in myotonia reported the insufficiency of good quality data to draw any conclusion about safety and efficacy of drugs [7]. Since then, however, placebo-controlled, randomized clinical trials (RCT) confirmed the benefits of mexiletine [8].
In PP, the carbonic anhydrase inhibitors (CAI), acetazolamide and dichlorphenamide, are frequently used to reduce weakness attack frequency [9]. In 2008, a Cochrane review of treatment for periodic paralysis reported one large study suggesting that dichlorphenamide is effective in the prevention of episodic weakness in both hypokalemic and hyperkalemic periodic paralyses [10]. The actual mechanism of action of the CAI in PP is unclear. In addition, thiazide diuretics can be used to maintain a low blood K + concentration to prevent hyperkalemic periodic paralysis. Conversely, potassium-sparing diuretics (e.g. eplerenone or spironolactone) can be used in hypokalemic periodic paralysis [8].
Congenital myasthenia and myopathy due to ion channel mutations are very rare diseases and there is little information regarding treatment [11].
The objectives of this systematic review were to identify the available evidence regarding the effects of pharmacological treatment of skeletal muscle ion channelopathies taking care of assessing the strength of the evidence, and to link treatments to genetic background whatever possible. This study was designed to open the way for defining a link between genotype and treatment, and to collect solid information that might stimulate the launch of clinical trials specifically addressing pharmacogenomics.
METHODS
The systematic review was performed following the guidelines of the Treatabolome methodology designed for writing FAIR-compliant systematic reviews of rare diseases’ treatments [12]. All steps were performed by two independent researchers (C.A. and S.V.). Two other researchers (J.F.D and B.F.) critically reviewed all the process and resolved disparities.
Types of studies and diseases considered
All types of human studies dealing with drug treatment of genetically diagnosed adults and children affected by NDM, PP, or Cav/Nav-related congenital myasthenia or congenital myopathies were considered for inclusion in the systematic review. The studies were classified by level of evidence and analyzed separately, giving emphasis to randomized controlled trials. Due to scarcity of data for rare diseases in general, nonrandomized trials, observational studies, cohort, series and case studies were then considered even when only qualitative outcomes were available. In addition, we analyzed preclinical studies, performed both in vivo or in vitro, which may add significant information related to mutation-dependent drug effects and may provide experimental support for the design of future human studies. Therapeutic interventions other than pharmacological interventions were not considered.
Outcome measures demonstrating positive or negative response to treatment were all considered, including measures of muscle function and performance (strength, ability and endurance), electromyography, clinical examination, frequency and severity of events, and patients filled-in questionnaires for quality of Life. For in vitro studies, we considered effects of drugs on ion currents generated by mutant channels and compared to wild-type.
Search methods for identification of studies
An electronic search of the literature was performed using the following databases:
– Pubmed (https://pubmed.ncbi.nlm.nih.gov/)
– The Cochrane Central Register of Controlled Trials (https://www.cochranelibrary.com/central)
– ClinicalTrials.gov (https://clinicaltrials.gov/)
– WHO International Clinical Trials Registry Platform (https://apps.who.int/trialsearch/default.aspx)
– European Clinical Trials Database (EudraCT) (https://eudract.ema.europa.eu)
The search was completed with an inspection of the personal databases of the authors. Finally, further relevant articles were extracted from the reference sections of the reviewed articles. Last search was performed on September 4, 2020. Only studies published in English, French or Italian language were included in the review.
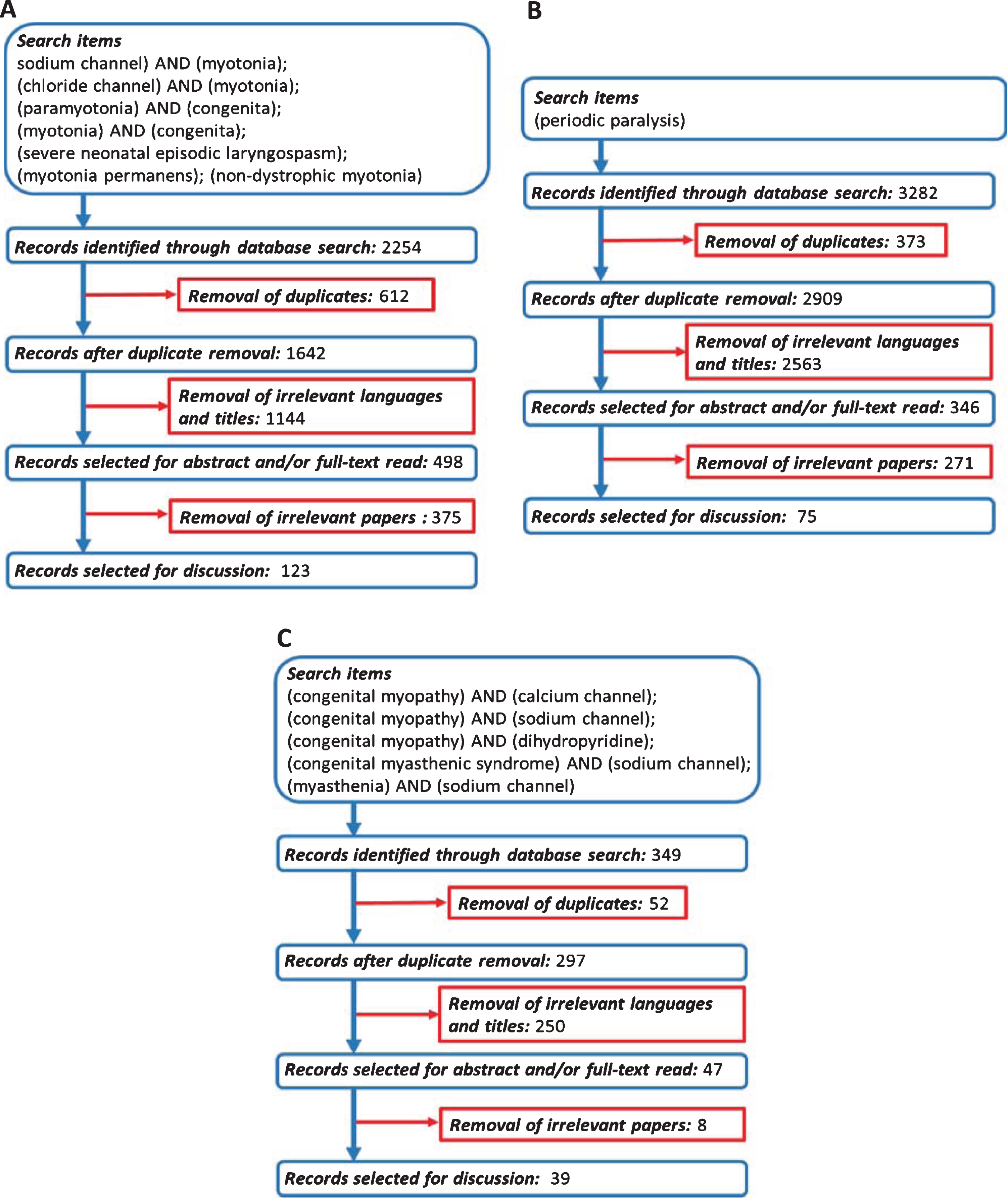
The authors independently screened titles of the publications identified through the searches, excluding those irrelevant. After this preliminary screening, the abstract and/or full-text was examined to assess further eligibility for inclusion. Search terms and database search results are presented in Fig. 2.
Fig. 2
Flowchart of search strategy and filtering steps. (A) Non-dystrophic myotonias. (B) Periodic paralyses. (C) Congenital myopathies related to SCN4A/CACNA1S genes.
Data extraction and analysis
Data from each included study were collected in a standardized form including study characteristics and design, genotype and clinical phenotype of study participants, number of participants, age distribution, treatment characteristics (dose, frequency, and duration), outcome measures, response to treatment (positive response, partial response, non-responsive, and side effects). Regarding preclinical studies, collected data included study characteristics and design, gene variant, treatment characteristics (dose in vivo or concentration in vitro), outcome measures, and response to treatment.
RESULTS AND DISCUSSION
Non-dystrophic Myotonia
The non-dystrophic myotonias (NDM) are genetic disorders characterized by skeletal muscle stiffness after voluntary contraction [1, 4, 13–15]. Disease onset typically occurs in the first two decades of life, with a few patients showing severe neonatal symptoms causing respiratory distress. Muscle stiffness occurs generally in absence of fixed weakness and wasting, but the disease can severely affect quality of life due to movement impairment, myalgia, or frequent falls. Prevalence is likely less than 1/100,000 [16]. The NDM encompass several disorders with distinct features but clinical overlap, which are myotonia congenita (MC), paramyotonia congenita (PMC), and sodium-channel myotonia (SCM) (Fig. 1).
MC is due to mutations in the CLCN1 gene encod-ing the chloride channel ClC-1 [17, 18]. The autosomal dominant form (Thomsen’s disease) is moderate to severe. The recessive form (Becker’s disease) is typically more severe and may present with transient weakness. Myotonia is often triggered by rapid movements after a period of rest, and is improved with exercise (warm-up phenomenon). Patients may develop muscle hypertrophy (herculean body), due to sustained involuntary muscle contraction. More than 250 CLCN1 mutations have been found in MC patients [19].
PMC and SCM are dominantly inherited allelic disorders caused by mutations in SCN4A gene encod-ing the voltage-dependent sodium channel Nav1.4 [20–22]. PMC is characterized by paradoxical myotonia (worsening with exercise) and sensitivity to cold and fasting. Some patients may present prolonged muscle weakness lasting several hours after sustained exercise, which is similar to that observed in the allelic disorder hyperkalemic periodic paralysis. In SCM, the symptoms are highly variable but prolonged weakness never occurs (pure myotonia). Some SCM can cause severe neonatal episodic laryngospasm (SNEL), which can eventually lead to death by respiratory arrest during an apnea episode [23–28]. Recently, myotonic SCN4A mutations have been associated with an increased risk of Sudden Infant Death Syndrome (SIDS), underscoring the life-threatening effects of myotonia in babies [29].
The NDM have been empirically treated with sodium channel blockers including mostly antiarrhythmics (e.g. quinidine, tocainide, mexiletine, fle-cainide) and anticonvulsants (e.g. phenytoin, carbamazepine) [30]. The mechanism of action of these drugs is merely symptomatic, consisting in the use-dependent inhibition of skeletal muscle sodium channels to reduce abnormal action potential firing. Indeed, the same drugs are used in both chloride and sodium channel myotonia. The antiarrhythmic mexiletine is the preferred drug today, because it has been more carefully studied in RCTs and observational studies, although no comparative test has never been published; it was recently appointed as orphan drug in NDM and was granted marketing authorization in 2019 by the European Medicine Agency [4]. In addition, the carbonic anhydrase inhibitor acetazolamide has been used in NDM and is often considered as a second choice after sodium channel blockers [31].
Randomized clinical trials (Class of Evidence I)
In 2012, the results of an international randomized, double-blind, placebo-controlled clinical trial of mexiletine in nondystrophic myotonia were published [8]. This RCT received much attention in the scientific community for having overcome many difficulties related to the orphan diseases with clinical and genetic heterogeneity [32]. Fifty-nine participants of both sex were enrolled. All had clinical myotonia but were genetically heterogeneous (34 had chloride channel mutations, 21 had sodium channel mutations, and 4 had no mutation identified). Mutations for dystrophic myotonia were excluded. The patients were randomized to receive either mexiletine or placebo for 4 weeks, followed by a week of washout, and then crossover to the other treatment for another 4 weeks. The mexiletine dose was 200 mg three times a day. Outcome measurements, including clinical assessment of eyelid and handgrip myotonia, electromyography, and SF-36 and INQOL questionnaires, were performed at baseline and at the end of each treatment period. In addition, an interactive voice response (IVR) diary was used to evaluate patient’s symptoms and adherence to treatment. The primary end-point was the severity score of stiffness reported by the patients via the IVR diary. Fifty-seven participants were included in the modified intention-to-treat analysis, whereas two were excluded for having failed to call the IVR system. Adherence to pills was very good, >90%. Significant improvement of the primary end-point and many secondary end-points was obtained with mexiletine. Two dropouts due to side effects occurred during mexiletine treatment. The most common side effect associated with mexiletine was gastrointestinal discomfort. One limitation of the RCT was the inclusion of participants with both chloride and sodium channel mutations to obtain necessary study power. A secondary analysis suggested major improvement in chloride-channel mutation carriers, but statistical power was not adequate.
Efficacy of mexiletine in NDM was further supported by aggregated N-of-1 trials conducted in a single Dutch center but involving international collaborations [33]. The aims of this study were to investigate effectiveness of mexiletine and to compare the results with the previous RCT. To allow comparison, drug regimen and outcome measures were similar. Thus, thirty genetically-confirmed myotonic patients were enrolled to receive 600 mg mexiletine daily and placebo for 4 weeks each, in randomized crossover periods with 1 week washout in between. By the end of the treatment sequence, a Bayesian analysis of the IVR scores allowed the calculation of the posterior probability (pp) of mexiletine to produce a meaningful improvement in each patient. If the value of pp was greater than 0.8, the patients were advised to abandon the trial and to start regular treatment with mexiletine; If pp was less than 0.2, the patient should discontinue the trial and mexiletine treatment; with pp in between 0.2 and 0.8, the patients were encouraged to continue the trial by repeating the randomized treatment sequence. At the completion of the trial, a Bayesian hierarchical model was used to analyze aggregated data. Twenty-seven participants completed the trial, while 3 dropouts were due to non-adherence to the protocol and one serious urticaria-like rash related to mexiletine. Effectiveness of mexiletine was statistically confirmed for the primary end-point (IVR-reported stiffness) and many secondary endpoints. In average, patients with chloride channel genotype showed a better response to mexiletine than carriers of sodium channel mutations regarding the primary end-point and several secondary end-points. Very common side effects were gastrointestinal disorders. Altogether, these results confirmed the results of the previous RCT and the value of N-of-1 trials for evaluating drugs in rare diseases. Three participants did not respond to mexiletine and discontinued the trial. Interestingly, all three non-responders carried sodium channel mutations (p.G1306V, p.R1448C, and p.L703P, Table 2). Noteworthy, patient with p.R1148C mutation had serum mexiletine levels in subtherapeutic range (0.17μg/ml compared to reference antiarrhythmic therapeutic range: 0.5–2.0μg/ml), which might explain the lack of response.
Table 2
Clinical reports of drug response in myotonic SCN4A mutation carriers
| Mutation | Reference | Response to drug (dose/day) | Number of individuals and other information |
| p.Ile215Thr (c.644T>C) | [46] | NR to PHE (nd), USE to QUI (nd), positive to MEX (nd) | (n = 1) |
| positive to MEX (400 mg) | (n = 1) | ||
| positive to MEX (nd) | (n = 1) | ||
| p.Gly241Val (c.722G>T) | [46] | NR to MEX (nd), PHE (nd), ACZ (nd), and CBZ (nd) | (n = 1) |
| p.Asn440Lys (c.1320T>G) | [47] | positive to ACZ (nd) and HCT (nd) | (n = 10, 1 kindred), myotonia with exercise induced weakness |
| p.Val445Met (c.1333G>A) | [48] | NR or USE with MEX (600 mg), QUI (975 mg), PHE (400 mg), CBZ (1,200 mg), TOC (1,200 mg), ACZ (500 mg), diazepam (10 mg), baclofen (80 mg), and cyclobenzaprine (40 mg) | (several individuals from 1 kindred) |
| positive to FLE (nd) | (n = 1 from the kindred as above) | ||
| [49] | positive to MEX (600 mg) | (n = 1) | |
| positive to PRC (3,000 mg) | (n = 1) | ||
| positive to QUI (600 mg) | (n = 1) | ||
| [50] | positive to MEX (300 mg) | (n = 1) moderate improvement | |
| [36] | positive to RAN (2,000 mg) | (n = 3) | |
| [51] | positive to MEX (nd) | (n = 1) | |
| p.Glu452Lys (nd) | [52] | NR to GAB (nd), positive to PHE (400 mg) | (n = 1) |
| p.Phe671Ser (nd) | [52] | NR to PHE (nd), positive to CBZ (nd) | (n = 1) |
| p.Leu689Phe (c.2065C>T) | [53] | positive to ACZ (250–500 mg) | (n = 1) |
| [54] | positive to PHE (nd) and ACZ (nd) | (n = 1) | |
| p.Ile693Leu (c.2077A>C) | [55] | NR to CBZ (nd), positive to PHE, MEX, or ACZ (nd) | (n = 1) |
| p.Ile693Thr (c.2078T>C) | [56] | positive to Mg2 + (3,600 mg) | (n = 1), aggravated symptoms with hypomagnesemia and hypocalcemia |
| [57] | USE with ACZ (5 mg/kg) | (n = 1) | |
| p.Leu703Pro (c. 2108T>C) | [53] | NR to CBZ (800 mg), positive to ACZ (250–1,000 mg) | (n = 1), headache at the highest ACZ doses |
| [33] | NR to MEX (600 mg) | (n = 1) | |
| p.Thr704Met (c. 2111C>T) | [58] | NR to ACZ (1,000 mg) | (n = 2), hyperPP+PMC |
| NR to ACZ (1,000 mg) and HCT (500 mg) | (n = 1), hyperPP+PMC | ||
| NR to ACZ (1,000 mg) and SLB (6 mg p.o. or 200μg nasally) | (n = 1), hyperPP+PMC | ||
| p.Leu796Val (c. 2389C>G) | [59] | positive to CBZ (36 mg/kg) | (n = 1), respiratory failure |
| [60] | positive to CBZ (20 mg/kg) | (n = 1), myotonic myopathy | |
| p.Arg1148Cys (nd) | [33] | NR to MEX (600 mg) | (n = 1), subtherapeutic plasma concentration |
| p.Pro1158Leu (c. 3473C>T) | [61] | NR to CBZ (nd), USE with MEX (200 mg), positive to FLE (70 mg) | (n = 1), myotonia permanens |
| p.Pro1158Ser (c. 3472C>T) | [62] | positive to ACZ (nd) | (n = 1), myotonia improvement but weakness worsening |
| p.Ile1160Val (c. 3478A>G) | [50] | positive to ACZ (500 mg) | (n = 1), significant improvement |
| p.Gly1292Asp (c.3875A>G) | [63] | NR to MEX (300 mg) | (n = 1) |
| p.Val1293Ile (c. 3877G>A) | [64] | positive to MEX (400 mg) | (n = 1) |
| [50] | positive to MEX (600 mg) | (n = 1), significant improvement | |
| [65] | USE with MEX (200 mg) | (n = 1) | |
| positive to MEX (400 mg) | (n = 1) | ||
| p.Asn1297Lys (c.3891C>A) | [23] | NR to CBZ (20 mg/kg), positive but unstained to MEX (17 mg/kg) | (n = 1), SNEL, dies at 20 months from cardiorespiratory arrest |
| p.Phe1298Cys (c.3893T>G) | [65] | USE with MEX (400 mg) | (n = 1), moderate improvement |
| p.Gly1306Ala (c.3917G>C) | [33] | positive to MEX (600 mg) | (n = 2) |
| [66] | NR to MEX, ACZ, and CBZ (nd) | (n = 1) | |
| p.Gly1306Glu (c.3917G>A) | [67] | NR to MEX (720 mg), oxcarbazepine (nd), and thiazide diuretics (nd), positive to ACZ (500 mg) | (n = 1) |
| [24] | positive to CBZ (40 mg/kg) | (n = 1), SNEL | |
| positive to MEX (15 mg/kg) | (n = 1), SNEL | ||
| [68] | PR to CBZ (600 mg), HQD (600 mg), TOC (1,200 mg), and MEX (800 mg), positive to FLE (200 mg) | (n = 2, mother and son), myotonia permanens | |
| [69] | positive to MEX+ACZ (nd) | (n = 1), SNEL | |
| positive to CBZ (12 mg(kg) | (n = 1), SNEL | ||
| [70] | positive to CBZ (20 mg/kg) | (n = 3), SNEL | |
| [26] | NR to CBZ (20 mg/kg), MEX (15 mg/kg), and ACZ (125 mg), positive to FLE (100 mg) | (n = 1), SNEL | |
| [71] | USE with FLE (200 mg) and HQD (300 mg), positive to MEX (1,200 mg) | (n = 1), SNEL Brugada syndrome with FLE and diarrhea with HQD | |
| [57] | positive to CBZ (200 mg*) | (n = 7/8) | |
| USE with CBZ (200 mg*) | (n = 1/8) | ||
| positive to ACZ (250 mg*) | (n = 4/6) | ||
| USE with ACZ (250 mg*) | (n = 2/6) | ||
| positive to PHE (nd) | (n = 1) | ||
| positive to MEX (400 mg) | (n = 1) | ||
| p.Gly1306Val (c.3917G>T) | [33] | positive to MEX (600 mg) | (n = 5) |
| NR to MEX (600 mg) | (n = 1), concomitant stable angina pectoris | ||
| [36] | NR to RAN (2,000 mg) | (n = 2) | |
| p.Thr1313Met (c.3938C>T) | [72] | NR to ACZ (250 mg), positive to MEX (850 mg) | (n = 2, 1 kindred) |
| [73] | positive to MEX (600–1,200 mg) | (n = 3, 1 kindred) | |
| [74] | NR to MEX (200 mg), USE with higher doses | (n = 1) | |
| [75] | USE with MEX (400 mg), positive to PRO (325 mg) | (n = 1) | |
| [50] | USE with MEX (nd) | (n = 1) | |
| [76] | USE with MEX (400 mg), positive to FLE (200 mg) | (n = 8, 1 kindred) | |
| [77] | positive to ACZ (500 mg) | (n = 1), paramyotonia improved but not persistent weakness | |
| NR to ACZ (250 mg) | (n = 1) | ||
| NR to MEX (150 mg) | (n = 1) | ||
| p.Leu1436Pro (nd) | [78] | NR to ACZ (nd) | (n = 2) |
| PR and USE to ACZ (nd) | (n = 1), allergic rash with ACZ | ||
| PR to CBZ (600 mg)+AMI (50 mg) | (n = 3), some improvement | ||
| p.Arg1448Cys (c.4342G>A) | [79] | positive on weakness, NR on stiffness ACZ (250 mg) | (n = 1) |
| [80] | NR to ACZ (500 mg), PR to MEX (400 mg) | (n = 1) | |
| [81] | positive to PYR (180 mg) and PHE (300 mg) | (n = 1), PYR resolved exercise-induced weakness and PHE reduced cold-induced stiffness | |
| [36] | positive to RAN (2,000 mg) | (n = 3) | |
| [82] | PR and USE with ACZ (500 mg) | (n = 1), PMC with HypoPP | |
| p.Ile1462Met (c.4386C>G) | [36] | positive to RAN (2,000 mg) | (n = 1) |
| p.Met1476Ile (c.4428G>A) | [83] | NR to QUI, PHE, CBZ, AMI, GAB, and ACZ | (n unknown) |
| positive to MEX (400 mg) | (n = 1) | ||
| p.Ala1481Asp (c.4442C>A) | [84] | NR to ACZ (1,000 mg) and PR to MEX (nd) | (n = 1) |
| p.Val1485Phe (nd) | [33] | positive to MEX (600 mg) | (n = 1) |
| p.Val1589Met (c.4765G>A) | [85] | NR to CBZ and PHE (nd), positive to ACZ (500 mg) | (n = 1) |
| [36] | positive to RAN (2,000 mg) | (n = 1) | |
| p.Met1592Val (c.4774A>G) | [86] | NR to ACZ (nd), PR with PHE i.v. (nd), positive to MEX (nd) | (n = 1) |
| p.Gln1633Glu (c.4897C>G) | [87] | positive to MEX (nd) | (n = 3, 1 kindred), myotonia permanens |
| p.Phe1705Ile (c.5113T>A) | [88] | PR to MEX (nd) | (n = 1) |
ACZ: acetazolamide; AMI: amitriptyline; CBZ: carbamazepine; FLE: flecainide; GAB: gabapentin; HCT: hydrochlorothiazide; HQD: hydroquinidine; hyperPP: hyperkalemic periodic paralysis; MEX: mexiletine; nd: not defined; NR: non responder; PHE: phenytoin; QUI: quinine; PMC: paramyotonia congenita; PR: partial response; PRC: procainamide; PRO: propafenone; PYR: pyridostigmine; RAN: ranolazine; SNEL: severe neonatal episodic laryngospasm; TOC: tocainide; USE: untolerated side effects; *mean value.
Following the first RCT, mexiletine has received orphan drug designation for the treatment of nondystrophic myotonia by the European Medicine Agency. Its use is however challenged by high costs or limited availability in some countries, side effects, and contraindications. This led a Danish group to perform a RCT of lamotrigine in NDM [34]. Lamotrigine is a sodium channel blocker indicated for treatment of epileptic seizures and bipolar disorder. The phase II randomized, double-blind, placebo-controlled, crossover-study of lamotrigine resembled that of mexiletine. The adult patients from the Danish registry with genetically confirmed NDM were all considered. Fifty-seven were excluded and 26 were finally included in the trial (14 with MC and 12 with PMC). They were randomized to receive either lamotrigine at increasing dose (from 0 to 300 mg a day) or placebo for 8 weeks, followed by a wash-out period of 1–3 weeks, and crossover period for 8 weeks. The primary outcome measure was the Myotonic Behavior Scale diary, a validated self-assessed questionnaire that consists in six statement to describe severity of stiffness, completed before treatment and during the treatment with 150 and 300 mg lamotrigine. Secondary outcome measures included eyelid and handgrip myotonia, leg myotonia assessed by the time-up-and-go test and the 14-Step-Stair test, the short form (SF-36) questionnaire, the creatine kinase level, and the use of escape medication (mexiletine). At the end, 5 participants were excluded from analysis due to loss of follow-up; notably, 1 patient developed reversible allergic reaction to lamotrigine. The most common side effect of lamotrigine was headache. Lamotrigine had significant effects on primary and many secondary outcomes. The estimated standardized effect sizes in the two RCT suggest similar efficacies of mexiletine and lamotrigine in NDM. There was no attempt to correlate drug effect with the specific mutations. In conclusion, the authors suggested that lamotrigine might be considered first-line in treatment-naive NDM patients and in patients intolerant to mexiletine.
Other human studies (class of evidence II-to-IV)
A number of studies have reported the effects of drugs on NDM patients with defined genotype, including open-label trials, retrospective case-series studies, and case report.
Two open-label studies (class IV level of evidence) were performed in a single center to test ranolazine on participants with genetically diagnosed PMC or myotonia congenita [35, 36]. Ranolazine is a sodium channel blocker indicated for angina pectoris. The rationale for its use in myotonia is based on the preferential inhibition of persistent sodium currents and the enhancement of sodium channel slow inactivation exerted by the drug [37–39]. Like mexiletine, ranolazine exerts frequency-dependent inhibition of sodium channels [40]. In addition, it is provided with more potential targets, including potassium channels. Experimental design was the same in both clinical studies, consisting in 4 weeks treatment with 500 mg bid for the two first weeks, and 1,000 mg bid the last two weeks. Thirteen participants had chloride channel mutations (12 with Thomsen’s disease, 1 with Becker’s disease), whereas 10 patients with sodium channel mutations were enrolled in the second study. Two patients did not take the highest dose, because of unpleasant side effects (migraine in one and insomnia, constipation, mental fog, and malaise in the other). The most common side effects were constipation and transient lightheadedness. In both studies, ranolazine improved subjective symptoms of stiffness, weakness, and pain, as well as clinical myotonia (Time Up-and-Go, eyelid and handgrip myotonia). There was only limited improvement of EMG myotonia. Although these studies lack of blinding and placebo controls, and do not compare ranolazine to any other drug, the authors suggested that ranolazine might be useful to patients not responding or intolerant to mexiletine and warrants investigation in a RCT.
One observational study performed in a single UK center included 63 patients, including 40 CLCN1 mutation carriers, 21 SCN4A mutation carriers, and 2 with mutations in both genes, who received mexiletine (up to 600 mg a day) for at least 6 months (up to ∼18 years) [41]. Efficacy was assessed from patient report and any possible side effects were collected. Available electrocardiograms were re-analyzed. The main information was occurrence of side effects in about half of the patients; the most common being dyspepsia (35 %) often requiring dyspeptic therapy and eventually leading to treatment withdraw. No serious adverse event was reported. Regarding efficacy, 12 patients were refractory to the drug, most of them carrying chloride channel mutations. In addition, CLCN1 mutation carriers required in average higher mexiletine doses compared to SCN4A mutations carriers, suggesting that the former are less sensitive to the drug. Such a result may appear in contrast to the observations made during the two mexiletine RCTs.
Another observational study was performed in two Italian centers [42]. Of the 112 NDM patients followed, 59 began mexiletine therapy, according to severity and individual needs and concerns (11 with SCN4A mutations and 48 with CLCN1 mutations). The patients affected by recessive myotonia congenita were the most prone to treatment (72%); 42% of the patients suffering from sodium channel myotonia required mexiletine; and only 5% of patients with dominant myotonia congenita require treatment. Patients under 12 took mexiletine at 8 mg/kg, while older patients took between 200 and 600 mg according to clinical severity and individual needs. Treatment efficacy was quantitatively assessed in 8 patients carrying CLCN1 mutations, showing significant benefit of mexiletine in reducing the transient CMAP depression and improving the chair test. Efficacy of treatment was not formally assessed in the other patients, but of the 59 patients who accepted to take mexiletine, 46 continued treatment, suggesting improvement of symptoms in 82 % of patients. Thirteen patients discontinued mexiletine because of side effects (n = 4), personal motivations (n = 6), or shift to flecainide (n = 3). The shift to flecainide was motivated by the hope to obtain better benefits, suggesting limited response to mexiletine in those patients, all three carrying a SCN4A mutation (see below). Again, dyspepsia was the most common side effect of mexiletine (42 %, requiring dose adjustment in 4 and dropout in other 4). A previous study by the same authors also demonstrated the efficacy of mexiletine in reducing not only muscle stiffness but also the transitory weakness characteristic of recessive myotonia congenita [43]. The latter was further confirmed in an independent study [44].
Recently a retrospective cohort study included 42 individuals with non-dystrophic myotonia followed in a large tertiary center in Ohio [45]. They reported that patients taking at least one medication for myotonia were ∼52% in CLCN1 mutation carriers and 80 % (n = 12) in SCN4A mutation carriers. The most common medication was mexiletine followed by ranolazine, then by acetazolamide and lamotrigine, with similar proportions in between SCN4A and CLCN1 mutation carriers. The study also evidenced that more than half of the patients have trialed several drugs before to find a satisfactory one, due to lack of response or untolerated side effects.
Altogether, these studies confirmed efficacy and safety of long-term use of mexiletine in NDM patients. However, they also point out to the occurrence of unpleasant side effects, side effect-related dropouts, and non-responders. There is no consensus as whether mexiletine would provide more benefits for CLCN1 or SCN4A mutation carriers, and specific RCTs with sufficient statistical power should be designed to test the hypothesis of a different response between the two genotypes. In addition, there are a number of evidences suggesting a further level of complexity, with some SCN4A mutations altering the sodium channel sensibility to mexiletine, which may influence the patient response (see below). Therefore, the type of mutation is likely to have an important impact on the choice of therapy in the precision medicine era.
Regarding the side effects, there is little information allowing a correlation with the genotype. Suetterlin and collaborators provided a table reporting the distribution of adverse events by genotype (SCN4A mutation carriers versus various subgroups of CLCN1 mutation carriers), suggesting little difference between the genotypes, although the limited number of patients does not allow drawing a definitive conclusion [41].
Non-responders may occur due to either pharmacokinetic mechanisms, such as altered drug metabol-ism or excretion, or pharmacodynamic mechanisms including alteration of molecular target or cellular signaling pathways. Altered metabolism or excretion of the drug may result in a subtherapeutic plasma drug concentration, which is rarely measured in the published studies. As said above, in the N-of-1 RCT, a non-responder had subtherapeutic mexiletine concentration, which might have caused the lack of response [33]. However, in the same study, another patient with subtherapeutic mexiletine concentration apparently responded to treatment. It is noteworthy that reference therapeutic concentration range has not been defined for any drug in myotonia; that of mexiletine refers to its use as a cardiac antiarrhythmic.
Regarding chloride channel myotonia, treatment with mexiletine and other sodium channel blockers is symptomatic. Inhibition of sodium currents reduces the propensity of myofibers to elicit action potentials, thereby contrasting myotonic runs of action potentials and muscle stiffness. The situation is more complex in sodium channel myotonia, because the myotonic mutations might influence the response of the channel to the drugs. This has been clearly demonstrated in vitro, as detailed in the next chapter. Whether this may be relevant to the clinical setting is less easy to demonstrate.
In Table 2, we collected all the information avai-lable from RCT, case-series, and case reports, regarding the response to treatment of specific SCN4A mutation carriers. In most cases, the response was positive. It should be noted however that not always the various treatment attempts before to reach the positive response are reported in the studies. Regarding the non-responders, two mutations captured our attention, p.G1306E and p.T1313M (Table 2).
The p.G1306E mutation is generally associated with a severe phenotype, called myotonia permanens, characterized by continuous stiffness associated with persistent myotonic activity, especially in facial, bulbar, neck and shoulder muscles [27, 67]. The mutation may also cause symptoms in early life, including stridor and severe laryngospasm, eventually leading to life-threatening acute hypoventilation and cyanosis [27, 28]. The p.G1306E carriers present an unusual response to treatment. Some of them, but not all, responded well to carbamazepine that is often used in the pediatric setting [24, 69, 70]. Yet, refractoriness to mexiletine and other drugs have been reported in various independent studies and it appeared that flecainide might be a better option for these patients [26, 27, 68].
The p.T1313M mutation is associated with PMC with or without episodes of hyperkalemic paralysis. Several independent studies reported non-tolerated side effects of mexiletine, which led to shift therapy to other sodium channel blockers, especially the class IC antiarrhythmics propafenone and flecainide [50, 75, 76]. Another T1313M carrier had only a partial response to 400 mg mexiletine [72]; Myotonia completely resolved with 850 mg a day, but such a dose also determined frequent fasciculation in limb and facial muscles.
Besides sodium channel blockers, the carbonic anhydrase inhibitor acetazolamide has been reported efficient in small cohorts of myotonic patients with sodium or chloride channel mutations [31, 53, 89–91]. A number of non-responders have been also reported (Table 2). Available data do not allow drawing a correlation with genotype.
It is worthy to note that reports have been published regarding patients carrying both CLCN1 and SCN4A mutations or in concomitance with myotonic dystrophy nucleotide repeats, which may increase further the complexity of treatment [3, 92–99].
Preclinical studies
Because sodium channel blockers are the preferred drugs against myotonia, one may expect that myotonic mutations in SCN4A may modify the sensibility of the channel to treatment. First evidence was provided by Fan and collaborators, who reported the different sensibility of PMC mutants to lidocaine [100]. Although lidocaine is not used in myotonia (because of intravenous way of administration), its orally-available derivatives tocainide and mexiletine have been largely used and, today, mexiletine remains the first choice. Further studies have reported altered sensitivity to mexiletine for a number of sodium channel variants (Table 3). In a few cases, the mutation may modify channel affinity by interacting directly or allosterically with the local anesthetic binding site situated in the channel pore [101]. However, in most cases, the altered sensitivity of the channel mutant is secondary to gating defects, due to the channel state-dependent affinity of the drugs; binding affinity of mexiletine for inactivated channels is by far greater than for resting channels [102, 103]. Thus, by modifying the relative time that the channel lasts in a determined state, a mutation may modify channel sensitivity to the drug. A gating defect that critically influences mexiletine effects is the shift of the voltage-dependence of fast inactivation; yet it is worth to note that other gating defects may contribute to altered drug sensitivity [65]. Interestingly, alteration of effects is rather specific to each individual drugs. Thus, p.G1306E and p.T1313M were both less sensitive to mexiletine compared to wild-type, but showed unaltered or even increased sensitivity to flecainide and propafenone [65, 103]. This offers a great opportunity for exploring other sodium channel blockers ready for repurposing in myotonia [37, 104–110].
Table 3
In vitro sensibility of SCN4A mutants to sodium channel blockers
| Mutation | Ref | Drug | Sensitivity* | Likely main mechanism / comments | |
| p.Val445Met | c.1333G>A | [101] | MEX | increased | increased inactivated state affinity |
| p.Ser804Phe | c.2411C>T | [101] | MEX | unchanged | |
| p.Ala1156Thr | c.3466G>A | [102] | MEX | unchanged | hyperPP phenotype |
| p.Pro1158Leu | c.3473C>T | [61] | MEX | reduced | Probably due to gating defect |
| p.Val1293Ile | c.3877G>A | [65, 101] | MEX | unchanged | |
| p.Asn1297Ser | c.3890A>G | [65] | MEX | reduced | Probably due to gating defect |
| FLE | unchanged | ||||
| p.Asn1297Lys | c.3891C>A | [65] | MEX | reduced | Probably due to gating defect |
| FLE | unchanged | ||||
| p.Phe1298Cys | c.3893T>G | [65] | MEX | reduced | Probably due to gating defect |
| FLE | unchanged | ||||
| p.Gly1306Glu | c.3917G>A | [102, 111] | MEX | reduced | Probably due to gating defect |
| [103] | FLE | unchanged | |||
| [105] | PIL | reduced | Probably due to gating defect | ||
| p.Ile1310Asn | c.3929T>A | [65] | MEX | reduced | Probably due to gating defect |
| FLE | increased | nd | |||
| p.Thr1313Met | c.3938C>T | [100] | LID | reduced | Right-shifted inactivation voltage dependence; reduced inactivated state affinity |
| [65] | MEX | reduced | Probably due to gating defect | ||
| FLE | increased | nd | |||
| PRO | unchanged | ||||
| p.Met1360Val | nd | [112] | MEX | unchanged | |
| p.Arg1448Cys | c.4342G>A | [100] | LID | increased | Probably due to gating defect |
| [102] | MEX | increased | Probably due to gating defect | ||
| [103] | FLE | increased | Probably due to gating defect | ||
| [105] | PIL | increased | Probably due to gating defect | ||
| [37] | RAN | increased | Probably due to gating defect | ||
| p.Arg1448His | c.4343G>A | [112] | MEX | increased | Probably due to gating defect |
| [37] | RAN | unchanged | |||
| p.Arg1448Pro | nd | [37] | RAN | increased | Probably due to gating defect |
| p.Phe1473Ser | nd | [111] | MEX | reduced | Probably due to gating defect |
| p.Met1476Ile | c.4428G>A | [113] | MEX | reduced | slower onset of mexiletine block and/or faster recovery from mexiletine block |
| p.Val1589Met | c.4765G>A | [101] | MEX | reduced | Reduced inactivated state affinity |
| p.Met1592Val | c.4774A>G | [101] | MEX | reduced | Reduced inactivated state affinity |
*compared to wild-type. FLE: flecainide; LID: lidocaine; MEX: mexiletine; nd: not defined; PIL: pilsicainide; PRO: propafenone; RAN: ranolazine.
Importantly, some studies suggest that in vitro results may be translated to the clinical setting. For instance, it is noteworthy that carriers of p.R1448 mutations generally respond very well to mexiletine treatment, in accord with the increased sensitivity of these mutants to mexiletine in vitro. A probable explanation is that, in the heterozygous myotonic patient, effects of mexiletine would rely essentially on the inhibition of R1448 channel mutants and leave wild-type channels quite undisturbed, thereby allowing a selective and safe therapeutic effect. Regarding the p.M1476I, carriers had little benefit from many sodium channel blockers except mexiletine [83], and this effect was attributed to the specific capacity of the drug to inhibit the conspicuous persistent sodium currents generated by the mutant channel [113]. Conversely, carriers of p.P1158L, p.G1306E, and p.T1313M mutations often obtained unsatisfactory response to mexiletine but responded well to flecainide or propafenone [26, 27, 61, 68, 75, 76], again in perfect line with the in vitro results. In these patients, effects of mexiletine would rely mainly on inhibition of wild-type channels with limited efficacy; increasing the dose to inhibit mutant channels would expose the patients to increased propensity of unpleasant and limiting side effects, which gives a plausible explanation for the unsatisfactory response in most of these patients.
Mexiletine is also the first choice in myotonia congenita, although it has no effect on chloride channels. More than 250 mutations are known in CLCN1 to cause myotonia by reducing the chloride conductance in sarcolemma, thereby favoring exacerbated excitability. Most of the mutations functionally characterized so far are predicted to reduce ClC-1 channel expression by defective cell trafficking or to reduce channel function through various gating defects [19, 114, 115]. Unfortunately, no direct activator of ClC-1 channel is known. Some studies aimed at deciphering the molecular interaction of known inhibitors with ClC-1 channels might provide some hints for the discovery of activators (gating modifiers) or pharmacological chaperones (trafficking correctors) [116–119]. Recently, two small molecules inhibiting ClC-1 ubiquitination showed in vitro effectiveness in correcting the impaired trafficking of misfolded ClC-1 protein [120, 121]. In addition, acetazolamide, which is used empirically in myotonia, was shown to increase chloride currents in cultured cells expressing wild-type ClC-1 channels, likely through intracellular pH modulation [122]. Whether this occurs in skeletal muscle fibers and in vivo remains to be demonstrated. Moreover, it appeared that myotonic CLCN1 mutations might abolish this effect [123]. Thus, much more efforts are needed to identify useful drugs acting on ClC-1 channels.
Animal models of rare diseases are invaluable tools for a better understanding of the pathogenesis and the discovery of new drugs. Regarding chloride channel myotonia, studies on the genetic models, such as the myotonic goat and adr mouse, and the pharmacologically-induced myotonic rat, have paved the way for human studies [6, 38, 39, 105–108, 124–131]. Regarding sodium channel myotonia, two mouse models carrying human mutations associated with myotonia and hyperPP are available [132, 133]. Up to now, no study has been performed to compare effects of drugs between chloride and sodium channel myotonia models.
Periodic paralysis
The primary periodic paralyses (PP) are genetically heterogeneous disorders characterized by episodes of flaccid paralysis [9] (Fig. 1). Hyperkalemic PP (hyperPP) and hypokalemic PP type 2 (hypoPP2) are due to mutations in SCN4A gene encoding the Nav1.4 sodium channel [134–138]. Hypokalemic PP type 1 (hypoPP1) is due to mutations in CACNA1S encoding the Cav1.1 calcium channel [139–141]. Andersen-Tawil syndrome (ATS) is a multisystem disorder due to mutations in KCNJ2 encoding the inwardly-rectifying Kir2.1 potassium channel [142, 143]. It should be noted that a number of patients presenting with primary PP have no mutation in these genes. Attacks of weakness can also occur in paramyotonia congenita. Cases of normokalemic PP have been reported and usually attributed to SCN4A mutations [144]. Besides the familial cases, a number of periodic paralysis syndromes have also established genetic risk, including sporadic periodic paralysis (SPP) and thyrotoxic periodic paralysis (TPP). Thyrotoxic hypokalemic periodic paralysis present with weakness attacks resembling to hypoPP [145]. A few cases have been linked to mutations in KCNJ18 gene or single nucleotide polymorphisms in various genes, including CACNA1S. Symptoms generally resolved with treatment of hyperthyroidism. Sporadic periodic paralysis is defined by the lack of mutation in CACNA1S, SCN4A, or KCNJ2 genes, together with normal thyroid function. Yet, it appears more and more evident that TPP and SPP are closely related diseases, sharing genetic susceptibility [146, 147]. ATS, SPP, and TPP were not considered in the systematic review.
Treatment options of primary PP include adaptation of lifestyle and diet to avoid triggers of attacks [1, 9]. Pharmacological treatment can be used to abort attacks or to reduce the frequency of attacks. In hypokalemic PP, oral or intravenous potassium supplementation may abort the attack. To reduce attack frequency, the carbonic anhydrase inhibitors (CAI), acetazolamide and dichlorphenamide, are the preferred drugs. Potassium-sparing diuretics (eplerenone or spironolactone) can be a useful additional therapy to a CAI or may be used as an alternative in non-responders or patients intolerant to CAI. Regarding hyperkalemic PP, acute treatment of severe attacks traditionally aims at normalizing the serum potassium concentration, using for instance an infusion of glucose and insulin [148, 149]. A small number of case studies also suggested that inhaled salbutamol may abort acute attack [150, 151]. The mechanism of action of both insulin and salbutamol likely involves stimulation of the Na+/K+ pump that, in turn, increases uptake of K+, decreases intracellular Na+, and hyperpolarizes muscle membrane [152, 153]. In addition, although no controlled trial has been published, a few case studies reported mixed results regarding the empiric use of intravenous calcium gluconate to abort paralysis in hyperPP [154, 155]. A case report reported that hypocalcemia induced by chemotherapy worsened paralytic attacks in a hyperPP patient [156]. Only a very few studies reported hypocalcemia during an attack in hyperPP patients [157], so it is unclear whether hypocalcemia is a common feature of hyperPP. Thus, further investigation is needed to elucidate the role of calcium supplementation in hyperPP. Chronic therapy consists in the use of CAI or thiazide diuretics. In ATS, the pharmacotherapy is mainly aimed at the prevention of cardiac events, while treatment of flaccid paralysis follows the recommendations for hypokalemic or hyperkalemic PP, depending on the blood level of potassium during attacks. Today, dichlorphenamide is the sole FDA-approved treatment for both hypokalemic and hyperkalemic PP (see below), whereas all other options are based on individual clinical expertise.
Randomized clinical trials (class of evidence I)
Although acetazolamide (ACZ) has long been used in PP [158, 159], there is no documented randomized trial. In a single-blind study, the more potent CAI dichlorphenamide (DCP) was shown effective in reducing persistent interattack weakness in patents refractory to acetazolamide [160]. Thus, multicenter, randomized, double blind, placebo-controlled, crossover trials of DCP have been performed in cohorts of hypokalemic and hyperkalemic PP. In the first two studies, patients received either DCP (50 mg bid) or placebo for 9 weeks, followed by 9 weeks washout, then shifted to the other treatment for a further 9 weeks [161]. The patients were provided home diaries for recording the severity and duration of the paralytic attacks.
Because most of hypoPP patients were already under treatment before the trial, a worsening of attacks was expected during the placebo period. Thus, the primary outcome variable was withdrawal of patients from the treatment phase due to an intolerable increase in attack frequency or severity. Secondary outcome variables were the rate of attack (average number per week), the severity-weighted attack rate, and the patients self-reported treatment phase. From the 39 hypoPP patients entering the treatment phase, 34 completed the study, due to loss of follow-up (n = 1), individual requests (n = 2), or side effects under DCP treatment (n = 2, dizziness and concentrating difficulty). The most common side effect occurring with DCP was paresthesia (42 %). More than 10 % of patients receiving DCP complained anorexia, diarrhea, dizziness, cognitive impairment, pruritus, or skin rash. The DCP treatment improved all the primary and secondary outcome measures.
In the hyperPP trial, the primary outcome measure was the rate of attack and secondary outcome variables were the severity-weighted attack rate and the preferred treatment phase. Thirty patients entered the treatment phase and 24 completed the study, due to loss of follow-up (n = 2), individual request (n = 2), or side effects during DCP treatment (n = 2, skin rash or memory loss). The side effects observed during DCP treatment were similar to the hypoPP trial. All the primary and secondary outcome measures improved with DCP.
A post-hoc analysis of this RCT concluded that DCP was similarly effective and tolerated in a small number of adolescents and in adults, although different types of adverse events occurred in the two groups [162].
Thus, these studies clearly demonstrated the effectiveness of DCP in reducing the rate and severity of paralytic attacks in both hypoPP and hyperPP. As outlined by the authors, the trials however did not address interattack weakness and a number of patients were not genetically diagnosed.
More recently, two other RCT were performed in parallel to assess short-term and long-term effects of DCP in hypoPP and hyperPP [163]. Thus, the trials were subdivided in two phases: the first one comparing DCP to placebo effects upon 9 weeks treatment and the second phase assessing long-term effects of DCP alone upon one-year follow-up. Records of paralytic attacks were collected through an interactive voice response diary. During the placebo-controlled, double-blinded phase, the primary outcome variable was the average number of attacks per week. Secondary outcome measures included severity-weighted attack rate, total attack duration per week, changes in muscle strength, lean body mass, and quality of life assessed with the short-form 36 questionnaire. In addition, the endpoint of eventual acute worsening was measured in hypoPP. Similar outcome measures were performed in various periods during the one-year uncontrolled extension phase.
In the hypoPP trial, 44 patients started the short-term phase. Three dropouts were due to misdiagnosis (n = 1), noncompliance (n = 1), and side effects in the DCP arm (n = 1, mental fog). Significant improvement of median attack rate, severity-weighted attack rate, and attack duration was measured in the DCP arm. From the short-term 36, physical function, role-physical, bodily pain, vitality, and social functioning were significantly improved with DCP. Dropouts due to acute worsening was observed only in the placebo arm (n = 5). Then, 41 patients started the one-year follow-up phase, of whom 10 withdrew due to side effects (n = 8) or disease worsening (n = 2). Thirty-one patients maintained the benefits of first phase DCP or improved compared to first-phase placebo.
In the hyperPP study, of the 21 patients starting the first phase, 3 dropouts occurred in the DCP arm due to individual request (n = 1) or skin rash (n = 2). A significant effect of DCP was obtained only for the median severity-weighted attack rate. Seventeen patients started the one-year follow-up; one withdrew due to adverse event. Compared to the first phase (both DCP and placebo arms), positive effects of DCP were significant on median attack rate, severity-weighted attack rate, and total weekly attack duration.
In the two phases of both trials, paresthesia was the most common side effect. In conclusion, these studies confirmed the benefits of DCP in hypoPP in the short-term (class I evidence) and suggested beneficial long-term effects over side effects (Class II evidence). The demonstration of DCP benefits in hyperPP was less evident, likely due to insufficient patient recruitment. Even more so, it was not possible to correlate DCP effectiveness to genetic background.
Other human studies (class of evidence II-to-IV)
HypoPP is genetically heterogeneous; about 60–70 % of patients carry mutations in the calcium channel (hypoPP1), while 10–20 % have sodium channel mutations (hypoPP2). Based on case reports, it has been hypothesized that hypoPP2 patients may not respond to acetazolamide [164, 165]. Yet, other studies report positive response in hypoPP2 [137, 166, 167]. Interestingly, a retrospective study was performed to evaluate the rate of response to ACZ in hypoPP and looked at a possible correlation with genotype [168]. The study included three cohorts, one at a UK center (n = 14), one at a Chinese center (n = 4), and one from literature review (n = 56). Considering the three cohorts together, statistical analysis suggested that carriers of CACN1AS mutations are more likely to respond positively to the drug compared to SCN4A mutation carriers. The response rate was 56 % in the former, compared to only 16 % in the later. In addition, most of the refractory hypoPP2 patients had a clinical worsening with the drug. The authors further observed that all the patients with substitutions of a voltage-sensor arginine residue by a glycine residue, whether in CACN1AS or SCN4A, were the less responsive to acetazolamide. This suggests that response to acetazolamide may not be gene specific (i.e. not CACNA1S vs SCN4A) but mutation specific (i.e. R to H substitutions respond more favorably than R to other amino acid substitutions). These observations advocate the design of specific randomized controlled trials to verify the sensibility of individual hypoPP mutations to CAI.
Preclinical studies
The mechanism of action of CAI in PP is not clearly defined. It was proposed that acidosis might mediate CAI effects through modulation of various metabolic processes [169]. The drugs can also induce kaliuresis, which might theoretically be especially helpful in hyperPP but counterproductive in hypoPP. Both ACZ and DCP were shown to open directly calcium-activated potassium channels in skeletal muscle, thereby favoring sarcolemma repolarization, which may contribute to their beneficial effects in PP [170]. Finally, ACZ was found to increase chloride currents in ClC-1 transfected channels [122, 123], which might play a role in preventing paralytic attacks.
Most of the hypoPP mutations correspond to substitution of an arginine residue in the S4 helices of the sodium or calcium channels [171]. These positively charged amino acids are critical to sense voltage changes and their substitution by non-charged residues cause various gating defects corresponding to a loss of function [138, 172–177]. Interestingly, a low pH is able to correct the molecular defect of SCN4A mutants when mutations substitute histidine for arginine, likely because histidine side chain will “recover” a positive charge at low pH [176]. Such result provides a molecular rationale for the more favorable response of patients carrying R-H mutations to CAI-induced acidosis, as reported above [168]. However, studies aimed at tested direct effects of CAI on voltage-gated calcium or sodium channels have not been reported.
In addition, another mechanism is likely involved in the pathogenesis of hypoPP, which is the creation of an aberrant gating pore [178–180]. Such gating pore has been found to occur in most hypoPP channel mutants [99, 181–186], and allows small leakage cationic currents at resting membrane potential that facilitates the paradoxical depolarization at low [K +] causing muscle weakness [15].
One possibility to increase treatment effectiveness would be to identify drugs able to inhibit the gating pore currents. Sokolov and collaborators showed that inorganic divalent (Ba2 +, Zn2 +) and trivalent (Gd3 +, La3 +, and Yb3 +) cations can exert voltage-dependent or independent inhibition of the gating pore currents created by one hypoPP2 mutation [187]. In addition, an aromatic guanidinium derivative, 1-(2,4-xylyl)guanidine, also inhibited gating pore currents without affecting the physiological sodium current, providing proof of the concept that small organic molecules may be selective for gating pore current inhibition. Natural toxins targeting sodium-channel voltage sensors may be useful tools to better understand gating pore function [188]. Recently, a spider venom toxin was shown to inhibit gating pore currents generated by the hypoPP2 mutation p.R222G, showing a way for drug discovery strategy [189].
In hyperPP, it is hypothesized that most SCN4A mutations impair channel slow inactivation and produce a persistent sodium current that depolarizes sarcolemma, which, in turn, inactivates sodium channels and renders the fiber inexcitable [190]. Thus, a selective inhibitor of the late sodium current might be useful in hyperPP, whereas more conventional sodium channel blockers may appear counterproductive. Only a few case studies reported the lack of effect of mexiletine or worsening with carbamazepine in hyperPP [191, 192]. Up to now, effects of selective late sodium current inhibitors in hyperPP have not been published.
Mouse models have been generated for hyperPP (carrying p.M1592V or p.I588V in SCN4A), hypoPP2 (carrying p.R669H in SCN4A), and hypoPP1 (carrying p.R528H in CACNA1S). These models well recapitulate the main symptoms of the diseases [132, 133, 193, 194]. Of note, studies in the hyperPP mouse model showed that hypocalcemia exacerbates paralysis, as in humans [133, 195]. They showed that in wild-type muscle fibers, hypocalcemia induced depolarization and loss of force only when external K + was rised to 10 mM. In the hyperPP muscle fibers, such effects occurred already in presence of 4.7 mM external K + . These results might provide an explanation for the beneficial effects of calcium supplementation in a subset of hyperPP individuals [196].
Recently, the loop diuretic bumetanide was shown to be effective at preventing paralytic attacks in the mouse models of hypoPP1 and hypoPP2, and to abort attacks in the hypoPP2 model [197, 198]. In contrast, no effect was found in the hyperPP mouse model. By inhibiting the Na-K-2Cl transporter in skeletal muscle fibers, bumetanide prevents intracellular accumulation of chloride ions and thus contrasts the paradoxical depolarization causing paralysis [199]. Human trials are needed to verify whether bumetanide may be effective in periodic paralysis, also because the drug is known to induce pronounced kaliuresis. A small cohort study showed limited effect of bumetanide in rescuing from paralytic attacks in hypoPP [200].
Congenital myopathies related to SCN4A/CACNA1S
Myopathy is generally not a predominant trait of gain-of-function muscle ion channelopathies, with only some rare exceptions [60, 77, 84, 201, 202]. Non-dystrophic myotonias can be associated with a Herculean appearance, due to continuous muscle contractions. The exacerbation of muscle activity also modifies the pattern of muscle fiber types, with a reduced expression of fast-twitch type 2B muscle fibers [203, 204]. Atrophy of specific muscles or groups of muscles have been also reported, but the causes are unknown.
Hyperkalemic and hypokalemic periodic paralyses can lead to late-onset permanent weakness with vacuolar myopathy and/or tubular aggregate myopathy [165, 205, 206]. Interestingly, acetazolamide was shown to prevent vacuolar myopathy in K+-depleted rats, a model of hypoPP, but whether this occurs in humans is unknown [207].
Loss-of-function mutations are rarer but generally more severe (Fig. 1). Congenital myasthenic syndrome has been reported in a few patients carrying two mutations in SCN4A [208–212]. The functional study of the mutant channels showed partial loss of function. Most patients did not respond to pyridostigmine treatment, while acetazolamide provided benefits in some but not all the patients [11].
Recently, a new clinical phenotype with life-threatening neonatal hypotonia and severe congenital myopathy was described in patients harboring recessive loss-of-function mutations in SCN4A [213]. This study reported 11 cases from 6 kindred, including 7 deaths during perinatal period and 4 adults presenting with congenital myopathy without myotonia. Several patients with variable myopathic traits have been since reported [214–216]. These cases likely represent a continuum of congenital myasthenia with the occurrence of more severe loss-of-function mutations with at least one null mutation (mutation that does not produce any functional protein) [217].
More complicated phenotypes combining myotonia, periodic paralysis, myasthenia, and/or myopathy have been described and expand the clinical spectrum of sodium channelopathies [60, 64, 77, 202, 218–220].
Whole exome sequencing of a cohort of congenital myopathic patients evidenced recessive mutations in CACNA1S in one severely affected patient [221]. Congenital myopathy with perinatal hypotonia and severe generalized permanent weakness was also described in 11 carriers of CACNA1S mutations determining reduced expression and/or abnormal cell localization of calcium channels [222]. Mutations were inherited either dominantly or recessively. Acetazolamide had positive effects on flaccid paralysis in one patient. Other cases have been since reported [223–226]. In addition, Cav1.1 dysfunction is the likely mechanism responsible for the disease in Native American myopathy and other congenital myopathies caused primarily by mutations in STAC3 [227–229]. Indeed, Stac3 protein (SH3 and cysteine-rich domain 3) is an essential component of the EC coupling apparatus and enhances Cav1.1 channel expression [230–232].
Because these myopathic diseases are genetically and phenotypically heterogeneous, they likely represent a big challenge for pharmacological treatment. Their extreme rarity makes RCT quite impossible, although there may be a possibility to perform N-of-1 trials. Interestingly, a two-case study suggests that acetazolamide may be beneficial in both SCN4A and CACNA1S related myopathies [233].
CONCLUSION
In the recent years, evidence-class 1 RCT have demonstrated the effectiveness of mexiletine and lamotrigine in NDM and of dichlorphenamide in hyperPP and hypoPP. However, these studies provided little information about the response to treatments of individual mutations or groups of mutations. In most cases, the drugs address the symptoms without interfering directly with the mutated channel defect. Thus, high dosages may be required to obtain full remission of the symptoms, exposing patients to higher risks of side effects, little compliance, and dropout. Second choice drugs are available, but they are often tested by chance in an empirical manner.
In the case of myotonia due to sodium channel mutations (SCM and PMC), some preclinical data and translational studies support the possibility to use a rational approach to choose a specific treatment for individual mutations or groups of mutations. For instance, flecainide may prove advantageous over mexiletine in patients carrying SCN4A mutations determining a left shift of inactivation voltage dependence, such as p.G1306E. Dedicated RCT comparing exploratory drugs to mexiletine in determined genetic background are needed to confirm such hypothesis.
In MC, much effort is needed in drug discovery and preclinical studies to identify new drugs able to correct the gating or trafficking defects induced by CLCN1 mutations.
In hyperPP, since the ultimate step of cellular pathomechanism may be inactivation of sodium channels, conventional sodium channel blockers may not be recommended; yet, drugs acting on specific gating properties of the mutant sodium channel would merit consideration. The RCT showed only a partial response to DCP compared to placebo. A trial comparing effectiveness and safety of DCP versus K+-wasting diuretics might be interesting.
In hypoPP, a major effort must be made to find drugs able to inhibit the gating-pore current produced by SCN4A and CACNA1S mutations.
Now that we have major information regarding the molecular mechanisms by which mutations alter ion channel function, the door is open to enter the new era of precision medicine in skeletal muscle channelopathies.
FUNDING
JFD received research grants for non-dystrophic myotonia from the Italian Telethon Foundation, French “Association contre les Myopathies”, and Bari University. BF received research grants provided by AFM-Telethon.
COMPETING INTERESTS
JFD is co-inventor of the international patent “safinamide for treating myotonia” (number WO 2019/22902S A1).
REFERENCES
[1] | Phillips L , Trivedi JR . Skeletal Muscle Channelopathies. Neurotherapeutics. (2018) ;15: (4):954–65. doi:10.1007/s13311-018-00678-0 |
[2] | Vivekanandam V , Männikkö R , Matthews E , Hanna MG . Improving genetic diagnostics of skeletal muscle channelopathies. Expert Rev Mol Diagn. (2020) ;20: (7):725–36. doi:10.1080/14737159.2020.1782195 |
[3] | Imbrici P , Liantonio A , Camerino GM , De Bellis M , Camerino C , Mele A , et al. Therapeutic approaches to genetic ion channelopathies and perspectives in drug discovery. Front Pharmacol. (2016) ;7: :121. doi:10.3389/fphar.2016.00121 |
[4] | Stunnenberg BC , LoRusso S , Arnold WD , Barohn RJ , Cannon SC , Fontaine B , et al. Guidelines on clinical presentation and management of nondystrophic myotonias. Muscle Nerve. 2020 Apr 8. doi: 10.1002/mus.26887. Online ahead of print. |
[5] | Desaphy J-F , Conte Camerino D , Franchini C , Lentini G , Tortorella V , De Luca A . Increased hindrance on the chiral carbon atom of mexiletine enhances the block of rat skeletal muscle Na+channels in a model of myotonia induced by ATX. Br J Pharmacol. (1999) ;128: (6):1165–74. doi: 10.1038/sj.bjp.0702901 |
[6] | De Luca A , Pierno S , Liantonio A , Desaphy J-F , Natuzzi F , Didonna MP , et al. New potent mexiletine and tocainide analogues evaluated in vivo and in vitro as antimyotonic agents on the myotonic ADR mouse. Neuromuscul Disord. (2004) ;14: (7):405–16. doi: 10.1016/j.nmd.2004.04.006 |
[7] | Trip J , Drost G , van Engelen BG , Faber CG . Drug treatment for myotonia. Cochrane Database Syst Rev. (2006) ;(1):CD004762. doi: 10.1002/14651858.CD004762.pub2 |
[8] | Statland JM , Bundy BN , Wang Y , Rayan DR , Trivedi JR , Sansone VA , et al. Mexiletine for symptoms and signs of myotonia in nondystrophic myotonia: A randomized controlled trial. JAMA. (2012) ;308: (13):1357–65. doi: 10.1001/jama.2012.12607 |
[9] | Statland JM , Fontaine B , Hanna MG , Johnson NE , Kissel JT , Sansone VA , et al. Review of the diagnosis and treatment of periodic paralysis. Muscle Nerve. (2018) ;57: (4):522–30. doi: 10.1002/mus.26009 |
[10] | Sansone V , Meola G , Links TP , Panzeri M , Rose MR . Treatment for periodic paralysis. Cochrane Database Syst Rev. (2008) ;(1):CD005045. doi: 10.1002/14651858.CD005045.pub2. |
[11] | Thompson R , Bonne G , Missier P , Lochmüller H . Targeted therapies for congenital myasthenic syndromes: Systematic review and steps towards a treatabolome. Emerg Top Life Sci. (2019) ;3: (1):19–37. doi: 10.1042/ETLS20180100 |
[12] | Atalaia A , Thompson R , Corvo A , Carmody L , Piscia D , Matalonga L , et al. Orphanet J Rare Dis. (2020) ;15: :206. doi:10.1186/s13023-020-01493-7 |
[13] | Suetterlin K , Männikkö R , Hanna MG . Muscle channelopathies: Recent advances in genetics, pathophysiology and therapy. Curr Opin Neurol. (2014) ;27: :583–90. doi: 10.1097/WCO.0000000000000127 |
[14] | Nicole S , Fontaine B . Skeletal muscle sodium channelopathies. Curr Opin Neurol. (2015) ;28: (5):508–14. doi: 10.1097/WCO.0000000000000238 |
[15] | Cannon SC . Channelopathies of skeletal muscle excitability. Compr Physiol. (2015) ;5: (2):761–90. doi:10.1002/cphy.c140062 |
[16] | Horga A , Raja Rayan DL , Matthews E , Sud R , Fialho D , Durran SC , et al. Prevalence study of genetically defined skeletal muscle channelopathies in England. Neurology. (2013) ;80: :1472–5. doi:10.1212/WNL.0b013e31828cf8d0 |
[17] | Koch MC , Steinmeyer K , Lorenz C , Ricker K , Wolf F , Otto M et al. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. (1992) ;257: (5071):797–800. doi:10.1126/science.1379744 |
[18] | George AL Jr , Crackower MA , Abdalla JA , Hudson AJ , Ebers GC . 1993. Molecular basis of Thomsen’s disease (autosomal dominant myotonia congenita). Nat Genet. (1993) ;3: (4):305–10. doi:10.1038/ng0493-305 |
[19] | Altamura C , Desaphy JF , Conte D , De Luca A , Imbrici P . Skeletal muscle ClC-1 chloride channels in health and diseases. Pflugers Arch. (2020) ;472: (7):961–75. doi:10.1007/s00424-020-02376-3 |
[20] | McClatchey AI , Van den Bergh P , Pericak-Vance MA , Raskind W , Verellen C , McKenna-Yasek D , et al. Tempera-ture-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell. (1992) ;68: (4):769–74. doi: 10.1016/0092-8674(92)90151-2 |
[21] | Ptacek LJ , George AL Jr , Barchi RL , Griggs RC , Riggs JE , Robertson M , et al. Mutations in an S4 segment of the adult skeletal muscle sodium channel cause paramyotonia congenita. Neuron. (1992) ;8: :891–7. doi:10.1016/0896-6273(92)90203-p |
[22] | Lerche H , Heine R , Pika U , George AL Jr , Mitrovic N , Browatzki M , et al. Human sodium channel myotonia: Slowed channel inactivation due to substitutions for a glycine within the III-IV linker. J Physiol. (1993) ;470: :13–22. doi:10.1113/jphysiol.1993.sp019843 |
[23] | Gay S , Dupuis D , Faivre L , Masurel-Paulet A , Labenne M , Colombani M , et al. Severe neonatal non-dystrophic myotonia secondary to a novel mutation of the voltage-gated sodium channel (SCN4A) gene. Am J Med Genet A. (2008) ;146A: :380–3. doi:10.1002/ajmg.a.32141 |
[24] | Lion-Francois L , Mignot C , Vicart S , Manel V , Sternberg D , Landrieu P , et al. Severe neonatal episodic laryngospasm due to de novo SCN4A mutations: A new treatable disorder. Neurology. (2010) ;75: (7):641–5. doi: 10.1212/WNL.0b013e3181ed9e96 |
[25] | Matthews E , Manzur AY , Sud R , Muntoni F , Hanna MG . Stridor as a neonatal presentation of skeletal muscle sodium channelopathy. Arch Neurol. (2011) ;68: (1):127–9. doi:10.1001/archneurol.2010.347 |
[26] | Portaro S , Rodolico C , Sinicropi S , Musumeci O , Valenzise M , Toscano A . Flecainide-responsive myotonia permanens with SNEL onset: A new case and literature review. Pediatrics. (2016) ;137: (4). doi:10.1542/peds.2015-3289 |
[27] | Lehmann-Horn F , D’Amico A , Bertini E , Lomonaco M , Merlini L , Nelson KR , et al. Myotonia permanens with Nav1.4-G1306E displays varied phenotypes during course of life. Acta Myol. (2017) ;36: (3):125–34. |
[28] | Cea G , Andreu D , Fletcher E , Ramdas S , Sud R , Hanna MG , et al. Sodium channel myotonia may be associated with high-risk brief resolved unexplained events. Version 2. Wellcome Open Res. (2020) ;5: :57. doi:10.12688/wellcomeopenres.15798.2 |
[29] | Männikkö R , Wong L , Tester DJ , Thor MG , Sud R , Kullmann DM , et al. Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: A case-control study. Lancet. (2018) ;391: (10129):1483–92. doi:10.1016/S0140-6736(18)30021-7 |
[30] | Heatwole CR , Statland JM , Logigian EL . The diagnosis and treatment of myotonic disorders. Muscle Nerve. (2013) ;47: (5):632–48. doi:10.1002/mus.23683 |
[31] | Griggs RC , Moxley RT 3rd , Riggs JE , Engel WK . Effects of acetazolamide on myotonia. Ann Neurol. (1978) ;3: (6):531–7. doi:10.1002/ana.410030614 |
[32] | Hoffman EP , Kaminski HJ . Mexiletine for treatment of myotonia: A trial triumph for rare disease networks. JAMA. (2012) ;308: (13):1377–8. doi:10.1001/jama.2012.12906 |
[33] | Stunnenberg BC , Raaphorst J , Groenewoud HM , Statland JM , Griggs RC , Woertman W , et al. Effect of mexiletine on muscle stiffness in patients with nondystrophic myotonia evaluated using aggregated N-of-1 trials. JAMA. (2018) ;320: (22):2344–53. doi:10.1001/jama.2018.18020 |
[34] | Andersen G , Hedermann G , Witting N , Duno M , Andersen H , Vissing J . The antimyotonic effect of lamotrigine in non-dystrophic myotonias: A double-blind randomized study. Brain. (2017) ;140: (9):2295–305. doi:10.1093/brain/awx192 |
[35] | Arnold WD , Kline D , Sanderson A , Hawash AA , Bartlett A , Novak KR , et al. Open-label trial of ranolazine for the treatment of myotonia congenita. Neurology. (2017) ;89: (7):710–3. doi:10.1212/WNL.0000000000004229 |
[36] | Lorusso S , Kline D , Bartlett A , Freimer M , Agriesti J , Hawash AA , et al. Open-label trial of ranolazine for the treatment of paramyotonia congenita. Muscle Nerve. (2019) ;59: (2):240–3. doi:10.1002/mus.26372 |
[37] | El-Bizri N , Kahlig KM , Shyrock JC , George AL Jr , Belardinelli L , Rajamani S . Ranolazine block of human Nav1.4 sodium channels and paramyotonia congenita mutants. Channels (Austin). (2011) ;5: (2):161–72. doi:10.4161/chan.5.2.14851 |
[38] | Novak KR , Norman J , Mitchell JR , Pinter MJ , Rich MM . Sodium channel slow inactivation as a therapeutic target for myotonia congenita. Ann Neurol. (2015) ;77: (2):320–32. doi:10.1002/ana.24331 |
[39] | Hawash AA , Voss AA , Rich MM . Inhibiting persistent inward sodium currents prevents myotonia. Ann Neurol. (2017) ;82: (3):385–95. doi:10.1002/ana.25017 |
[40] | Wang GK , Calderon J , Wang SY . State- and use-dependent block of muscle Nav1.4 and neuronal Nav1.7 voltage-gated Na+channel isoforms by ranolazine. Mol Pharmacol. (2008) ;73: (3):940–8. doi: 10.1124/mol.107.041541 |
[41] | Suetterlin KJ , Bugiardini E , Kaski JP , Morrow JM , Matthews E , Hanna MG , et al. Long-term safety and efficacy of mexiletine for patients with skeletal muscle channelopathies. JAMA Neurol. (2015) ;72: (12):1531–3. doi:10.1001/jamaneurol.2015.2338 |
[42] | Modoni A , D’Amico A , Primiano G , Capozzoli F , Desaphy J-F , Lo Monaco M . Long-term safety and usefulness of mexiletine in a large cohort of patients affected by non-dystrophic myotonias. Front. Neurol. (2020) ;11: :300. doi:10.3389/fneur.2020.00300 |
[43] | Lo Monaco M , D’Amico A , Luigetti M , Desaphy JF , Modoni A . Effect of mexiletine on transitory depression of compound motor action potential in recessive myotonia congenita. Clin Neurophysiol. (2015) ;126: (2):399–403. doi: 10.1016/j.clinph.2014.06.008 |
[44] | Ginanneschi F , Mignarri A , Lucchiari S , Ulzi G , Comi GP , Rossi A , et al. Neuromuscular excitability changes produced by sustained voluntary contraction and response to mexiletine in myotonia congenita. Neurophysiol Clin. (2017) ;47: :247–52. doi:10.1016/j.neucli.2017.01.003 |
[45] | Meyer AP , Roggenbuck J , LoRusso A , Kissel J , Smith RM , Kline D , et al. Genotype-Phenotype Correlations and Characterization of Medication Use in Inherited Myotonic Disorders. Front Neurol. (2020) ;11: :593. doi:10.3389/fneur.2020.00593 |
[46] | Pagliarani S , Lucchiari S , Scarlato M , Redaelli E , Modoni A , Magri F , et al. Sodium Channel Myotonia Due to Novel Mutations in Domain I of Nav1.4. Front Neurol. (2020) ;11: :255. doi:10.3389/fneur.2020.00255 |
[47] | Lehmann-Horn F , Orth M , Kuhn M , Jurkat-Rott K . A novel N440K sodium channel mutation causes myotonia with exercise-induced weakness–exclusion of CLCN1 exon deletion/duplication by MLPA. Acta Myol. (2011) ;30: (2):133–7. |
[48] | Rosenfeld J , Sloan-Brown K , George AL Jr. . A novel muscle sodium channel mutation causes painful congenital myotonia. Ann Neurol. (1997) ;42: :811–4. doi:10.1002/ana.410420520 |
[49] | Trip J , Faber CG , Ginjaar HB , van Engelen BG , Drost G . Warm-up phenomenon in myotonia associated with the V445M sodium channel mutation. J Neurol. (2007) ;254: (2):257–8. doi:10.1007/s00415-006-0353-2 |
[50] | Al-Ghamdi F , Darras BT , Ghosh PS . Spectrum of nondystrophic skeletal muscle channelopathies in children. Pediatr Neurol. (2017) ;70: :26–33. doi:10.1016/j.pediatrneurol.2017.02.006 |
[51] | Matsumoto N , Nishimoto R , Matsuoka Y , Takeda Y , Morimatsu H . Anesthetic management of a patient with sodium-channel myotonia: A case report. JA Clin Rep. (2019) ;5: (1):77. doi:10.1186/s40981-019-0300-8 |
[52] | Dupré N , Chrestian N , Bouchard JP , Rossignol E , Brunet D , Sternberg D , et al. Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscul Disord. (2009) ;19: (5):330–4. doi: 10.1016/j.nmd.2008.01.007 |
[53] | Markhorst JM , Stunnenberg BC , Ginjaar IB , Drost G , Erasmus CE , Sie LTL . Clinical experience with longterm acetazolamide treatment in children with nondystrophic myotonias: A three-case report. Pediatr Neurol. (2014) ;51: :537–41. doi:10.1016/j.pediatrneurol.2014.05.027 |
[54] | Hata T , Nagasaka T , Koh K , Tsuchiya M , Ichinose Y , Nan H et al. Pathological findings in a patient with non-duystrophic myotonia with a mutation of the SCN4A gene; a cse report. BMC Neurology. (2019) ;19: :125. doi:10.1186/s12883-019-1360-0 |
[55] | Yoshinaga H , Sakoda S , Good JM , Takahashi MP , Kubota T , Arikawa-Hirasawa E , et al. A novel mutation in SCN4A causes severe myotonia and school-age-onset paralytic episodes. J Neurol Sci. (2012) ;315: (1-2):15–9. doi: 10.1016/j.jns.2011.12.015 |
[56] | Mankodi A , Grunseich C , Skov M , Cook L , Aue G , Purev E , et al. Divalent cation-responsive myotonia and muscle paralysis in skeletal muscle sodium channelopathy. Neuromuscul Disord. (2015) ;25: (11):908–12. doi:10.1016/j.nmd.2015.08.007 |
[57] | Avila-Smirnow D , Vargas Leal CP , Beytía Reyes MLA , Cortés Zepeda R , Escobar RG , Kleinsteuber Saa K , et al. Non-dystrophic myotonia Chilean cohort with predominance of the SCN4A Gly1306Glu variant. Neuromuscul Disord. (2020) ;30: (7):554–61. doi:10.1016/j.nmd.2020.04.006 |
[58] | Brancati F , Valente EM , Davies NP , Sarkozy A , Sweeney MG , LoMonaco M , et al. Severe infantile hyperkalaemic periodic paralysis and paramyotonia congenita: Broadening the clinical spectrum associated with the T704M mutation in SCN4A. J Neurol Neurosurg Psychiatry. (2003) ;74: (9):1339–41. doi:10.1136/jnnp.74.9.1339 |
[59] | Pechmann A , Eckenweiler M , Schorling D , Stavropoulou D , Lochmüller H , Kirschner J . De novo variant in SCN4A causes neonatal sodium channel myotonia with general muscle stiffness and respiratory failure. Neuromuscul Disord. (2019) ;29: (11):907–9. doi:10.1016/j.nmd.2019.09.001 |
[60] | Elia N , Nault T , McMillan HJ , Graham GE , Huang L , Cannon SC . Myotonic myopathy with secondary joint and skeletal anomalies from the c.2386C>G, p.L769V mutation in SCN4A. Front Neurol. (2020) ;11: :77. doi:10.3389/fneur.2020.00077 |
[61] | Desaphy J-F , Carbonara R , D’Amico A , Modoni A , Roussel J , Imbrici P , et al. Translational approach to address therapy in myotonia permanens due to a new SCN4A mutation. Neurology. (2016) ;86: (22):2100–8. doi:10.1212/WNL.0000000000002721 |
[62] | Sugiura Y , Aoki T , Sugiyama Y , Hida C , Ogata M , Yamamoto T . Temperature-sensitive sodium channelo-pathy with heat-induced myotonia and cold-induced paralysis. Neurology. (2000) ;54: (11):2179–81. doi:10.1212/wnl.54.11.2179 |
[63] | Kokunai Y , Goto K , Kubota T , Fukuoka T , Sakoda S , Ibi T , et al. A sodium channel myotonia due to a novel SCN4A mutation accompanied by acquired autoimmune myasthenia gravis. Neurosci Lett. (2012) ;519: (1):67–72. doi:10.1016/j.neulet.2012.05.023 |
[64] | Magot A , David A , Sternberg D , Péréon Y . Focal and abnormally persistent paralysis associated with congenital paramyotonia. BMJ Case Rep. (2014) ;2014: :bcr2014204430. doi:10.1136/bcr-2014-20443 |
[65] | Farinato A , Altamura C , Imbrici P , Maggi L , Bernasconi P , Mantegazza R , et al. Pharmacogenetics of myotonic hNav1.4 sodium channel mutants situated near the inactivation gate. Pharmacol Res. (2019) ;141: :224–35. doi: 10.1016/j.phrs.2019.01.004 |
[66] | Torbergsen T , Jurkat-Rott K , Stålberg EV , Løseth S , Hødneø A , Lehmann-Horn F . Painful cramps and giant myotonic discharges in a family with the Nav1.4-G1306A mutation. Muscle Nerve. (2015) ;52: (4):680–3. doi:10.1002/mus.24672 |
[67] | Colding-Jørgensen E , Duno M , Vissing J . Autosomal dominant monosymptomatic myotonia permanens. Neurology. (2006) ;67: :153–5. doi:10.1212/01.wnl.0000223838.88872.da |
[68] | Desaphy J-F , Modoni A , Lomonaco M , Camerino DC . Dramatic improvement of myotonia permanens with flecainide: A two-case report of a possible bench-to-bedside pharmacogenetics strategy. Eur J Clin Pharmacol. (2013) ;69: (4):1037–9. doi:10.1007/s00228-012-1414-3 |
[69] | Caietta E , Milh M , Sternberg D , Lépine A , Boulay C , McGonigal A , et al. Diagnosis and outcome of SCN4A-related severe neonatal episodic laryngospasm (SNEL): 2 new cases. Pediatrics. (2013) ;132: (3):e784–7. doi:10.1542/peds.2012-3065 |
[70] | Singh RR , Tan SV , Hanna MG , Robb SA , Clarke A , Jungbluth H . Mutations in SCN4A: A rare but treatable cause of recurrent life-threatening laryngospasm. Pediatrics. (2014) ;134: (5):e1447–50. doi:10.1542/peds.2013-3727 |
[71] | Cavalli M , Fossati B , Vitale R , Brigonzi E , Ricigliano VAG , Saraceno L , et al. Flecainide-induced Brugada syndrome in a patient with skeletal muscle sodium channelopathy: A case report with critical therapeutical implications and review of the literature. Front Neurol. (2018) ;9: :385. doi:10.3389/fneur.2018.00385 |
[72] | Jackson CE , Barohn RJ , Ptacek LJ . Paramyotonia congenita: Abnormal short exercise test, and improvement after mexiletine therapy. Muscle Nerve. (1994) ;17: (7):763–8. doi:10.1002/mus.880170710 |
[73] | Kinoshita M , Sasaki R , Nagano T , Matsuda A , Nakamura S , Takahama M , et al. Thr11313Met mutation in skeletal muscle sodium channels in a Japanese family with paramyotonia Congenita. Intern Med. (2003) ;42: :856–61. doi:10.2169/internalmedicine.42.856 |
[74] | Fukudome T , Izumoto H , Goto H , Matsuo H , Yoshimura T , Sakoda SI , et al. Paramyotonia congenita due to a de novo mutation: A case report, Muscle Nerve. (2003) ;28: :232–5. doi:10.1002/mus.10396 |
[75] | Alfonsi E , Merlo IM , Tonini M , Ravaglia S , Brugnoni R , Gozzini A , et al. Efficacy of propafenone in paramyotonia congenita. Neurology. (2007) ;68: (13):1080–1. doi:10.1212/01.wnl.0000257825.29703.e8 |
[76] | Terracciano C , Farina O , Esposito T , Lombardi L , Napolitano F , Blasiis P , et al. Successful long-term therapy with flecainide in a family with paramyotonia congenita. J Neurol Neurosurg Psychiatry. (2018) ;89: (11):1232–4. doi: 10.1136/jnnp-2017-317615 |
[77] | Taminato T , Mori-Yoshimura M , Miki J , Sasaki R , Sato N , Oya Y , et al. Paramyotonia Congenita with Persistent Distal and Facial Muscle Weakness: A Case Report with Literature Review. J Neuromuscul Dis. (2020) ;7: (2):193–201. doi:10.3233/JND-190440 |
[78] | Bissay V , Keymolen K , Lissens W , Laureys G , Schmedding E , De Keyser J . Late onset painful cold-aggravated myotonia: Three families with SCN4A L1436P mutation. Neuromuscul Disord. (2011) ;21: (8):590–3. doi:10.1016/j.nmd.2011.05.006 |
[79] | Plassart E , Eymard B , Maurs L , Hauw JJ , Lyon-Caen O , Fardeau M et al. Paramyotonia congenita: Genotype to phenotype correlations in two families and report of a new mutation in the sodium channel gene. J Neurol Sci. (1996) ;142: (1-2):126–33. doi:10.1016/0022-510x(96)00173-6 |
[80] | Sallansonnet-Froment M , Bounolleau P , De Greslan T , Ricard D , Taillia H , Renard JL . Paramyotonie congénitale d’Eulenburg [Eulenburg’s paramyotonia congenita]. Rev Neurol (Paris). (2007) ;163: (11):1083–90. doi:10.1016/s0035-3787(07)74181-4 |
[81] | Khadilkar SV , Singh RK , Mansukhani KA , Urtizberea JA , Sternberg D . Relief from episodic weakness with pyridostigmine in paramyotonia congenita: A family study. Muscle Nerve. (2010) ;41: (1):133–7. doi:10.1002/mus.21434 |
[82] | van Osch T , Stunnenberg BC , Sternberg D , Kerklaan BJ . Prolonged attacks of weakness with hypokalemia in SCN4A-related paramyotonia congenita. Muscle Nerve. (2018) ;58: (4):E27–8. doi:10.1002/mus.26190 |
[83] | Rossignol E , Mathieu J , Thiffault I , Tétreault M , Dicaire MJ , Chrestian N , et al. A novel founder SCN4A muta-tion causes painful cold-induced myotonia in French-Canadians. Neurology. (2007) ;69: (20):1937–41. doi:10.1212/01.wnl.0000290831.08585.2c |
[84] | Schoser BG , Schröder JM , Grimm T , Sternberg D , Kress W . A large German kindred with cold-aggravated myotonia and a heterozygous A1481D mutation in the SCN4A gene. Muscle Nerve. (2007) ;35: (5):599–606. doi:10.1002/mus.20733 |
[85] | Ferriby D , Stojkovic T , Sternberg D , Hurtevent JF , Hurtevent JP , Vermersch P . A new case of autosomal dominant myotonia associated with the V1589M missense mutation in the muscle sodium channel gene and its phenotypic classification. Neuromuscul Disord. (2006) ;16: (5):321–4. doi:10.1016/j.nmd.2006.01.015 |
[86] | Rempe T , Subramony SH . “Status myotonicus” in Nav1.4-M1592V channelopathy. Neuromuscul Disord. (2020) ;30: (5):424–6. doi:10.1016/j.nmd.2020.03.002 |
[87] | Kubota T , Kinoshita M , Sasaki R , Aoike F , Takahashi MP , Sakoda S , et al. New mutation of the Na channel in the severe form of potassium-aggravated myotonia. Muscle Nerve. (2009) ;39: (5):666–73. doi:10.1002/mus.21155 |
[88] | Wu FF , Gordon E , Hoffman EP , Cannon SC . A C-terminal skeletal muscle sodium channel mutation associated with myotonia disrupts fast inactivation. J Physiol. (2005) ;565: (Pt 2):371–80. doi:10.1113/jphysiol.2005.082909 |
[89] | Benstead TJ , Camfield PR , King DB . Treatment of paramyotonia congenita with acetazolamide. Can J Neurol Sci. (1987) ;14: (2):156–8. doi:10.1017/s0317167100026305 |
[90] | Trudell RG , Kaiser KK , Griggs RC . Acetazolamide-responsive myotonia congenita. Neurology. (1987) ;37: (3):488–91. doi:10.1212/wnl.37.3.488 |
[91] | Moreira SD , Barreto R , Roriz JM . Becker myotonia - a recently identified mutation in iberian descendants with apparent acetazolamide-responsive phenotype. Muscle Nerve. (2015) ;51: :9334. doi:10.1002/mus.24534 |
[92] | Sun C , Van Ghelue M , Tranebjærg L , Thyssen F , Nilssen Ø , Torbergsen T . Myotonia congenita and myotonic dystrophy in the same family: Coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet. (2011) ;80: :574–80. doi:10.1111/j.1399-0004.2010.01616.x |
[93] | Cardani R , Giagnacovo M , Botta A , Rinaldi F , Morgante A , Udd B et al. Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2. J Neurol. (2012) ;259: :2090–9. doi:10.1007/s00415-012-6462-1 |
[94] | Furby A , Vicart S , Camdessanché JP , Fournier E , Chabrier S , Lagrue E et al. Heterozygous CLCN1 mutations can modulate phenotype in sodium channel myotonia. Neuromuscul Disord. (2014) ;24: :953–9. doi:10.1016/j.nmd.2014.06.439 |
[95] | Bugiardini E , Rivolta I , Binda A , Soriano Caminero A , Cirillo F , Cinti A et al. SCN4A mutation as modifying factor of myotonic dystrophy type 2 phenotype. Neuromuscul Disord. (2015) ;25: :301–7. doi:10.1016/j.nmd.2015.01.006 |
[96] | Kato H , Kokunai Y , Dalle C , Kubota T , Madokoro Y , Yuasa H et al. A case of non-dystrophic myotonia with concomitant mutations in the SCN4A and CLCN1 genes. J Neurol Sci. (2016) ;369: :254–8. doi:10.1016/j.jns.2016.08.030 |
[97] | Maggi L , Ravaglia S , Farinato A , Brugnoni R , Altamura C , Imbrici P , et al. Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics. (2017) ;18: (4):219–25. doi:10.1007/s10048-017-0525-5 |
[98] | Binda A , Renna LV , Bosè F , Brigonzi E , Botta A , Valaperta R , et al. SCN4A as modifier gene in patients with myotonic dystrophy type 2. Sci Rep. (2018) ;8: (1):11058. doi:10.1038/s41598-018-29302-z |
[99] | Thor MG , Vivekanandam V , Sampedro-Castañeda M , Tan SV , Suetterlin K , Sud R , et al. Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation. Sci Rep. (2019) ;9: (1):17560. doi:10.1038/s41598-019-54041-0 |
[100] | Fan Z , George AL Jr , Kyle JW , Makielski JC . Two human paramyotonia congenita mutations have opposite effects on lidocaine block of Na+channels expressed in a mammalian cell line. J Physiol. (1996) ;496: (Pt 1):275–86. doi: 10.1113/jphysiol.1996.sp021684 |
[101] | Takahashi MP , Cannon SC . Mexiletine block of disease-associated mutations in S6 segments of the human skeletal muscle Na(+) channel. J Physiol. (2001) ;537: (Pt 3):701–14. doi:10.1111/j.1469-7793.2001.00701.x |
[102] | Desaphy J-F , De Luca A , Tortorella P , De Vito D , George AL Jr , Conte Camerino D . Gating of myotonic Na channel mutants defines the response to mexiletine and a potent derivative. Neurology. (2001) ;57: :1849–57. doi:10.1212/wnl.57.10.1849 |
[103] | Desaphy J-F , De Luca A , Didonna MP , George AL Jr , Conte Camerino D . Different flecainide sensitivity of hNav1.4 channels and myotonic mutants explained by state-dependent block. J Physiol. (2004) ;554: :321–34. doi: 10.1113/jphysiol.2003.046995 |
[104] | Desaphy J-F , Dipalma A , Costanza T , Bruno C , Lentini G , Franchini C , et al. Molecular determinants of state-dependent block of voltage-gated sodium channels by pilsicainide. Br J Pharmacol. (2010) ;160: (6):1521–33. doi: 10.1111/j.1476-5381.2010.00816.x |
[105] | De Bellis M , Carbonara R , Roussel J , Farinato A , Massari A , Pierno S , et al. Increased sodium channel use-dependent inhibition by a new potent analogue of tocainide greatly enhances in vivo antimyotonic activity. Neuropharmacology. (2017) ;113: (Pt A):206–16. doi: 10.1016/j.neuropharm.2016.10.013 |
[106] | Desaphy J-F , Costanza T , Carbonara R , Conte Camerino D . In vivo evaluation of antimyotonic efficacy of β-adrenergic drugs in a rat model of myotonia. Neuropharmacology. (2013) ;65: :21–7. doi:10.1016/j.neuropharm.2012.09.006 |
[107] | Desaphy J-F , Carbonara R , Costanza T , Conte Camerino D . Preclinical evaluation of marketed sodium channel blockers in a rat model of myotonia discloses promising antimyotonic drugs. Exp Neurol. (2014) ;255: :96–102. doi: 10.1016/j.expneurol.2014.02.023 |
[108] | Desaphy J-F , Farinato A , Altamura C , De Bellis M , Imbrici P , Tarantino N , et al. Safinamide potential in nondystrophic myotonias: Inhibition of skeletal muscle voltage-gated sodium channels and skeletal muscle hyperexcitability in vitro and in vivo. Exp Neurol. 2020;113287, 2020. doi: 10.1016/j.expneurol.2020.113287 |
[109] | Farinato A , Altamura C , Desaphy JF . Effects of benzothiazolamines on voltage-gated sodium channels. Handb Exp Pharmacol. (2018) ;246: :233–50. doi:10.1007/164 2017_46 |
[110] | Skov M , de Paoli FV , Nielsen OB , Pedersen TH . The anti-convulsants lacosamide, lamotrigine, and rufinamide reduce myotonia in isolated human and rat skeletal muscle. Muscle Nerve. (2017) ;56: :136–42. doi:10.1002/mus.25452 |
[111] | Fleischhauer R , Mitrovic N , Deymeer F , Lehmann-Horn F , Lerche H . Effects of temperature and mexiletine on the F1473S Na+channel mutation causing paramyotonia congenita. Pflugers Arch. (1998) ;436: :757–65. doi:10.1007/s004240050699 |
[112] | Weckbecker K , Würz A , Mohammadi B , Mansuroglu T , George AL Jr , Lerche H , et al. Different effects of mexiletine on two mutant sodium channels causing paramyotonia congenita and hyperkalemic periodic paralysis. Neuromuscul Disord. (2000) ;10: (1):31–9. doi:10.1016/s0960-8966(99)00060-7 |
[113] | Zhao J , Duprè N , Puymirat J , Chahine M . Biophysical characterization of M1476I, a sodium channel founder mutation associated with cold-induced myotonia in French Canadians. J Physiol. (2012) ;590: (11):2629–44. doi:10.1113/jphysiol.2011.223461 |
[114] | Altamura C , Lucchiari S , Sahbani D , Ulzi G , Comi GP , D’Ambrosio P , et al. The analysis of myotonia congenita mutations discloses functional clusters of amino acids within the CBS2 domain and the C-terminal peptide of the ClC-1 channel. Hum Mutat. (2018) ;39: (9):1273–83. doi: 10.1002/humu.23581 |
[115] | Jeng CJ , Fu SJ , You CY , Peng YJ , Hsiao CT , Chen TY , et al. Defective Gating and Proteostasis of Human ClC-1 Chloride Channel: Molecular Pathophysiology of Myotonia Congenita. Front Neurol. (2020) ;11: :76. doi:10.3389/fneur.2020.00076 |
[116] | Estévez R , Schroeder BC , Accardi A , Jentsch TJ , Pusch M . Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. (2003) ;38: :47–59. doi:10.1016/s0896-6273(03)00168-5 |
[117] | Altamura C , Mangiatordi GF , Nicolotti O , Sahbani D , Farinato A , Leonetti F , et al. Mapping ligand-binding pockets in ClC-1 channels through an integrated in silico and experimental approach using anthracene-9-carboxylic acid and niflumic acid. Br J Pharmacol. (2018) ;175: (10):1770–80. doi:10.1111/bph.14192 |
[118] | Altamura C , Ivanova EA , Imbrici P , Conte E , Camerino GM , Dadali EL , et al. Pathomechanisms of a CLCN1 mutation found in a Russian family suffering from Becker’s myotonia. Front Neurol. (2020) ;11: :1019. doi:10.3389/fneur.2020.01019 |
[119] | Wang K , Preisler SS , Zhang L , Cui Y , Missel JW , Grønberg C , et al. Structure of the human ClC-1 chloride channel. PLoS Biol. (2019) ;17: (4):e3000218. doi:10.1371/journal.pbio.3000218 |
[120] | Chen YA , Peng YJ , Hu MC , Huang JJ , Chien YC , Wu JT , et al. The Cullin 4A/B-DDB1-Cereblon E3 ubiquitin ligase complex mediates the degradation of CLC-1 chloride channels. Sci Rep. (2015) ;5: :10667. doi:10.1038/srep10667 |
[121] | Peng YJ , Huang JJ , Wu HH , Hsieh HY , Wu CY , Chen SC , et al. Regulation of CLC-1 chloride channel biosynthesis by FKBP8 and Hsp90b. Sci Rep. (2016) ;6: :32444. doi: 10.1038/srep32444 |
[122] | Eguchi H , Tsujino A , Kaibara M , Hayashi H , Shirabe S , Taniyama K , et al. Acetazolamide acts directly on the human skeletal muscle chloride channel. Muscle Nerve. (2006) ;34: (3):292–7. doi:10.1002/mus.20585 |
[123] | Desaphy J-F , Gramegna G , Altamura C , Dinardo MM , Imbrici P , George AL Jr , et al. Functional characterization of ClC-1 mutations from patients affected by recessive myotonia congenital presenting with different clinical phenotypes. Exp. Neurol. (2013) ;248: :530–40. doi:10.1016/j.expneurol.2013.07.018 |
[124] | Bryant SH , Morales-Aguilera A . Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J Physiol. (1971) ;219: (2):367–83. doi:10.1113/jphysiol.1971.sp009667 |
[125] | Beck CL , Fahlke C , George AL Jr. . Molecular basis for decreased muscle chloride conductance in the myotonic goat. Proc Natl Acad Sci USA. (1996) ;93: (20):11248–52. doi:10.1073/pnas.93.20.11248 |
[126] | Camerino DC , Pierno S , De Luca A , Bryant SH . Antimyotonic effects of tocainide enantiomers on skeletal muscle fibers of congenitally myotonic goats. Neuromuscul Disord. (2000) ;10: (3):160–4. doi:10.1016/s0960-8966(99)00101-7 |
[127] | Mehrke G , Brinkmeier H , Jockusch H . The myotonic mouse mutant ADR: Electrophysiology of the muscle fiber. Muscle Nerve. (1988) ;11: (5):440–6. doi:10.1002/mus.880110505 |
[128] | Steinmeyer K , Klocke R , Ortland C , Gronemeier M , Jockusch H , Gründer S , Jentsch TJ . Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature. (1991) ;354: (6351):304–8. |
[129] | Gronemeier M , Condie A , Prosser J , Steinmeyer K , Jentsch TJ , Jockusch H . Nonsense and missense mutations in the muscular chloride channel gene Clc-1 of myotonic mice. J Biol Chem. (1994) ;269: (8):5963–7. |
[130] | Hoppe K , Chaiklieng S , Lehmann-Horn F , Jurkat-Rott K , Wearing S , Klingler W . Preclinical pharmacological in vitro investigations on low chloride conductance myotonia: Effects of potassium regulation. Pflugers Arch. (2020) ;472: (10):1481–94. |
[131] | Dupont C , Denman KS , Hawash AA , Voss AA , Rich MM . Treatment of myotonia congenita with retigabine in mice. Exp Neurol. (2019) ;315: :52–59. doi:10.1016/j.expneurol.2019.02.002 |
[132] | Hayward LJ , Kim JS , Lee MY , Zhou H , Kin JW , Misra K et al. Targeted mutation of mouse skeletal muscle sodium channel produces myotonia and potassium-sensitive weakness. J Clin Invest. (2008) ;118: :1437–49. doi: 10.1172/JCI32638 |
[133] | Corrochano S , Männikkö R , Joyce PI , McGoldrick P , Wettstein J , Lassi G , et al. Novel mutations in human and mouse SCN4A implicate AMPK in myotonia and periodic paralysis. Brain. (2014) ;137: (Pt 12):3171–85. doi:10.1093/brain/awu292 |
[134] | Fontaine B , Khurana TS , Hoffman EP , Bruns GA , Haines JL , Trofatter JA , et al. Hyperkalemic periodic paralysis and the adult muscle sodium channel alpha-subunit gene. Science. (1990) ;250: (4983):1000–2. doi:10.1126/science.2173143 |
[135] | Ptacek LJ , George AL Jr , Griggs RC , Tawil R , Kallen RG , Barchi RL , Robertson M , Leppert MF . Identification of a mutation in the gene causing hyperkalemic periodic paralysis. Cell. (1991) ;67: (5):1021–7. doi:10.1016/0092-8674(91)90374-8. |
[136] | Rojas CV , Wang JZ , Schwartz LS , Hoffman EP , Powell BR , Brown RH Jr. . A Met-to-Val mutation in the skeletal muscle Nav channel alpha-subunit in hyperkalaemic periodic paralysis. Nature. (1991) ;354: (6352):387–9. doi: 10.1038/354387a0 |
[137] | Bulman DE , Scoggan KA , van Oene MD , Nicolle MW , Hahn AF , Tollar LL , et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. (1999) ;53: (9):1932–6. doi:10.1212/wnl.53.9.1932 |
[138] | Jurkat-Rott K , Mitrovic N , Hang C , Kouzmekine A , Iaizzo P , Herzog J , et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci. USA. (2000) ;97: (17):9549–54. doi:10.1073/pnas.97.17.9549 |
[139] | Fontaine B , Vale-Santos J , Jurkat-Rott K , Reboul J , Plassart E , Rime CS , et al. Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nat. Genet. (1994) ;6: (3):267–72. doi:10.1038/ng0394-267 |
[140] | Jurkat-Rott K , Lehmann-Horn F , Elbaz A , Heine R , Gregg RG , Hogan K , et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. (1994) ;3: (8):1415–9. doi:10.1093/hmg/3.8.1415 |
[141] | Ptacek LJ , Tawil R , Griggs RC , Engel AG , Layzer RB , Kwiecinski H , et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. (1994) ;77: (6):863–8. doi:10.1016/0092-8674(94)90135-x |
[142] | Tawil R , Ptacek LJ , Pavlakis SG , DeVivo DC , Penn AS , Ozdemir C , et al. Andersen’s syndrome: Potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann Neurol. (1994) ;35: (3):326–30. doi: 10.1002/ana.410350313 |
[143] | Plaster NM , Tawil R , Tristani-Firouzi M , Canun S , Bendahhou S , Tsunoda A , et al. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. (2001) ;105: (4):511–9. doi: 10.1016/s0092-8674(01)00342-7 |
[144] | Vicart S , Sternberg D , Fournier E , Ochsner F , Laforet P , Kuntzer T , et al. New mutations of SCN4A cause a potassium-sensitive normokalemic periodic paralysis. Neurology. (2004) ;63: (11):2120–7. doi:10.1212/01.wnl.0000145768.09934.ec |
[145] | Salih M , van Kinschot CMJ , Peeters RP , de Herder WW , Duschek EJJ , van der Linden J , et al. Thyrotoxic periodic paralysis: An unusual presentation of hyperthyroidism. Neth J Med. (2017) ;75: (8):315–20. |
[146] | Nakaza M , Kitamura Y , Furuta M , Kubota T , Sasaki R , Takahashi MP . Analysis of the genetic background associated with sporadic periodic paralysis in Japanese patients. J Neurol Sci. (2020) ;412: :116795. doi:10.1016/j.jns.2020.116795 |
[147] | Cannon SC . Further evidence for shared genetic susceptibility in both sporadic and thyrotoxic periodic paralysis. J Neurol Sci. (2020) ;412: :116794. doi:10.1016/j.jns.2020.116794 |
[148] | Charles G , Zheng C , Lehmann-Horn F , Jurkat-Rott K , Levitt J . Characterization of hyperkalemic periodic paralysis: A survey of genetically diagnosed individuals. J Neurol. (2013) ;260: (10):2606–13. doi:10.1007/s00415-013-7025-9 |
[149] | Fialho D , Griggs RC , Matthews E . Periodic paralysis. Handb Clin Neurol. (2018) ;148: :505–20. doi:10.1016/B978-0-444-64076-5.00032-6 |
[150] | Wang P , Clausen T . Treatment of attacks in hyperkalaemic familial periodic paralysis by inhalation of salbutamol. Lancet. (1976) ;1: (7953):221–3. doi:10.1016/s0140-6736(76)91340-4 |
[151] | Hanna MG , Stewart J , Schapira AH , Wood NW , Morgan-Hughes JA , Murray NM . Salbutamol treatment in a patient with hyperkalaemic periodic paralysis due to a mutation in the skeletal muscle sodium channel gene (SCN4A). J Neurol Neurosurg Psychiatry. (1998) ;65: (2):248–50. doi: 10.1136/jnnp.65.2.248 |
[152] | Flatman JA , Clausen T . Combined effects of adrenaline and insulin on active electrogenic Na+-K+transport in rat soleus muscle. Nature. (1979) ;281: (5732):580–1. doi: 10.1038/281580a0 |
[153] | Clausen T , Nielsen OB , Clausen JD , Pedersen TH , Hayward LJ . Na+,K+-pump stimulation improves contractility in isolated muscles of mice with hyperkalemic periodic paralysis. J Gen Physiol. (2011) ;138: (1):117–30. doi: 10.1085/jgp.201010586 |
[154] | Gamstorp I , Hauge M , Helweglarsen HF , Mjones H , Sagild U . Adynamia episodica hereditaria: A disease clinically resembling familial periodic paralysis but characterized by increasing serum potassium during the paralytic attacks. Am J Med. (1957) ;23: (3):385–90. doi:10.1016/0002-9343(57)90318-2 |
[155] | Samaha FJ . Hyperkalemic Periodic Paralysis. A Genetic Study, Clinical Observations, and Report of a New Method of Therapy. Arch Neurol. (1965) ;12: :145–54. doi:10.1001/archneur.1965.00460260035004 |
[156] | Mankodi AC , Grunseich M , Skov L , Cook G , Aue E , Purev D , et al. Divalent cation responsive myotonia and muscle paralysis in skeletal muscle sodium channelopathy. Neuromuscul Disord. (2015) ;25: :908–12. doi:10.1016/j.nmd.2015.08.007 |
[157] | Dyken ML , Timmons GD . Hyperkalemic periodic paralysis with hypocalcemic episode. Absence of an autosomal dominant pedigree pattern. Archiv Neurol. (1963) ;9: :508–17. doi:10.1001/archneur.1963.00460110076008 |
[158] | Resnick JS , Engel WK , Griggs RC , Stam AC . Acetazolamide prophylaxis in hypokalemic periodic paralysis. N Engl J Med. (1969) ;278: :582–6. doi:10.1056/NEJM196803142781102 |
[159] | Griggs RC , Engel WK , Resnick JS . Acetazolamide treatment of hypokalemic periodic paralysis: Prevention of attacks and improvement of persistent weakness. Ann Intern Med. (1970) ;73: :39–48. doi:10.7326/0003-4819-73-1-39 |
[160] | Dalakas MC , Engel WK . Treatment of “permanent” muscle weakness in familial periodic paralysis. Muscle Nerve. (1983) ;6: :182–6. doi:10.1002/mus.880060303 |
[161] | Tawil R , McDermott MP , Brown R Jr , Shapiro BC , Ptacek LJ , McManis PG , et al. Randomized trials of dichlorphenamide in the periodic paralyses: Working Group on Periodic Paralysis. Ann Neurol. (2000) ;47: :46–53. |
[162] | Ciafaloni E , Cohen F , Griggs R . Efficacy and safety of dichlorphenamide for primary periodic paralysis in adolescents compared with adults. Pediatr Neurol. (2019) ;101: :43–46. doi:10.1016/j.pediatrneurol.2019.07.019 |
[163] | Sansone VA , Burge J , McDermott MP , Smith PC , Herr B , Tawil R , et al. Randomized, placebo-controlled trials of dichlorphenamide in periodic paralysis. Neurology. (2016) ;86: (15):1408–16. doi:10.1212/WNL.0000000000002416 |
[164] | Bendahhou S , Cummins TR , Griggs RC , Fu Y-H , Ptácek LJ . Sodium channel inactivation defects are associated with acetazolamide exacerbated hypokalemic periodic paralysis. Ann Neurol. (2001) ;50: :417–20. doi:10.1002/ana.1144 |
[165] | Sternberg D , Maisonobe T , Jurkat-Rott K , Nicole S , Launay E , Chauveau D , et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. (2001) ;124: :1091–9. doi:10.1093/brain/124.6.1091 |
[166] | Davies NP , Eunson LH , Samuel M , Hanna MG . Sodium channel gene mutations in hypokalemic periodic paralysis: An uncommon cause in the UK. Neurology. (2001) ;57: :1323–5. doi:10.1212/wnl.57.7.1323 |
[167] | Venance SL , Jurkat-Rott K , Lehmann-Horn F , Tawil R . SCN4A-associated hypokalemic periodic paralysis merits a trial of acetazolamide. Neurology. (2004) ;63: (10):1977. doi:10.1212/01.wnl.0000143068.99794.5b |
[168] | Matthews E , Portaro S , Ke Q , Sud R , Haworth A , Davis MB , et al. Acetazolamide efficacy in hypokale-mic periodic paralysis and the predictive role of genotype. Neurology. (2011) ;77: (22):1960–4. doi:10.1212/WNL.0b013e31823a0cb6 |
[169] | Vroom FW , Jarrell MA , Maren TH . Acetazolamide treatment of hypokalemic periodic paralysis: Probable mechanism of action. Arch Neurol. (1975) ;32: :385–92. doi: 10.1001/archneur.1975.00490480051006 |
[170] | Tricarico D , Mele A , Conte Camerino D . Carbonic anhydrase inhibitors ameliorate the symptoms of hypokalaemic periodic paralysis in rats by opening the muscular Ca2+-activated-K+channels. Neuromuscul Disord. (2006) ;16: (1):39–45. doi:10.1016/j.nmd.2005.10.005 |
[171] | Matthews E , Labrum R , Sweeney MG , Sud R , Haworth A , Chinnery PF , et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. (2009) ;72: (18):1544–7. doi:10.1212/01.wnl.0000342387.65477.46 |
[172] | Lapie P , Goudet C , Nargeot J , Fontaine B , Lory P . Electrophysiological properties of the hypokalaemic periodic paralysis mutation (R528H) of the skeletal muscle alpha 1s subunit as expressed in mouse L cells. FEBS Lett. (1996) ;382: (3):244–8. doi:10.1016/0014-5793(96)00173-1 |
[173] | Morrill JA , Brown RH Jr , Cannon SC . Gating of the L-type Ca channel in human skeletal myotubes: An activation defect caused by the hypokalemic periodic paralysis mutation R528H. J Neurosci. (1998) 15;18: (24):10320–34. doi: 10.1523/JNEUROSCI.18-24-10320.1998 |
[174] | Morrill JA , Cannon SC . Effects of mutations causing hypokalaemic periodic paralysis on the skeletal muscle L-type Ca2+channel expressed in Xenopus laevis oocytes. J Physiol. (1999) ;520: :321–36. doi:10.1111/j.1469-7793.1999.00321.x |
[175] | Struyk AF , Scoggan KA , Bulman DE , Cannon SC . The human skeletal muscle Na channel mutation R669H associated with hypokalemic periodic paralysis enhances slow inactivation. J Neurosci. (2000) ;20: (23):8610–7. doi: 10.1523/JNEUROSCI.20-23-08610.2000 |
[176] | Kuzmenkin A , Muncan V , Jurkat-Rott K , Hang C , Lerche H , Lehmann-Horn F , et al. Enhanced inactivation and pH sensitivity of Na(+) channel mutations causing hypokalaemic periodic paralysis type II. Brain. (2002) ;125: (Pt 4):835–43. doi:10.1093/brain/awf071 |
[177] | Carle T , Lhuillier L , Luce S , Sternberg D , Devuyst O , Fontaine B , et al. Gating defects of a novel Na+channel mutant causing hypokalemic periodic paralysis. Biochem Biophys Res Commun. (2006) ;348: (2):653–61. doi:10.1016/j.bbrc.2006.07.101 |
[178] | Sokolov S , Scheuer T , Catterall WA . Gating pore current in an inherited ion channelopathy. Nature. (2007) ;446: (7131):76–8. doi:10.1038/nature05598 |
[179] | Sokolov S , Scheuer T , Catterall WA . Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc Natl Acad Sci USA. (2008) ;105: :19980–5. doi:10.1073/pnas.0810562105 |
[180] | Struyk AF , Cannon SC . A Na+channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J Gen Physiol. (2007) ;130: (1):11–20. doi:10.1085/jgp.200709755 |
[181] | Struyk AF , Markin VS , Francis D , Cannon SC . Gating pore currents in DIIS4 mutations of NaV1.4 associated with periodic paralysis: Saturation of ion flux and implications for disease pathogenesis. J Gen Physiol. (2008) ;132: :447–64. doi:10.1085/jgp.200809967 |
[182] | Francis DG , Rybalchenko V , Struyk A , Cannon SC . Leaky sodium channels from voltage sensor mutations in periodic paralysis, but not paramyotonia. Neurology. (2011) ;76: (19):1635–41. doi:10.1212/WNL.0b013e318219fb57 |
[183] | Groome JR , Lehmann-Horn F , Fan C , Wolf M , Winston V , Merlini L , Jurkat-Rott K . Nav1.4 mutations cause hypokalaemic periodic paralysis by disrupting IIIS4 movement during recovery. Brain. (2014) ;137: (Pt4):998–1008. doi:10.1093/brain/awu015 |
[184] | Mi W , Rybalchenko V , Cannon SC . Disrupted coupling of gating charge displacement to Na+current activation for DIIS4 mutations in hypokalemic periodic paralysis. J Gen Physiol. (2014) ;144: (2):137–45. doi:10.1085/jgp.201411199 |
[185] | Fuster C , Perrot J , Berthier C , Jacquemond V , Allard B . Elevated resting H+ current in the R1239H type 1 hypokalaemic periodic paralysis mutated Ca2 + channel. J Physiol. (2017) ;595: (20):6417–28. doi:10.1113/JP274638 |
[186] | Bayless-Edwards L , Winston V , Lehmann-Horn F , Arinze P , Groome JR , Jurkat-Rott K . NaV1.4 DI-S4 periodic paralysis mutation R222W enhances inactivation and promotes leak current to attenuate action potentials and depolarize muscle fibers. Sci Rep. (2018) ;8: (1):10372. doi: 10.1038/s41598-018-28594-5 |
[187] | Sokolov S , Scheuer T , Catterall WA . Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J Gen Physiol. (2010) ;136: :225–36. doi:10.1085/jgp.201010414 |
[188] | Xiao Y , Blumenthal K , Cummins TR . Gating-pore currents demonstrate selective and specific modulation of individual sodium channel voltage-sensors by biological toxins. Mol Pharmacol. (2014) ;86: :159–67. doi:10.1124/mol.114.092338 |
[189] | Männikkö R , Shenkarev ZO , Thor MG , Berkut AA , Myshkin MY , Paramonov AS , et al. Spider toxin inhibits gating pore currents underlying periodic paralysis. Proc Natl Acad Sci U S A. (2018) ;115: (17):4495–500. doi: 10.1073/pnas.1720185115 |
[190] | Cannon SC . Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol. (2018) ;246: :309–30. doi:10.1007/164_2017_52 |
[191] | Ricker K , Rohkamm R , Böhlen R . Adynamia episodica and paralysis periodica paramyotonica. Neurology. (1986) ;36: (5):682–6. doi:10.1212/wnl.36.5.682 |
[192] | Zukić S , Sinanović O , Alečković-Halilović M , Hodžić R , Kovačević L , Hodžić S . Hyperkalemic periodic paralysis aggravated by voltage - gate sodium channel blocker antiepileptic drug? Med Hypotheses. (2020) ;139: :109683. doi:10.1016/j.mehy.2020.109683 |
[193] | Wu F , Mi W , Burns DK , Fu Y , Gray HF , Struyk AF , et al. A sodium channel knockin mutant (NaV1.4-R669H) mouse model of hypokalemic periodic paralysis. J Clin Invest. (2011) ;121: :4082–94. doi:10.1172/JCI57398 |
[194] | Wu F , Mi W , Hernandez-Ochoa EO , Burns DK , Fu Y , Gray HF , et al. A calcium channel mutant mouse model of hypokalemic periodic paralysis. J Clin Invest. (2012) ;122: :4580–91. doi:10.1172/JCI66091 |
[195] | Uwera F , Ammar T , McRae C , Hayward LJ , Renaud JM . Lower Ca2+enhances the K+-induced force depression in normal and HyperKPP mouse muscles. J Gen Physiol. (2020) ;152: (7):e201912511. doi:10.1085/jgp.201912511 |
[196] | Cannon SC . A role for external Ca2+in maintaining muscle contractility in periodic paralysis. J Gen Physiol. (2020) ;152: (7):e202012615. doi:10.1085/jgp.202012615 |
[197] | Wu F , Mi W , Cannon SC . Bumetanide prevents transient decreases in muscle force in murine hypokalemic periodic paralysis. Neurology. (2013) ;80: :1110–6. doi:10.1212/WNL.0b013e3182886a0e |
[198] | Wu F , Mi W , Cannon SC . Beneficial effects of bumetanide in a CaV1.1-R528H mouse model of hypokalaemic periodic paralysis. Brain. (2013) ;136: (Pt 12):3766–74. doi:10.1093/brain/awt280 |
[199] | Geukes Foppen RJ , van Mil HG , Siegenbeek van Heukelom J . Effects of chloride transport on bistable behaviour of the membrane potential in mouse skeletal muscle. J Physiol. (2002) ;542: :181–91. doi:10.1113/jphysiol.2001.013298 |
[200] | Scalco R , Morrow J , Manole A , Skorupinska I , Ricciardi F , Matthews E , et al. RCT of 2 mg bumetanide for hypokalaemic periodic paralysis. Neuromuscular Dis. (2019) ;29: (suppl. 1):S69. |
[201] | Nagamitsu S , Matsuura T , Khajavi M , Armstrong R , Gooch C , Harati Y , et al. A “dystrophic” variant of autosomal recessive myotonia congenita caused by novel mutations in the CLCN1 gene. Neurology. (2000) ;55: (11):1697–703. doi:10.1212/wnl.55.11.1697 |
[202] | Fusco C , Frattini D , Salerno GG , Canali E , Bernasconi P , Maggi L . New phenotype and neonatal onset of sodium channel myotonia in a child with a novel mutation of SCN4A gene. Brain Dev. (2015) ;37: (9):891–3. doi:10.1016/j.braindev.2015.02.004 |
[203] | Crews J , Kaiser KK , Brooke MH . Muscle pathology of myotonia congenita. J Neurol Sci. (1976) ;28: (4):449–57. doi:10.1016/0022-510x(76)90116-7. |
[204] | Salviati G , Biasia E , Betto R , Danieli Betto D . Fast to slow transition induced by experimental myotonia in rat EDL muscle. Pflugers Arch. (1986) ;406: (3):266–72. doi: 10.1007/BF00640912 |
[205] | Links TP , Zwarts MJ , Wilmink JT , Molenaar WM , Oosterhuis HJ . Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. (1990) ;113: (pt 6):1873–89. doi:10.1093/brain/113.6.1873 |
[206] | Bradley WG , Taylor R , Rice DR , Hausmanowa-Petruzewicz I , Adelman LS , Jenkison M , et al. Progressive myopathy in hyperkalemic periodic paralysis. Arch Neurol. (1990) ;47: (9):1013–7. doi:10.1001/archneur.1990.00530090091018 |
[207] | Tricarico D , Lovaglio S , Mele A , Rotondo G , Mancinelli E , Meola G , Camerino DC . Acetazolamide prevents vacuolar myopathy in skeletal muscle of K(+) -depleted rats. Br J Pharmacol. (2008) ;154: (1):183–90. doi:10.1038/bjp.2008.42 |
[208] | Tsujino A , Maertens C , Ohno K , Shen XM , Fukuda T , Harper CM , Cannon SC , Engel AG . Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA. (2003) ;100: :7377–82. doi:10.1073/pnas.1230273100 |
[209] | Arnold WD , Feldman DH , Ramirez S , He L , Kassar D , Quick A , Klassen TL , Lara M , Nguyen J , Kissel JT , Lossin C , Maselli RA . Defective fast inactivation recovery of Nav1.4 in congenital myasthenic syndrome. Ann Neurol. (2015) ;77: :840–50. doi:10.1002/ana.24389 |
[210] | Habbout K , Poulin H , Rivier F , Giuliano S , Sternberg D , Fontaine B , et al. A recessive Nav1.4 mutation underlies congenital myasthenic syndrome with periodic paralysis. Neurology. (2016) ;86: :161–9. doi:10.1212/WNL.0000000000002264 |
[211] | Elia N , Palmio J , Castañeda MS , Shieh PB , Quinonez M , Suominen T , et al. Myasthenic congenital myopathy from recessive mutations at a single residue in Na(V)1.4. Neurology. (2019) ;92: (13):e1405–15. doi:10.1212/WNL.0000000000007185 |
[212] | Echaniz-Laguna A , Biancalana V , Nadaj-Pakleza A , Fournier E , Matthews E , Hanna MG , et al. Homozygous C-terminal loss-of-function NaV1.4 variant in a patient with congenital myasthenic syndrome. J Neurol Neurosurg Psychiatry. (2020) ;91: (8):898–900. doi:10.1136/jnnp-2020-323173 |
[213] | Zaharieva IT , Thor MG , Oates EC , van Karnebeek C , Hendson G , Blom E , et al. Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or ‘classical’ congenital myopathy. Brain. (2016) ;139: (Pt 3):674–91. doi: 10.1093/brain/awv352 |
[214] | Gonorazky HD , Marshall CR , Al-Murshed M , Hazrati LN , Thor MG , Hanna MG , et al. Congenital myopathy with “corona” fibres, selective muscle atrophy, and craniosynostosis associated with novel recessive mutations in SCN4A. Neuromuscul Disord. (2017) ;27: (6):574–80. doi: 10.1016/j.nmd.2017.02.001 |
[215] | Mercier S , Lornage X , Malfatti E , Marcorelles P , Letournel F , Boscher C , et al. Expanding the spectrum of congenital myopathy linked to recessive mutations in SCN4A. Neurology. (2017) ;88: (4):414–6. doi:10.1212/WNL.0000000000003535 |
[216] | Sloth CK , Denti F , Schmitt N , Bentzen BH , Fagerberg C , Vissing J , et al. Homozygosity for SCN4A Arg1142Gln causes congenital myopathy with variable disease expression. Neurol Genet. (2018) ;4: (5):e267. doi:10.1212/NXG.0000000000000267 |
[217] | Wu F , Mi W , Fu Y , Struyk A , Cannon SC . Mice with a NaV1.4 sodium channel null allele have latent myasthenia, without susceptibility to periodic paralysis. Brain. (2016) ;139: (Pt 6):1688–99. doi:10.1093/brain/aww070 |
[218] | Bednarz M , Stunnenberg BC , Kusters B , Kamsteeg EJ , Saris CG , Groome J , et al. A novel Ile1455Thr variant in the skeletal muscle sodium channel alpha-subunit in a patient with a severe adult-onset proximal myopathy with electrical myotonia and a patient with mild paramyotonia phenotype. Neuromuscul Disord. (2017) ;27: (2):175–82. doi:10.1016/j.nmd.2016.09.023 |
[219] | Luo S , Sampedro Castañeda M , Matthews E , Sud R , Hanna MG , Sun J , et al. Hypokalaemic periodic paralysis and myotonia in a patient with homozygous mutation p.R1451L in Nav1.4. Sci Rep. (2018) ;8: (1):9714. doi: 10.1038/s41598-018-27822-2 |
[220] | Waldrop M , Amornvit J , Pierson CR , Boue DR , Sahenk Z . A Novel De Novo Heterozygous SCN4a Mutation Causing Congenital Myopathy, Myotonia and Multiple Congenital Anomalies. J Neuromuscul Dis. (2019) ;6: (4):467–73. doi: 10.3233/JND-190425 |
[221] | Hunter JM , Ahearn ME , Balak CD , Liang WS , Kurdoglu A , Corneveaux JJ , et al. Novel pathogenic variants and genes for myopathies identified by whole exome sequencing. Mol Genet Genomic Med. (2015) ;3: (4):283–301. doi: 10.1002/mgg3.142 |
[222] | Schartner V , Romero NB , Donkervoort S , Treves S , Munot P , Pierson TM , et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. (2017) ;133: (4):517–33. doi:10.1007/s00401-016-1656-8 |
[223] | Edizadeh M , Vazehan R , Javadi F , Dehdahsi S , Fadaee M , Faraji Zonooz M , et al. De novo mutation in CACNA1S gene in a 20-year-old man diagnosed with metabolic myopathy. Arch Iran Med. (2017) ;20: (9):617–20. |
[224] | Anandan C , Cipriani MA , Laughlin RS , Niu Z , Milone M . Rhabdomyolysis and fluctuating asymptomatic hyperCKemia associated with CACNA1S variant. Eur J Neurol. (2018) ;25: (2):417–9. doi:10.1111/ene.13528 |
[225] | Nicolau S , Liewluck T , Tracy JA , Laughlin RS , Milone M . Congenital myopathies in the adult neuromuscular clinic: Diagnostic challenges and pitfalls. Neurol Genet. (2019) ;5: (4):e341. doi:10.1212/NXG.0000000000000341 |
[226] | Yiş U , Hiz S , Güneş S , Diniz G , Baydan F , Töpf A , et al. Dihydropyridine receptor congenital myopathy in a consangineous Turkish family. J Neuromuscul Dis. (2019) ;6: (3):377–84. doi:10.3233/JND-190383 |
[227] | Horstick EJ , Linsley JW , Dowling JJ , Hauser MA , McDonald KK , Ashley-Koch A et al. Stac3 is a component of the excitation-contraction coupling machinery and mutated in Native American myopathy. Nat Commun. (2013) ;4: :1952. doi:10.1038/ncomms2952 |
[228] | Waldrop MA , Boue DR , Sites E , Flanigan KM , Shell R . Clinico-pathological Conference: A hypotonic newborn with cleft palate, micrognathia and bilateral club feet. Pediatr Neurol. (2017) ;74: :11–4. doi:10.1016/j.pediatrneurol.2017.01.026 |
[229] | Zaharieva IT , Sarkozy A , Munot P , Manzur A , O’Grady G , Rendu J , et al. STAC3 variants cause a congenital myopathy with distinctive dysmorphic features and malignant hyperthermia susceptibility. Hum Mutat. (2018) ;39: (12):1980–94. doi:10.1002/humu.23635 |
[230] | Linsley JW , Hsu IU , Groom L , Yarotskyy V , Lavorato M , Horstick EJ , et al. Congenital myopathy results from misregulation of a muscle Ca2+channel by mutant Stac3. Proc Natl Acad Sci U S A. (2017) ;114: (2):E228–36. doi:10.1073/pnas.1619238114 |
[231] | Polster A , Nelson BR , Papadopoulos S , Olson EN , Beam KG . Stac proteins associate with the critical domain for excitation-contraction coupling in the II-III loop of CaV1.1. J Gen Physiol. (2018) ;150: (4):613–24. doi:10.1085/jgp.201711917 |
[232] | Wu F , Quinonez M , DiFranco M , Cannon SC . Stac3 enhances expression of human Ca(V)1.1 in Xenopus oocytes and reveals gating pore currents in HypoPP mutant channels. J Gen Physiol. (2018) ;150: (3):475–89. doi:10.1085/jgp.201711962 |
[233] | Matthews E , Hartley L , Sud R , Hanna MG , Muntoni F , Munot P . Acetazolamide can improve symptoms and signs in ion channel-related congenital myopathy. J Neurol Neurosurg Psychiatry. (2019) ;90: (2):243–5. doi:10.1136/jnnp-2017-317849 |

