
Novel Pathogenic Variants in a French Cohort Widen the Mutational Spectrum of GNE Myopathy
Abstract
Background: GNE myopathy is a rare autosomal recessively inherited muscle disease resulting from mutations in the gene encoding GNE (UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase), a key enzyme in sialic acid biosynthesis. 154 different pathogenic variants have been previously associated with GNE myopathy.
Objective: Describe novel pathogenic variants associated with GNE myopathy in a large French cohort.
Methods: We analyzed mutational data from 32 GNE myopathy index patients. Novel, as well as previously published pathogenic variants, were examined for possible deleterious effects on splicing.
Results: We describe 13 novel pathogenic variants in GNE, identified in the first large French cohort reported to date. We also find that 6 published pathogenic variants might have a previously unrecognized deleterious effect on splicing.
Conclusions: Novel pathogenic GNE variants described here raise the total number of different pathogenic variants reported to 167, complementing the recently published GNE mutation update. Our novel findings on possible splice-disrupting effects by several variants suggest that the pathogenicity mechanism of these variants could be reinterpreted, expanding our knowledge about the GNE mutational spectrum.
INTRODUCTION
GNE myopathy (MIM # 605820) is a rare auto-somal recessive myopathy caused by mutations inthe gene encoding a key enzyme in sialic acid biosynthesis, UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE) [1, 2]. This disorder was initially described as a distal myopathy in Japanese [3], and as a quadriceps sparing myopathy in Iranian Jews [4], associated with rimmed vacuoles on muscle biopsy. However, the phenotype and histopathological features seem to be more heterogeneous than initially expected, as limb-girdle presentation [5] as well as cases with histological inflammation [6, 7] or without rimmed vacuoles [8] associated to GNE myopathy have been reported.
A recent overview of the GNE mutational spectrum published by Celeste et al. [9] added 7 novel pathogenic variants to the 147 different pathogenic variants associated with GNE myopathy already reported in the literature. Here, we describe 13 novel pathogenic GNE variants associated with GNE myopathy identified in the first large French cohort analyzed to date, raising the total number of different pathogenic variants associated with GNE myopathy to 167. We also analyzed the possible deleterious effects on splicing of the newly discovered as well as all previously published missense variants. Bioinformatics analysis indicates that 6 previously published missense variants were likely to have medium to severe deleterious effects on splicing, allowing possible re-interpretation of the cause of pathogenicity of these variants.
MATERIALS AND METHODS
Patients and genetic analysis
We analyzed mutational data from 32 GNE myopathy index patients diagnosed genetically in the Department of Medical Genetics in Marseille, the reference center for genetic diagnosis of this disorder in France. Following informed consent, genomic DNA was extracted from blood samples. Mutational analysis was done using PCR amplification of GNE coding exons and flanking intronic boundaries (primer sequences available on request) followed by direct Sanger sequencing on a 3500XL Genetic Analyzer ® (Applied Biosystems-Life Technologies, Carlsbad, CA). We used the new hGNE2 reference nomenclature (GenBank accession numbers: Protein: NP_001121699.1; mRNA: NM_001128227.2), as recommended by the latest publication on GNE myopathy nomenclature [9, 10].
Pathogenicity predictions
We used the following predictive algorithms to analyze the pathogenicity of the novel variants associated with GNE myopathy: UMD-Predictor (http://www.umd-hts.eu) [11], SIFT (Sort Intolerant From Tolerant human Protein; http://sift.jcvi.org) [12] and PolyPhen-2 (Polymorphism Phenotyping v2; http://genetics.bwh.harvard.edu/pph2) [13]. Possible deleterious effects of novel and published missense variants on splicing were assessed using the latest version of Human Splicing Finder (HSF 3.0, http://www.umd.be/HSF3), a predictive tool based on a combination of different matrices for auxiliary sequence prediction, and Position Weight Matrices to assess the strength of 5’ and 3’ splice sites and branch points [14]. HSF has been shown previously to precisely predict the effect of mutations affecting 5’ and 3’ splice sites and branch points, and/or activating cryptic splice sites [14, 15]. Correct prediction of activating or inactivating effects of mutations on exonic splicing enhancers (ESE) and exonic splicing enhancers (ESS) has also been validated [14], though it remains critical to exactly determine the precise effects generated by affecting these cis-acting elements, such as identifying the motif/protein couple implicated.
Based on these prediction algorithms (UMD-Predictor, SIFT, PolyPhen-2 and HSF) we classified the overall pathogenicity of the variants into 3 groups of severity (severe, medium and mild) as detailed in Table 1. Allele frequencies were evaluated using the 1000G (1000 Genomes, http://www.1000genomes.org[genomes.org]) and ESP (Exome Sequencing Project, https://esp.gs.washington.edu/drupal) database.
In vitro minigene splicing assay
Functional analysis of possibly abnormal splicing caused by c.717T>G was done using an in vitro minigene splicing assay [16]. The variant c.717T>G was selected for specific functional analysis using the in vitro minigene splicing assay because of the discrepancy between a predicted “benign” missense change at the amino-acid level versus a predicted deleterious effect on splicing. According to previously described methods [15], GNE Exon 5 and approximately 150 bp of flanking intronic sequences were amplified from a control DNA sample and then cloned in the pCAS2 vector. The c.717T>G substitution was subsequently introduced using site-directed mutagenesis and analyzed for specific splicing abnormalities following transfection in HEK 293 cells. Forty-eight hours after transfection, transcriptional analysis was performed and the effect on splicing identified using RT-PCR and further confirmed using direct Sanger sequencing [15].
RESULTS
13 novel pathogenic GNE variants were identified in patients with GNE myopathy
Among the 32 patients with genetic diagnosis of GNE myopathy, 11 presented with homozygous and 21 with compound heterozygous pathogenic variants (Supplementary Table). Of these, 13 pathogenic GNE variants (12 missense and 1 nonsense variants) were novel and were not present in 15190 alleles from exome sequence databases. Using different pathogenicity prediction programs (UMD-predictor, SIFT, PolyPhen-2, and HSF) combined, 10 out of the 12 novel missense GNE variants were found to have severe predicted pathogenic effects (Table 1). Absence of these variants in control datasets (1000G and ESP databases, data not shown), as well as predicted pathogenicity effects, suggest that they are indeed the likely cause of GNE myopathy in patients analyzed in this study.
Possible effects on splicing of GNE missense variants
Prediction algorithms used for pathogenicity assessment of variants based on their effect on protein sequence do not take into account possible effects of these variants on splicing of the GNE gene. Celeste et al. have recently reported possible splice effects for 5 exonic and 7 intronic variants. However, since only missense variants within 5 basepairs of a splice junction were examined in this study, 119 other missense variants remained unexplored [9]. As splice-disrupting variants can be found deeper in exons, we expanded these results by analyzing all published pathogenic missense variants as well as the 13 novel variants from our cohort (Table 1). Using the HSF prediction tool [14], we classified GNE variants in 4 groups based on their effects on splicing (probable, possible, uncertain, not affected). Interestingly, we found that 6 of the published pathogenic variants (NM_001128227.2: c.98A>G, c.179T>G, c.271A>G, c.731A>T, c.1394A>G, c.2098G>A), and 5 out of the 13 novel pathogenic variants reported here, were likely to have a previously unreported potential splicing effect (Table 1).
Moreover, 2 pathogenic variants (c.271A>G and c.715G>A) [9] with mild predicted effects on protein, and even a supposed polymorphism (c.717T>G) [9] were predicted to have a possible effect on splicing, suggesting that they might be more deleterious than previously thought. Since we encountered the c.717T>G variant in one patient (P15), we analyzed in detail its possible deleterious effect on splicing using an in vitro minigene splicing assay [15, 16]. For this variant, the corresponding amino-acid change (p.Asp239Glu) is classified as “benign” (PolyPhen-2 [13] pathogenicity score 0.245), while in silico analysis using Human Splicing Finder [14] predicts the activation of a cryptic donor site and the inactivation of an exonic splicing enhancer (ESE). In vitro analysis using the minigene splicing assay confirms that the c.717T>G variant activates a cryptic donor site resulting in the deletion of 145 bp in the 3’ extremity of exon 5. The abnormally spliced transcript (Fig. 1) is probably deleterious as it leads to a frameshift mutation introducing a premature translation termination codon in the amino-acid sequence (Asp239GlufsX9).
DISCUSSION
In this study, we describe 13 novel pathogenic variants associated with GNE myopathy, raising the total number of published pathogenic GNE variants to date to 167. We also examined all currently known pathogenic GNE missense variants for predicted deleterious effects on splicing to get a better understanding of the pathogenicity range of the GNE mutational spectrum. Taken together, a total of 9.7% (13 out of 134) among all pathogenic GNE missense variants associated to GNE myopathy are predicted to have a deleterious (possible or probable) effect on splicing. This result is in accordance with previous reports underlining the importance of deleterious effects on splicing caused by exonic substitutions [17], but has not been pointed out to date in the context of GNE myopathy. Splice effect prediction should be taken into account for mutational data interpretation in genetic diagnosis, as efficient correlation between predictive data and confirmation at the transcriptional level has been shown [14]. However, it is important to keep in mind that even though in silico tools for splicing defect prediction are useful for genetic diagnosis, they have inherent limitations depending on the underlying algorithms [18] and warrant functional confirmation if possible. Importantly, in silico tools for splicing defect prediction may be of interest for selecting variants for further functional transcriptional analyzes. In particular, substitutions predicted to cause benign or moderately severe missense changes at the amino-acid level (as evaluated in example using PolyPhen-2 [13]), might have a severe predicted deleterious effect on splicing. In our cohort, this was the case for the variant c.717T>G, predicted to cause a “benign” amino-acid change while possibly activating a cryptic splice-donor site, a fact that was further confirmed using a functional minigene splicing assay.
Most of the reported pathogenic GNE missense variants are considered to have a qualitative functional, and not quantitative, deleterious effect [9]. However, several mutations including, as expected, nonsense or frameshifting mutations, have been shown to cause GNE mRNA [19] or protein instability [20] resulting in severely decreased levels of expression and therefore implicated as causative of quantitative pathogenic effects. We emphasize that, as for numerous other genetic diseases [17], a proportion of substitutions predicted to cause deleterious missense changes at the amino-acid level in fact cause deleterious changes at the mRNA level due to abnormal splicing, affecting quantitatively or qualitatively the GNE enzyme function. Moreover, variants predicted as “possibly pathogenic” or even apparently “benign”, based on missense pathogenicity prediction algorithms such as PolyPhen-2 [13] could likewise have deleterious effects due to abnormal splicing. Given the variability of the onset and the evolution of GNE myopathy [21, 22], intra-individual variability in splicing efficiency and of nonsense-mediated mRNA decay [23, 24] could be a possible explanation for the observed clinical heterogeneity. Another possible component of this heterogeneity could be the association of disease-causing mutations in trans. Indeed, the potential pathogenicity of different variants in the GNE gene may implicate variant combinations modulating the clinical presentation and evolution of GNE myopathy.
Altogether, our study contributes novel GNE mutational data from the first large French cohort analyzed to date, in a context of recent advances in therapeutic trials for this disease [25], for which the importance of providing an accurate genetic diagnosis for inclusion of patients becomes even more important.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We sincerely thank Christophe Pécheux, Ghadi Raï, Christophe Béroud and the referring neurologists, in particular Pascal Laforêt, Bruno Eymard, Françoise Darcel, Laila Bastaki, Satish Khadilkar, Meena A. Kannan, Shahram Attarian and Jean Pouget, for their contribution to this work. We sincerely thank the patients for their implication.
We sincerely thank the APHM, INSERM and Aix Marseille University for supporting this work.
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-150074.
REFERENCES
1 | Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T(2001) The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathyNat Genet29: 18387 |
2 | Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y(2002) Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathyNeurology59: 1116891693 |
3 | Nonaka I, Sunohara N, Ishiura S, Satoyoshi E(1981) Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formationJ Neurol Sci. juill51: 1141155 |
4 | Argov Z, Yarom R(1984) “Rimmed vacuole myopathy” sparing the quadriceps: A unique disorder in Iranian JewsJ Neurol Sci64: 13343 |
5 | Park Y-E, Kim H-S, Choi E-S, Shin J-H, Kim S-Y, Son E-H(2012) Limb-girdle phenotype is frequent in patients with myopathy associated with GNE mutationsJ Neurol Sci321: 1-27781 |
6 | Tanboon J, Rongsa K, Pithukpakorn M, Boonyapisit K, Limwongse C, Sangruchi T(2014) A Novel Mutation of the GNE Gene in Distal Myopathy with Rimmed Vacuoles: A Case with InflammationCase Rep Neurol6: 15559 |
7 | Kannan MA, Challa S, Urtizberea AJ, Krahn M, Jabeen AS, Borgohain R(2012) Distal myopathy with rimmed vacuoles and inflammation: A genetically proven caseNeurol India60: 6631634déc |
8 | Weihl CC, Miller SE, Zaidman CM, Pestronk A, Baloh RH, Al-Lozi M(2011) Novel GNE mutations in two phenotypically distinct HIBM2 patientsNeuromusculDisord21: 2102105févr |
9 | Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MCV, Leoyklang P(2014) Mutation Update for GNE Gene Variants Associated with GNE MyopathyHum Mutat Aug35: 8915926 |
10 | Huizing M, Carrillo-Carrasco N, Malicdan MCV, Noguchi S, Gahl WA, Mitrani-Rosenbaum S(2014) GNE myopathy: New name and new mutation nomenclatureNeuromusculDisord NMD24: 5387389mai |
11 | Frédéric MY, Lalande M, Boileau C, Hamroun D, Claustres M, Béroud C(2009) UMD-predictor, a new prediction tool for nucleotide substitution pathogenicity-application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2 Hum Mutat30: 6952959juin |
12 | Ng PC(2003) SIFT: Predicting amino acid changes that affect protein functionNucleic Acids Res31: 13381238141 juill |
13 | Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P(2010) A method and server for predicting damaging missense mutationsNat Methods7: 4248249avr |
14 | Desmet F-O, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C(2009) Human Splicing Finder: An online bioinformatics tool to predict splicing signalsNucleic Acids Res37: 9e67e67 |
15 | Kergourlay V, Raï G, Blandin G, Salgado D, Béroud C, Lévy N(2014) Identification of Splicing Defects Caused by Mutations in the Dysferlin GeneHum Mutat35: 1215321541déc |
16 | Gaildrat P, Killian A, Martins A, Tournier I, Frébourg T, Tosi M(2010) Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variantsMethods Mol Biol653: 249257 |
17 | Cartegni L, Chew SL, Krainer AR(2002) LISTENING TO SILENCE AND UNDERSTANDING NONSENSE: EXONIC MUTATIONS THAT AFFECT SPLICINGNat Rev Genet3: 42852981 avr |
18 | Jian X, Boerwinkle E, Liu X(2014) In silico tools for splicing defect prediction: A survey from the viewpoint of end usersGenet Med16: 7497503 |
19 | Broccolini A, Ricci E, Cassandrini D, Gliubizzi C, Bruno C, Tonoli E(2004) Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion-body myopathyHum Mutat23: 6632632juin |
20 | Penner J, Mantey LR, Elgavish S, Ghaderi D, Cirak S, Berger M(2006) Influence of UDP-GlcNAc 2-Epimerase/ManNAc Kinase Mutant Proteins on Hereditary Inclusion Body Myopathy † Biochemistry (Mosc)45: 929682977mars |
21 | Béhin A, Dubourg O, Laforêt P, Pêcheux C, Bernard R, Lévy N(2008) [Distal myopathy due to mutations of GNE gene: Clinical spectrum and diagnosis]RevNeurol (Paris)164: 5434443mai |
22 | Ikeda-Sakai Y, Manabe Y, Fujii D, Kono S, Narai H, Omori N(2012) Novel Mutations of the GNE Gene in Distal Myopathy with Rimmed Vacuoles Presenting with Very Slow ProgressionCase Rep Neurol4: 2120125 |
23 | Resta N, Susca FC, Di Giacomo MC, Stella A, Bukvic N, Bagnulo R(2006) A homozygous frameshift mutation in the ESCO2 gene: Evidence of intertissue and interindividual variation in Nmd efficiencyJ Cell Physiol209: 16773 |
24 | Kerr TP, Sewry CA, Robb SA, Roberts RG(2001) Long mutant dystrophins and variable phenotypes: Evasion of nonsense-mediated decay?Hum Genet109: 4402407 |
25 | A Phase 2 Study to Evaluate the Dose and Pharmacodynamic Efficacy of Sialic Acid-Extended Release (SA-ER) Tablets in Patients With GNE Myopathy or Hereditary Inclusion Body Myopathy; Ultragenyx Pharmaceutical; ClinicalTrials.gov: NCT01517880 |
Figures and Tables
Fig.1
Transcriptional analysis of the GNE variant c.717T>G using an in vitro minigene splicing assay. Transfection into HEK 293 cells was performed both using a wild type (Exon 5 WT) construction and the mutant pCAS2 construction (Exon 5 c.717T>G). Forty-eight hours after transfection, transcriptional analysis was performed and the effect on splicing identified using RT-PCR was further confirmed using Sanger sequencing. A. RT-PCR analysis identifies the presence of an abnormally spliced transcript (arrow) in HEK 293 cells transfected with the mutant pCAS2 construction (Exon 5 c.717T>G), as compared to cells transfected with the wild type (Exon 5 WT) construction. B. Sequence analysis of the different transcripts identifies for the abnormally spliced transcript a 145 bp deletion of the 3’ extremity of exon 5, correlating with the activation of a cryptic splice-donor site caused by the c.717T>G variant (indicated by *).
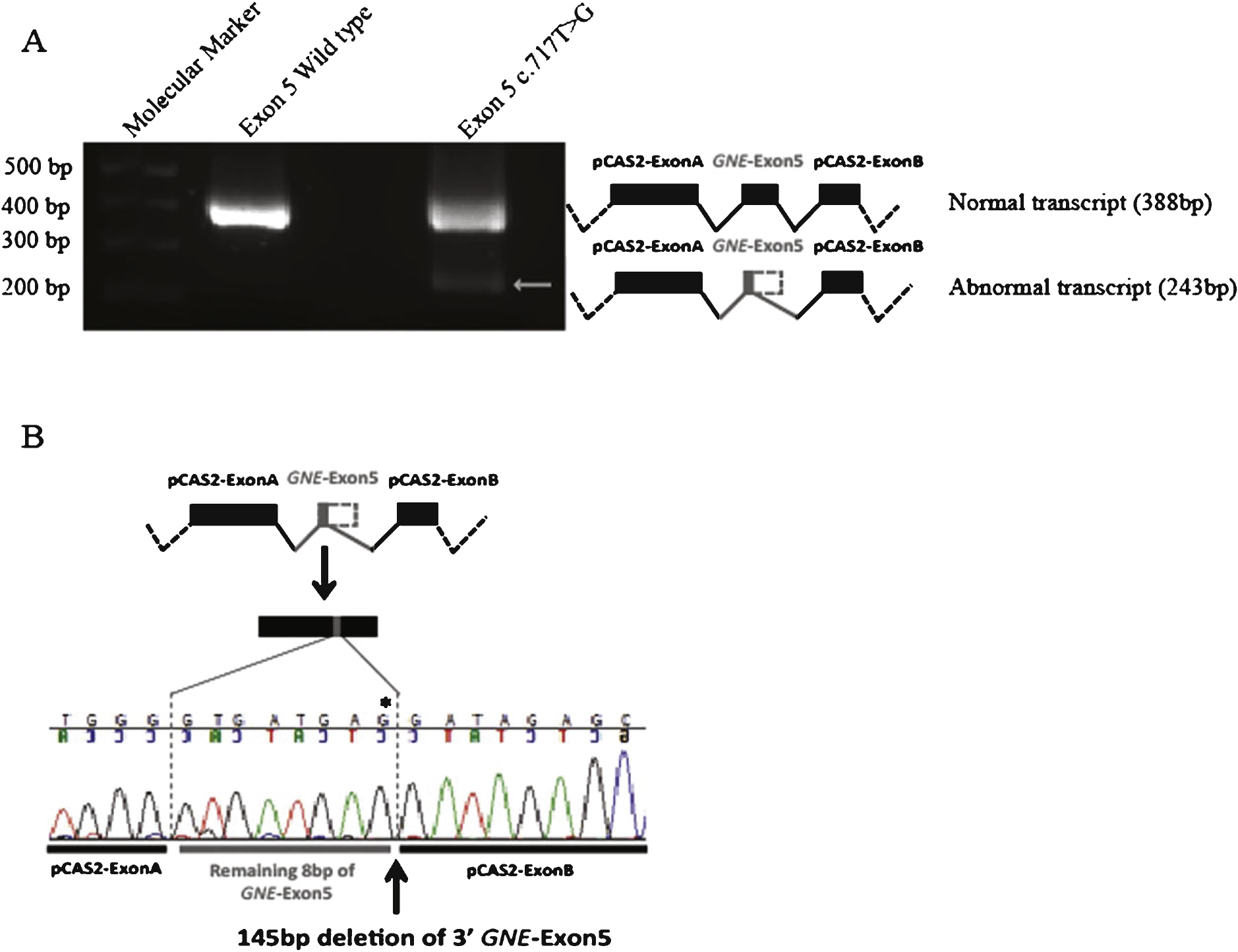
Table 1
Summary of the novel pathogenic variants and published variants associated with GNE myopathy predicted to have an effect on splicing
| Nucleotide Substitution | GNE exona | Amino-acid Substitution (GNE protein domainb) | UMD-predictor | SIFT | Poly-Phen 2 | Severity predictionc | Splicing prediction (HSF)d | |
| mRNA Variant 1 NM_001128227.2 | hGNE2 NP_001121699.1 | |||||||
| Novel pathogenic variants | ||||||||
| c.268C>T | 4 | p.Arg90X (ep) | Severe | |||||
| c.537T>G | 4 | p.His179Gln (ep) | Pathogenic | Damaging | Probably Damaging | Severe | Possible | |
| c.679G>T | 4 | p.Asp227Tyr (ep) | Pathogenic | Damaging | Probably Damaging | Severe | Possible | |
| c.752A>C | 5 | p.His251Pro (ep) | Pathogenic | Damaging | Probably Damaging | Severe | Not affected | |
| c.860C>T | 5 | p.Ala287Val (ep-AR) | Pathogenic | Tolerated | Probably Damaging | Severe | Probable | |
| c.946G>C | 6 | p.Asp316His (ep-AR) | Pathogenic | Damaging | Possibly Damaging | Severe | Possible | |
| c.1183G>A | 8 | p.Gly395Arg (ep) | Pathogenic | Damaging | Probably Damaging | Severe | Uncertain | |
| c.1218A>C | 8 | p.Lys406Asn (ep) | Pathogenic | Tolerated | Possibly Damaging | Medium | Not affected | |
| c.1535G>C | 10 | p.Arg512Pro (kin) | Pathogenic | Tolerated | Possibly Damaging | Medium | Not affected | |
| c.1768G>A | 11 | p.Gly590Arg (kin) | Pathogenic | Damaging | Probably Damaging | Severe | Probable | |
| c.1835G>T | 11 | p.Cys612Phe (kin) | Pathogenic | Damaging | Probably Damaging | Severe | Not affected | |
| c.1850G>A | 11 | p.Cys617Tyr (kin) | Pathogenic | Damaging | Probably Damaging | Severe | Not affected | |
| c.2045T>G | 13 | p.Leu682Arg (kin) | Pathogenic | Damaging | Possibly Damaging | Severe | Not affected | |
| Published variants with predicted deleterious effects on splicing | Initial splicing prediction by Celeste and al. | |||||||
| c.98A>G | 3 | p.Glu33Gly (ep) | Pathogenic | Damaging | Probably Damaging | Severe | Possible | No splicing prediction |
| c.179T>G | 3 | p.Met60Arg (ep) | Pathogenic | Damaging | Probably Damaging | Severe | Probable | No splicing prediction |
| c.271A>G | 4 | p.Met91Val (ep) | Pathogenic | Tolerated | Benign | Mild | Possible | No splicing prediction |
| c.709G>A | 4 | p.Gly237Ser (ep) | Pathogenic | Tolerated | Possibly Damaging | Medium | Probable | Medium splice effect |
| c.715G>A | 5 | p.Asp239Asn (ep) | Probably pathogenic | Tolerated | Benign | Mild | Possible | Mild splice effect |
| c.717T>G | 5 | p.Asp239Glu (ep) | Probably pathogenic | Tolerated | Benign | Milde | Probable | No splicing prediction |
| c.731A>T | 5 | p.Asp244Val (ep) | Pathogenic | Tolerated | Possibly Damaging | Medium | Possible | No splicing prediction |
| c.1394A>G | 9 | p.Tyr465Cys (kin) | Pathogenic | Tolerated | Possibly Damaging | Medium | Possible | No splicing prediction |
| c.2098G>A | 13 | p.Gly700Arg (kin) | Pathogenic | Damaging | Probably Damaging | Severe | Probable | No splicing prediction |
aGNE exon: GNE exons were numbered as in Celeste et al. (2014). bGNE protein domain: - ep: UDP-GlcNAc 2-epimerase domain; - ep-AR: Allosteric region; - kin: ManNAc kinase domain. cSeverity was evaluated by combining the scores of the 3 protein effect prediction programs: - Severe: 3 severe scores; 2 severe and 1 medium; 2 severe and 1 mild. - Medium: 1 severe, 1 medium and 1 mild score. - Mild: 2 mild scores and 1 severe; 2 mild scores and 1 medium. d4 groups of splicing effect prediction were defined: - Probable: strong splicing effect due to broken DS (donor site) or AS (acceptor site) or/and new DS/AS creation or/and strong possibility of broken ESE (Exonic Splicing Enhancer) site. - Possible: medium splicing effect predicted due to new DS/AS or/and medium possibility of broken ESE site. - Uncertain: mild splicing effect due to new DS/AS or/and low possibility of broken ESE site. - Not affected: weak or no splicing effect predicted. eConsidered in Celeste et al. (2014) as “(likely) non-disease causing Single Nucleotide Polymorphism (SNP)”. In Bold: Severe and Medium predicted severity.

