
Aluminum and Amyloid-β in Familial Alzheimer’s Disease
Abstract
Genetic predispositions associated with metabolism of the amyloid-β protein precursor underlie familial Alzheimer’s disease; a form of dementia characterized by early disease onset and elevated levels of cortical amyloid-β. Human exposure to aluminum is linked to the etiology of Alzheimer’s disease and recent research measured a high content of aluminum in brain tissue in familial Alzheimer’s disease. To elaborate upon this finding, we have obtained brain tissues from a Colombian cohort of donors with familial Alzheimer’s disease. We have used established methods to measure the aluminum content of these tissues and we have compared the data with a recently measured dataset for control brain tissues. We report significantly higher levels of aluminum in brain tissues in donors with familial Alzheimer’s disease than in control tissues from donors without neurological impairment or neurodegeneration. We have used aluminum-specific fluorescence microscopy along with complementary imaging for amyloid-β to demonstrate a very high degree of co-localization of these two risk factors in brain tissue in familial Alzheimer’s disease. Aluminum and amyloid-β were co-located in senile plaques as well as vasculature, the latter resembling cerebral amyloid angiopathy. Aluminum was also found separately from amyloid-β in intracellular compartments including glia and neuronal axons. The research has identified an arguably unique association between high brain aluminum content and amyloid-β and allows postulation that genetic predispositions defining familial Alzheimer’s disease underlie this relationship.
INTRODUCTION
An association between aluminum and amyloid-β in Alzheimer’s disease has been postulated for 40 years [1]. It has not been without controversy [2] though a consensus does now support the co-localization if not co-deposition of these two major risk factors in Alzheimer’s disease [3]. Familial Alzheimer’s disease, fAD, is characterized by genetic mutations affecting the expression and metabolism of the amyloid-β protein precursor (AβPP), leading to early onset of disease [4, 5]. One fAD mutation is PS1-E280A (Glu280Ala), a mutation that occurs in a significant cohort of individuals in Colombia. The mutation results in elevated cortical levels of amyloid-β, early disease onset (<50 years of age) and an aggressive disease etiology [6].
Previous research on brain tissue from 12 donors diagnosed as fAD [7] demonstrated significant accumulations of aluminum with 11 of the 12 brains having at least one tissue sample where the concentration of aluminum was defined as pathologically concerning (≥3.00 μg/g dry wt.). Aluminum-specific fluorescence microscopy [8] confirmed the presence of aluminum in these tissues and suggested that the majority of deposits of aluminum were extracellular associated with neuronal and cellular debris. A tentative suggestion was made that aluminum and amyloid-β were co-located in a senile plaque-like structure. Herein we have measured aluminum in brain tissue in Colombian donors carrying the fAD mutation PS1-E280A and compared the data with data for control brains. We have also used aluminum-specific fluorescence microscopy to investigate interrelationships between aluminum and amyloid-β in fAD.
MATERIALS AND METHODS
Human brain tissues
Brain tissues, frozen and fixed, from donors who carried the PS1-E280A mutation were obtained from the brain bank of the Universidad de Antioquia, Medellin, Colombia and research was carried out following ethical approval by Keele University (ERP 2391). The consultant neuropathologist at the London Neurodegenerative Diseases Brain Bank chose the control brain tissues for us and their detailed data are presented elsewhere (Exley and Clarkson, unpublished).
Measurement of aluminum in fAD brain tissues
The aluminum content of tissues was measured by an established and fully validated method [9] that herein is described only briefly. Samples of cortex, between 0.6 and 2.0 g in weight, were thawed at room temperature and cut using a stainless-steel blade into sections approximately 0.3–0.5 g in weight. Tissues were dried for 48 h, to a constant weight, in an incubator at 37°C. Dry and thereafter weighed tissues were digested in a microwave (MARS Xpress CEM Microwave Technology Ltd.) in a mixture of 1 mL 15.8M HNO3 (Fisher Analytical Grade) and 1 mL 30% w/v H2O2 (BDH Aristar). The resulting digests were clear with no fatty residues and, upon cooling, were made up to 5 mL volume using ultrapure water (cond. <0.067 μS/cm). Total aluminum was measured in each sample by transversely heated graphite furnace atomic absorption spectrometry (TH GFAAS) using matrix-matched standards and an established analytical program alongside previously validated quality assurance data [9]. The latter included method blanks, detailed descriptions of which have been published recently [10].
Microtomy
All chemicals were from Sigma Aldrich, UK unless otherwise stated. Paraffin-embedded brain tissue blocks mounted on Tissue Tek® Uni-cassettes® (Sakura Finetek Europe B.V., Netherlands), were cooled on wet ice for 10 min and sectioned at 5 μm via the use of a rotary RM2025 microtome using Surgipath DB80 LX low-profile stainless-steel microtome blades (both from Leica Microsystems, UK). Adjacent and numbered serial sections were floated out on ultrapure water (cond. <0.067 μS/cm) at 45°C and lifted onto SuperFrost® Plus slides (Thermo Scientific, UK). Sections were subsequently dried at ambient temperature overnight. Dried sections were heated at 62°C for 20 min and allowed to cool to ambient temperature, immediately prior to use.
Deparaffinization and rehydration of paraffin-embedded tissue sections
Tissue sections were deparaffinized and rehydrated through 250 mL of the following reagents: Histo-Clear (National Diagnostics, US) for 3 min, fresh Histo-Clear for 1 min, 100% v/v ethanol (HPLC grade used throughout) for 2 min and 95, 70, 50, and 30% v/v ethanol for 1 min. Slides were finally rehydrated via immersion into ultrapure water for 35 s. Sections were agitated throughout processing. For fluorescence microscopy, rehydrated tissue sections were subsequently outlined with a hydrophobic PAP pen allowing for staining in moisture chambers.
Lumogallion staining
Lumogallion (4-chloro-3-(2,4-dihydroxyphenyla zo)-2-hydroxybenzene-1-sulphonic acid, TCI Europe N.V., Belgium) staining was performed as described elsewhere [11]. Briefly, lumogallion prepared at 1 mM in 50 mM PIPES, pH 7.4 was added to PAP-outlined sections in moisture chambers for 45 min. Autofluorescence of the non-stained sections made use of the PIPES buffer only. Following staining, sections were rinsed in the same PIPES buffer and washed for 30 s in ultrapure water, prior to mounting with Fluoromount™.
Amyloid-β staining
Congo red and thioflavin S (ThS) staining was performed as previously described [12]. Briefly, Congo red staining was performed via immersion of rehydrated tissue sections into 0.5% w/v Congo red in 50% v/v ethanol for 5 min, rinsing in 0.2% w/v potassium hydroxide in 80% v/v ethanol for 3 s and washing in ultrapure water for 30 s. Sections were mounted with Faramount (Agilent Dako, UK). For ThS staining, rehydrated sections were stained in moisture chambers with ca 0.075% w/v ThS in 50% v/v ethanol for 8 min, twice rinsed for 10 s in 80% v/v ethanol and washed in ultrapure water for 30 s. Sections were mounted with Fluoromount™.
Fluorescence microscopy
Fluorescence, brightfield, and polarized light microscopy were performed via use of an Olympus BX50 fluorescence microscope (mercury source), equipped with a BX-FLA reflected light attachment. Lumogallion fluorescence was collected through an Olympus U-MNIB3 fluorescence filter cube (excitation filter: 470–495 nm, dichromatic mirror: 505 nm, longpass emission filter: 510 nm) and ThS fluorescence collected by use of an Olympus U-MWBV2 cube (excitation filter: 400–440 nm, dichromatic mirror: 455 nm, longpass emission filter: 475 nm). Polarized light illumination was achieved by use of a U-POT drop-in polarizer and a U-ANT transmitted light analyzer (both from Olympus, UK). Images were acquired by use of a ColorView III charged coupled device (CCD) camera and the CellD software suite (Olympus, SiS Imaging Solutions, GmbH). Fluorescence channels were merged using Photoshop (Adobe Systems Inc., USA).
Statistical analyses
Data for aluminum content of tissues were skewed and were not normally distributed. For descriptive summary statistics, the median and interquartile-range were calculated for each donor, each lobe, sex and group. For all test statistics and models, aluminum content data were log transformed. Due to unbalanced groups (fAD donors = 12, control donors = 20) and unbalanced numbers of samples per donor and lobe, mixed effect models including random effects for donors and lobe were used [13].
First, a model was calculated for the fAD group to analyze differences between the factors lobes, gender, and associations with the covariate age. The model contains all these factors as main terms without interactions. Additional random effects were nested and included the factors number of samples, nested within lobe and donor.
A second model included the factor group, to analyze differences between the fAD and the control groups. We considered a p-value smaller than 0.05 to be statistically significant. To obtain pairwise differences between lobes post hoc tests with Tukey correction were performed with the function glht from the R package multcomp. For all analysis RStudio Version 1.1.463 © 2009–2018 were used.
RESULTS
Aluminum content of fAD brain tissues
The aluminum content of brain tissue across all donors and all brain regions ranged from 0.30 to 33.48 μg/g dry wt. (Table 1). The majority of tissues (45 out of 83) had an aluminum content above 1.99 μg/g dry wt. with 14, 24, 10, and 35 tissues having contents in the range <1.00, 1.00–1.99, 2.00–2.99, and ≥3.00 μg/g dry wt. respectively. The median (IQR) aluminum content for each lobe was 2.04 (1.09–5.67), 2.19 (1.44–5.14), 2.11 (1.04–4.88), and 2.31 (1.02–6.48) for occipital, temporal, frontal, and parietal, respectively. There were no significant differences in aluminum content between lobes. There was no significant relationship between age of donor and aluminum content. There was a significant difference in aluminum content (median and IQR) between genders (Supplementary Figure 1) with females (4.68; 3.22–6.95) having a higher content than males (1.62; 1.04–3.86) (p = 0.01951).
Table 1
Aluminum content (μg/g dry wt.) of brain tissues from Colombian donors with a diagnosis of familial Alzheimer’s disease
| Case ID | Gender | Age | Brain Region – Lobe | |||
| Occipital | Temporal | Frontal | Parietal | |||
| 225 | Male | 48 | 2.21 | 3.07 | 1.20 | 1.02 |
| 1.20 | 0.81 | 11.81 | ||||
| 4.78 | ||||||
| 213 | Male | 55 | 8.54 | 1.40 | 1.05 | 1.82 |
| 2.71 | 2.19 | 1.22 | 2.67 | |||
| 1.05 | 2.31 | |||||
| 244 | Male | 55 | 1.60 | 5.35 | 2.33 | 6.48 |
| 193 | Male | 48 | 0.41 | 1.03 | 1.10 | 0.64 |
| 0.54 | 1.47 | 0.27 | 0.53 | |||
| 0.83 | 1.02 | |||||
| 189 | Female | 52 | 1.10 | 5.83 | 0.70 | 4.90 |
| 3.22 | ||||||
| 163 | Female | 68 | 5.06 | 2.36 | 1.89 | 4.62 |
| 3.90 | 3.34 | |||||
| 2.19 | ||||||
| 178 | Male | 59 | 0.30 | 1.49 | 0.88 | 0.44 |
| 1.54 | 0.49 | |||||
| 0.86 | ||||||
| 023 | Male | 61 | 2.04 | 1.29 | ||
| 088 | Female | 50 | 3.57 | 7.42 | 3.49 | |
| 0.62 | ||||||
| 237 | Male | 53 | 1.07 | 1.92 | 4.12 | 1.72 |
| 1.35 | 8.01 | 10.27 | 1.88 | |||
| 1.63 | ||||||
| 77 | Female | 54 | 6.27 | 5.68 | 4.68 | 9.73 |
| 9.38 | 42.50 | 6.95 | ||||
| 22.17 | ||||||
| 31.30 | ||||||
| 204 | Male | 52 | 31.16 | 4.92 | 16.44 | 2.05 |
| 33.48 | 6.88 | 9.83 | ||||
| 5.47 | ||||||
Data are for all tissue samples measured with 1–4 replicates per lobe.
Comparison with control brain tissues
The aluminum content (median and IQR) of fAD brain tissues (2.19; 1.10–5.41) was significantly higher (p < 0.001) than control tissues (0.60; 0.35–0.98; Supplementary Figure 2).
Imaging of aluminum and amyloid-β in fAD
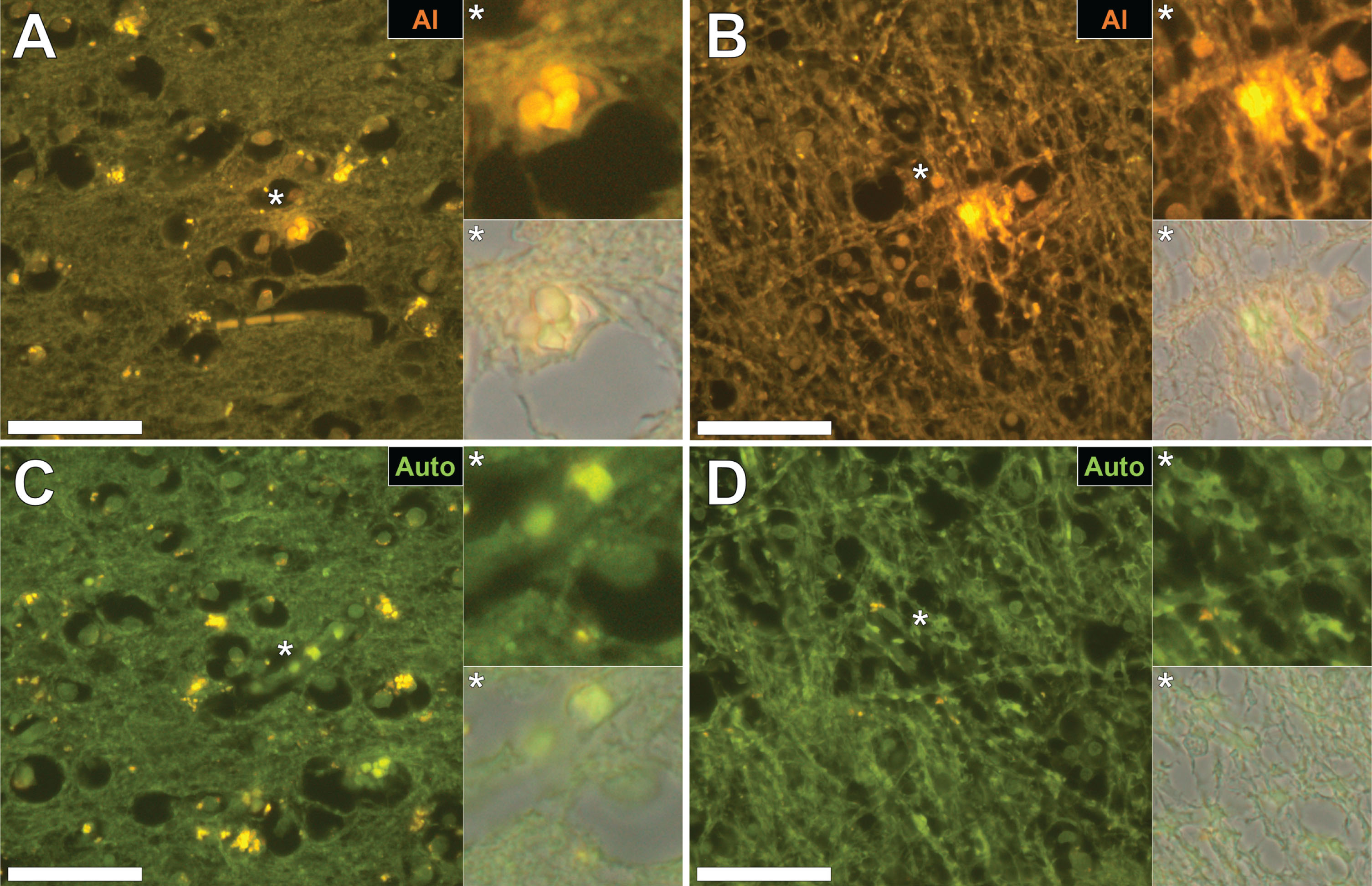
Aluminum-specific fluorescence microscopy confirmed the presence of significant numbers of deposits of aluminum in brain tissue from all donors across all four lobes and in grey and white matter. Approximately two thirds of aluminum deposits were identified in grey matter. The majority of all deposits of tissue aluminum were shown to be extracellular and in most cases (71 out of 89), as confirmed using both ThS and Congo red, were co-located with amyloid-β. Tissue aluminum emitted characteristic orange fluorescence and was often senile plaque-like in size and appearance (Figs. 1A, 2A). Complementary staining of both same and serial sections using ThS and/or Congo red identified these aluminum-rich deposits as being additionally composed of amyloid-β (Figs. 1B, 2B) with merged images demonstrating intimate associations between amyloid and the metal (Figs. 1C, 2C). Congo red in combination with polarized light confirmed the presence of β-sheet amyloid-β in these plaque-like structures (Figs. 1D, 2D). In some structures, aluminum appeared to be associated with a core of amyloid-β (Fig. 3A–C) surrounded by a ‘nest’ of ThS-positive amyloid threads (Fig. 3C, D). There was also evidence of classical cerebral amyloid angiopathy where blood vessel walls stained positively for both aluminum and amyloid-β with the latter showing characteristic apple-green birefringence under polarized light (Fig. 4A–D). Occasional ThS-positive deposits were identified as spherulites by their classical Maltese cross signature under polarized light (Fig. 4C, D). Occasionally aluminum was identified in intracellular locations in tissues including in glia-like cells (Fig. 5A) and in neuronal axons (Fig. 5B).
Fig.1
Aluminum and amyloid-β deposition in a senile plaque in the occipital cortex of a donor with familial Alzheimer’s disease (fAD). A) Aluminum is identified using lumogallion as an orange fluorescence emission. B) Thioflavin S (ThS) staining of an adjacent serial section revealed a green fluorescence emission characteristic of amyloid-β. C) Merging of fluorescence channels reveals co-localization of aluminum and amyloid-β. D) Congo red staining confirmed the presence of amyloid-β in the same plaque, producing a red hue under bright-field illumination and apple-green birefringence (magnified insert denoted by an asterisk) under polarized light. Magnification: X 400, scale bars = 50 μm.
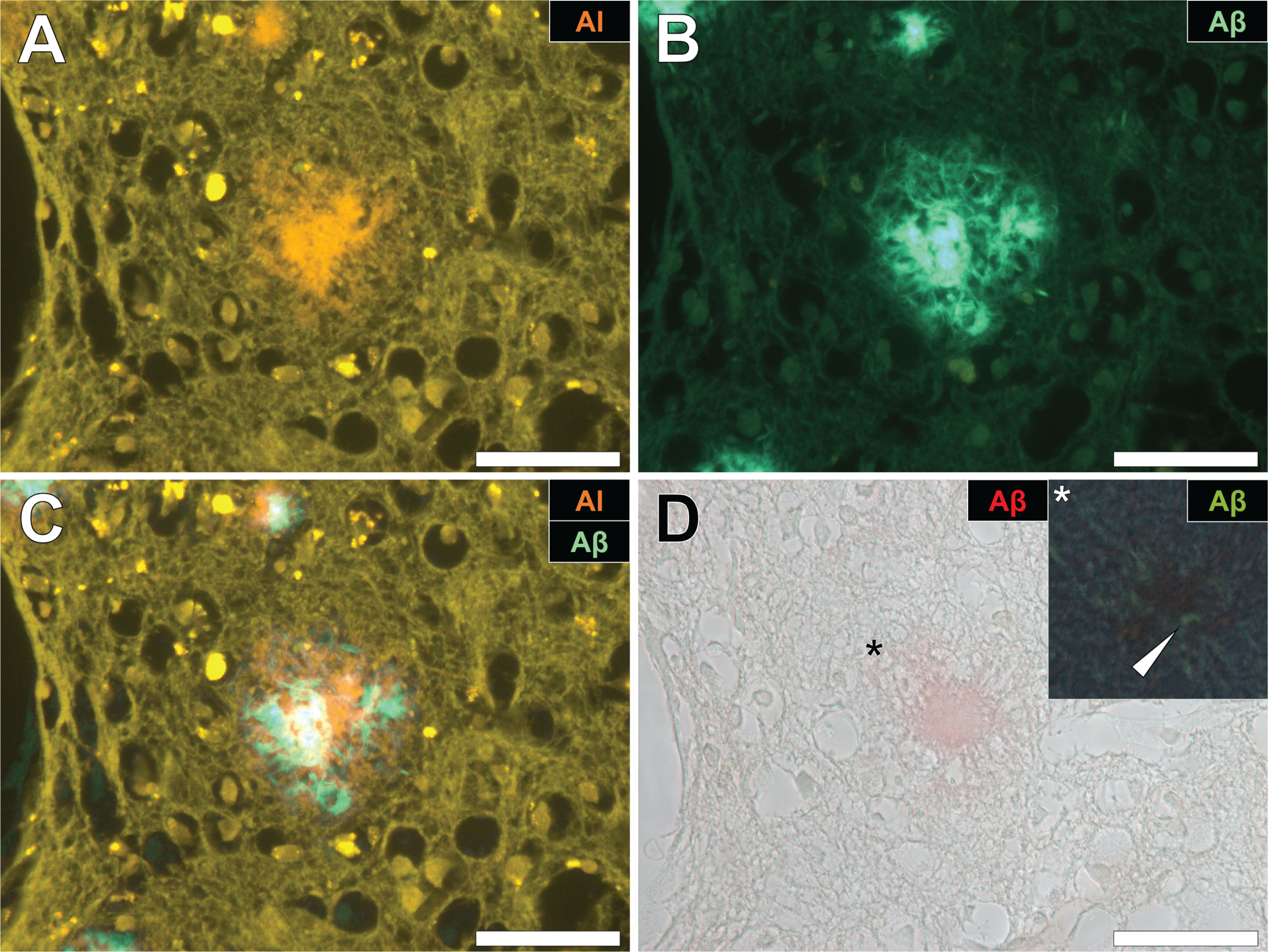
Fig.2
Aluminum and amyloid-β co-located in a senile plaque in the grey-white matter interface of the parietal lobe of a donor with fAD. A) Aluminum was detected via an orange fluorescence emission upon lumogallion staining. B) ThS-reactive fluorescence (green) indicative of amyloid-β in the identical senile plaque. C) Merging of fluorescence channels reveals co-localization of aluminum and amyloid-β. D) Congo red staining of the same tissue region revealed apple-green birefringence in the form of a Maltese-Cross diffraction pattern or spherulite (magnified insert), under polarized light. Magnification: X 400, scale bars = 50 μm.
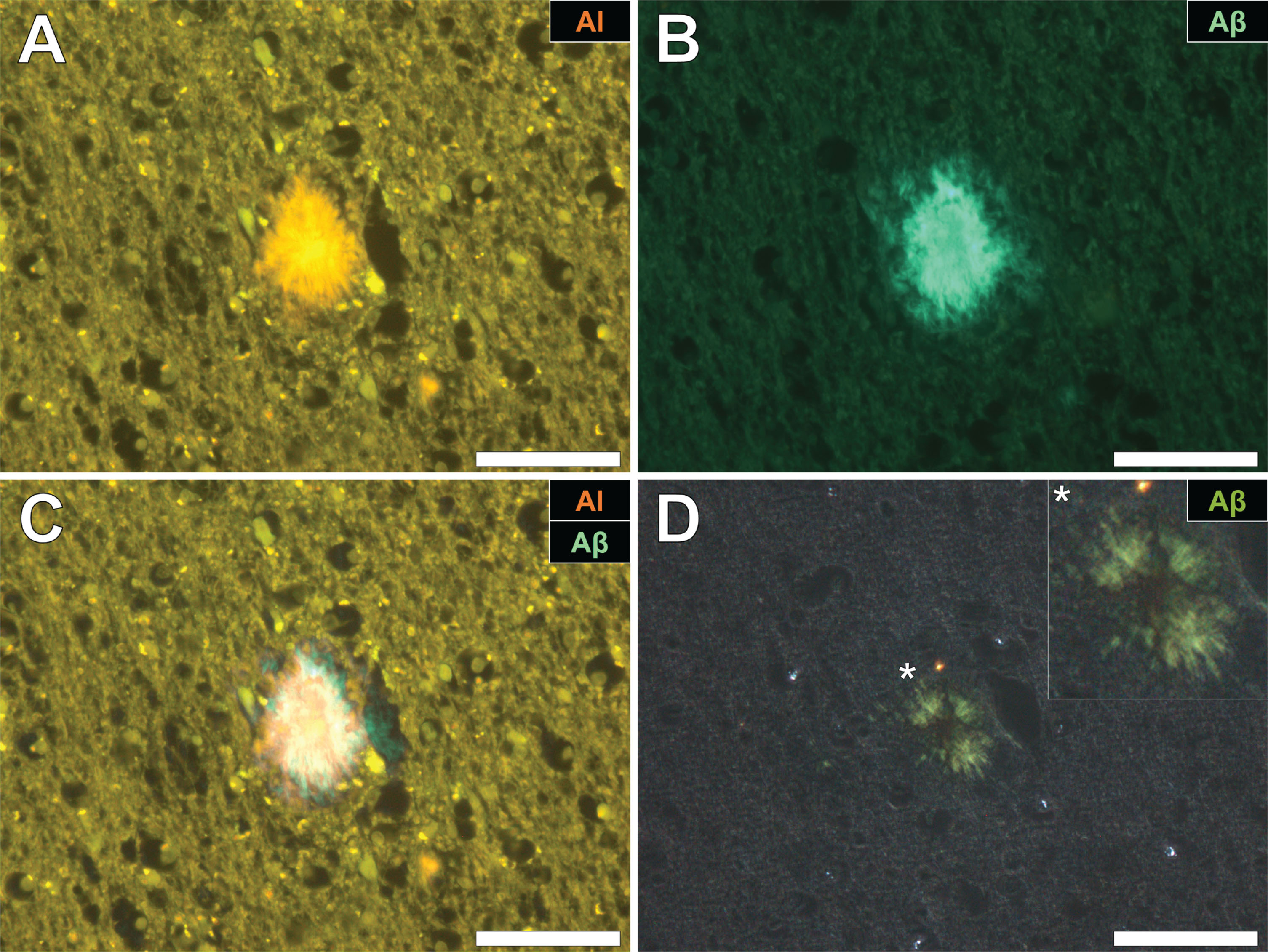
Fig.3
Aluminum deposition in a senile plaque in the temporal cortex of a donor with fAD. A) Autofluorescence of the non-stained section revealed a weak green fluorescence emission of a plaque-like structure. B) Aluminum identified as orange fluorescence upon lumogallion staining. C) ThS staining showed the presence of a senile plaque via an intense green fluorescence emission. D) Merging of the lumogallion and ThS fluorescence channels identified aluminum at the core of the senile plaque with ThS reactive threads of amyloid-β identified at its periphery (white arrows). Magnified inserts are denoted by asterisks in the respective fluorescence micrographs. Magnification: X 400, scale bars = 50 μm.
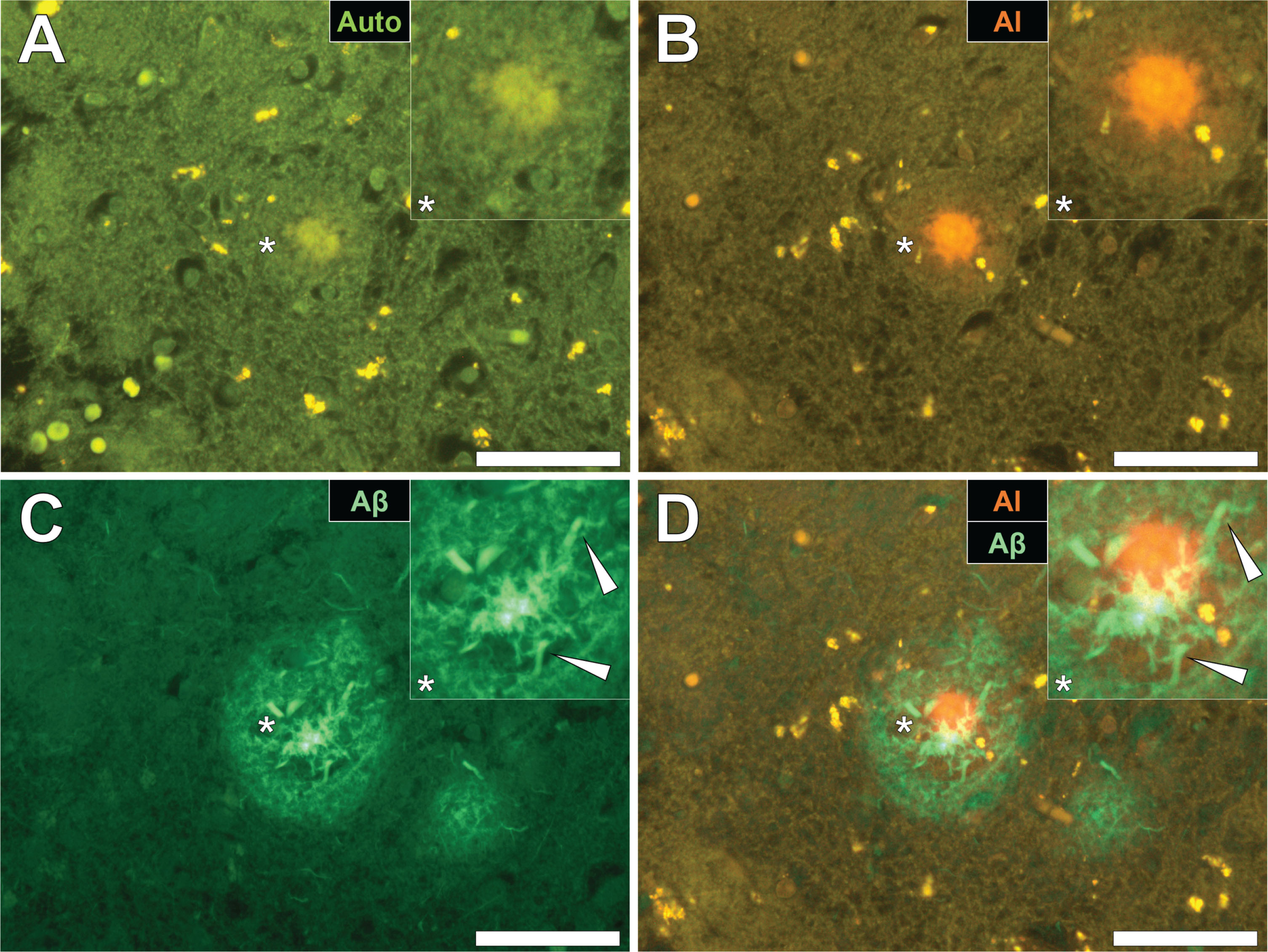
Fig.4
Cerebral amyloid angiopathy (CAA) in a blood vessel in the frontal cortex of a donor with fAD. A) Autofluorescence revealed a weak green fluorescence emission of the vessel wall. B) Lumogallion revealed the presence of aluminum (orange fluorescence) in the same vessel. C) Positive ThS-staining identifies amyloid-β in the vessel wall. D) Apple-green birefringence when stained with Congo red under polarized light is indicative of amyloid-β in a β sheet conformation. White arrows highlight mineralized deposits appearing as spherulites, producing a Maltese-Cross diffraction pattern. Magnification: X 400, scale bars = 50 μm.
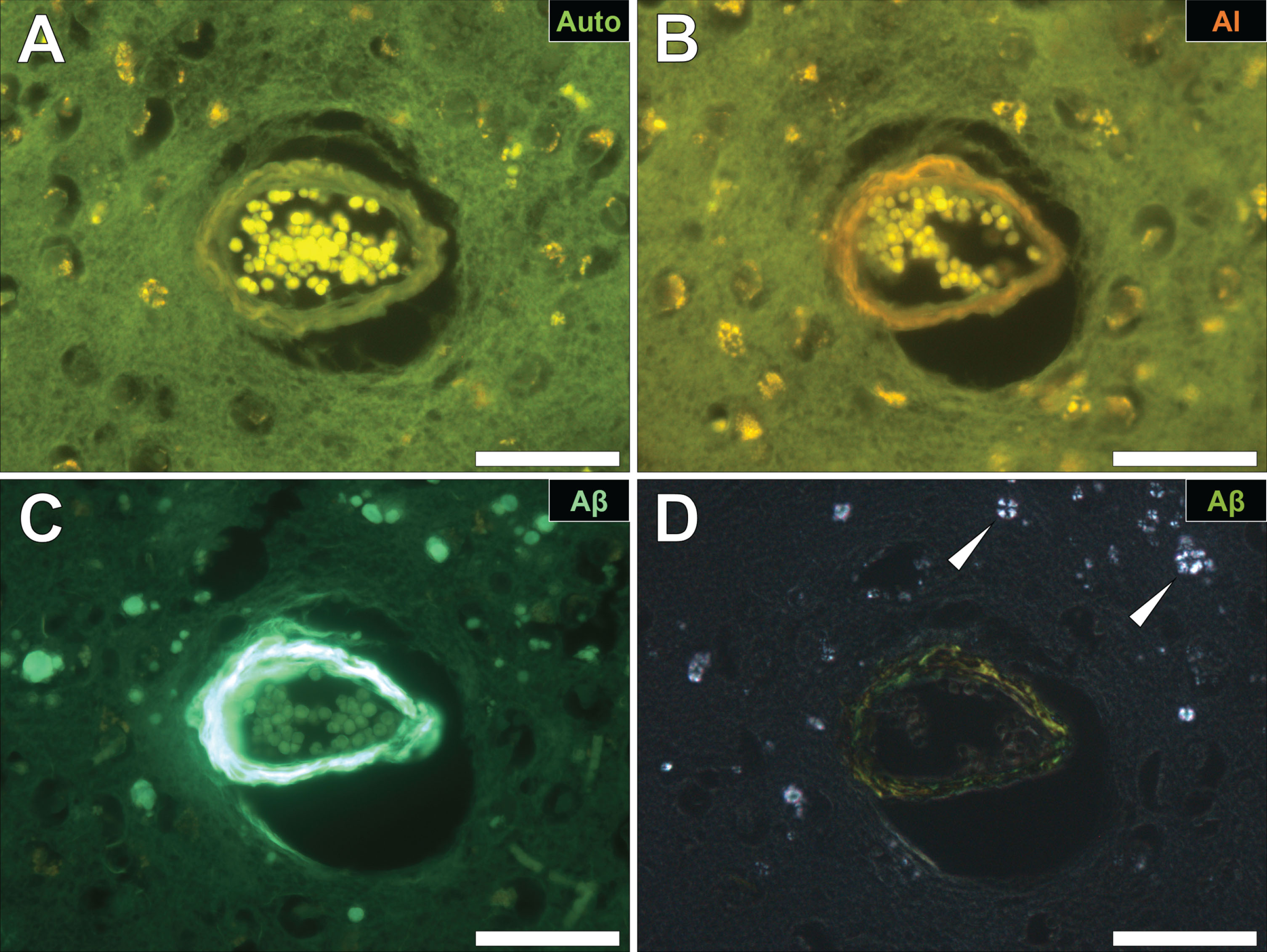
Fig.5
Intracellular aluminum in glial cells in the occipital cortex (A) and in neuronal axons in white matter of the parietal lobe (B) of donors with fAD. C, D) Autofluorescence of unstained serial sections confirming the presence of aluminum. Magnified inserts are denoted by asterisks in the respective fluorescence micrographs. Magnification: X 400, scale bars = 50 μm.
DISCUSSION
The aluminum content of brain tissue from donors with the PS1-E280A fAD mutation was universally high with 42% of tissues having a concentration of aluminum above 3.00 μg/g dry wt., a content considered as pathologically significant [10]. Aluminum content in brain tissue in fAD was significantly higher than brain tissues from donors with neither clinical nor neuropathological diagnosis of neurodegenerative disease (Exley and Clarkson, unpublished). The new data confirm unequivocally the previous observation of very high brain aluminum content in fAD [7]. Aluminum-specific fluorescence microscopy identified aluminum in all brain tissues investigated (Figs. 1–5). The majority of all deposits of aluminum were extracellular and predominantly co-located with amyloid-β in primarily senile plaques and occasionally the vasculature. While the predominance of aluminum-amyloid deposits and the frequency with which aluminum and amyloid-β were observed together suggested their co-deposition, detailed scrutiny of the images favored their co-localization. For example, with either aluminum or amyloid-β acting as a nidus for the precipitation of the other. Deposits of both aluminum and amyloid were identified in the absence of each other. The former were found in a number of intracellular environments including in microglia-like cells and associated with neuronal axons. The presence of aluminum in axons in fAD brain tissues might imply an association with tau protein [14] though such remains to be confirmed.
This is the second study confirming significantly high brain aluminum content in fAD but it is the first to demonstrate an unequivocal association between the location of aluminum and amyloid-β in fAD. It shows that two prominent risk factors in the etiology of AD [15, 16] are intimately interwoven in the neuropathology of fAD. While there is a long history suggesting a role for aluminum in the provenance of senile plaques [17], the observation herein of the co-localization of aluminum and amyloid-β in vasculature has not previously been seen in either cerebral amyloid angiopathy [18] or autism spectrum disorder (Mold and Exley, unpublished). The association of aluminum and amyloid-β in brain tissue in neurodegenerative disease is not inevitable and so raises the question, what is different about fAD? The two distinct populations of fAD studied, previously [7] and herein, share a single characteristic in being prone to elevated levels of cortical amyloid-β early in life. This is attributed to genetic mutations associated with the metabolic machinery processing AβPP [1, 4, 5]. However, are the elevated levels of cortical amyloid-β, as opposed to cerebral spinal fluid amyloid-β [19], the direct consequence of enzymatic processing of AβPP or, perhaps, the indirect effect of elevated levels of brain aluminum? Might genetic predispositions underlying fAD be responsible for increased uptake and retention of aluminum in brain tissue with elevated amyloid-β being an indirect or even direct consequence of such. One could envisage increased amyloid-β in brain tissue as either an ameliorative response to high brain aluminum content or simply adventitious, aluminum acting as a nidus for the precipitation of amyloid-β. Either way it does seem indisputable that aluminum and amyloid-β are inextricably linked in the neuropathology of fAD.
ACKNOWLEDGMENTS
MM is a Children’s Medical Safety Research Institute (CMSRI; a charity based in Washington DC, USA) Research Fellow. Many thanks to the families involved with donation of tissues to the brain bank of the Universidad de Antioquia, Medellin, Colombia.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-1140r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-191140.
REFERENCES
[1] | Duckett S , Galle P ((1980) ) Electron microscope-microprobe studies of aluminium in the brains of cases of Alzheimer’s disease and aging patients. J Neuropathol Exp Neurol 39: , 350. |
[2] | Landsberg JP , McDonald B , Watt F ((1992) ) Absence of aluminium in neuritic plaque cores in Alzheimer’s disease. Nature 360: , 65–68. |
[3] | Beauchemin D , Kisilevsky R ((1998) ) A method based on ICP-MS for the analysis of Alzheimer’s amyloid plaques. Anal Chem 70: , 1026–1029. |
[4] | Goate A , Chartierharlin MC , Mullan M , Brown J , Crawford F ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s-disease. Nature 349: , 704–706. |
[5] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[6] | Lopera F , Ardilla A , Martínez A , Madrigal L , Arango-Viana JC , Lemere CA , Arango-Lasprilla JC , Hincapíe L , Arcos-Burgos M , Ossa JE , Behrens IM , Norton J , Lendon C , Goate AM , Ruiz-Linares A , Rosselli M , Kosik KS ((1997) ) Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA 277: , 793–799. |
[7] | Mirza A , King A , Troakes C , Exley C ((2017) ) Aluminium in brain tissue in familial Alzheimer’s disease. J Trace Elem Med Biol 40: , 30–36. |
[8] | Mirza A , King A , Troakes C , Exley C ((2016) ) The identification of aluminum in human brain tissue using lumogallion and fluorescence microscopy. J Alzheimers Dis 54: , 1333–1338. |
[9] | House E , Esiri M , Forster G , Ince PG , Exley C ((2012) ) Aluminium, iron and copper in human brain tissues donated to the medical research council’s cognitive function and ageing study. Metallomics 4: , 56–65. |
[10] | Exley C , Mold MJ ((2019) ) Aluminium in human brain tissue: How much is too much? J Biol Inorg Chem 24: , 1279–1282. |
[11] | Mold M , Umar D , King A , Exley C ((2018) ) Aluminium in brain tissue in autism. J Trace Elem Med Biol 46: , 76–82. |
[12] | Mold M , Cottle J , Exley C ((2019) ) Aluminium in brain tissue in epilepsy: A case report from Camelford. Int J Environ Res Public Health 16: , 2129. |
[13] | Zuur AF , Ineo EN , Walker NJ , Saveliev AA , Smith GM ((2009) ), Mixed Effects Models and Extensions in Ecology with R. Springer-Verlag, New York. |
[14] | Good PF , Perl DP , Bierer LM , Schmeidler J ((1992) ) Selective accumulation of aluminium and iron in the neurofibrillary tangles of Alzheimer’s disease: A laser microprobe (LAMMA) study. Ann Neurol 31: , 286–292. |
[15] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease - the amyloid cascade hypothesis. Science 256: , 184–185. |
[16] | Exley C ((2017) ) Aluminum should now be considered a primary etiological factor in Alzheimer’s disease. J Alzheimers Dis Rep 1: , 23–25. |
[17] | Exley C ((2005) ) The aluminium-amyloid cascade hypothesis and Alzheimer’s disease. In Alzheimer’s Disease: Cellular and Molecular Aspects of Amyloid Beta, HarrisR, FahrenholzF, eds. Subcellular Biochemistry Volume 38, pp. 225–234. |
[18] | Mold M , Cottle J , King A , Exley C ((2019) ) Intracellular aluminium in inflammatory and glial cells in cerebral amyloid angiopathy: A case report. Int J Environ Res Public Health 16: , E1459. |
[19] | Fleisher AS , Chen K , Quiroz YT , Jakimovich LJ , Gutierrez Gomez M , Langois CM , Langbaum JB , Roontiva A , Thiyyagura P , Lee W , Ayutyanont N , Lopez L , Moreno S , Muñoz C , Tirado V , Acosta-Baena N , Fagan AM , Giraldo M , Garcia G , Huentelman MJ , Tariot PN , Lopera F , Reiman EM ((2015) ) Associations between biomarkers and age in the presenilin 1 E280A autosomal dominant Alzheimer disease kindred: A cross-sectional study. JAMA 72: , 316–324. |

