
Trehalose Improves Cognition in the Transgenic Tg2576 Mouse Model of Alzheimer’s Disease
Abstract
This study assessed the therapeutic utility of the autophagy enhancing stable disaccharide trehalose in the Tg2576 transgenic mouse model of Alzheimer’s disease (AD) via an oral gavage of a 2% trehalose solution for 31 days. Furthermore, as AD is a neurodegenerative condition in which the transition metals, iron, copper, and zinc, are understood to be intricately involved in the cellular cascades leading to the defining pathologies of the disease, we sought to determine any parallel impact of trehalose treatment on metal levels. Trehalose treatment significantly improved performance in the Morris water maze, consistent with enhanced learning and memory. The improvement was not associated with significant modulation of full length amyloid-β protein precursor or other amyloid-β fragments. Trehalose had no effect on autophagy as assessed by western blot of the LC3-1 to LC3-2 protein ratio, and no alteration in biometals that might account for the improved cognition was observed. Biochemical analysis revealed a significant increase in the hippocampus of both synaptophysin, a synaptic vesicle protein and surrogate marker of synapses, and doublecortin, a reliable marker of neurogenesis. The growth factor progranulin was also significantly increased in the hippocampus and cortex with trehalose treatment. This study suggests that trehalose might invoke a suite of neuroprotective mechanisms that can contribute to improved cognitive performance in AD that are independent of more classical trehalose-mediated pathways, such as Aβ reduction and activation of autophagy. Thus, trehalose may have utility as a potential AD therapeutic, with conceivable implications for the treatment of other neurodegenerative disorders.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disorder manifesting as progressive cortical and hippocampal cell loss resulting in cognitive decline. It is the leading form of dementia in the elderly being both sporadic and familial in nature, and to date there is no effective therapy to arrest the cognitive decline. The two major hallmarks of AD pathology are the accumulations of the ∼4 kD amyloid-β peptide (Aβ), the cleavage product of the larger amyloid-β protein precursor (AβPP), and the accumulation of intracellular neurofibrillary tangles which consist of hyperphosphorylated microtubule associated protein tau. Recently, a study in the APP/PS1 (APP KM670/671NL (Swedish), PSEN1 L166P) transgenic mouse model of AD that contains the human transgenes for both APP bearing the Swedish mutation and Presenillin1 containing the L166P mutation, indicated that the stable disaccharide, trehalose dihydrate, effectively alleviated cognitive decline as evidenced by a reduced latency to locate the hidden platform in the Morris water maze, concomitant with reduced Aβ plaques in the hippocampus of the trehalose treated mice [1]. While these data were encouraging, the utility of trehalose as a potential therapeutic for AD was clouded by the delivery method, which involved the use of invasive intracerebral injections. Given these data, we elected to examine the therapeutic utility of trehalose in the well characterized and widely used Tg2576 (APP KM670/671L) transgenic mouse model of AD using a more physiologically relevant and translatable oral delivery method. The Tg2576 overexpresses a mutant form of APP (695 isoform) resulting in elevated levels of Aβ and an age-associated accumulation of amyloid plaques concomitant with cognitive deficits.
Trehalose has demonstrated therapeutic utility in many other neurodegenerative mouse models. Briefly, trehalose has been shown to suppress inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage in rabbits [2], induce chaperone molecules along with autophagy in a mouse model of Lewy body disease [3], ameliorate dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation [4], delay the progression of amyotrophic lateral sclerosis by enhancing autophagy in motorneurons [5], alleviate polyglutamine-mediated pathology in a mouse model of Huntington disease [6], and upregulate progranulin expression in human and mouse models of progranulin haploinsufficiency [7]. Given the breadth of positive data regarding trehalose in animal models of neurodegeneration, the potential for the use of trehalose as a human therapeutic for AD has been hypothesized [8, 9], although this yet to be thoroughly investigated.
Finally, the metal ions, iron (Fe) [10–12], zinc (Zn) [13, 14], and copper (Cu) [15–17], are considered key factors in the pathogenic accumulation of Aβ into amyloid plaques as well as to the loss of tau function and neurofibrillary tangle formation. This concept arose more than two decades ago and has been the subject of much scientific review [18–21], prompting discussion around the potential of metal modulating therapies [22–26], leading to investigation of the therapeutic efficacy of metal modulating compounds in transgenic animal models of AD [27–30] and in human clinical trials [31]. As such, and because the effect of trehalose in other models suggests a potential impact on metals (e.g., effects on protein aggregation, oxidative stress, cognition, etc.), then in this study we also sought to determine whether trehalose treatment impacted metal levels, or indeed whether there was any correlation between metal levels and outcomes following trehalose treatment.
METHODS
All procedures were carried out in accordance with protocols approved by the Howard Florey Animal Ethics Committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes as described by the National Health and Medical Research Council of Australia.
Compound
Trehalose (Sigma) has a generous safety profile in rodents and humans [32, 33] and was posted as “Generally Regarded as Safe” (GRAS) for human consumption by the U.S. Federal Drug Administration (FDA) in October 2000. It is used as a food additive, and is also an excipient in many pharmaceuticals, making trehalose a safe, natural and pharmaceutically accepted product.
Animals
Transgenic Tg2576 and wild-type mice were treated daily with either a 2% trehalose solution (0.1 ml/10 g bodyweight; Tg2576; n = 15, average age 16.2±3 months of age, and wild-type; n = 10, average age 16.4±3 months of age) or standard suspension vehicle (SSV; 0.9% NaCl, 0.5% Na-carboxymethylcellulose, 0.5% benzyl alcohol and 0.4% Tween 80) (Tg2576; n = 12, 16±3 months of age, and wildtype; n = 10, 16.2±3 months of age) via oral gavage. Mice were acquired from Charles River Laboratories, Wilmington, Massachusetts, USA. All mice used in this study were re-genotyped after completion of the study to ensure accuracy of transgenic background.
Behavioral assessment
The Morris water maze was used to assess the effect of trehalose on spatial learning and memory function. The pre-training acclimation day of the water maze was performed on day 23 of dosing, followed by six days of place discrimination training of four 90 second trials per day, conducted on days 24–29 of dosing. The probe trial was performed 24 hours after training on dosing day 30 to assess retention of the task. Mice were culled on the following day one hour after dosing. Data was processed using the Ethovision automated tracking system prior to statistical analysis.
Western blotting analysis
The cortex and hippocampus of one hemisphere was homogenized in 15 volumes of ice-cold PBS containing Complete Protease Inhibitor Cocktail tablets (Roche Applied Science, Indianapolis, IN, USA) and subsequently centrifuged (100,000xg) for 30 min at 4°C. Supernatant was removed to yield the soluble fraction. The remaining pellet underwent further extraction via vigorous agitation (30 min) in the above-mentioned homogenization buffer containing 2% (vol/vol) Triton X100. Insoluble material was pelleted via centrifugation (20,000 g, 20 min), and the supernatant retained as the membrane fraction. Protein concentrations were determined using the Pierce BCA protein assay (Pierce Biotechnology, Rockford, IL, USA), and were used to ensure equal protein loading (10 μg) on the gel. Samples were prepared for PAGE by the addition of 4x protein sample loading buffer (LICOR, Lincoln, Nebraska, USA) and 10x NuPAGE sample reducing agent (to a final 1x concentration). Samples were heated to 70°C for 10 min, loaded onto Bolt 4–12% Bis-Tris Plus gels (Invitrogen-Life Technologies, Grand Island, NY, USA) along with Odyssey One-Color protein molecular weight markers (LICOR, Lincoln, Nebraska, USA, Cat LCR928-4000) and run at 125 V for 60 min in appropriately diluted Bolt MES SDS 20x running buffer (Invitrogen-Life Technologies, Grand Island, NY, USA). Gels were transferred to Immobilon–PF, PVDF membrane (Millipore) using the Invitrogen Bolt wet-gel Transfer Device (Invitrogen-Life Technologies, Grand Island, NY, USA) at 15 V for 60 min in appropriately diluted 20x Bolt transfer buffer (Invitrogen-Life Technologies, Grand Island, NY, USA). Membranes were blocked in Tris-buffered saline with tween 20 (TBST) containing 5% skim milk powder and then incubated with primary antibody overnight at 4°C (Doublecortin (DCX) antibody diluted 1 : 1000, Cell Signaling Technology, Danvers, MA, USA Cat # 4604; Synaptophysin antibody diluted 1 : 1000, Millipore, MA, USA, Cat # AB9272; GAPDH diluted 1 : 10000 Millipore, MA, USA, Cat # MAB374; Pro-granulin antibody diluted 1 : 1000 Invitrogen, MA, USA, Cat # PA5-46995; 4G8 diluted 1 : 500 Biolegend CA, USA Cat # SIG-39229; 22C11 (in-house) antibody diluted 1 : 500). Blots were rinsed in TBST and incubated with appropriate secondary antibody at room temperature for 1 h (IRDye800 Goat anti-mouse Cat # LCR926-32210; IRdye800 Goat anti-Rabbit Cat # LCR926-32211; IRDye680 Goat anti-mouse Cat # LCR926-68070; IRDye680 Goat anti-Rabbit Cat # LCR926-68071; IRDye800 Goat anti-rat Cat # 92632219, LI-COR Biosciences, Lincoln, Nebraska, USA), followed by further rinsing and imaged using a LI-COR Odyssey Imaging system (LI-COR Biosciences, Lincoln NE, USA), and analyzed with Image Studio Lite software (LI-COR Biosciences, Lincoln NE, USA). Sample data were normalized to total protein loaded and to the GAPDH loading control and then expressed as arbitrary units based upon densitometric quantitation.
LA-ICP-MS imaging
Analysis was performed using a New Wave Research UP213 laser ablation system with a two-volume large format cell (ablation area 15×15 cm). This system was hyphenated to an Agilent Technologies 7500ce ICP-MS fitted with ‘cs’ lenses for enhanced sensitivity. Quantitative data was obtained by representative ablation of matrix-matched tissue standards produced according to the protocol previously reported [34]. Hydrogen was used as a reaction gas at a flow rate of 3 mL min–1 to remove potential interferences arising from trace oxygen impurities in the argon gas used, particularly 40Ar16O+ on 56Fe+ [35]. The specificity of hydrogen for reducing interference on Fe was the main determinant of its selection; non-specific collision gases such as helium reduce signal to noise ratios and do not attenuate interferences effectively in LA-ICP-MS [36]. We have recently shown that matrix-based interference formation at biologically-relevant concentrations are negligible using these experimental conditions [37]. Measured masses included m/z = 56 for Fe, 62 for Cu, and 66 for Zn with an integration time of 0.08 seconds per mass. Limits of detection (LOD) and quantification (LOQ) were 0.55 μg g–1 and 1.58 μg g–1 for Fe; 1.20 μg g–1 and 1.89 μg g–1 for Cu; and 0.39 μg g–1 and 1.08 μg g–1 for Zn. A representative image (Fig. 1) reconstruction is provided for Fe, Cu, and Zn.
Fig.1
Representative LA-ICPMS schematic of SSV (top row) and trehalose (bottom row) treated Tg2576 brain sections showing Fe, Cu, and Zn. Scale bar = 1 mm.
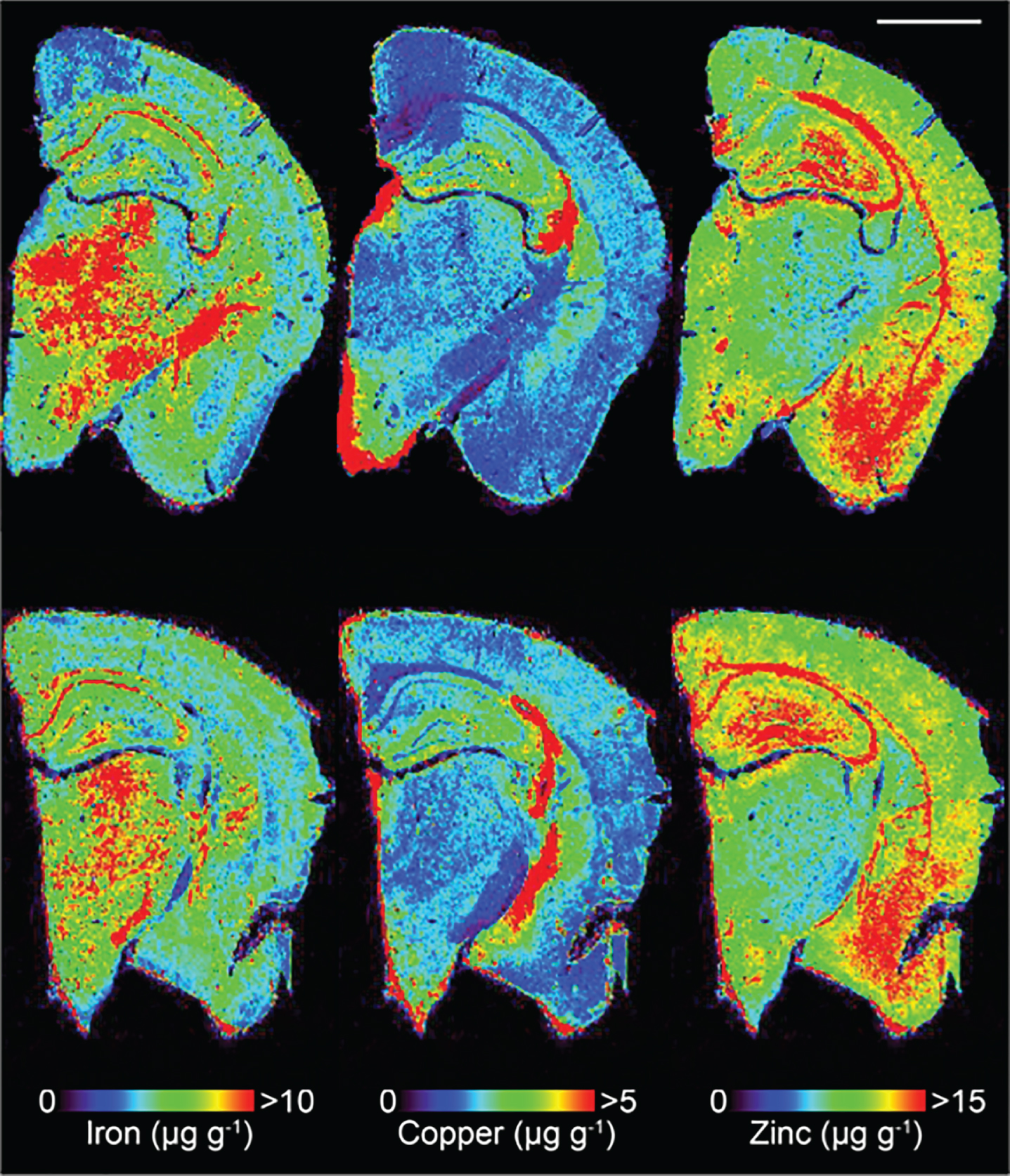
Image construction and region of interest selection
Lines of ablation were spaced apart the same distance as the laser beam diameter. A beam diameter of 30 μm was used, traversing the section at a speed of 120 μm s–1, laser fluence of 0.3 J cm–2 and repetition rate of 20 Hz. The ICP-MS was configured to collect 4 data points per second [35] thus the resultant images had pixel dimensions was equivalent to 30×30 μm2 (total pixel area = 900 μm2). Images were produced by reducing multiple ablation lines into ASCII data files via a Python script for importing into ENVI 6.0 (Exelis Visual Information Solutions, Boulder, CO, USA), from which regions of interest (ROIs) were extracted and statistically analyzed. One brain slice hemisphere per animal underwent ablation and subsequent analysis. Additionally, the hippocampus was manually extracted as an individual ROI using the Zn map [38] for analysis without adjacent cortical tissue, and hippocampal areas CA1, CA2 and CA3 were also subsequently manually extracted.
Statistical analysis
Statistical analysis was carried out in Prism 6.0 h (Graph-Pad, La Jolla, CA, USA). Analysis was carried out using either a two-tailed t test or a two-way repeated measures ANOVA as appropriate, with significance recognized as p < 0.05.
RESULTS
Behavior
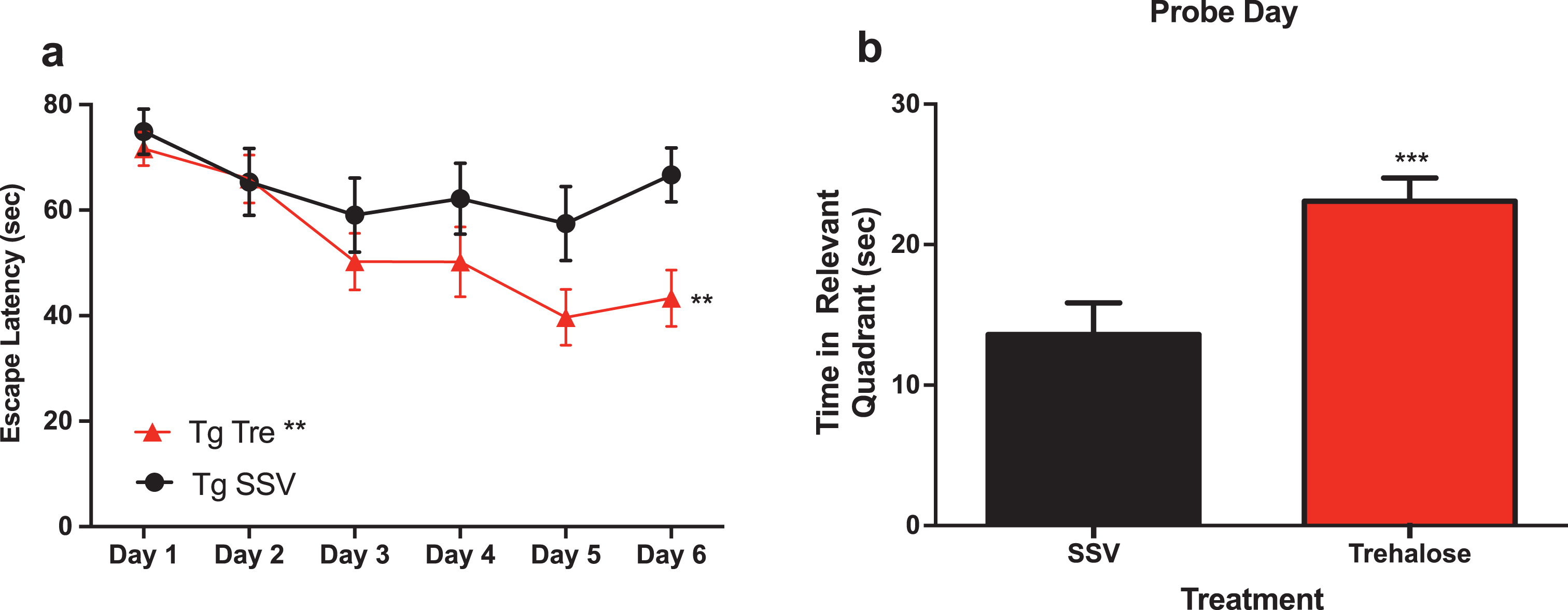
A two-way repeated measure analysis with a post-hoc Bonferroni’s analysis of the water maze revealed that trehalose treated mice (n = 15) demonstrated a significantly (p < 0.0081) enhanced task acquisition (learning) across the trial, as compared to the vehicle control group (n = 12) (Fig. 2a). In addition, trehalose-treated mice performed significantly (p < 0.0003) better in the probe trial (Fig. 2b), indicating an improvement in memory recall. An absence of any significant difference in swim speed suggests that overall, there is a robust improvement in cognition for the trehalose treated mice. No significant differences were observed between wild-type SSV treated mice and Tg2576 SSV treated mice, most likely as a result of the advanced age of the animals and the anticipated confound of normal age-related cognitive deficits. Further studies in younger mice would help delineate the potential differential effect on disease versus aging-related pathways in relation to cognitive performance. Indeed, it remains possible that trehalose may impact normal aging, as wild-type trehalose treated mice showed a trend toward better performance as compared to wildtype SSV treated and Tg2576 SSV treated mice, although this did not reach statistical significance.
Fig.2
Trehalose treatment improves cognitive performance of Tg2576 mice in the Morris water maze. Two-way repeated measures ANOVA (++p < 0.0081) revealed a significant decrease in escape latency for Tg2576 trehalose treated mice compared to Tg2576 SSV treated litter mate controls over the course of the trial (Fig. 1a). A Bonferroni’s post-hoc analysis further revealed a significant decrease in latency for trehalose treated mice on day six (*p = 0.0233). Two-tailed t-test revealed a significant (***p < 0.0003) increase in time spent in the relevant quadrant for trehalose treated Tg2576 mice compared to SSV treated Tg2576 control littermates on probe day (Fig. 3b).
Metal analyses
Assessment of Fe concentrations via LA-ICPMS revealed no significant difference between trehalose (n = 7) and SSV (n = 7) treated mice, in the cortex (Fig. 3a), hippocampus (Fig. 3b) or in the CA1 (Fig. 3c), CA2 (Fig. 3d) or CA3 (Fig. 3e) regions of the hippocampus.
Fig.3
Trehalose does not alter metal levels in Tg2576 mice. Fe was unaltered between trehalose and SSV treated animals in all areas assessed (Fig. 3a-e). Similarly, Zn was unaltered between trehalose and SSV treated animals in all areas assessed (Fig. 3f-j). Cu also revealed non-significant changes between trehalose and SSV treated animals in all areas assessed (Fig. 3k-o).
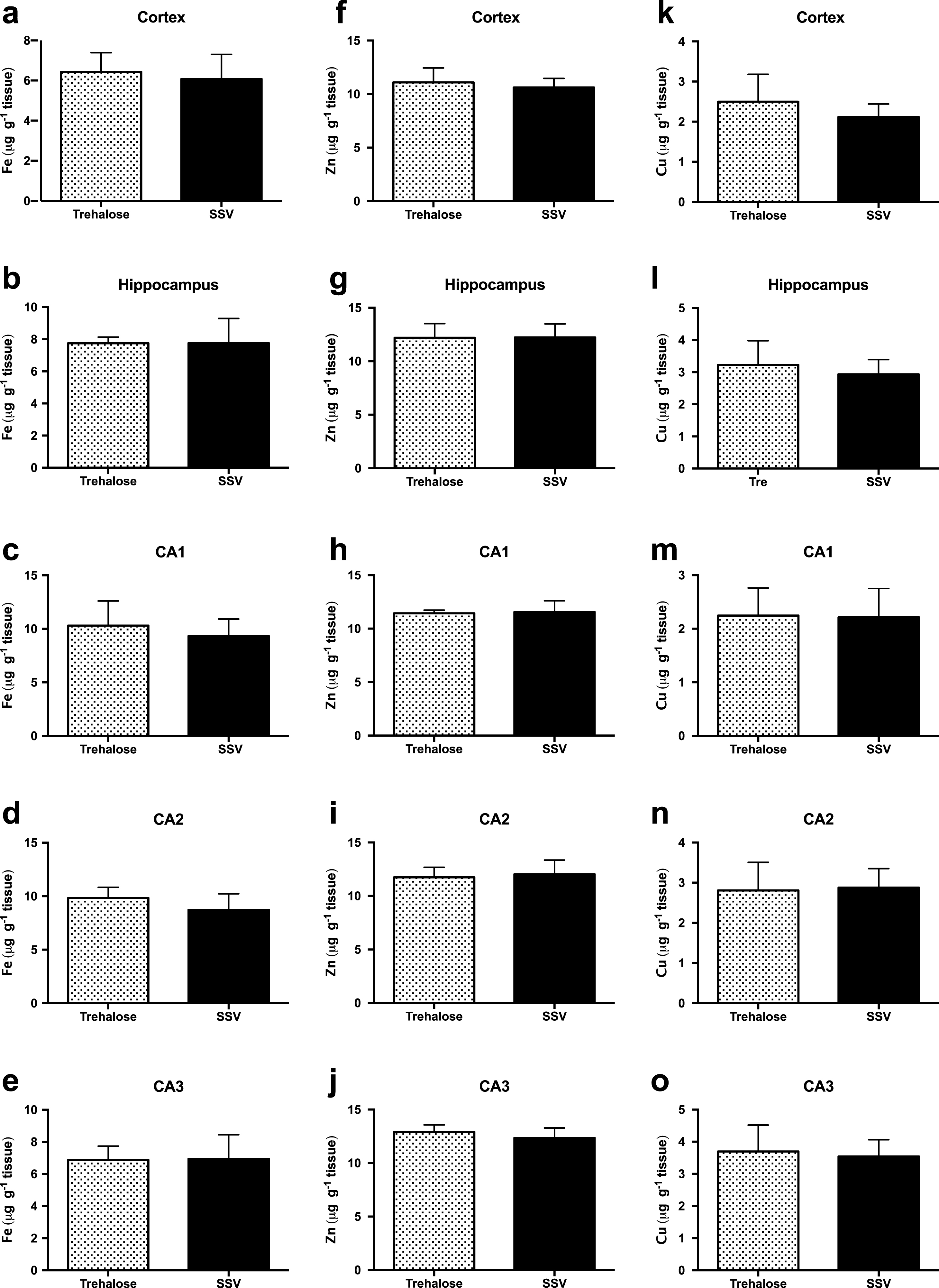
Similarly, Zn concentrations also revealed no significant difference between trehalose (n = 7) and SSV (n = 7) treated mice, in the cortex (Fig. 3f), hippocampus (Fig. 3g) or in the CA1 (Fig. 3h), CA2 (Fig. 3i), or CA3 (Fig. 3j) regions of the hippocampus. Furthermore, Cu concentrations were also unchanged for trehalose (n = 7) and SSV (n = 7) treated mice, in the cortex (Fig. 3k), hippocampus (Fig. 3l) or in the CA1 (Fig. 3m), CA2 (Fig. 3n), or CA3 (Fig. 3o) regions of the hippocampus. Analysis also confirmed no significant elevation or reduction in Fe, Zn, or Cu between Tg2576 and wild-type mice.
Western blotting
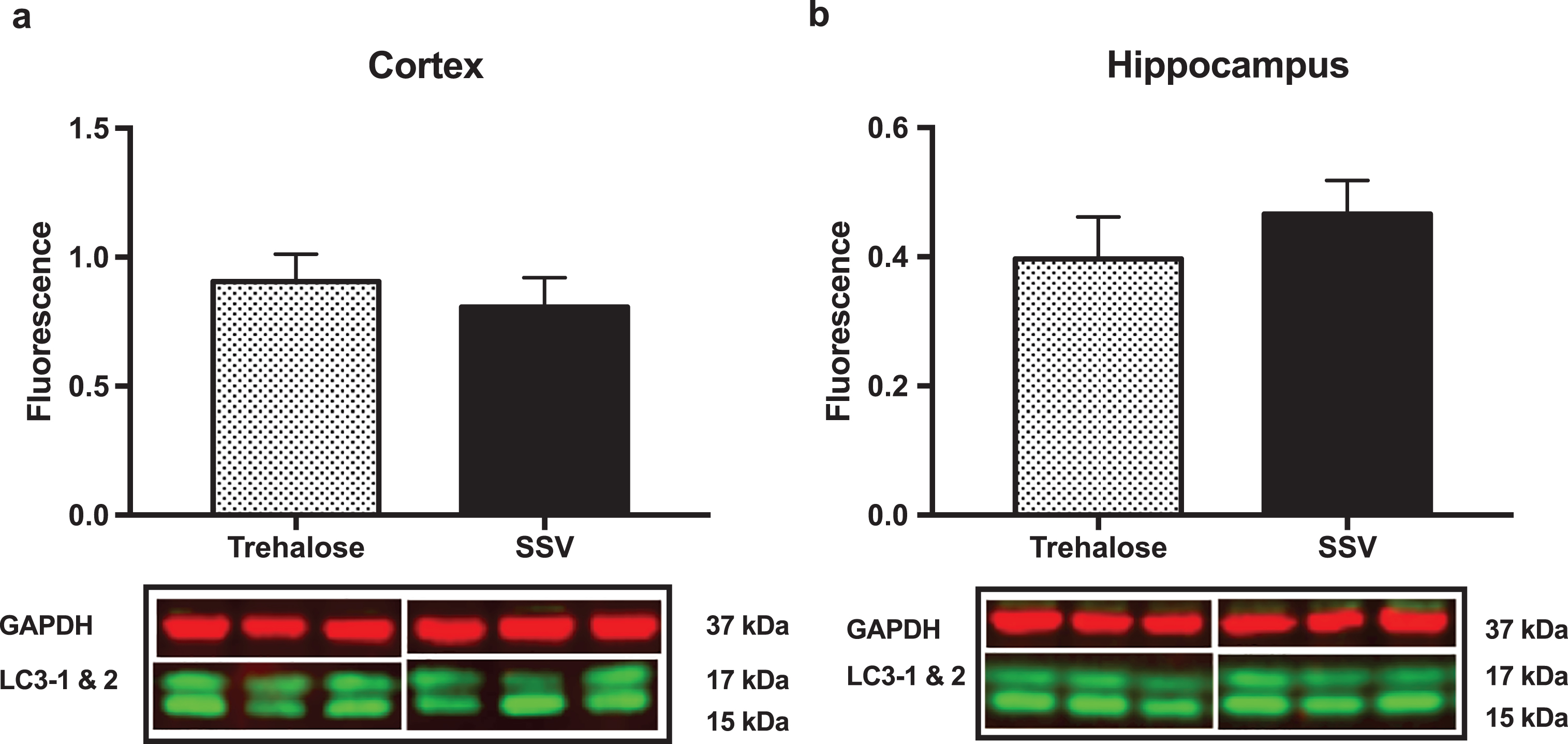
Results indicated a significant increase (+48%) in DCX (vehicle, 0.15±0.009, n = 8; trehalose 0.24±0.040, n = 8; p = 0.0401; Fig. 4a) and synaptophysin (+147%) (vehicle, 0.22±0.122, n = 8; trehalose 1.46±0.162, n = 8; p = 0.0001; Fig. 4b) protein expression in the hippocampus only of trehalose treated mice. Additionally, a significant increase in progranulin protein expression was observed in the trehalose treated group in both the cortex (+43%) (vehicle, 3.81±0.62, n = 8; trehalose 5.95±0.13, n = 8; p = 0.0052; Fig. 4c) and hippocampus (+33%) (vehicle, 4.00±0.53, n = 8; trehalose, 5.62±0.17, n = 8; p = 0.0126; Fig. 4d), when compared with the SSV control group. Soluble and membrane fractions of cortex (Soluble; vehicle, 0.55±0.02, n = 8; trehalose, 0.52±0.02, n = 8; p = 0.3466; Fig 5a. Membrane; vehicle 1.51±0.16, n = 8, trehalose, 1.49±0.10, n = 8; p = 0.9377; Fig. 5c) and hippocampus (Soluble; vehicle 0.39±0.04, n = 8; trehalose, 0.42±0.01, n = 8; p = 0.5767; Fig. 5b. Membrane; vehicle 1.07±0.05, n = 8; trehalose, 1.12±0.06, n = 8; p = 0.5649; Fig. 5d) were also assessed for AβPP levels; however, no significant differences were observed. Similarly, there were no differences observed in any Aβ fragments following trehalose treatment (data not shown). Autophagy was assessed via LC3 (Fig. 6); however, there was no significant difference in the ratio between LC3-1 and LC3-2 for trehalose treated animals when compared to controls in either the cortex (vehicle 0.80±0.04, n = 8; trehalose, 0.90±0.04, n = 8; p = 0.15) or the hippocampus (vehicle, 0.46±0.02, n = 8; trehalose, 0.39±0.02, n = 8; p = 0.10).
Fig.4
Trehalose significantly increases synaptophysin, Doublecortin (DCX) and Progranulin. A significant increase in DCX protein levels was observed in the hippocampus of the trehalose treated mice (*p = 0.0401) (a). Similarly, a significant increase in synaptophysin levels was also observed in the hippocampus (****p = 0.0001) (b). Progranulin protein expression was also shown to be significantly elevated in both the cortex (**p = 0.0052) (c) and the hippocampus (*p = 0.0126) (d).
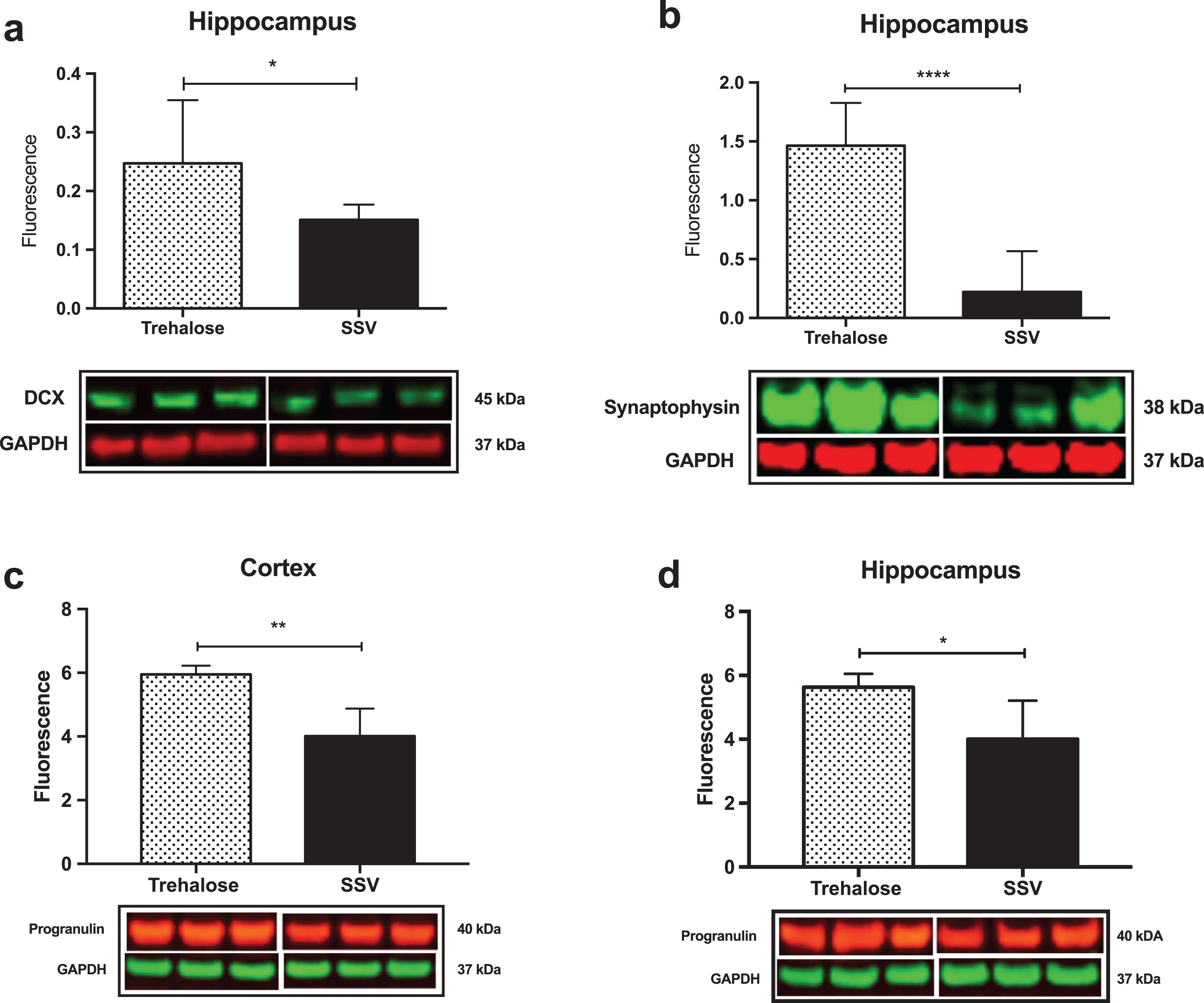
Fig.5
Trehalose does not alter AβPP expression. Trehalose did not significantly alter the expression of AβPP in the soluble fraction of cortex (a) or hippocampus (b), nor in the membrane fraction of cortex (c) or hippocampus (d).
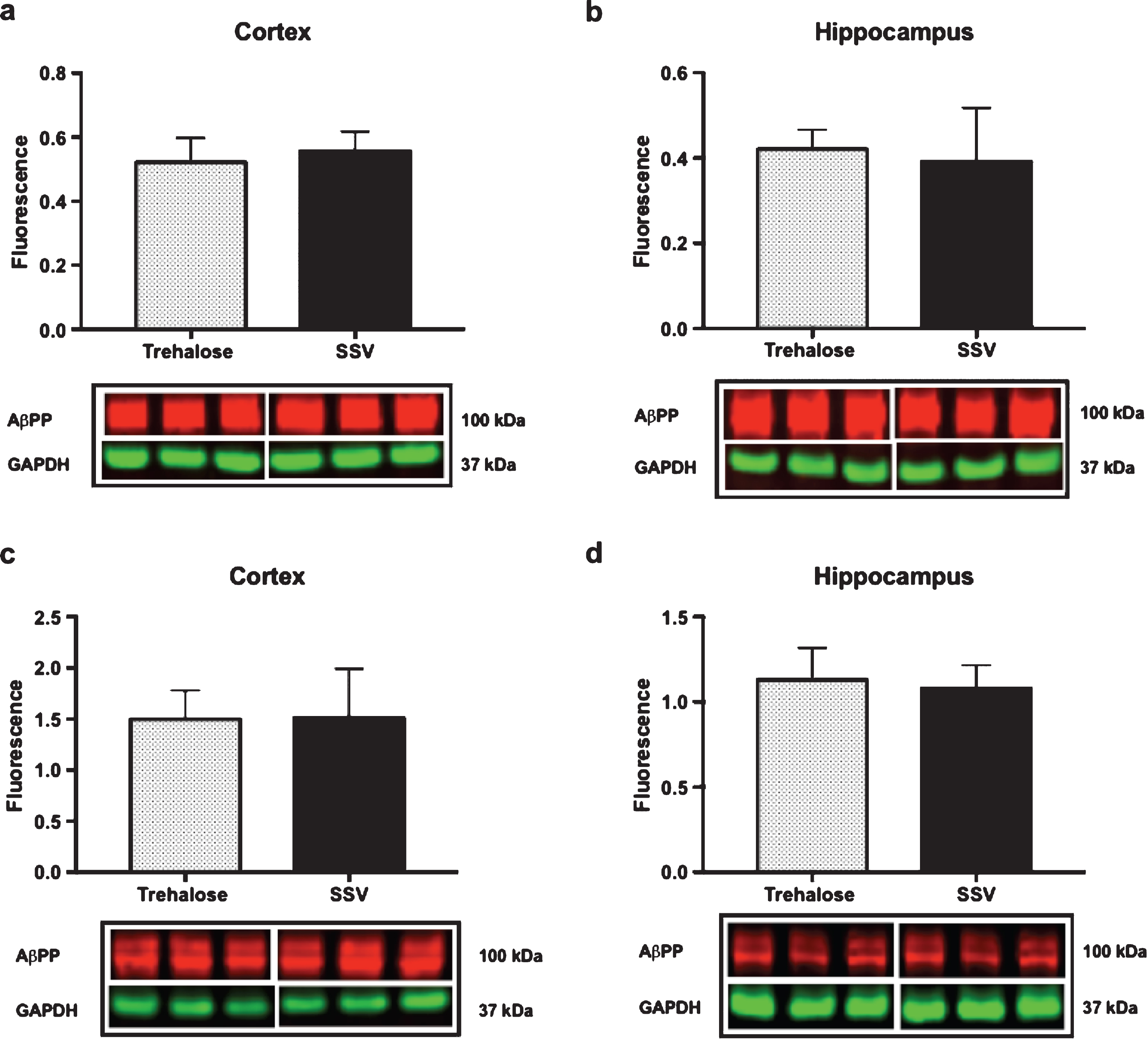
Fig.6
Trehalose does not alter LC3-1. Ratio of LC3-1 to LC3-2 revealed no significant difference between the trehalose treated group when compared to their SSV treated littermates.
DISCUSSION
The data presented herein demonstrates that trehalose treatment enhanced the cognitive performance of the Tg2576 mouse model of AD in the Morris water maze. Trehalose treated mice performed significantly better during the learning phase of the trial, and importantly, demonstrated enhanced memory retention on the probe day of the trial when compared to SSV treated littermate controls. Our data concurs with previously published work in which an Amyloid Precursor Protein/Presenilin 1 (APP/PS1) transgenic mouse model of AD was utilized to assess the therapeutic efficacy of intracerebral injections of trehalose [1]. Importantly, however, this study recapitulates the data in a different transgenic mouse model of AD using the more physiologically and therapeutically relevant route of oral administration. Moreover, this study also identifies biochemical pathways modulated by trehalose that may play a role in cognition, an observation that has not previously been reported in animal studies using a trehalose-treatment paradigm. These findings both support the use of trehalose in the treatment of AD, and also present the potential for trehalose to be utilized in other neurodegenerative disorders.
Due to the key role played by metals in both the pathogenesis of AD and in learning and memory [29, 39], we initially performed an analysis of the spatial distribution and concentration of Fe, Zn, and Cu in the brains of both trehalose treated and untreated mice. Many other studies have shown metal modulation to have beneficial pathological and physical outcomes [27–30, 40], and so this would allow us to assess whether there was any correlation between these metals and the observed behavioral improvement in the trehalose-treated mice. To assess metal concentrations, we utilized LA-ICPMS imaging of brain slices, which has previously been used to effectively discriminate brain metal differences in both human AD [41] and aged mouse tissue [40].
The Tg2576 mice have previously been shown to have an age-dependent reduction in brain Cu and Zn levels, with males showing an age-related reduction in Fe while aged females showed an increase in Fe [42]; observations which were accredited to the overexpression of AβPP and Aβ. Our results indicated no significant change for Fe, Cu, or Zn concentrations, in any region assessed between the trehalose treated and non-treated groups (Fig. 3a-o). A possible explanation as to the discrepancy between these results could be that the abovementioned study utilized a bulk tissue lysate analysis, whereas our study is regionally defined. Our data also revealed no significant differences in metal concentrations between wild-type and Tg2576 mice, which may reflect the age of the wild-type mice used in this study, as metal accumulation in the aging mouse brain has been previously noted in the literature [42].
Nevertheless, we found the lack of metal modulation to be perplexing as trehalose has been demonstrated to induce autophagy and reduce pathological metal associated protein aggregates in many cell models [43–47] and affect behavioral improvements in mouse models of neurodegenerative disease presumably via the same mechanism [4–6]. We therefore assessed autophagy by evaluating LC3-2 protein expression via western blotting, whereby upon autophagic induction, LC3 is converted from a cytosolic form (LC3-1) to a phosphatidylethanolamine-conjugated form (LC3-2), and thereby subsequently recruited to membranes of autophagasomes [48]. We revealed no significant difference between the LC3-1 to LC3-2 ratio between trehalose treated and non-treated groups (Fig. 6), indicating that the beneficial behavioral effect we observed with trehalose was unlikely to be due to an autophagy-mediated effect on protein aggregates. Given this, it is possible that the lack of any reduction in metal may be due to the absence of autophagy mediated degradation of protein aggregates that are implicated in the dyshomeostasis of metals observed in AD. Zinc, for example, has been shown to accumulate in the pathological extracellular plaques of AD [49–52], and can bind Aβ causing rapid aggregation and precipitation of the Aβ peptide [53]. Trehalose has been shown to inhibit aggregation and neurotoxicity of Aβ in cell culture [46], and significantly reduce the number of amyloid deposits in APP/PS1 mice [1]. Had autophagy been induced and affected a change in Aβ aggregation in this study, it is likely that we would have detected a change in the localized zinc concentrations.
A recent publication by Tien et al. [54], for example, revealed that trehalose altered the subcellular trafficking and metabolism of AβPP. We therefore assessed the protein expression levels of AβPP due to its contribution to Aβ formation in the Tg2576 mouse and human AD. Our results indicated no significant differences in AβPP protein expression levels in soluble and membrane extracted fractions from the cortex or hippocampus of trehalose treated and non-treated mice using the anti-AβPP antibody 22C11 (Fig. 5a-d). Further analysis of the membrane fractions with the Aβ antibodies, WO2 and 4G8, also revealed no significant alteration in any Aβ species (data not shown). It should be noted, however, that the results of Tien et al. [53] coincided with robust LC3-II protein accumulation, indicating enhanced autophagy, which was not observed in this current study.
We therefore hypothesized that trehalose may be indirectly increasing synaptic activity. Accordingly, we assessed the pre-synaptic vesicle protein synaptophysin, which is a surrogate marker for the number of synapses, via western blot and revealed a significant increase in synaptophysin expression in the hippocampus, but not the cortex, of trehalose treated mice (Fig. 4b). Many murine studies have linked increased synaptophysin expression with enhanced spatial memory [55–57]. Moreover, loss of this pre-synaptic protein in the hippocampus correlates with the cognitive decline in AD [58].
We also probed for doublecortin, the endogenous early neuronal marker and surrogate indicator of neurogenesis [59], and revealed a significant increase in DCX expression in the hippocampus of the trehalose treated mice (Fig. 4a) that was not observed in the cortex, nor in the vehicle treated tissue. The hippocampus is the most susceptible brain region in AD, whereby cell death and associated degeneration of the tissue is likely to be the underlying cause of memory deficits [60, 61]. However, the role of neurogenesis in AD and in the hippocampus of transgenic murine models of AD has previously resulted in controversial and contraindicative results [62–65]. Nevertheless, decreased neurogenesis correlating with an increase in Aβ burden has been shown in a transgenic mouse model of AD [62], and the potential of modulating hippocampal neurogenesis as a therapeutic approach for AD has been proposed [66].
We also assessed the secreted growth factor Progranulin, a known regulator of neuronal growth and survival [67, 68]. We hypothesized that trehalose may upregulate progranulin expression as it has been demonstrated to do so in human and mouse models of progranulin haploinsufficiency [7]. Western blot analysis revealed a significant increase in progranulin in both the hippocampus (Fig. 4c) and cortex (Fig. 4d) of trehalose treated mice. Interestingly, progranulin has previously been shown to be protective against Aβ toxicity [69], and progranulin missense mutations contribute to the risk for clinically diagnosed AD [70]. The observation that trehalose can elevate progranulin protein expression in an AD transgenic mouse model, therefore, suggests another possible mechanism of action of this compound.
Conclusion
Our results demonstrate that trehalose administration can improve cognitive outcomes in the Tg2576 transgenic mouse model of AD. Given that trehalose is FDA “GRAS”, has a generous safety profile, and is currently used as an excipient in many pharmaceutical formulations for human use, we believe that trehalose could be a viable candidate for further pharmacological investigation as a potential therapeutic option for patients with AD, either as a monotherapy or in conjunction with other treatment alternatives.
ACKNOWLEDGMENTS
D.J.H. is supported by an Australian Research Council Linkage Project with Agilent Technologies (LP140100095). DJH is also supported by a National Health and Research Council Industry Career Development Fellowship (GNT1122981) in partnership with Agilent Technologies. In addition, the Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. The Elemental Bio-imaging Facility receives material support from Agilent Technologies.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0322r1).
REFERENCES
[1] | Du J , Liang Y , Xu F , Sun B , Wang Z ((2013) ) Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol 65: , 1753–1756. |
[2] | Echigo R , Shimohata N , Karatsu K , Yano F , Kayasuga-Kariya Y , Fujisawa A , Ohto T , Kita Y , Nakamura M , Suzuki S , Mochizuki M , Shimizu T , Chung UI , Sasaki N ((2012) ) Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J Transl Med 10: , 80. |
[3] | Tanji K , Miki Y , Maruyama A , Mimura J , Matsumiya T , Mori F , Imaizumi T , Itoh K , Wakabayashi K ((2015) ) Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochem Biophys Res Commun 465: , 746–752. |
[4] | Rodriguez-Navarro JA , Rodriguez L , Casarejos MJ , Solano RM , Gomez A , Perucho J , Cuervo AM , Garcia de Yebenes J , Mena MA ((2010) ) Trehalose ameliorates dopaminergic and tau pathology in parkindeleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 39: , 423–438. |
[5] | Castillo K , Nassif M , Valenzuela V , Rojas F , Matus S , Mercado G , Court FA , van Zundert B , Hetz C ((2013) ) Trehalose delays theprogression of amyotrophic lateral sclerosis by enhancingautophagy in motoneurons. Autophagy 9: , 1308–1320. |
[6] | Tanaka M , Machida Y , Niu S , Ikeda T , Jana NR , Doi H , Kurosawa M , Nekooki M , Nukina N ((2004) ) Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 10: , 148–154. |
[7] | Holler CJ , Taylor G , McEachin ZT , Deng Q , Watkins WJ , Hudson K , Easley CA , Hu WT , Hales CM , Rossoll W , Bassell GJ , Kukar T ((2016) ) Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: A novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener 11: , 46. |
[8] | Brouns F ((2007) ) Can trehalose help alleviate Huntington or Alzheimer disease? Curr Top Nutraceutical Res 5: , 51–53. |
[9] | Emanuele E ((2014) ) Can trehalose prevent neurodegeneration? Insights from experimental studies. Curr Drug Targets 15: , 551–557. |
[10] | Belaidi AA , Bush AI ((2016) ) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J Neurochem 139: (Suppl 1), 179–197. |
[11] | Hare D , Ayton S , Bush A , Lei P ((2013) ) A delicate balance: Iron metabolism and diseases of the brain. Front Aging Neurosci 5: , 34. |
[12] | Smith MA , Harris PL , Sayre LM , Perry G ((1997) ) Iron accumulation inAlzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94: , 9866–9868. |
[13] | Craddock TJ , Tuszynski JA , Chopra D , Casey N , Goldstein LE , Hameroff SR , Tanzi RE ((2012) ) The zinc dyshomeostasis hypothesis ofAlzheimer’s disease., e. PLoS One 7: , 33552. |
[14] | Watt NT , Whitehouse IJ , Hooper NM ((2010) ) The role of zinc in Alzheimer’s disease. Int J Alzheimers Dis 2011: , 971021. |
[15] | Donnelly PS , Xiao Z , Wedd AG ((2007) ) Copper and Alzheimer’s disease. Curr Opin Chem Biol 11: , 128–133. |
[16] | Gaggelli E , Kozlowski H , Valensin D , Valensin G ((2006) ) Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem Rev 106: , 1995–2044. |
[17] | Waggoner DJ , Bartnikas TB , Gitlin JD ((1999) ) The role of copper inneurodegenerative disease. Neurobiol Dis 6: , 221–230. |
[18] | Adlard PA , Bush AI ((2006) ) Metals and Alzheimer’s disease. J Alzheimers Dis 10: , 145–163. |
[19] | Barnham KJ , Bush AI ((2008) ) Metals in Alzheimer’s and Parkinson’s diseases. Curr Opin Chem Biol 12: , 222–228. |
[20] | Brown DR ((2011) ) Metals in neurodegenerative disease. Metallomics 3: , 226–228. |
[21] | Greenough MA , Camakaris J , Bush AI ((2013) ) Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 62: , 540–555. |
[22] | Adlard PA , Bush AI ((2012) ) Metal chaperones: A holistic approach to the treatment of Alzheimer’s disease. Front Psychiatry 3: , 15. |
[23] | Ayton S , Lei P , Bush AI ((2015) ) Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 12: , 109–120. |
[24] | Barnham KJ , Bush AI ((2014) ) Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev 43: , 6727–6749. |
[25] | Bush AI , Tanzi RE ((2008) ) Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 5: , 421–432. |
[26] | Zatta P , Drago D , Bolognin S , Sensi SL ((2009) ) Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol Sci 30: , 346–355. |
[27] | Adlard PA , Bica L , White AR , Nurjono M , Filiz G , Crouch PJ , Donnelly PS , Cappai R , Finkelstein DI , Bush AI ((2011) ) Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer’s disease. PLoS One 6: , e17669. |
[28] | Adlard PA , Cherny RA , Finkelstein DI , Gautier E , Robb E , Cortes M , Volitakis I , Liu X , Smith JP , Perez K , Laughton K , Li QX , Charman SA , Nicolazzo JA , Wilkins S , Deleva K , Lynch T , Kok G , Ritchie CW , Tanzi RE , Cappai R , Masters CL , Barnham KJ , Bush AI ((2008) ) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59: , 43–55. |
[29] | Adlard PA , Parncutt J , Lal V , James S , Hare D , Doble P , Finkelstein DI , Bush AI ((2015) ) Metal chaperones prevent zinc-mediated cognitive decline. Neurobiol Dis 81: , 196–202. |
[30] | Cherny RA , Atwood CS , Xilinas ME , Gray DN , Jones WD , McLean CA , Barnham KJ , Volitakis I , Fraser FW , Kim Y , Huang X , Goldstein LE , Moir RD , Lim JT , Beyreuther K , Zheng H , Tanzi RE , Masters CL , Bush AI ((2001) ) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30: , 665–676. |
[31] | Lannfelt L , Blennow K , Zetterberg H , Batsman S , Ames D , Harrison J , Masters CL , Targum S , Bush AI , Murdoch R , Wilson J , Ritchie CW , PBT2-201-EURO study group ((2008) ) Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol 7: , 779–786. |
[32] | Liu M , Zhang M , Ye H , Lin S , Yang Y , Wang L , Jones G , Trang H ((2013) ) Multiple toxicity studies of trehalose in mice by intragastric administration. Food Chem 136: , 485–490. |
[33] | Richards AB , Krakowka S , Dexter LB , Schmid H , Wolterbeek AP , Waalkens-Berendsen DH , Shigoyuki A , Kurimoto M ((2002) ) Trehalose: A review of properties, history of use and human tolerance, and results of multiple safety studies. Food Chem Toxicol 40: , 871–898. |
[34] | Hare DJ , Lear J , Bishop D , Beavis A , Doble PA ((2013) ) Protocol forproduction of matrix-matched brain tissue standards for imaging bylaser ablation-inductively coupled plasma-mass spectrometry. Anal Methods 5: , 1915–1921. |
[35] | Lear J , Hare DJ , Fryer F , Adlard PA , Finkelstein DI , Doble PA ((2012) ) High-resolution elemental bioimaging of Ca, Mn, Fe, Co, Cu, and Zn employing LA-ICP-MS and hydrogen reaction gas. Anal Chem 84: , 6707–6714. |
[36] | Mason PRD , Kraan WJ ((2002) ) Attenuation of spectral interferences during laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) using an rf only collision and reaction cell. J Anal At Spectrom 17: , 858–867. |
[37] | Hare DJ , Fryer F , Paul B , Bishop DP , Doble PA ((2016) ) Characterisation of matrix-based polyatomic interference formation in laser ablation-inductively coupled plasma-mass spectrometry using dried micro-droplet ablation and its relevance for bioimaging. Anal Methods 8: , 7552–7556. |
[38] | Hare DJ , Lee JK , Beavis AD , van Gramberg A , George J , Adlard PA , Finkelstein DI , Doble PA ((2012) ) Three-dimensional atlas of iron, copper, and zinc in the mouse cerebrum and brainstem. Anal Chem 84: , 3990–3997. |
[39] | Adlard PA , Parncutt JM , Finkelstein DI , Bush AI ((2010) ) Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J Neurosci 30: , 1631–1636. |
[40] | Adlard PA , Sedjahtera A , Gunawan L , Bray L , Hare D , Lear J , Doble P , Bush AI , Finkelstein DI , Cherny RA ((2014) ) A novel approach to rapidly prevent age-related cognitive decline. Aging Cell 13: , 351–359. |
[41] | Hare DJ , Raven EP , Roberts BR , Bogeski M , Portbury SD , McLean CA , Masters CL , Connor JR , Bush AI , Crouch PJ , Doble PA ((2016) ) Laser ablation-inductively coupled plasma-mass spectrometry imaging of white and gray matter iron distribution in Alzheimer’s disease frontal cortex. Neuroimage 137: , 124–131. |
[42] | Maynard CJ , Cappai R , Volitakis I , Cherny RA , White AR , Beyreuther K , Masters CL , Bush AI , Li QX ((2002) ) Overexpression of Alzheimer’s disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem 277: , 44670–44676. |
[43] | Beranger F , Crozet C , Goldsborough A , Lehmann S ((2008) ) Trehalose impairs aggregation of PrPSc molecules and protects prion-infected cells against oxidative damage. Biochem Biophys Res Commun 374: , 44–48. |
[44] | Casarejos MJ , Solano RM , Gomez A , Perucho J , de Yebenes JG , Mena MA ((2011) ) The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem Int 58: , 512–520. |
[45] | Kruger U , Wang Y , Kumar S , Mandelkow EM ((2012) ) Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging 33: , 2291–2305. |
[46] | Liu R , Barkhordarian H , Emadi S , Park CB , Sierks MR ((2005) ) Trehalose differentially inhibits aggregation and neurotoxicity of beta-amyloid 40 and 42. Neurobiol Dis 20: , 74–81. |
[47] | Sarkar S , Davies JE , Huang Z , Tunnacliffe A , Rubinsztein DC ((2007) ) Trehalose, a novel mTOR-independent autophagy enhancer,accelerates the clearance of mutant huntingtin andalpha-synuclein. J Biol Chem 282: , 5641–5652. |
[48] | Otomo C , Metlagel Z , Takaesu G , Otomo T ((2013) ) Structure of the human ATG12∼ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 20: , 59–66. |
[49] | Lovell MA , Robertson JD , Teesdale WJ , Campbell JL , Markesbery WR ((1998) ) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158: , 47–52. |
[50] | Miller LM , Wang Q , Telivala TP , Smith RJ , Lanzirotti A , Miklossy J ((2006) ) Synchrotron-based infrared and X-ray imaging showsfocalized accumulation of Cu and Zn co-localized with beta-amyloiddeposits in Alzheimer’s disease. J Struct Biol 155: , 30–37. |
[51] | Stoltenberg M , Bush AI , Bach G , Smidt K , Larsen A , Rungby J , Lund S , Doering P , Danscher G ((2007) ) Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. Neuroscience 150: , 357–369. |
[52] | Suh SW , Jensen KB , Jensen MS , Silva DS , Kesslak PJ , Danscher G , Frederickson CJ ((2000) ) Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res 852: , 274–278. |
[53] | Bush AI , Pettingell WH , Multhaup G , d Paradis M , Vonsattel JP , Gusella JF , Beyreuther K , Masters CL , Tanzi RE ((1994) ) Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 265: , 1464–1467. |
[54] | Tien NT , Karaca I , Tamboli IY , Walter J ((2016) ) Trehalose alters subcellular trafficking and the metabolism of the Alzheimer-associated amyloid precursor protein. J Biol Chem 291: , 10528–10540. |
[55] | Frick KM , Fernandez SM ((2003) ) Enrichment enhances spatial memory and increases synaptophysin levels in aged female mice. Neurobiol Aging 24: , 615–626. |
[56] | Frick KM , Fernandez SM , Bulinski SC ((2002) ) Estrogen replacement improves spatial reference memory and increases hippocampal synaptophysin in aged female mice. Neuroscience 115: , 547–558. |
[57] | Mook-Jung I , Hong HS , Boo JH , Lee KH , Yun SH , Cheong MY , Joo I , Huh K , Jung MW ((2001) ) Ginsenoside Rb1 and Rg1 improve spatial learning and increase hippocampal synaptophysin level in mice. J Neurosci Res 63: , 509–515. |
[58] | Reddy PH , Mani G , Park BS , Jacques J , Murdoch G , Whetsell W Jr. , Kaye J , Manczak M ((2005) ) Differential loss of synapticproteins in Alzheimer’s disease: Implications for synapticdysfunction. J Alzheimers Dis 7: , 103–117; discussion 173-180. |
[59] | Couillard-Despres S , Winner B , Schaubeck S , Aigner R , Vroemen M , Weidner N , Bogdahn U , Winkler J , Kuhn HG , Aigner L ((2005) ) Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21: , 1–14. |
[60] | Barber R , McKeith IG , Ballard C , Gholkar A , O’Brien JT ((2001) ) A comparison of medial and lateral temporal lobe atrophy in dementia with Lewy bodies and Alzheimer’s disease: Magnetic resonance imaging volumetric study. Dement Geriatr Cogn Disord 12: , 198–205. |
[61] | Thompson PM , Hayashi KM , De Zubicaray GI , Janke AL , Rose SE , Semple J , Hong MS , Herman DH , Gravano D , Doddrell DM , Toga AW ((2004) ) Mapping hippocampal and ventricularchange in Alzheimer disease. Neuroimage 22: , 1754–1766. |
[62] | Dong H , Goico B , Martin M , Csernansky CA , Bertchume A , Csernansky JG ((2004) ) Modulation of hippocampal cell proliferation, memory, and amyloid plaque deposition in APPsw (Tg2576) mutant mice by isolation stress. Neuroscience 127: , 601–609. |
[63] | Jin K , Galvan V , Xie L , Mao XO , Gorostiza OF , Bredesen DE , Greenberg DA ((2004) ) Enhanced neurogenesis in Alzheimer’s diseasetransgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A 101: , 13363–13367. |
[64] | Krezymon A , Richetin K , Halley H , Roybon L , Lassalle JM , Frances B , Verret L , Rampon C ((2013) ) Modifications of hippocampal circuits and early disruption of adult neurogenesis in the tg2576 mouse model of Alzheimer’s disease. PLoS One 8: , e76497. |
[65] | Taniuchi N , Niidome T , Goto Y , Akaike A , Kihara T , Sugimoto H ((2007) ) Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport 18: , 1801–1805. |
[66] | Chuang TT ((2010) ) Neurogenesis in mouse models of Alzheimer’s disease. Biochim Biophys Acta 1802: , 872–880. |
[67] | Gass J , Lee WC , Cook C , Finch N , Stetler C , Jansen-West K , Lewis J , Link CD , Rademakers R , Nykjaer A , Petrucelli L ((2012) ) Progranulin regulates neuronal outgrowth independent of sortilin. Mol Neurodegener 7: , 33. |
[68] | Toh H , Chitramuthu BP , Bennett HP , Bateman A ((2011) ) Structure, function, and mechanism of progranulin; the brain and beyond. J Mol Neurosci 45: , 538–548. |
[69] | Minami SS , Min SW , Krabbe G , Wang C , Zhou Y , Asgarov R , Li Y , Martens LH , Elia LP , Ward ME , Mucke L , Farese RV Jr , Gan L ((2014) ) Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med 20: , 1157–1164. |
[70] | Brouwers N , Sleegers K , Engelborghs S , Maurer-Stroh S , Gijselinck I , van der Zee J , Pickut BA , Van den Broeck M , Mattheijssens M , Peeters K , Schymkowitz J , Rousseau F , Martin JJ , Cruts M , De Deyn PP , Van Broeckhoven C ((2008) ) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71: , 656–664. |

