
Synaptic Membrane Synthesis in Rats Depends on Dietary Sufficiency of Vitamin C, Vitamin E, and Selenium: Relevance for Alzheimer’s Disease
Abstract
Chronic consumption of a diet enriched with nutritional precursors of phospholipids, including uridine and the polyunsaturated fatty acids, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), was shown previously to enhance levels of brain phospholipids and synaptic proteins in rodents. Vitamin C, vitamin E, and selenium may directly affect the breakdown or synthesis of membrane phospholipids. The present study investigated the necessity of antioxidants for the effectiveness of supplementation with uridine plus DHA and EPA (as fish oil) in rats. Rats were randomized to four treatment groups and received, for 6 weeks, one of four experimental diets, i.e., a diet low in antioxidants, a diet high in antioxidants, a diet low in antioxidants supplemented with DHA+EPA+uridine, or a diet high in antioxidants supplemented with DHA+EPA+uridine. On completion of dietary treatment, rats were sacrificed, and brain levels of phospholipids, synaptic proteins, and two enzymes involved in phospholipid synthesis (choline-phosphate cytidylyltransferase, PCYT1A, and choline/ethanolamine phosphotransferase, CEPT1) were analyzed. Levels of phospholipids, the pre- and post-synaptic proteins Synapsin-1 and PSD95, and the enzymes PCYT1A and CEPT1 were significantly enhanced by combined supplementation of DHA+EPA+uridine and antioxidants and not enhanced by supplementation of DHA+EPA+uridine with insufficient antioxidant levels. Our data suggest that dietary vitamin C, vitamin E, and selenium are essential for the phospholipid precursors’ effects on increasing levels of membrane phospholipids and synaptic proteins, the indirect indicators of synaptogenesis. Their concomitant supply may be relevant in Alzheimer’s disease patients, because the disease is characterized by synapse loss and lower plasma and brain levels of phospholipid precursors and antioxidants.
INTRODUCTION
Phospholipids are the main constituents of brain membranes, and their syntheses require that precursors are taken up into the brain from the circulation and interact via the Kennedy pathway [1]. These precursors include choline, uridine, and polyunsaturated fatty acids (PUFAs) like docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) that are already in the circulation and can also be obtained from nutritional sources.
Supplementation with uridine and DHA for several weeks resulted in enhanced levels of phosphatidylcholine (PC), as well as other membrane phospholipids, in brains of healthy adult rodents [2, 3]. The same effect was observed when DHA was substituted with another omega-3 PUFA, EPA, but not the omega-6 PUFA arachidonic acid (AA) [4]. The enhancement in levels of membrane phospholipids is associated with increased levels of synaptic membranes, evidenced by enhanced levels of Synapsin-1 and PSD95, specific pre- and post-synaptic proteins, respectively, as well as structural changes reflected by the increased number of dendritic spines [3, 5]. These findings suggested that formation of new synapses, i.e., synaptogenesis, is determined, at least in part, by the availability of phospholipid precursors to the brain. These increases are relevant for conditions that are characterized by a loss of synapses and lower plasma and brain levels of phospholipid precursors, such as Alzheimer’s disease (AD) [6–10].
Phospholipid synthesis thus depends strongly on the availability of the rate-limiting phospholipid precursors; however, other nutrients may enhance the effects of these precursors. Vitamin C, vitamin E, and selenium may directly affect the breakdown or synthesis of neuronal membranes. The most important way antioxidants act to stabilize cell membranes is by counteracting oxidative stress, which in turn leads to lipid peroxidation particularly in neuronal membranes [11, 12]. Extensive lipid peroxidation causes altered membrane composition, loss of membrane fluidity, reductions in membrane potential, and increased ionic permeability, eventually resulting in cell death. Vitamin C, vitamin E, and selenium (as part of the enzyme glutathione peroxidase) can prevent lipid peroxidation and, in turn, protect phospholipid precursors (i.e., DHA and EPA) and lipid-containing membrane components from peroxidation [12]. Vitamin E has an established role in terminating the fatty acid free-radicals chain reaction; each PUFA that is oxidized damages approximately three other PUFA molecules [13]. In addition, selenium may stimulate membrane synthesis by increasing the activity of cholinephosphotransferase [14], a key enzyme in the Kennedy pathway that catalyzes the formation of PC from diacylglycerol and cytidine diphosphate (CDP)-choline. Vitamin C contributes to optimal collagen synthesis [15], a process involved in neurite outgrowth and synapse formation [16]; vitamin E is a structural component of neuronal membranes [17], primarily associated with PC [18, 19] and concentrated at soma-neurite junctions, suggesting its role in neuronal connectivity [20]; and selenium is a constituent of selenoprotein P, which is required for normal synaptic functioning [21].
Previous supplementation studies reporting effects of DHA, EPA, and uridine on brain phospholipid levels were performed with standard rodent diets, containing sufficient levels (i.e., requirements according to the National Research Council report on the nutrient requirements of laboratory animals, [22]) of the antioxidants, vitamin E and selenium, and no added vitamin C as the latter is not essential in most rodents [22]. Hence, possible necessity of antioxidants for increasing phospholipid levels with phospholipid precursors is unknown, and supplementation of these precursors is possibly not as effective in conditions with lower plasma and/or brain levels of the antioxidants, e.g., in AD [23–26].
To obtain a proof-of-principle, the effects of DHA, EPA, and uridine supplementation were reassessed in the current study using diets that were either nearly devoid of vitamin C, vitamin E, and selenium or supplemented with high levels of these antioxidants.
MATERIAL AND METHODS
Animals
Male Wistar Albino rats at 3-4 months of age (300–350 g) were obtained from Experimental Animals Breeding and Research Center, Uludag University Medical School, Bursa, Turkey and were individually housed (to ensure a precise measurement of body weight and food intake) in Euro Type III H cages containing tunnels, balls, and nestlets as enrichment and wood shaving as the bedding in a temperature (22–24°C) and humidity (60%) controlled room with free access to standard rat chow and water under a 12/12 h light/dark cycle. The experimental protocol was approved by the Animal Care and Use Committee of Uludag University, Bursa, Turkey (Approval ID: 2013-17/04). All experiments conformed to the “National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996” and the “Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes”. All efforts were made to minimize the number of animals used and the individual level of discomfort.
Diets
Four diets with varying levels of vitamin C, vitamin E, and selenium plus DHA, EPA, and uridine were formulated: 1) Antioxidant (AOX) low diet; 2) AOX high diet; 3) AOX low+DHA+EPA+uridine diet; and 4) AOX high+DHA+EPA+uridine diet. Diets were AIN-93 M based [27], isoenergetic, and identical with respect to their protein, carbohydrate, and fiber contents. The vitamin mix (AIN-93-VX) and mineral mix (AIN-93M-MX) [27] were prepared without vitamin E and selenium, and these were subsequently supplemented accordingly. The vitamin mix (AIN-93-VX) does not contain vitamin C.
The AOX low diets contained no added vitamin E, vitamin C, and selenium. Due to the intrinsic content of vitamin E and selenium in the raw materials, the minimal levels of these nutrients in the AOX low diets were 3.85 mg vitamin E (RRR-alpha-tocopherol)/kg diet and 0.07 mg selenium/kg diet, which equals, respectively, 21% and 47% of the nutrient requirements of rats [22]. A complete depletion of dietary vitamin E and selenium is also not desirable, because this would induce complications. The AOX high diets contained 1600 mg vitamin E (RRR-alpha-tocopherol)/kg diet, 1600 mg vitamin C/kg diet, and 1.2 mg selenium/kg diet. This equals 8889% and 800% of the nutrient requirements of rats for vitamin E and selenium, respectively [22].
These low and high levels of antioxidants were used to create distinctive low and high antioxidant availability, increasing the potential impact of the antioxidants in this proof-of-principle experiment.
The DHA+EPA+uridine supplemented diets contained 32.0 g fish oil/kg diet, providing 8.0 g DHA/kg diet (0.80%) and 1.8 g EPA/kg diet (0.18%). These diets also contained 10.0 g uridine-5’-monophosphate (UMP) disodium salt 24–27% H2O/kg diet (1%). The AOX low diets did not contain any fish oil, DHA, EPA, or added uridine.
Compositions of the diets are listed in Table 1. Diets were manufactured by Ssniff Spezialdiäten (Soest, Germany). All diets were stored at –20°C until use, in order to prevent lipid oxidation.
Table 1
Detailed compositions of the experimental diets
| Diets | ||||
| Ingredients (g/100 g diet) | AOX low | AOX high | AOX low | AOX high |
| DHA+EPA | DHA+EPA | |||
| +uridine | +uridine | |||
| Cornstarch, pre-gelatinized | 35.59 | 35.11 | 34.59 | 34.11 |
| Caseine | 14.00 | 14.00 | 14.00 | 14.00 |
| Maltodextrin, 10 DE | 15.50 | 15.50 | 15.50 | 15.50 |
| Sucrose | 10.00 | 10.00 | 10.00 | 10.00 |
| Dextrose | 10.00 | 10.00 | 10.00 | 10.00 |
| Soy oil | 1.90 | 1.90 | ||
| Coconut oil | 0.90 | 0.90 | 0.10 | 0.10 |
| Corn oil | 2.20 | 2.20 | 1.70 | 1.70 |
| Fish oil | 3.20 | 3.20 | ||
| Cellulose powder | 5.00 | 5.00 | 5.00 | 5.00 |
| Mineral and trace element premix (AIN-93M-MX) without selenium | 3.50 | 3.50 | 3.50 | 3.50 |
| Vitamin mix (AIN-93-VX) without vitamin E | 1.00 | 1.00 | 1.00 | 1.00 |
| L-cystine | 0.180 | 0.180 | 0.180 | 0.180 |
| Choline (choline chloride, 50%) | 0.230 | 0.230 | 0.230 | 0.230 |
| Tert-butylhydroquinone | 0.0008 | 0.0008 | 0.0008 | 0.0008 |
| Uridine 5′-monophosphate (UMP disodium salt, 24–27% H2O) | 1.000 | 1.000 | ||
| Selenium (sodium selenite pentahydrate, 100%) | 0.00038 | 0.00038 | ||
| Vitamin E (all rac α-tocopherol acetate, 50% pure, 500 IU/kg) | 0.4788 | 0.4788 | ||
| Ascorbic acid (100%) | 0.160 | 0.160 | ||
| Sum | 100.0 | 100.0 | 100.0 | 100.0 |
The AOX low diets contain no added vitamin E, vitamin C, and selenium and are therefore not listed as ingredients of the AOX low diets. However, the AOX low diets do contain some vitamin E (3.85 mg RRR-alpha-tocopherol/kg diet) and selenium (0.07 mg/kg diet) due to the intrinsic content of vitamin E and selenium in the raw materials. This intrinsic content also adds up to the added vitamin E and selenium (in the table as all rac α-tocopherol acetate and sodium selenite pentahydrate) in the AOX high diets.
Experimental design
All rats (n = 40) were initially put, for two weeks, on the AOX low diet without DHA+EPA+uridine. Rats were then randomized to four groups (n = 10 per group) and received, for 6 weeks, one of the four experimental diets. Food intake and body weight of each rat were measured daily and weekly, respectively.
Sample preparation
On completion of dietary treatments, rats were sacrificed under ketamine (80 mg/kg) and xylazine (10 mg/kg) anesthesia. Brains were removed immediately and frozen at –80°C until they were homogenized in 50 volumes of ice-cold saline on the day of assay. Trunk blood was collected in a subset of rats (n = 4 per group) into heparinized tubes, centrifuged at 5000 rpm for 10 min, and plasmas were separated and frozen for further analyses of the lipid peroxidation product malondialdehyde (MDA) and fatty acid contents.
Brain phospholipid measurement
Brain phospholipids were extracted according to the Folch method [28] and measured as described previously [4]. Briefly, aliquots of brain homogenates were mixed with chloroform plus methanol mixture (2 : 1 v/v), vortexed and allowed to stand overnight at +4°C (18–20 h). Next day, aliquots (0.1 and 0.4 mL) of the lower (organic) phase were vacuum-dried. Residues of 0.1 mL aliquots were assayed for total phospholipids [4, 29]. Residues of 0.4 mL aliquots of the lower phase were reconstituted in 40μl methanol and subjected to thin-layer chromatography using silica G plates (Adsorbosil Plus-1, Alltech Associates, Deerfield, IL, USA), and a solvent system consisting of chloroform/ethanol/triethylamine/water (30 : 34 : 30 : 8) as the mobile phase [4, 29]. Bands for individual phospholipid classes were scraped off the plates, extracted into methanol, vacuum-dried, and assayed for phosphorus content. Aliquots of homogenates were assayed for total protein using a bicinchoninic acid reagent (Perkin Elmer, Norwalk, CT, USA). Phospholipid levels were expressed as nmol/mg protein.
Analyses of synaptic proteins
Synaptic proteins were assayed by western blot as described previously [2–4]. Briefly, aliquots of brain homogenates were mixed with equal volumes of Laemmi loading buffer and boiled prior to gel electrophoresis. Equal amounts of protein were loaded and separated using SDS-PAGE (4–20%; Bio-Rad, Hercules, CA, USA) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA), which were then rinsed in TBST buffer and incubated overnight in TBST solution containing the primary antibody of interest (goat anti-PSD-95 and rabbit anti-synapsin-1; Abcam, Cambridge, MA, USA). The next day, blots were incubated with the appropriate peroxidase-linked secondary antibody, followed by visualization of protein-antibody complexes using the enhanced chemiluminescence system (Millipore, Billerica, MA, USA), and digital images were developed using a Licor C-Digit blot scanner (LI-COR Biotechnology, Lincoln, NE, USA). Immunoreactive bands were compared densitometrically using the blot scanner’s software. Membranes were stripped off using a stripping buffer (Thermo Fisher Scientific, Rockford, IL, USA), and then incubated with β-tubulin antibody (mouse anti-βIII-tubulin; Sigma-Aldrich, St. Louis, MO, USA) used as the loading control.
Plasma and brain fatty acid analyses
Plasma and brain total lipid fatty acid composition was detected using gas chromatography (GC) as described previously [30]. Total lipids were extracted from plasma and brain homogenates using a modified Bligh and Dyer protocol [31] by adding methanol, 1% EDTA solution, and dichloromethane to 100μL of plasma or 300μL of brain homogenate. After vortexing for 5 min, samples were centrifuged at 1750×g for 10 min, and the organic phase (dichloromethane and lipids) was collected. The dichloromethane layer was dried using a SpeedVac® concentrator. Next, 2.0 mL methanol and 40μL concentrated sulphuric acid (2 v/v%) were added to the dried extract and samples were heated at 100°C for 60 min [32]. After cooling, 2 mL hexane and 0.5 mL 2.5 mol/L sodium hydroxide solution were added. Samples were subsequently vortexed and centrifuged for 5 min at 1750×g, after which the upper layer was collected and dried using a SpeedVac®. Dried samples were subsequently dissolved in 125μL iso-octane and analyzed by GC (Shimadzu Corporation, Kyoto, Japan) using flame ionization detection with a CP-SIL88 column (50 m×0.25 mm id. 0.20μm film thickness; Agilent Technologies, Inc., Santa Clara, CA, USA). Fatty acids were identified based on retention time using an external reference standard. An internal standard was used for absolute quantification of fatty acids in plasma and brain homogenate.
Plasma and brain malondialdehyde assay
Plasma and brain MDA concentration was determined by fluorometric HPLC, as previously described [33]. Plasma and brain homogenate samples were mixed with thiobarbituric acid (TBA) in a sodium acetate buffer solution (pH = 3.5). After heating and centrifugation, the supernatant containing the formed TBA-MDA adduct was separated in a reversed-phase column and quantified by fluorescence detection (excitation λ= 515 nm; emission λ= 553 nm).
Analyses of Kennedy pathway enzymes
Brain levels of two Kennedy pathway enzymes, namely the choline-phosphate cytidylyltransferase (PCYT1A) and choline/ethanolamine phosphotransferase (CEPT1), were analyzed in brain homogenates using commercial ELISA kits according to the instructions of the manufacturer (Shanghai YeHua Biological Technology, Pudong District, Shanghai, China).
Statistics
Statistical analyses were performed using SPSS software (version 19, SPSS Inc., Chicago, IL, USA). Data were expressed as mean±standard error of means (SEM). p-values less than 0.05 were considered significant. All data were analyzed using one-way analysis of variance (ANOVA) followed by post-hoc Tukey test, except for body weight and food intake, which were analyzed by ANOVA with repeated measures followed by Tukey test.
RESULTS
Body weight and food intake
No significant difference was observed between treatment groups with respect to food intake (F(3,36) = 0.97, p = 0.42) and body weight (F(3,36) = 1.14, p = 0.35) during the course of the study (data not shown). Overall, average body weight and food intake of all experimental groups during the study period were 383 g and 19.8 g/day, respectively.
Brain phospholipid levels
Dietary intervention affected the contents of total phospholipids (F(3,36) = 5.70, p = 0.003) in brains of rats (Fig. 1A). Specifically, compared with the AOX low diet (366±16 nmol/mg protein) and the AOX high diet (363±18 nmol/mg protein), consumption of the AOX high+DHA+EPA+uridine diet (458±18 nmol/mg protein) for 6 weeks enhanced levels of total phospholipids in the brain significantly by 25.1% (p = 0.006) and 26.1% (p = 0.005), respectively. Levels of brain total phospholipids in rats fed the AOX high+DHA+EPA+uridine diet also were higher compared to those fed the AOX low+DHA+EPA+uridine diet (392±22 nmol/mg protein), but the effect did not reach statistical significance (p = 0.075). Total phospholipid levels did not differ between rats fed the AOX low, AOX high, and AOX low+DHA+EPA+uridine diets.
Fig.1
Levels of total and individual phospholipids in rat brain tissue homogenates after 6 weeks of dietary intervention. Data were expressed as mean±SEM and analyzed by one-way ANOVA followed by post-hoc Tukey test. Bars that do not share a common letter significantly differ (p < 0.05).
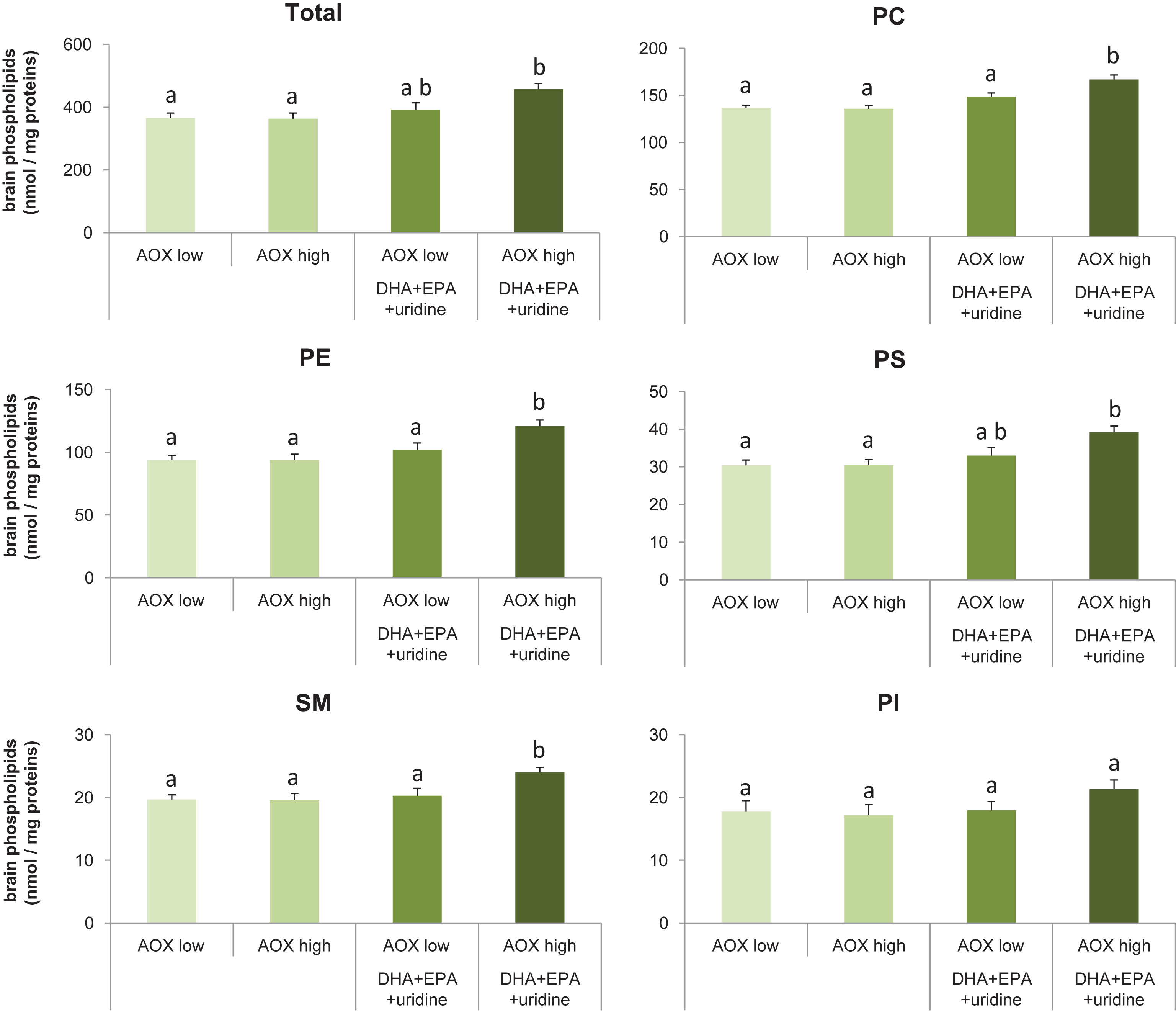
Brain levels of individual phospholipids (Fig. 1B-F) such as PC (F(3,36) = 14.62, p < 0.001), phosphatidylethanolamine (PE; F(3,36) = 7.60, p < 0.001), phosphatidylserine (PS; F(3,36) = 6.21, p = 0.002), and sphingomyelin (SM; F(3,36) = 4.92, p = 0.006) were significantly affected by dietary intervention, whereas phosphatidylinositol levels did not differ among groups (F(3,36) = 1.39, p = 0.26). Post-hoc analyses showed that in rats receiving the AOX high+DHA+EPA+uridine diet, PC levels were increased by 21.9% (p < 0.001), 22.8% (p < 0.001), or 12.1% (p = 0.008); PE levels were increased by 28.7% (p = 0.001), 28.7% (p = 0.001), or 18.6% (p = 0.032); and SM levels were increased by 21.8% (p = 0.013), 22.4% (p = 0.011), or 18.2% (p = 0.04) compared with those receiving the AOX low, AOX high, or AOX low+DHA+EPA+uridine diet, respectively. AOX high+DHA+EPA+uridine diet also significantly increased brain PS levels by 28.5% compared with either the AOX low (p = 0.003) or AOX high diet (p = 0.004) but not the AOX low+DHA+EPA+uridine diet (p = 0.055). PC, PE, and PS levels did not differ between the AOX low, AOX high, and AOX low+DHA+EPA+uridine groups.
Levels of synaptic proteins
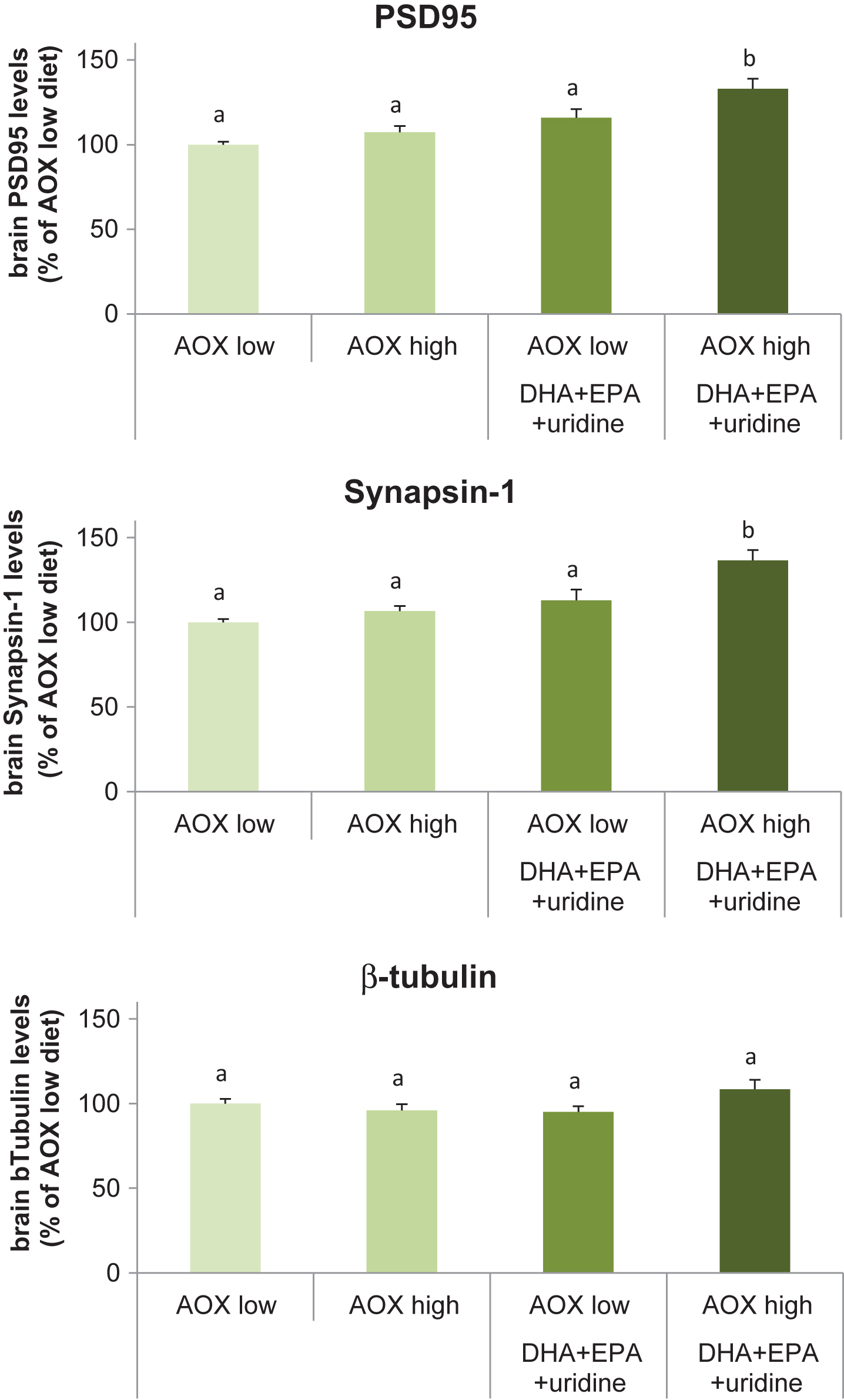
Dietary intervention affected the rat brain levels of the pre- and post-synaptic proteins, synapsin-1 (F(3,36) = 11.42, p < 0.001) and PSD95 (F(3,36) = 10.10, p < 0.001), respectively (Fig. 2A and 2B). Levels of β-tubulin, the structural protein assayed as a loading control, did not differ significantly among groups (F(3,36) = 2.34, p = 0.089) (Fig. 2C). Compared with the AOX low diet, levels of Synapsin-1 was significantly enhanced (by 36.6%; p < 0.001) in rats receiving the AOX high+DHA+EPA+uridine diet. Synapsin-1 levels were also significantly higher in rats fed with this diet compared with those receiving the AOX high (p < 0.001) and AOX low+DHA+EPA+uridine (p = 0.006) diet. Synapsin-1 levels did not differ between the AOX low, AOX high, and AOX low+DHA+EPA+uridine diets. Supplementation with AOX high+DHA+EPA+uridine diet increased the levels of PSD95 by 32.9% compared with the AOX low diet (p < 0.001), and PSD95 levels in rats fed with this diet were significantly higher than those in rats supplemented with AOX high (p = 0.001) and AOX low+DHA+EPA+uridine (p = 0.048) diets. PSD95 levels did not differ between rats fed the AOX low, AOX high, and AOX low+DHA+EPA+uridine diets.
Fig.2
Levels of PSD95, Synapsin-1, and β-tubulin in rat brain tissue homogenates after 6 weeks of dietary intervention. Data were expressed as percentage of the AOX low diet group, mean±SEM, and analyzed by one-way ANOVA followed by post-hoc Tukey test. Bars that do not share a common letter significantly differ (p < 0.05).
Plasma and brain fatty acids profile
Plasma concentrations (Table 2A) of AA (F(3,12) = 14.29, p < 0.001), DHA (F(3,12) = 25.38, p < 0.001), EPA (F(3,12) = 38.11, p < 0.001), total saturated fatty acids (SFA; F(3,12) = 6.76, p = 0.006), total monounsaturated fatty acids (MUFA; F(3,12) = 4.49, p = 0.025), total PUFA (F(3,12) = 7.45, p = 0.004), total omega-6 PUFA (F(3,12) = 25.38, p < 0.001), total omega-3 PUFA (F(3,12) = 40.10, p < 0.001), and total fatty acids (F(3,12) = 7.07, p = 0.005) were altered by dietary intervention. Changes were also found for brain concentrations (Table 2B) of AA (F(3,36) = 11.85, p < 0.001), DHA (F(3,36) = 7.03, p < 0.001), EPA (F(3,36) = 229.17, p < 0.001), total omega-6 PUFA (F(3,36) = 11.85, p < 0.001), and total omega-3 PUFA (F(3,36) = 8.18, p < 0.001). In general, compared with rats fed the diets lacking DHA+EPA+uridine, plasma and brain concentrations of AA and total omega-6 PUFA were decreased while concentrations of DHA, EPA, and total omega-3 PUFA were increased significantly in rats supplemented with the DHA+EPA+uridine-containing diets, irrespective of the antioxidant content. In addition, concentrations of total SFA, total MUFA, total PUFA, and total fatty acids were only significantly reduced in plasma samples of rats fed the AOX high+DHA+EPA+uridine diet compared to the diets without DHA+EPA+uridine.
Table 2
A) Plasma fatty acid quantitative concentrations in rats
| Plasma concentration (mg/L) | AOX low | AOX high | AOX low | AOX high |
| DHA+EPA | DHA+EPA | |||
| +uridine | +uridine | |||
| DHA (22 : 6n3) | 40.7±4.9a | 35.2±4.2a | 167.5±23.8b | 121.0±6.5b |
| EPA (20 : 5n3) | 7.0±0.5a | 6.7±0.5a | 90.9±11.1b | 74.5±9.1b |
| AA (20 : 4n6) | 478.2±42.7a | 406.3±40.5a | 225.1±36.9b | 204.8±16.1b |
| Total SFA | 588.5±34.8a | 634.1±48.7a | 499.5±53.2a,b | 384.0±27.7b |
| Total MUFA | 351.4±30.6a,b | 389.4±65.4a | 258.9±47.5a,b | 184.6±10.9b |
| Total PUFA | 1042.6±75.1a | 1034.9±64.6a | 828.5±70.9a,b | 683.3±37.0b |
| Total n6 | 966.9±69.1a | 960.3±58.0a | 545.1±48.2b | 469.5±24.8b |
| Total n3 | 71.3±6.1a | 68.0±5.4a | 281.9±29.7b | 212.3±13.4b |
| n6/n3 ratio | 13.6±0.4a | 14.2±0.4a | 2.0±0.2b | 2.2±0.1b |
| Total FA | 1987.3±135.3a | 2063.8±170.8a | 1591.5±162.9a,b | 1255.9±74.3b |
| B) Brain fatty acid quantitative concentrations in rats | ||||
| Brain concentration | AOX low | AOX high | AOX low | AOX high |
| (mg/L brain homogenate) | DHA+EPA | DHA+EPA | ||
| +uridine | +uridine | |||
| DHA (22 : 6n3) | 70.3±2.5a,c | 67.4±2.3a | 80.4±2.3b | 79.5±2.7b,c |
| EPA (20 : 5n3) | 0.0a | 0.0a | 0.4±0.02b | 0.4±0.02b |
| AA (20 : 4n6) | 60.4±1.6a | 58.7±1.5a | 50.9±1.4b | 51.2±1.3b |
| Total SFA | 255.9±4.3a | 251.2±4.5a | 254.1±4.1a | 255.7±2.6a |
| Total MUFA | 161.2±2.5a | 158.3±3.1a | 161.8±3.2a | 168.9±1.8a |
| Total PUFA | 168.8±5.3a | 164.3±5.4a | 163.7±4.4a | 164.0±4.6a |
| Total n6 | 92.6±2.8a | 90.9±3.0a | 76.4±2.1b | 77.4±1.9b |
| Total n3 | 76.2±2.5a | 73.3±2.3a | 87.3±2.4b | 86.5±2.7b |
| n6/n3 ratio | 1.22±0.01a | 1.24±0.01a | 0.88±0.01b | 0.90±0.01b |
| Total FA | 603.6±11.7a | 591.8±12.6a | 596.9±10.8a | 606.5±6.8a |
Quantitative concentrations of fatty acids in rat plasma (A) and brain homogenates (B) after 6 weeks of dietary intervention. Data were expressed as mean±SEM and analyzed by one-way ANOVA followed by post-hoc Tukey test. Different letters indicate mean values were significantly different (p < 0.05).
Plasma and brain MDA levels
Levels of MDA in the plasma (F(3,12) = 1.52, p = 0.26) or brain (F(3,36) = 1.47, p = 0.24) of rats did not differ significantly among treatment groups (Table 3).
Table 3
Levels of MDA in the plasma and brain
| AOX low | AOX high | AOX low | AOX high | |
| DHA+EPA | DHA+EPA | |||
| +uridine | +uridine | |||
| Plasma MDA (nmol/mL plasma) | 1.4±0.2 | 1.4±0.1 | 2.0±0.3 | 1.7±0.3 |
| Brain MDA (nmol/mL homogenate) | 13.2±0.4 | 12.7±1.1 | 11.5±0.6 | 11.5±0.6 |
MDA levels in plasma samples and rat brain tissue homogenates after 6 weeks of dietary intervention. Data were expressed as mean±SEM and analyzed by one-way ANOVA followed by post-hoc Tukey test. No significant difference was found among groups with regard to MDA levels either in the plasma or brain.
Kennedy pathway enzymes
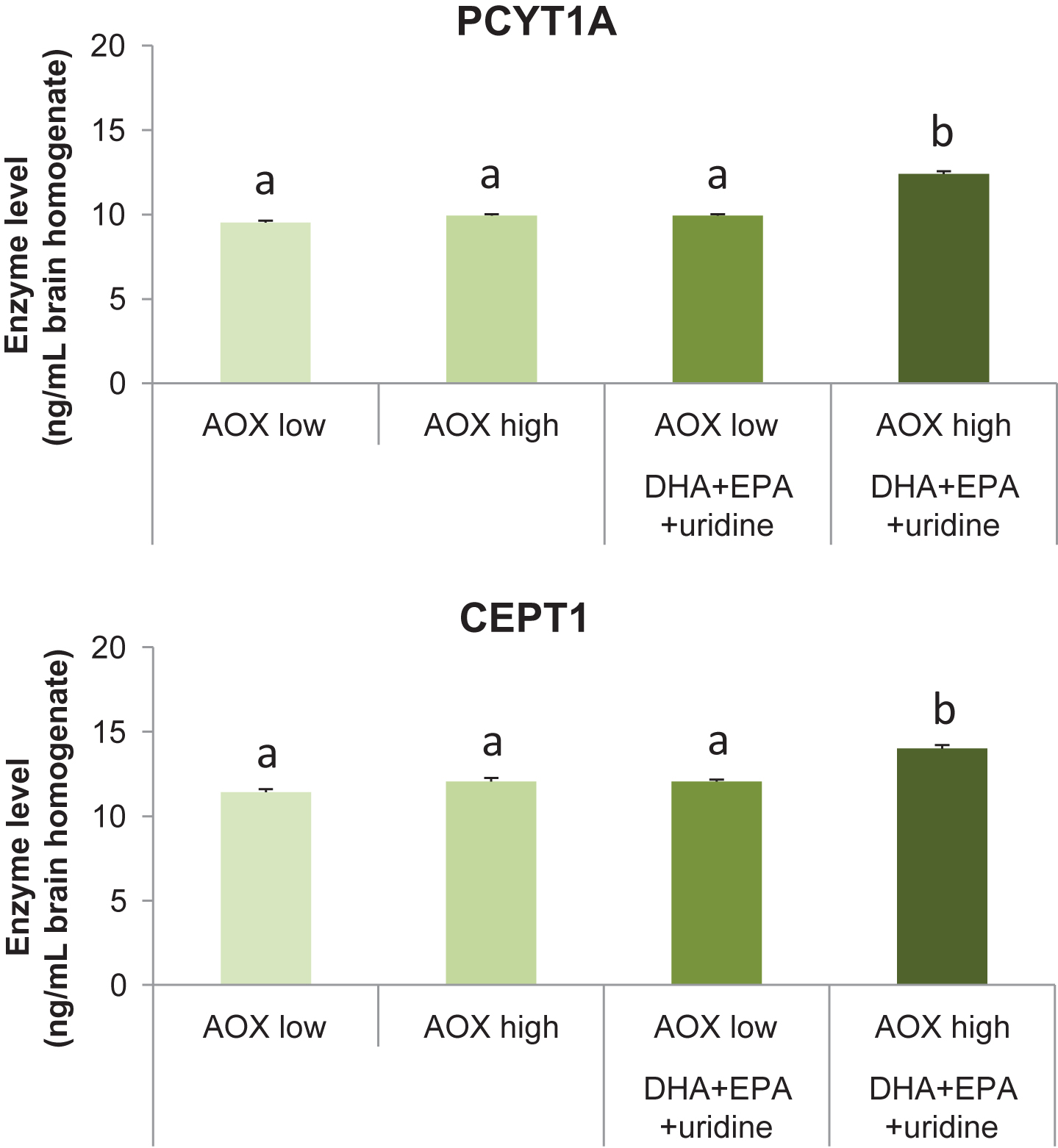
Dietary intervention affected the levels of the Kennedy pathway enzymes PCYT1A (F(3,36) = 122.62, p < 0.001) and CEPT1 (F(3,36) = 38.70, p < 0.001) in the rat brain (Fig. 3A and 3B). Levels of PCYT1A were increased significantly in rats receiving the AOX high+DHA+EPA+uridine diet (12.40±0.16 ng/mL) compared with those receiving the AOX low (9.52±0.12 ng/mL), AOX high (9.92±0.09 ng/mL), and AOX low+DHA+EPA+uridine (9.93±0.09 ng/ml) diets (p < 0.001 for all). PCYT1A levels did not differ between the AOX low, AOX high, and AOX low+DHA+EPA+uridine groups. Consumption of AOX high+DHA+EPA+uridine (14.01±0.20 ng/ml) diet enhanced the levels of CEPT1 significantly compared with AOX low (11.42±0.18 ng/ml), AOX high (12.05±0.21 ng/ml), and AOX low+DHA+EPA+uridine (12.06±0.11 ng/ml) diet (p < 0.001 for all). CEPT1 levels did not differ between rats fed the AOX low, AOX high, and AOX low+DHA+EPA+uridine diets.
Fig.3
Levels of PCYT1A and CEPT1 in rat brain tissue homogenates after 6 weeks of dietary intervention. Data were expressed as mean±SEM and analyzed by one-way ANOVA followed by post-hoc Tukey test. Bars that do not share a common letter significantly differ (p < 0.05).
DISCUSSION
The present data show that brain levels of phospholipids and synaptic proteins are significantly enhanced when rats are supplemented with DHA, EPA, and uridine in the presence of sufficient amounts of vitamin C, vitamin E, and selenium. These data indicate that the frequently reported increase of markers of synaptogenesis by dietary neuronal membrane precursors depends on adequate intake of vitamin C, vitamin E, and selenium.
A substantial amount of the synthesis of phospholipids is controlled by the availability of nutrients, such as uridine [34], the PUFAs DHA and EPA [35], choline [36], and their interaction via the Kennedy pathway [1]. As such, supplementing adult gerbils with uridine in the diet plus DHA [2, 3] or EPA [4] administered by gavage increases the quantity of phospholipids in the brain. Concomitant increases in pre- and post-synaptic protein [2–4] levels and increases in dendritic spine densities [3, 5] indicate that the administration of phospholipid precursors in particular raises levels of membranes associated with synapses.
In the above mentioned previous studies, a uridine-fortified standard rat chow (Teklad Global 16% Protein Rodent Diet, Evigo, former Harlan, Madison, WI, USA) was used that contained 110 IU vitamin E/kg diet (corresponding to 73.8 mg RRR-α-tocopherol/kg diet) and 0.23 mg selenium/kg diet, which is respectively 4.1 and 1.5-fold those of the estimated nutrient requirements of rats (27 IU/kg for vitamin E and 0.15 mg/kg for selenium; [22]). Despite the fact that vitamin C is not part of a normal rat diet like the Teklad Global 16% Protein Rodent Diet, it is a potentially beneficial dietary constituent for rats [22]. Hence, in these previous studies the content of these antioxidants in the diets was equal or above levels recommended for rats, mice, and gerbils [22]. Possible contribution of antioxidants to membrane levels was not investigated, while they may directly affect the breakdown or synthesis of neuronal membranes through several mechanisms, including preventing lipid peroxidation, activating Kennedy pathway enzymes, and affecting the neuronal membranes directly (reviewed in [37]).
The present study was undertaken to investigate potential necessity of antioxidants for the effectiveness of supplementation with uridine and fish oil containing DHA and EPA, with regard to synaptic membrane levels in rats. A dietary background of vitamin C, vitamin E, and selenium inadequacy was used to obtain a proof-of-principle of the concerted action of the antioxidants and DHA+EPA+uridine. Such a lower nutrient status is relevant for conditions associated with insufficiencies of these nutrients. For instance, people with AD have lower plasma levels of vitamin C [23], vitamin E [23], selenium [24], uridine [8], and DHA [7] compared with control subjects. All of these nutrients, except for selenium, are also lower in the AD brain or cerebrospinal fluid [9, 10, 25, 26]. The lower nutritional status in AD might result from disease-related changes in nutrient intake and metabolic alterations. In addition to lower blood and brain levels of the phospholipid precursors and antioxidants, AD patients experience synapse loss [6], which has been linked to the breakdown of membrane phospholipids [38, 39]. Hence, AD patients may benefit from dietary concomitant supplementation phospholipid precursors and antioxidants to support synaptic membrane formation and counteract the consequences of synapse loss.
This study showed that levels of total and individual phospholipids (except for phosphatidylinositol) and levels of specific pre- and post-synaptic proteins Synapsin-1 and PSD9 were enhanced by about 22–36% in rats receiving the DHA+EPA+uridine diet that contained the antioxidants, as compared with the diet low in antioxidants without DHA+EPA+uridine. On the contrary, no significant difference was observed with regard to levels of phospholipids and synaptic proteins if the DHA+EPA+uridine-containing diet lacked the antioxidants. These results replicate the previously observed effects of supplementation of DHA or EPA (300 mg/kg body weight, by gavage) and uridine (0.5 g/100 g diet) using standard chow containing sufficient levels of antioxidants [2–4].
The mechanisms by which quantities of synaptic membranes were increased by DHA+EPA+uridine diet that also included the antioxidants probably involve the substrate saturation of enzymes in the Kennedy pathway of phospholipid synthesis by DHA/EPA and uridine [2, 40], as well as above-mentioned effects of antioxidants on preventing membrane degradation [12] and stimulating Kennedy pathway enzymes [14]. The action of antioxidants are of crucial importance, considering that the newly-formed membranes by DHA+EPA-containing diets contain relatively high amounts of DHA and EPA, the long-chain omega-3 PUFAs that are highly vulnerable to oxidation [41, 42].
In addition to the presumably higher precursor saturation of the Kennedy pathway enzymes, levels of PCYT1A, the enzyme that combines cytidine triphosphate and phosphocholine to form CDP-choline in the rate-limiting step of PC synthesis [1], and those of CEPT1, the enzyme that catalyzes the formation of PC or PE from the combination of diacylglycerol and CDP-choline or CDP-ethanolamine [1], respectively, were found to be increased significantly in rats receiving the antioxidants in combination with DHA+EPA+uridine in their diet. These data suggest a cooperative effect of the antioxidants and phospholipid precursors in enhancing the operation of Kennedy pathway. Accordingly, quantities of membranes are enhanced on the one hand by providing more precursors and on the other hand by preventing oxidation with antioxidants, which in turn might result in larger amounts of Kennedy pathway enzymes in synapses where these enzymes are retained.
These data also show that quantitative plasma and brain concentrations of AA and total omega-6 PUFA were decreased while concentrations of DHA, EPA, and total omega-3 PUFA were increased in rats supplemented with the DHA+EPA-containing diets, mainly irrespective of the level of antioxidants. These data are in good accord with previous studies reporting lowered plasma omega-6 PUFA levels and increased omega-3 PUFA levels by supplementation with diets rich in DHA and EPA both in rodents [43] and humans [44]. In addition, the observed decrease in plasma total fatty acid (and plasma total SFA, MUFA, and PUFA) concentration is in line with well-known plasma triglyceride-lowering effect of DHA and/or EPA [45]. Remarkably, the current results suggest a synergistic effect of antioxidants on lowering total fatty acid content.
Levels of MDA, a lipid peroxidation product especially derived from PUFAs and a reliable marker of oxidative stress [46], did not differ significantly among treatment groups. Plasma MDA tended to increase in rats receiving the DHA+EPA-containing diets, whereas an opposite trend could be seen for brain MDA. Unexpectedly, antioxidant supplementation had no observable effect on MDA [47], but perhaps MDA levels were too low to induce a further reduction.
In conclusion, data obtained in the present study show that levels of brain membrane phospholipids and synaptic proteins were enhanced in rats supplemented with DHA+EPA+uridine when the diet also contained sufficient quantities of the antioxidants vitamin C, vitamin E, and selenium. The data indicate that the frequently reported increase of markers of synaptogenesis by dietary neuronal membrane precursors depends on adequate intake of vitamin C, vitamin E, and selenium and that formation of new synapses requires the concerted action of phospholipid precursors and the antioxidants. This would be particularly relevant for (early) AD, where there is an increased need for synapse formation while levels of plasma and brain phospholipid precursors and antioxidants are low.
ACKNOWLEDGMENTS
The present research has been partly funded by Nutricia Research.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0081r1).
REFERENCES
[1] | Kennedy EP , Weiss SB ((1956) ) The function of cytidine coenzymes in the biosynthesis of phospholipides. J Biol Chem 222: , 193–214. |
[2] | Wurtman RJ , Ulus IH , Cansev M , Watkins CJ , Wang L , Marzloff G ((2006) ) Synaptic proteins and phospholipids are increased in gerbil brain by administering uridine plus docosahexaenoic acid orally. Brain Res 1088: , 83–92. |
[3] | Sakamoto T , Cansev M , Wurtman RJ ((2007) ) Oral supplementation with docosahexaenoic acid and uridine-5’-monophosphate increases dendritic spine density in adult gerbil hippocampus. Brain Res 1182: , 50–59. |
[4] | Cansev M , Wurtman RJ ((2007) ) Chronic administration of docosahexaenoic acid or eicosapentaenoic acid, but not arachidonic acid, alone or in combination with uridine, increases brain phosphatide and synaptic protein levels in gerbils. Neuroscience 148: , 421–431. |
[5] | Cansev M , Marzloff G , Sakamoto T , Ulus IH , Wurtman RJ ((2009) ) Giving uridine and/or docosahexaenoic acid orally to rat dams during gestation and nursing increases synaptic elements in brains of weanling pups. Dev Neurosci 31: , 181–192. |
[6] | de Wilde MC , Overk CR , Sijben JW , Masliah E ((2016) ) Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement 12: , 633–644. |
[7] | Lin PY , Chiu CC , Huang SY , Su KP ((2012) ) A meta-analytic review of polyunsaturated fatty acid compositions in dementia. J Clin Psychiatry 73: , 1245–1254. |
[8] | Olde Rikkert MG , Verhey FR , Sijben JW , Bouwman FH , Dautzenberg PL , Lansink M , Sipers WM , van Asselt DZ , van Hees AM , Stevens M , Vellas B , Scheltens P ((2014) ) Differences in nutritional status between very mild Alzheimer’s disease patients and healthy controls. J Alzheimers Dis 41: , 261–271. |
[9] | Czech C , Berndt P , Busch K , Schmitz O , Wiemer J , Most V , Hampel H , Kastler J , Senn H ((2012) ) Metabolite profiling of Alzheimer’s disease cerebrospinal fluid. PLoS One 7: , e31501. |
[10] | Söderberg M , Edlund C , Kristensson K , Dallner G ((1991) ) Fatty acid composition of brain phospholipids in aging and in Alzheimer’s disease. Lipids 26: , 421–425. |
[11] | Machlin LJ , Bendich A ((1987) ) Free radical tissue damage: Protective role of antioxidant nutrients. FASEB J 1: , 441–445. |
[12] | Gutteridge JM ((1995) ) Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem 41: , 1819–1828. |
[13] | Chow CK ((1979) ) Nutritional influence on cellular antioxidant defense systems. Am J Clin Nutr 32: , 1066–1081. |
[14] | Liu SY , Tardi PG , Choy PC , Man RY ((1993) ) Effects of selenium supplement on the de novo biosynthesis of glycerolipids in the isolated rat heart. Biochim Biophys Acta 1170: , 307–313. |
[15] | Switzer BR , Summer GK ((1972) ) Collagen synthesis in human skin fibroblasts: Effects of ascorbate, -ketoglutarate and ferrous ion on proline hydroxylation. J Nutr 102: , 721–728. |
[16] | Fox MA , Sanes JR , Borza DB , Eswarakumar VP , Fässler R , Hudson BG , John SW , Ninomiya Y , Pedchenko V , Pfaff SL , Rheault MN , Sado Y , Segal Y , Werle MJ , Umemori H ((2007) ) Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 129: , 179–193. |
[17] | Lucy JA ((1972) ) Functional and structural aspects of biological membranes: A suggested structural role for vitamin E in the control of membrane permeability and stability. Ann N Y Acad Sci 203: , 4–11. |
[18] | Quinn PJ ((2004) ) Is the distribution of alpha-tocopherol in membranes consistent with its putative functions? Biochemistry (Mosc) 69: , 58–66. |
[19] | Atkinson J , Harroun T , Wassall SR , Stillwell W , Katsaras J ((2010) ) The location and behavior of alpha-tocopherol in membranes. Mol Nutr Food Res 54: , 641–651. |
[20] | Monroe EB , Jurchen JC , Lee J , Rubakhin SS , Sweedler JV ((2005) ) Vitamin E imaging and localization in the neuronal membrane. J Am Chem Soc 127: , 12152–12153. |
[21] | Peters MM , Hill KE , Burk RF , Weeber EJ ((2006) ) Altered hippocampus synaptic function in selenoprotein P deficient mice. Mol Neurodegener 1: , 12. |
[22] | National Research Council ((1995) ) Nutrient requirements of laboratory animals, National Academic Press, Washington. |
[23] | Lopes da Silva S , Vellas B , Elemans S , Luchsinger J , Kamphuis P , Yaffe K , Sijben J , Groenendijk M , Stijnen T ((2014) ) Plasma nutrient status of patients with Alzheimer’s disease: Systematic review and meta-analysis. Alzheimers Dement 10: , 485–502. |
[24] | Loef M , Schrauzer GN , Walach H ((2011) ) Selenium and Alzheimer’s disease: A systematic review. J Alzheimers Dis 26: , 81–104. |
[25] | Glasø M , Nordbø G , Diep L , Bøhmer T ((2004) ) Reduced concentrations of several vitamins in normal weight patients with late-onset dementia of the Alzheimer type without vascular disease. J Nutr Health Aging 8: , 407–413. |
[26] | Jiménez-Jiménez FJ , de Bustos F , Molina JA , Benito-León J , Tallón-Barranco A , Gasalla T , Ortí-Pareja M , Guillamón F , Rubio JC , Arenas J , Enríquez-de-Salamanca R ((1997) ) Cerebrospinal fluid levels of alpha-tocopherol (vitamin E) in Alzheimer’s disease. J Neural Transm (Vienna) 104: , 703–710. |
[27] | Reeves PG , Nielsen FH , Fahey GC Jr ((1993) ) AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J Nutr 123: , 1939–1951. |
[28] | Folch J , Lees M , Sloane Stanley GH ((1957) ) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: , 497–509. |
[29] | Svanborg A , Svennerholm L , Myrén I , Soomägi MR , Claesson B , Rohman S ((1961) ) Plasma total lipid, cholesterol, triglycerides, phospholipids and free fatty acids in a healthy Scandinavian population. Acta Med Scand 169: , 43–49. |
[30] | van Wijk N , Balvers M , Cansev M , Maher TJ , Sijben JW , Broersen LM ((2016) ) Dietary crude lecithin increases systemic availability of dietary docosahexaenoic acid with combined intake in rats. Lipids 51: , 833–846. |
[31] | Bligh EG , Dyer WJ ((1959) ) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: , 911–917. |
[32] | Christie WW ((1993) ) Preparation of ester derivatives of fatty acids for chromatographic analysis In Advances in Lipid Methodology, Volume 2, Christie WW , ed. Oily Press, Dundee, pp. 69–111. |
[33] | Fukunaga K , Yoshida M , Nakazono N ((1998) ) A simple, rapid, highly sensitive and reproducible quantification method for plasma malondialdehyde by high-performance liquid chromatography. Biomed Chromatogr 12: , 300–303. |
[34] | Cansev M ((2006) ) Uridine and cytidine in the brain: Their transport and utilization. Brain Res Rev 52: , 389–397. |
[35] | Mitchell RW , On NH , Del Bigio MR , Miller DW , Hatch GM ((2011) ) Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J Neurochem 117: , 735–746. |
[36] | Pardridge WM , Cornford EM , Braun LD , Oldendorf WH ((1979) ) Transport of choline and choline analogues through the blood–brain barrier. In Nutrition and the Brain. Raven Press, New York, pp. 25–34. |
[37] | van Wijk N , Broersen LM , de Wilde MC , Hageman RJ , Groenendijk M , Sijben JW , Kamphuis PJ ((2014) ) Targeting synaptic dysfunction in Alzheimer’s disease by administering a specific nutrient combination. J Alzheimers Dis 38: , 459–479. |
[38] | Nitsch RM , Blusztajn JK , Pittas AG , Slack BE , Growdon JH , Wurtman RJ ((1992) ) Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci U S A 89: , 1671–1675. |
[39] | Prasad MR , Lovell MA , Yatin M , Dhillon H , Markesbery WR ((1998) ) Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res 23: , 81–88. |
[40] | Wurtman RJ , Cansev M , Sakamoto T , Ulus IH ((2009) ) Use of phosphatide precursors to promote synaptogenesis. Annu Rev Nutr 29: , 59–87. |
[41] | Benzie IF ((1996) ) Lipid peroxidation: A review of causes, consequences, measurement and dietary influences. Int J Food Sci Nutr 47: , 233–261. |
[42] | Shahidi F , Zhong Y ((2010) ) Lipid oxidation and improving the oxidative stability. Chem Soc Rev 39: , 4067–4079. |
[43] | Engström K , Saldeen AS , Yang B , Mehta JL , Saldeen T ((2009) ) Effect of fish oils containing different amounts of EPA, DHA, and antioxidants on plasma and brain fatty acids and brain nitric oxide synthase activity in rats. Ups J Med Sci 114: , 206–213. |
[44] | Woodman RJ , Mori TA , Burke V , Puddey IB , Watts GF , Beilin LJ ((2002) ) Effects of purified eicosapentaenoic and docosahexaenoic acids on glycemic control, blood pressure, and serum lipids in type 2 diabetic patients with treated hypertension. Am J Clin Nutr 76: , 1007–1015. |
[45] | Agren JJ , Hänninen O , Julkunen A , Fogelholm L , Vidgren H , Schwab U , Pynnönen O , Uusitupa M ((1996) ) Fish diet, fish oil and docosahexaenoic acid rich oil lower fasting and postprandial plasma lipid levels. Eur J Clin Nutr 50: , 765–771. |
[46] | Giera M , Lingeman H , Niessen WM ((2012) ) Recent advancements in the LC- and GC-based analysis of malondialdehyde (MDA): A brief overview. Chromatographia 75: , 433–440. |
[47] | Goldfarb AH , Bloomer RJ , McKenzie MJ ((2005) ) Combined antioxidant treatment effects on blood oxidative stress after eccentric exercise. Med Sci Sports Exerc 37: , 234–239. |

